Prenatal Exposure to Valproic Acid Affects Microglia and Synaptic Ultrastructure in a Brain-Region-Specific Manner in Young-Adult Male Rats: Relevance to Autism Spectrum Disorders

 ,
,  ,
, 
Abstract
1. Introduction
2. Results
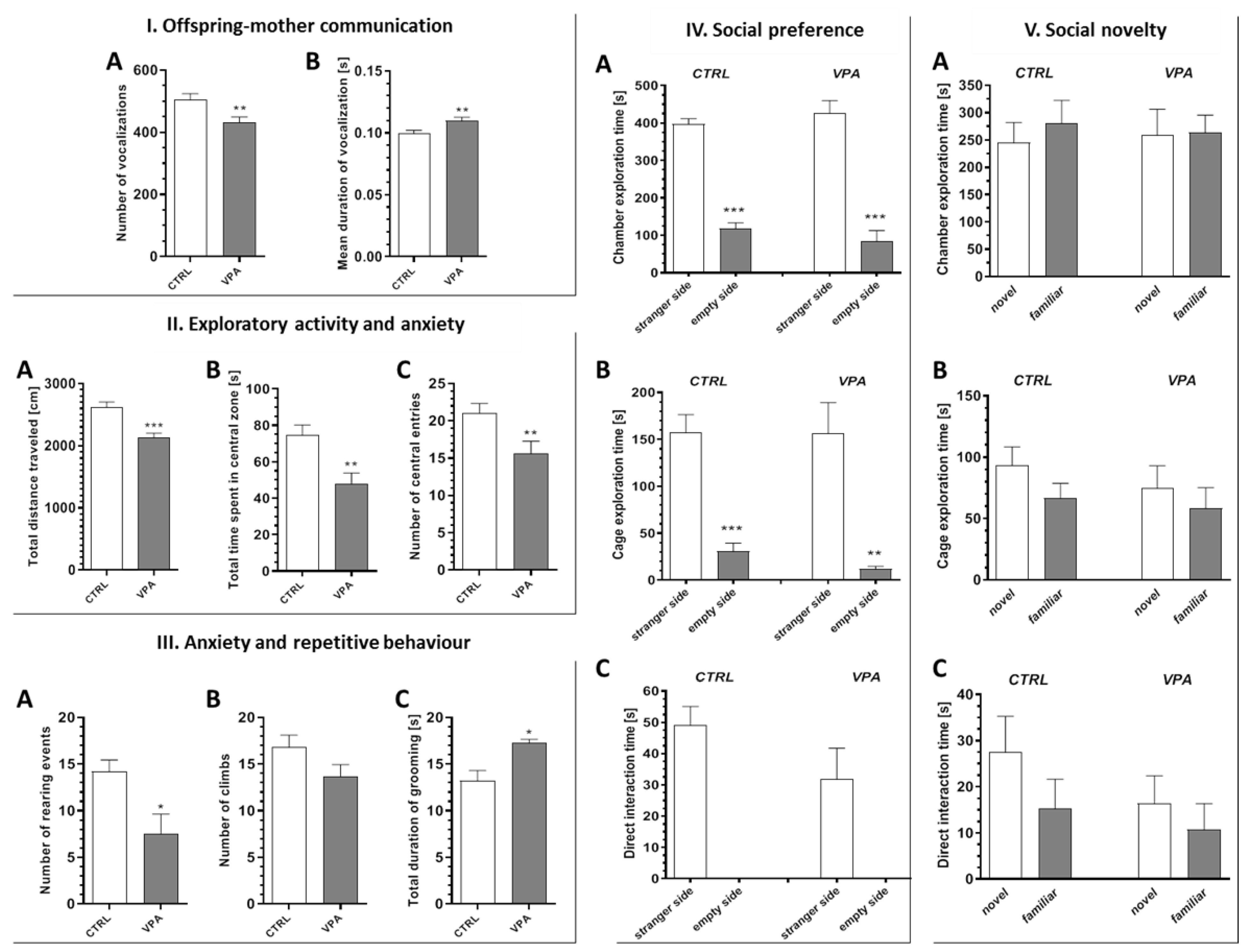
2.1. Prenatal Exposure to VPA Caused the Manifestation of Some Autistic-Like Behaviour in Young-Adult Offspring
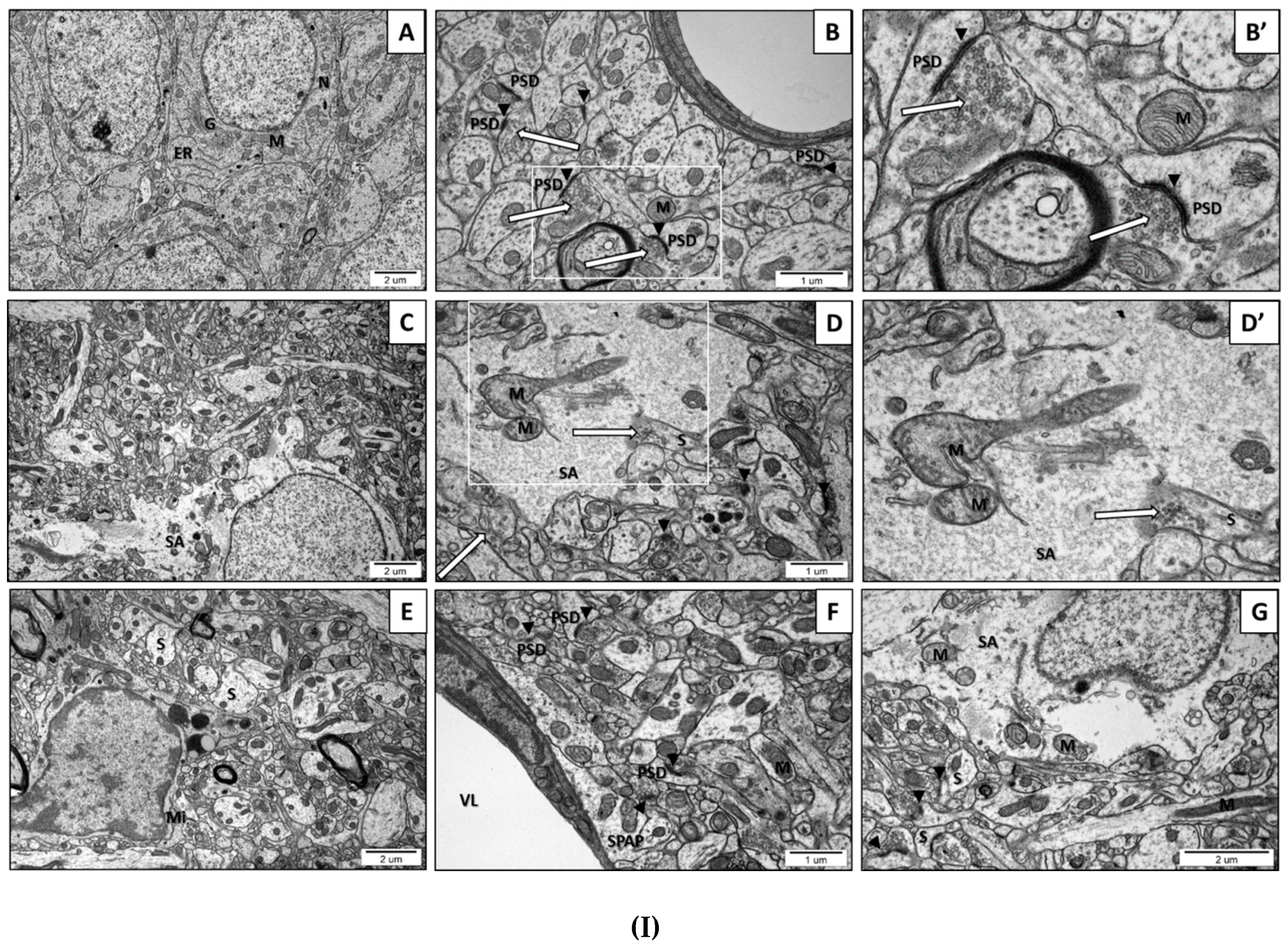
