Genetic Susceptibility to Chronic Liver Disease in Individuals from Pakistan


Abstract
:1. Introduction
2. Results
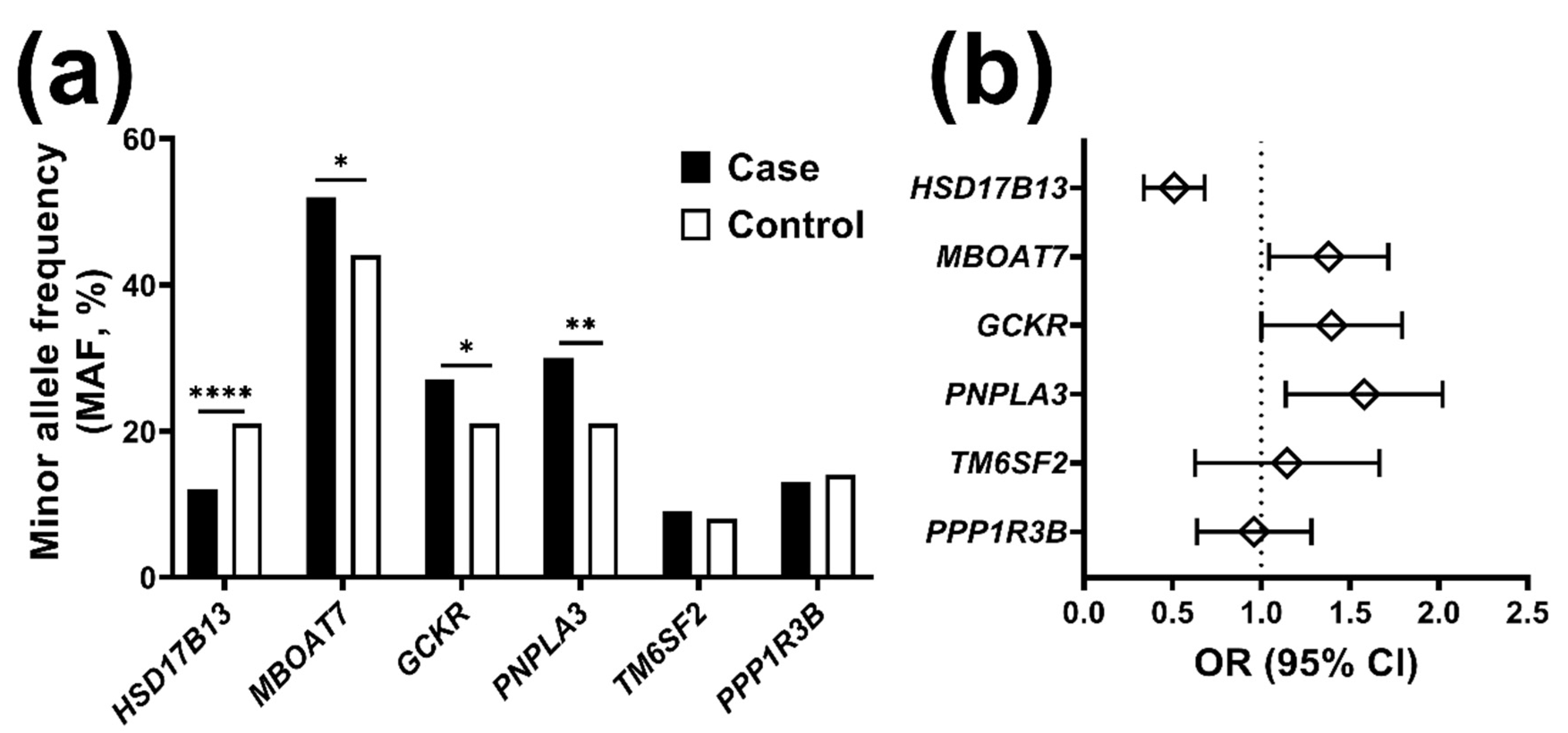
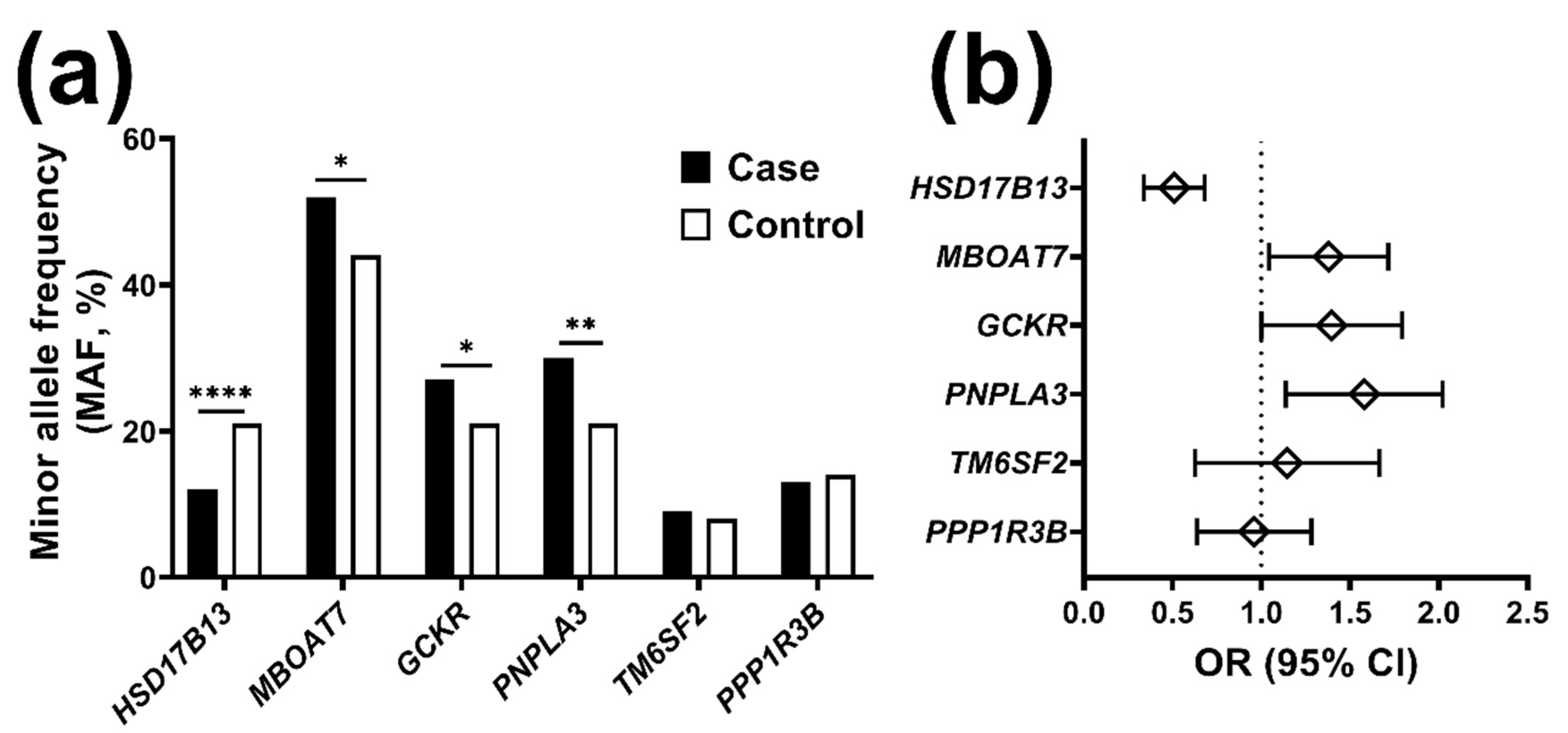
2.1. The Frequency of Risk Alleles for Liver Disease Is Higher in Individuals with Chronic Liver Disease
2.2. The HSD17B13 Minor Allele Reduces the Risk for Chronic Liver Disease
3. Discussion
4. Materials and Methods
4.1. Study Cohort
4.2. DNA Extraction and Genotyping
4.3. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ALT | Alanine transaminase |
| AST | Aspartate transaminase |
| BMI | Body mass index |
| CGKR | Glucokinase regulator |
| CI | Confidence interval |
| FFPE | Formalin-Fixed Paraffin-Embedded |
| HCC | Hepatocellular carcinoma |
| HDL | High-density lipoproteins |
| HSD17B13 | Hydroxysteroid 17-Beta Dehydrogenase 13 |
| LDL | Low-density lipoproteins |
| MBOAT7 | Membrane Bound O-Acyltransferase Domain Containing 7 |
| OR | Odds ratio. |
| PNPLA3 | Patatin-like phospholipase domain-containing protein 3 |
| PPP1R3B | Protein Phosphatase 1 Regulatory Subunit 3B |
| TM6SF2 | Transmembrane 6 Superfamily Member 2 |
References
- Rinella, M.E. Nonalcoholic fatty liver disease: A systematic review. JAMA 2015, 313, 2263–2273. [Google Scholar] [CrossRef] [PubMed]
- Lavanchy, D. Evolving epidemiology of hepatitis C virus. Clin. Microbiol. Infect. 2011, 17, 107–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajarizadeh, B.; Grebely, J.; Dore, G.J. Epidemiology and natural history of HCV infection. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.S.; Khan, S.; Rahim, H.; Chishti, K.A.; Khan, A.G. Prevalence of non alcoholic fatty liver and non alcoholic Steatohepatitis in Peshawar Cantonment, Khyber Pakhtunkhwa, Pakistan. Pak. J. Pharm. Sci. 2018, 31, 193–198. [Google Scholar]
- Valenti, L.V.C.; Baselli, G.A. Genetics of Nonalcoholic Fatty Liver Disease: A 2018 Update. Curr. Pharm. Des. 2018, 24, 4566–4573. [Google Scholar] [CrossRef] [PubMed]
- Romeo, S.; Sanyal, A.; Valenti, L. Leveraging Human Genetics to Identify Potential New Treatments for Fatty Liver Disease. Cell Metab. 2020, 31, 35–45. [Google Scholar] [CrossRef]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [Green Version]
- Mancina, R.M.; Dongiovanni, P.; Petta, S.; Pingitore, P.; Meroni, M.; Rametta, R.; Borén, J.; Montalcini, T.; Pujia, A.; Wiklund, O.; et al. The MBOAT7-TMC4 Variant rs641738 Increases Risk of Nonalcoholic Fatty Liver Disease in Individuals of European Descent. Gastroenterology 2016. [Google Scholar] [CrossRef] [Green Version]
