Multiple Sulfatase Deficiency: A Disease Comprising Mucopolysaccharidosis, Sphingolipidosis, and More Caused by a Defect in Posttranslational Modification

 , , and
, , and 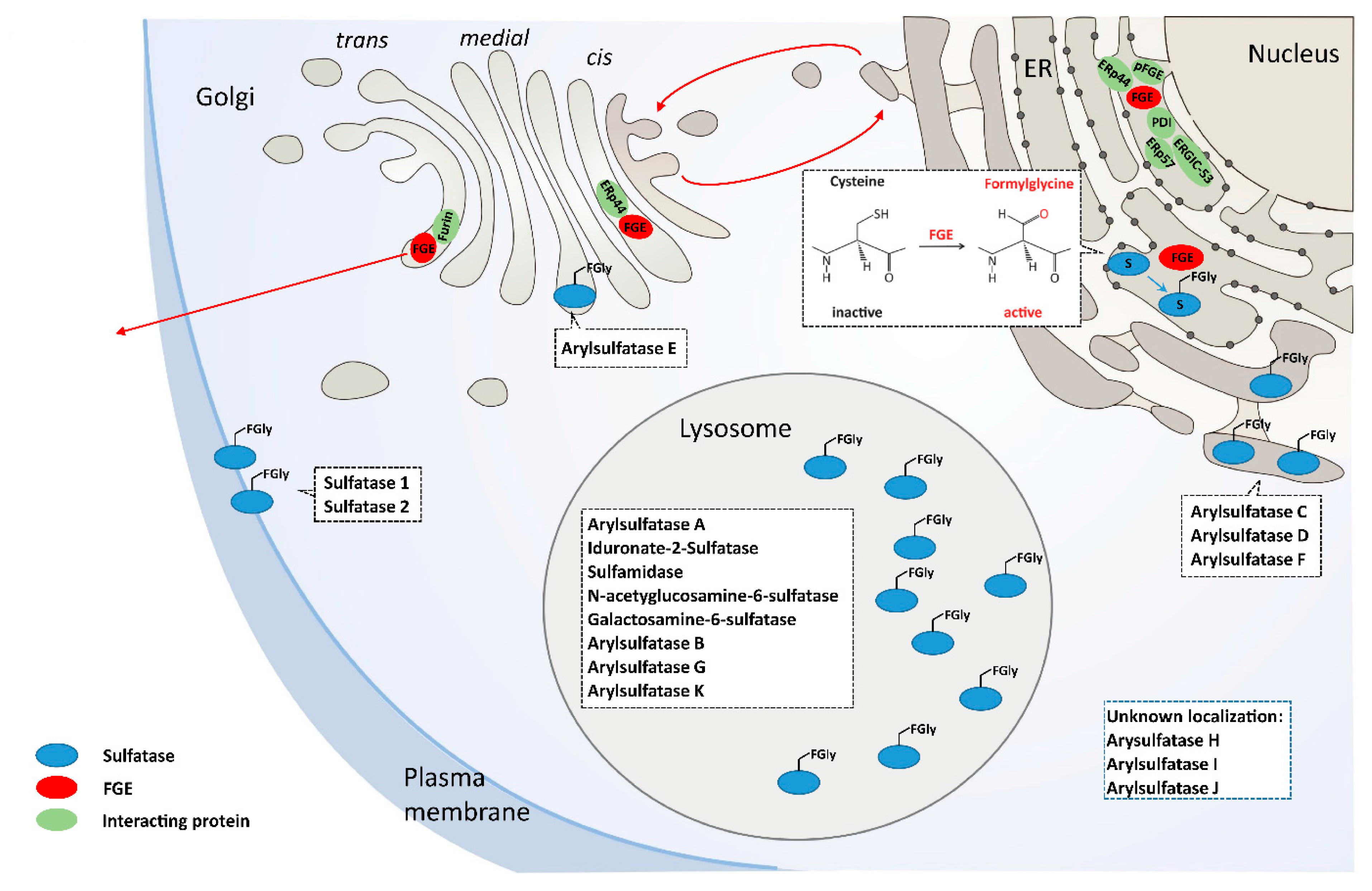{kind=link}
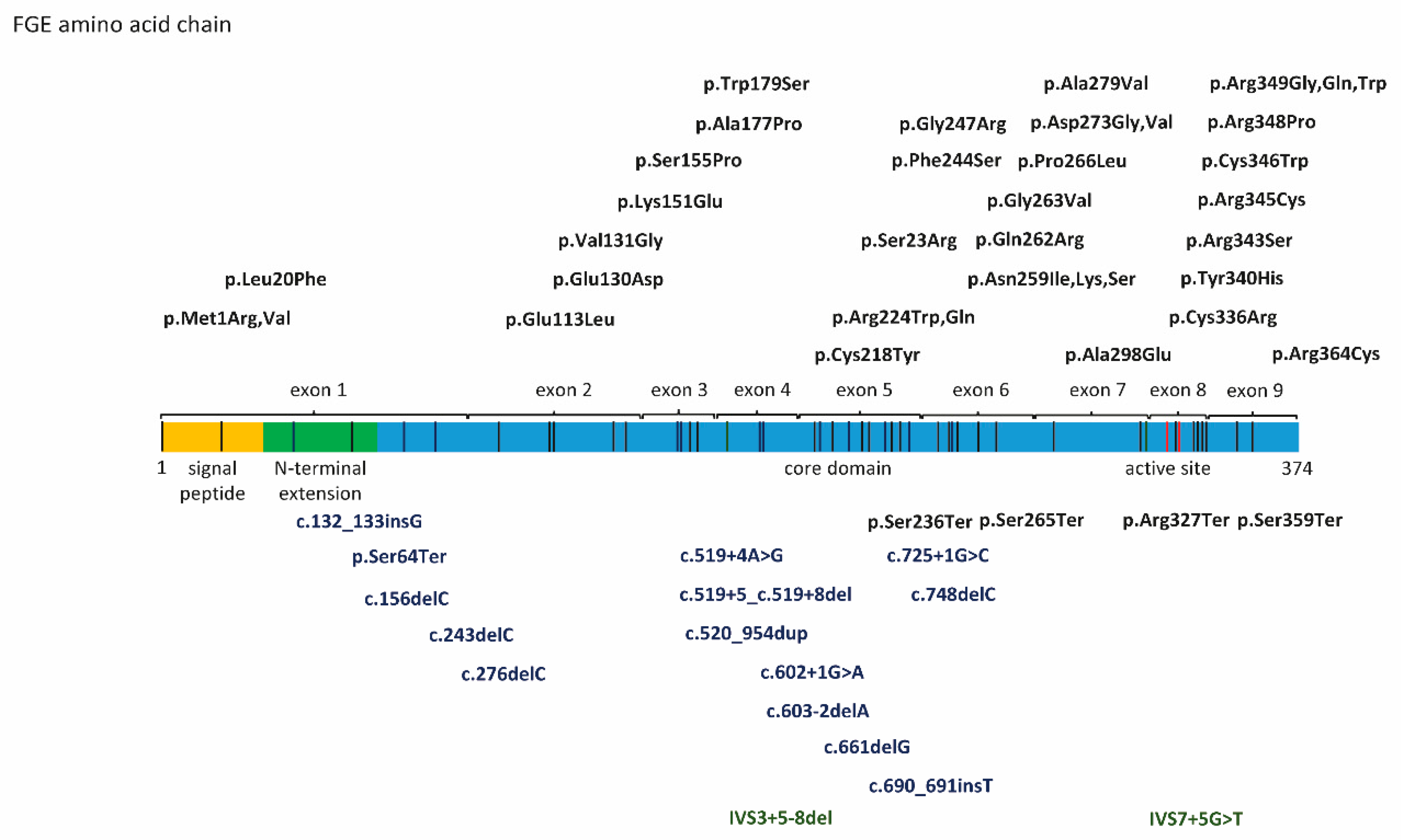{kind=link}
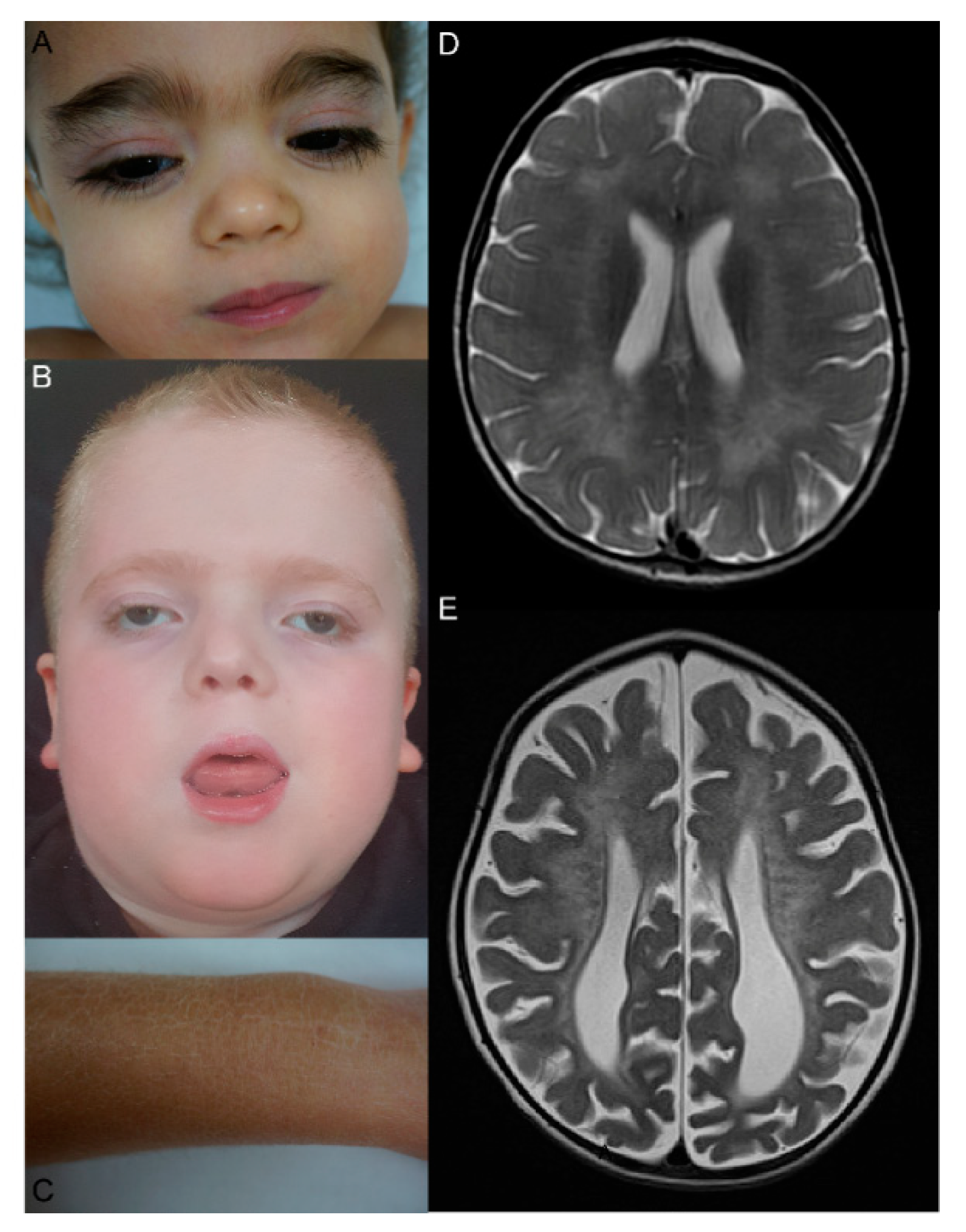{kind=link}
Abstract
Share and Cite
Schlotawa, L.; Adang, L.A.; Radhakrishnan, K.; Ahrens-Nicklas, R.C. Multiple Sulfatase Deficiency: A Disease Comprising Mucopolysaccharidosis, Sphingolipidosis, and More Caused by a Defect in Posttranslational