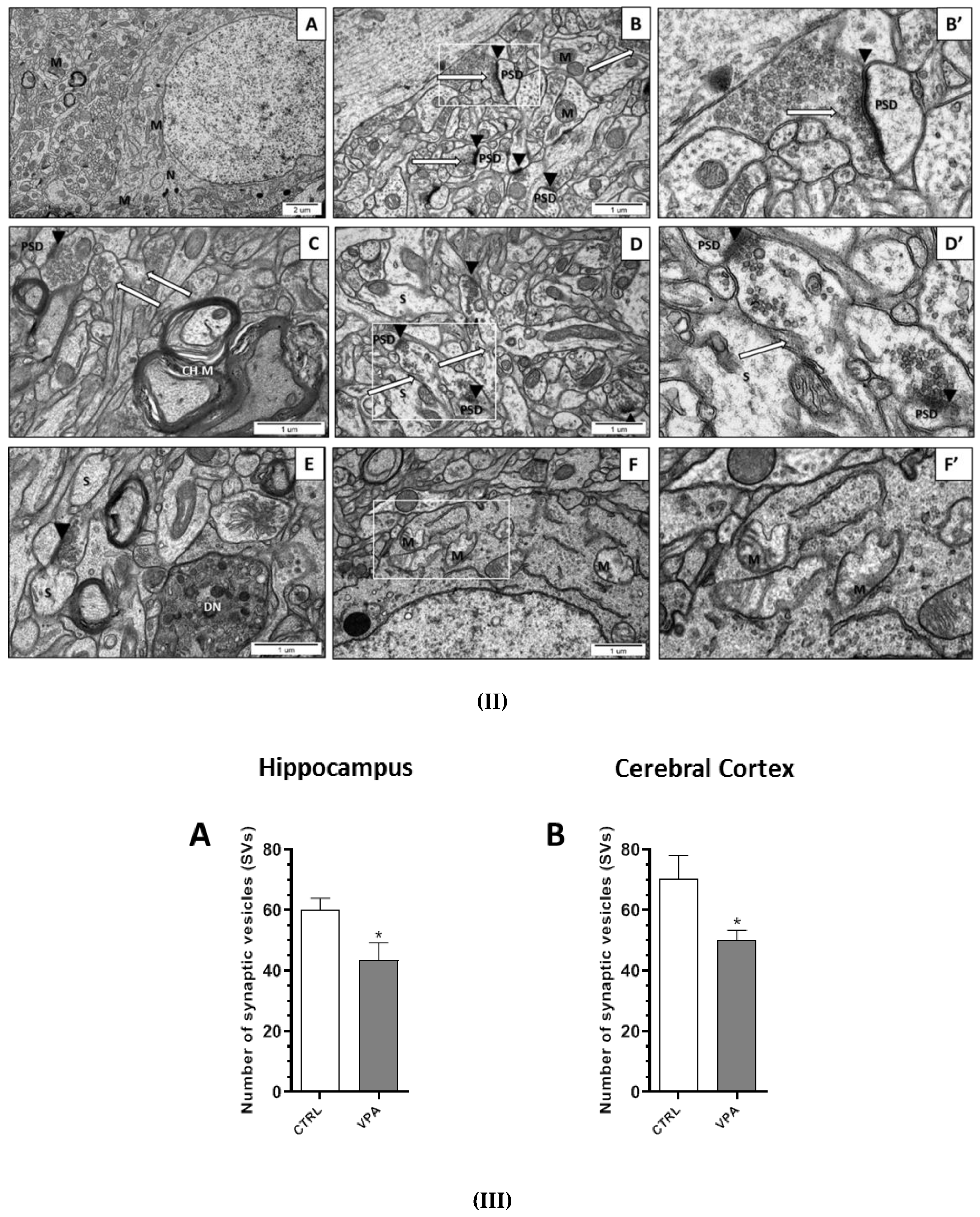
2.2. Prenatal Exposure to VPAInduced Ultrastructural Changes in the Synapses of Young-Adult Offspring. The Features of Synaptic Pathology in TEM Analysis