- Buch, S.; Stickel, F.; Trépo, E.; Way, M.; Herrmann, A.; Nischalke, H.D.; Brosch, M.; Rosendahl, J.; Berg, T.; Ridinger, M.; et al. A genome-wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol-related cirrhosis. Nat. Genet. 2015. [Google Scholar] [CrossRef]
- Viitasalo, A.; Eloranta, A.M.; Atalay, M.; Romeo, S.; Pihlajamäki, J.; Lakka, T.A. Association of MBOAT7 gene variant with plasma ALT levels in children: The PANIC study. Pediatr. Res. 2016, 80, 651–655. [Google Scholar] [CrossRef]
- Speliotes, E.K.; Yerges-Armstrong, L.M.; Wu, J.; Hernaez, R.; Kim, L.J.; Palmer, C.D.; Gudnason, V.; Eiriksdottir, G.; Garcia, M.E.; Launer, L.J.; et al. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet. 2011, 7, e1001324. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Petta, S.; Maglio, C.; Fracanzani, A.L.; Pipitone, R.; Mozzi, E.; Motta, B.M.; Kaminska, D.; Rametta, R.; Grimaudo, S.; et al. Transmembrane 6 superfamily member 2 gene variant disentangles nonalcoholic steatohepatitis from cardiovascular disease. Hepatology 2015, 61, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Kozlitina, J.; Smagris, E.; Stender, S.; Nordestgaard, B.G.; Zhou, H.H.; Tybjaerg-Hansen, A.; Vogt, T.F.; Hobbs, H.H.; Cohen, J.C. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2014, 46, 352–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abul-Husn, N.S.; Cheng, X.; Li, A.H.; Xin, Y.; Schurmann, C.; Stevis, P.; Liu, Y.; Kozlitina, J.; Stender, S.; Wood, G.C.; et al. A Protein-Truncating HSD17B13 Variant and Protection from Chronic Liver Disease. N. Engl. J. Med. 2018, 378, 1096–1106. [Google Scholar] [CrossRef]
- Ma, Y.; Belyaeva, O.V.; Brown, P.M.; Fujita, K.; Valles, K.; Karki, S.; de Boer, Y.S.; Koh, C.; Chen, Y.; Du, X.; et al. 17-Beta Hydroxysteroid Dehydrogenase 13 Is a Hepatic Retinol Dehydrogenase Associated With Histological Features of Nonalcoholic Fatty Liver Disease. Hepatology 2019, 69, 1504–1519. [Google Scholar] [CrossRef]
- Stender, S.; Smagris, E.; Lauridsen, B.K.; Kofoed, K.F.; Nordestgaard, B.G.; Tybjaerg-Hansen, A.; Pennacchio, L.A.; Dickel, D.E.; Cohen, J.C.; Hobbs, H.H. Relationship between Genetic Variation at PPP1R3B and Liver Glycogen and Triglyceride Levels. Hepatology 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dongiovanni, P.; Meroni, M.; Mancina, R.M.; Baselli, G.; Rametta, R.; Pelusi, S.; Männistö, V.; Fracanzani, A.L.; Badiali, S.; Miele, L.; et al. Protein phosphatase 1 regulatory subunit 3B gene variation protects against hepatic fat accumulation and fibrosis in individuals at high risk of nonalcoholic fatty liver disease. Hepatol. Commun. 2018, 2, 666–675. [Google Scholar] [CrossRef] [Green Version]