Modification. Int. J. Mol. Sci. 2020, 21, 3448. https://doi.org/10.3390/ijms21103448
Schlotawa L, Adang LA, Radhakrishnan K, Ahrens-Nicklas RC. Multiple Sulfatase Deficiency: A Disease Comprising Mucopolysaccharidosis, Sphingolipidosis, and More Caused by a Defect in Posttranslational Modification. International Journal of Molecular Sciences. 2020; 21(10):3448. https://doi.org/10.3390/ijms21103448
Chicago/Turabian StyleSchlotawa, Lars, Laura A. Adang, Karthikeyan Radhakrishnan, and Rebecca C. Ahrens-Nicklas. 2020. "Multiple Sulfatase Deficiency: A Disease Comprising Mucopolysaccharidosis, Sphingolipidosis, and More Caused by a Defect in Posttranslational Modification" International Journal of Molecular Sciences 21, no. 10: 3448. https://doi.org/10.3390/ijms21103448
APA StyleSchlotawa, L., Adang, L. A., Radhakrishnan, K., & Ahrens-Nicklas, R. C. (2020). Multiple Sulfatase Deficiency: A Disease Comprising Mucopolysaccharidosis, Sphingolipidosis, and More Caused by a Defect in Posttranslational Modification. International Journal of Molecular Sciences, 21(10), 3448. https://doi.org/10.3390/ijms21103448