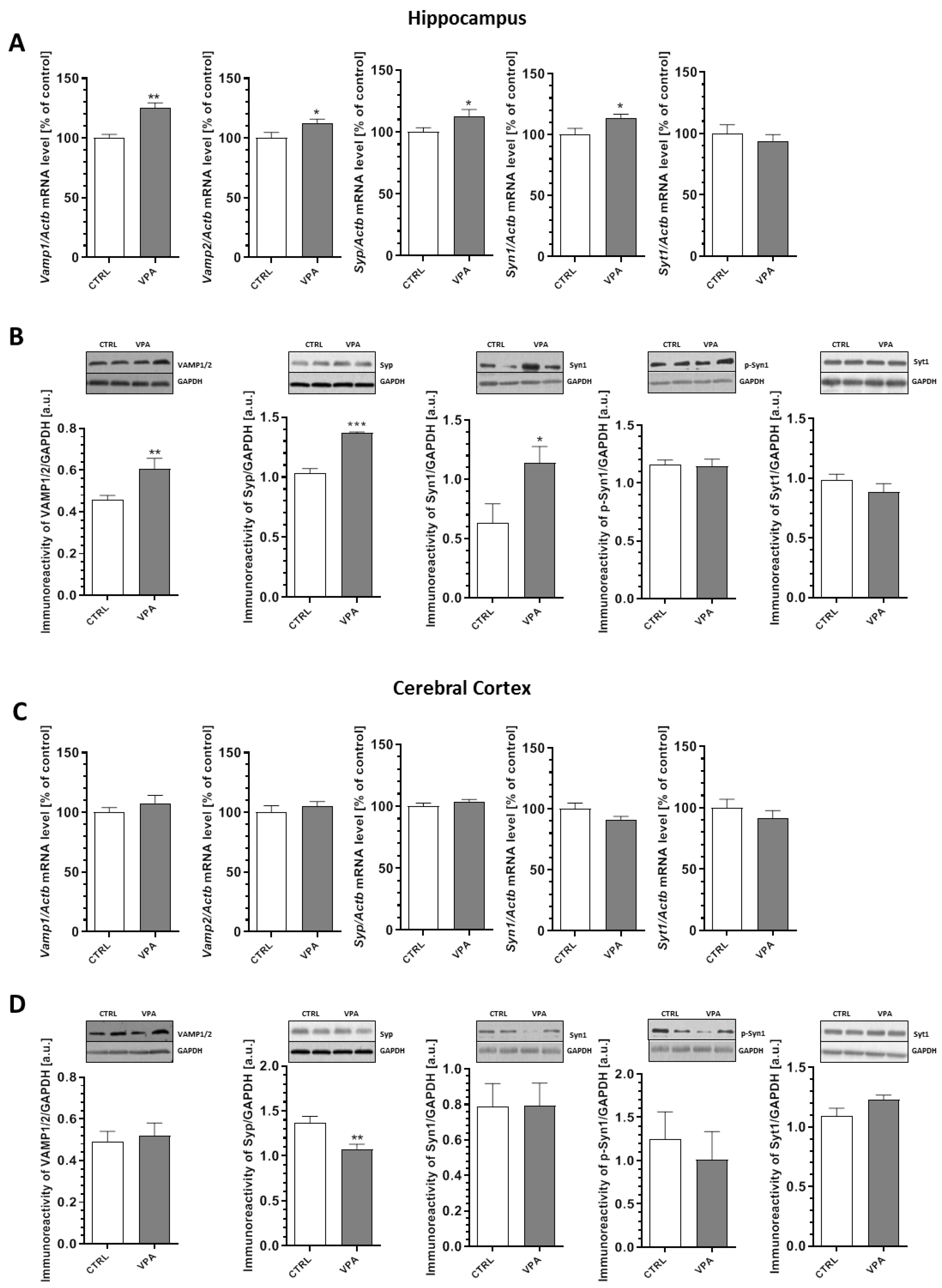
2.3. Prenatal Exposure to VPA Altered the Levels of the Presynaptic Proteins: Synaptic Vesicle Proteins (v-SNARE) And Proteins Associated with The Presynaptic Membrane (t-SNARE)