- Pirazzi, C.; Adiels, M.; Burza, M.A.; Mancina, R.M.; Levin, M.; Stahlman, M.; Taskinen, M.-R.; Orho-Melander, M.; Perman, J.; Pujia, A.; et al. Patatin-like phospholipase domain-containing 3 (PNPLA3) I148M (rs738409) affects hepatic VLDL secretion in humans and in vitro. J. Hepatol. 2012, 57, 1276–1282. [Google Scholar] [CrossRef]
- Pingitore, P.; Pirazzi, C.; Mancina, R.M.; Motta, B.M.; Indiveri, C.; Pujia, A.; Montalcini, T.; Hedfalk, K.; Romeo, S. Recombinant PNPLA3 protein shows triglyceride hydrolase activity and its I148M mutation results in loss of function. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2014, 1841, 574–580. [Google Scholar] [CrossRef] [Green Version]
- He, S.; McPhaul, C.; Li, J.Z.; Garuti, R.; Kinch, L.; Grishin, N.V.; Cohen, J.C.; Hobbs, H.H. A sequence variation (I148M) in PNPLA3 associated with nonalcoholic fatty liver disease disrupts triglyceride hydrolysis. J. Biol. Chem. 2010, 285, 6706–6715. [Google Scholar] [CrossRef] [Green Version]
- Pirazzi, C.; Valenti, L.; Motta, B.M.; Pingitore, P.; Hedfalk, K.; Mancina, R.M.; Burza, M.A.; Indiveri, C.; Ferro, Y.; Montalcini, T.; et al. PNPLA3 has retinyl-palmitate lipase activity in human hepatic stellate cells. Hum. Mol. Genet. 2014, 23, 4077–4085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caddeo, A.; Jamialahmadi, O.; Solinas, G.; Pujia, A.; Mancina, R.M.; Pingitore, P.; Romeo, S. MBOAT7 is anchored to endomembranes by six transmembrane domains. J. Struct. Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, K.; Epand, R.M. Enrichment of phosphatidylinositols with specific acyl chains. Biochim. Biophys. Acta 2014, 1838, 1501–1508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, Y.; Shimanaka, Y.; Caddeo, A.; Kubo, T.; Mao, Y.; Kubota, T.; Kubota, N.; Yamauchi, T.; Mancina, R.M.; Baselli, G.; et al. LPIAT1/MBOAT7 depletion increases triglyceride synthesis fueled by high phosphatidylinositol turnover. Gut 2020. [Google Scholar] [CrossRef]
- Veiga-da-Cunha, M.; Van Schaftingen, E. Identification of fructose 6-phosphate- and fructose 1-phosphate-binding residues in the regulatory protein of glucokinase. J. Biol. Chem. 2002, 277, 8466–8473. [Google Scholar] [CrossRef] [Green Version]
- Orho-Melander, M.; Melander, O.; Guiducci, C.; Perez-Martinez, P.; Corella, D.; Roos, C.; Tewhey, R.; Rieder, M.J.; Hall, J.; Abecasis, G.; et al. Common missense variant in the glucokinase regulatory protein gene is associated with increased plasma triglyceride and C-reactive protein but lower fasting glucose concentrations. Diabetes 2008, 57, 3112–3121. [Google Scholar] [CrossRef] [Green Version]
- Saxena, R.; Voight, B.F.; Lyssenko, V.; Burtt, N.P.; de Bakker, P.I.; Chen, H.; Roix, J.J.; Kathiresan, S.; Hirschhorn, J.N.; Daly, M.J.; et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science 2007, 316, 1331–1336. [Google Scholar] [CrossRef]
- Holmen, O.L.; Zhang, H.; Fan, Y.; Hovelson, D.H.; Schmidt, E.M.; Zhou, W.; Guo, Y.; Zhang, J.; Langhammer, A.; Lochen, M.L.; et al. Systematic evaluation of coding variation identifies a candidate causal variant in TM6SF2 influencing total cholesterol and myocardial infarction risk. Nat. Genet. 2014, 46, 345–351. [Google Scholar] [CrossRef]
- Mahdessian, H.; Taxiarchis, A.; Popov, S.; Silveira, A.; Franco-Cereceda, A.; Hamsten, A.; Eriksson, P.; van’t Hooft, F. TM6SF2 is a regulator of liver fat metabolism influencing triglyceride secretion and hepatic lipid droplet content. Proc. Natl. Acad. Sci. USA 2014, 111, 8913–8918. [Google Scholar] [CrossRef] [Green Version]
- Prill, S.; Caddeo, A.; Baselli, G.; Jamialahmadi, O.; Dongiovanni, P.; Rametta, R.; Kanebratt, K.P.; Pujia, A.; Pingitore, P.; Mancina, R.M.; et al. The TM6SF2 E167K genetic variant induces lipid biosynthesis and reduces apolipoprotein B secretion in human hepatic 3D spheroids. Sci. Rep. 2019, 9, 11585. [Google Scholar] [CrossRef] [Green Version]
- Su, W.; Wang, Y.; Jia, X.; Wu, W.; Li, L.; Tian, X.; Li, S.; Wang, C.; Xu, H.; Cao, J.; et al. Comparative proteomic study reveals 17β-HSD13 as a pathogenic protein in nonalcoholic fatty liver disease. Proc. Natl. Acad. Sci. USA 2014, 111, 11437–11442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pothineni, N.V.; Delongchamp, R.; Vallurupalli, S.; Ding, Z.; Dai, Y.; Hagedorn, C.H.; Mehta, J.L. Impact of hepatitis C seropositivity on the risk of coronary heart disease events. Am. J. Cardiol. 2014, 114, 1841–1845. [Google Scholar] [CrossRef] [Green Version]
- Arain, S.Q.; Talpur, F.N.; Channa, N.A.; Ali, M.S.; Afridi, H.I. Serum lipid profile as a marker of liver impairment in hepatitis B Cirrhosis patients. Lipids Health Dis. 2017, 16, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghadir, M.R.; Riahin, A.A.; Havaspour, A.; Nooranipour, M.; Habibinejad, A.A. The relationship between lipid profile and severity of liver damage in cirrhotic patients. Hepat. Mon. 2010, 10, 285–288. [Google Scholar] [PubMed]
- Kozlitina, J.; Stender, S.; Hobbs, H.H.; Cohen, J.C. HSD17B13 and Chronic Liver Disease in Blacks and Hispanics. N. Engl. J. Med. 2018, 379, 1876–1877. [Google Scholar] [CrossRef] [PubMed]
- Kallwitz, E.; Tayo, B.O.; Kuniholm, M.H.; Daviglus, M.; Zeng, D.; Isasi, C.R.; Cotler, S.J. Association of HSD17B13 rs72613567:TA with non-alcoholic fatty liver disease in Hispanics/Latinos. Liver Int. 2020. [Google Scholar] [CrossRef]
- Yang, J.; Trépo, E.; Nahon, P.; Cao, Q.; Moreno, C.; Letouzé, E.; Imbeaud, S.; Bayard, Q.; Gustot, T.; Deviere, J.; et al. A 17-Beta-Hydroxysteroid Dehydrogenase 13 Variant Protects From Hepatocellular Carcinoma Development in Alcoholic Liver Disease. Hepatology 2019, 70, 231–240. [Google Scholar] [CrossRef]
- Stickel, F.; Lutz, P.; Buch, S.; Nischalke, H.D.; Silva, I.; Rausch, V.; Fischer, J.; Weiss, K.H.; Gotthardt, D.; Rosendahl, J.; et al. Genetic variation in HSD17B13 reduces the risk of developing cirrhosis and hepatocellular carcinoma in alcohol misusers. Hepatology 2019. [Google Scholar] [CrossRef]
- Scheiner, B.; Stättermayer, A.F.; Schwabl, P.; Bucsics, T.; Paternostro, R.; Bauer, D.; Simbrunner, B.; Schmidt, R.; Marculescu, R.; Ferlitsch, A.; et al. Impact of HSD17B13 rs72613567 genotype on hepatic decompensation and mortality in patients with portal hypertension. Liver Int. 2020, 40, 393–404. [Google Scholar] [CrossRef] [Green Version]
- Ferenci, P.; Pfeiffenberger, J.; Stättermayer, A.F.; Stauber, R.E.; Willheim, C.; Weiss, K.H.; Munda-Steindl, P.; Trauner, M.; Schilsky, M.; Zoller, H. HSD17B13 truncated variant is associated with a mild hepatic phenotype in Wilson’s Disease. JHEP Rep. 2019, 1, 2–8. [Google Scholar] [CrossRef] [Green Version]
- Stender, S.; Romeo, S. HSD17B13 as a promising therapeutic target against chronic liver disease. Liver Int. 2020. [Google Scholar] [CrossRef] [PubMed]
- Mancina, R.M.; Matikainen, N.; Maglio, C.; Söderlund, S.; Lundbom, N.; Hakkarainen, A.; Rametta, R.; Mozzi, E.; Fargion, S.; Valenti, L.; et al. Paradoxical Dissociation between Hepatic Fat Content and De Novo Lipogenesis Due to PNPLA3 Sequence Variant. J. Clin. Endocrinol. Metab. 2015, 100, E821–E825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondul, A.; Mancina, R.M.; Merlo, A.; Dongiovanni, P.; Rametta, R.; Montalcini, T.; Valenti, L.; Albanes, D.; Romeo, S. PNPLA3 1148M Variant Influences Circulating Retinol in Adults with Nonalcoholic Fatty Liver Disease or Obesity. J. Nutr. 2015, 145, 1687–1691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pingitore, P.; Dongiovanni, P.; Motta, B.M.; Meroni, M.; Lepore, S.M.; Mancina, R.M.; Pelusi, S.; Russo, C.; Caddeo, A.; Rossi, G.; et al. PNPLA3 overexpression results in reduction of proteins predisposing to fibrosis. Hum. Mol. Genet. 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruschi, F.V.; Claudel, T.; Tardelli, M.; Caligiuri, A.; Stulnig, T.M.; Marra, F.; Trauner, M. The PNPLA3 I148M variant modulates the fibrogenic phenotype of human hepatic stellate cells. Hepatology 2017, 65, 1875–1890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindén, D.; Ahnmark, A.; Pingitore, P.; Ciociola, E.; Ahlstedt, I.; Andréasson, A.C.; Sasidharan, K.; Madeyski-Bengtson, K.; Zurek, M.; Mancina, R.M.; et al. Pnpla3 silencing with antisense oligonucleotides ameliorates nonalcoholic steatohepatitis and fibrosis in Pnpla3 I148M knock-in mice. Mol. Metab. 2019, 22, 49–61. [Google Scholar] [CrossRef]
- Colombo, M.; Pelusi, S. Towards Precision Medicine in Nonalcoholic Fatty Liver Disease with PNPLA3 as a Therapeutic Target. Gastroenterology 2019, 157, 1156–1157. [Google Scholar] [CrossRef]
- Sookoian, S.; Pirola, C.J. Meta-analysis of the influence of I148M variant of patatin-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology 2011, 53, 1883–1894. [Google Scholar] [CrossRef]
- Kitamoto, T.; Kitamoto, A.; Yoneda, M.; Hyogo, H.; Ochi, H.; Nakamura, T.; Teranishi, H.; Mizusawa, S.; Ueno, T.; Chayama, K.; et al. Genome-wide scan revealed that polymorphisms in the PNPLA3, SAMM50, and PARVB genes are associated with development and progression of nonalcoholic fatty liver disease in Japan. Hum. Genet. 2013. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Xing, C.; Tian, Z.; Ku, H.C. Genetic variant I148M in PNPLA3 is associated with the ultrasonography-determined steatosis degree in a Chinese population. BMC Med. Genet. 2012, 13, 113. [Google Scholar] [CrossRef] [Green Version]
- Kanth, V.V.; Sasikala, M.; Rao, P.N.; Steffie Avanthi, U.; Rao, K.R.; Nageshwar Reddy, D. Pooled genetic analysis in ultrasound measured non-alcoholic fatty liver disease in Indian subjects: A pilot study. World J. Hepatol. 2014, 6, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Ching-Yeung Yu, B.; Kwok, D.; Wong, V.W. Magnitude of Nonalcoholic Fatty Liver Disease: Eastern Perspective. J. Clin. Exp. Hepatol. 2019, 9, 491–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.C.; Chang, P.F.; Chang, M.H.; Ni, Y.H. Genetic variants in GCKR and PNPLA3 confer susceptibility to nonalcoholic fatty liver disease in obese individuals. Am. J. Clin. Nutr. 2014, 99, 869–874. [Google Scholar] [CrossRef] [Green Version]
- Popescu, C.I.; Dubuisson, J. Role of lipid metabolism in hepatitis C virus assembly and entry. Biol. Cell 2009, 102, 63–74. [Google Scholar] [CrossRef]
- Barba, G.; Harper, F.; Harada, T.; Kohara, M.; Goulinet, S.; Matsuura, Y.; Eder, G.; Schaff, Z.; Chapman, M.J.; Miyamura, T.; et al. Hepatitis C virus core protein shows a cytoplasmic localization and associates to cellular lipid storage droplets. Proc. Natl. Acad. Sci. USA 1997, 94, 1200–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perlemuter, G.; Sabile, A.; Letteron, P.; Vona, G.; Topilco, A.; Chrétien, Y.; Koike, K.; Pessayre, D.; Chapman, J.; Barba, G.; et al. Hepatitis C virus core protein inhibits microsomal triglyceride transfer protein activity and very low density lipoprotein secretion: A model of viral-related steatosis. FASEB J. 2002, 16, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Agnello, V.; Chung, R.T.; Kaplan, L.M. A role for hepatitis C virus infection in type II cryoglobulinemia. N. Engl. J. Med. 1992, 327, 1490–1495. [Google Scholar] [CrossRef]
- Fukuhara, T.; Ono, C.; Puig-Basagoiti, F.; Matsuura, Y. Roles of Lipoproteins and Apolipoproteins in Particle Formation of Hepatitis C Virus. Trends Microbiol. 2015, 23, 618–629. [Google Scholar] [CrossRef]
- Zhu, C.; Zhang, R.; Liu, D.; Mukhtar, M.M.; Liu, W.; Peng, G.; Wang, K.; Hao, Q.; Xu, Y.; Liu, F.; et al. Association of functional polymorphism of ApoB promoter with hepatitis C virus infection. Clin. Chim. Acta 2009, 401, 124–127. [Google Scholar] [CrossRef]
- Harada, R.; Kimura, M.; Sato, Y.; Taniguchi, T.; Tomonari, T.; Tanaka, T.; Tanaka, H.; Muguruma, N.; Shinomiya, H.; Honda, H.; et al. APOB codon 4311 polymorphism is associated with hepatitis C virus infection through altered lipid metabolism. BMC Gastroenterol. 2018, 18, 24. [Google Scholar] [CrossRef] [Green Version]
- Gu, Q.L.; Han, Y.; Lan, Y.M.; Li, Y.; Kou, W.; Zhou, Y.S.; Hai, X.J.; Yan, B.; Ci, C.H. Association between polymorphisms in the APOB gene and hyperlipidemia in the Chinese Yugur population. Braz. J. Med. Biol. Res. 2017, 50, e6613. [Google Scholar] [CrossRef] [Green Version]
- Ghani, R.A.; Saqlain, M.; Zafar, M.M.; Jabeen, S.; Naqvi, S.M.; Raja, G.K. Identification of Metabolic risk phenotypes predisposing to Non-Alcoholic Fatty Liver Disease in a Pakistani Cohort. Pak. J. Med. Sci. 2017, 33, 121–126. [Google Scholar] [CrossRef]
- Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; Abecasis, G.R.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Miller, S.A.; Dykes, D.D.; Polesky, H.F. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988, 16, 1215. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
| Characteristic | Case | Control | p-Value |
|---|---|---|---|
| N | 587 | 68 | |
| Age, Years | 48 ± 11 | 46 ± 12 | 0.166 |
| Male gender, n (%) | 476 (81) | 42 (62) | 2.1 × 10−4 |
| BMI, Kg/m2 | 26 ± 5 | 26 ± 4 | 0.290 |
| Cholesterol, mg/dL | 88 ± 40 | 167 ± 35 | 2.3 × 10−45 |
| LDL, mg/dL | 51 ± 32 | 96 ± 41 | 1.9 × 10−23 |
| HDL, mg/dL | 19 ± 12 | 40 ± 9 | 1.2 × 10−44 |
| Triglyceride, mg/dL | 69 (54–100) | 154 (120–191) | 1.7 × 10−25 |
| ALT, IU/L | 66 (54–84) | 28 (22–34) | 7.1 × 10−73 |
| AST, IU/L | 61 (47–78) | 29 (23–32) | 2.7 × 10−64 |
| NASH/Viral disease, n (%) | 62/522 (11/89) | - | - |
| HCC/Cirrhosis, n (%) | 197/390 (34/66) | - | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raja, A.M.; Ciociola, E.; Ahmad, I.N.; Dar, F.S.; Naqvi, S.M.S.; Moaeen-ud-Din, M.; Raja, G.K.; Romeo, S.; Mancina, R.M. Genetic Susceptibility to Chronic Liver Disease in Individuals from Pakistan. Int. J. Mol. Sci. 2020, 21, 3558. https://doi.org/10.3390/ijms21103558
Raja AM, Ciociola E, Ahmad IN, Dar FS, Naqvi SMS, Moaeen-ud-Din M, Raja GK, Romeo S, Mancina RM. Genetic Susceptibility to Chronic Liver Disease in Individuals from Pakistan. International Journal of Molecular Sciences. 2020; 21(10):3558. https://doi.org/10.3390/ijms21103558
Chicago/Turabian StyleRaja, Asad Mehmood, Ester Ciociola, Imran Nazir Ahmad, Faisal Saud Dar, Syed Muhammad Saqlan Naqvi, Muhammad Moaeen-ud-Din, Ghazala Kaukab Raja, Stefano Romeo, and Rosellina Margherita Mancina. 2020. "Genetic Susceptibility to Chronic Liver Disease in Individuals from Pakistan" International Journal of Molecular Sciences 21, no. 10: 3558. https://doi.org/10.3390/ijms21103558
APA StyleRaja, A. M., Ciociola, E., Ahmad, I. N., Dar, F. S., Naqvi, S. M. S., Moaeen-ud-Din, M., Raja, G. K., Romeo, S., & Mancina, R. M. (2020). Genetic Susceptibility to Chronic Liver Disease in Individuals from Pakistan. International Journal of Molecular Sciences, 21(10), 3558. https://doi.org/10.3390/ijms21103558