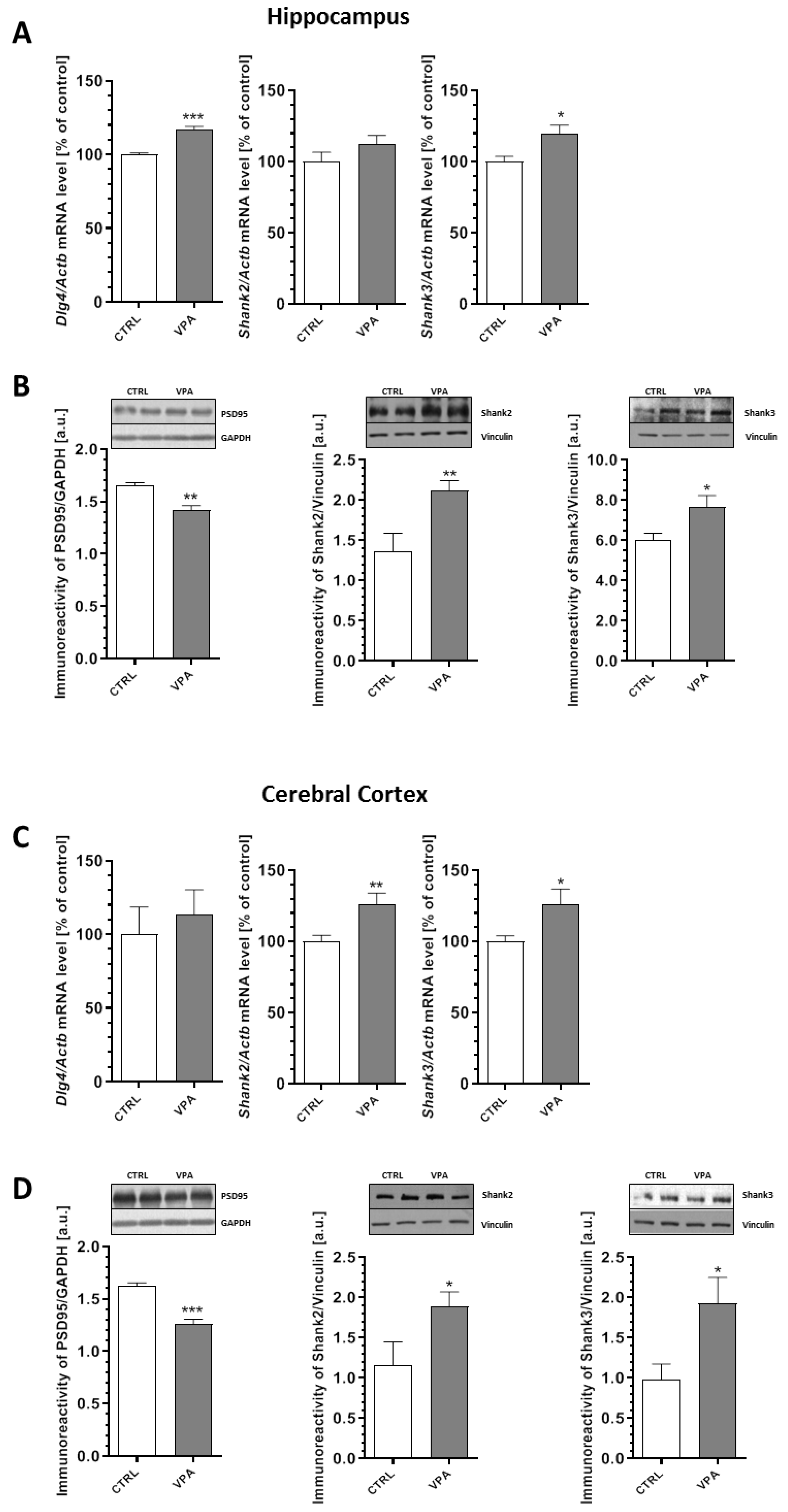
2.4. Prenatal Exposure to VPA Evoked Changes in the Level of Postsynaptic Density (PSD) Proteins and Synaptic Cell Adhesion Molecules
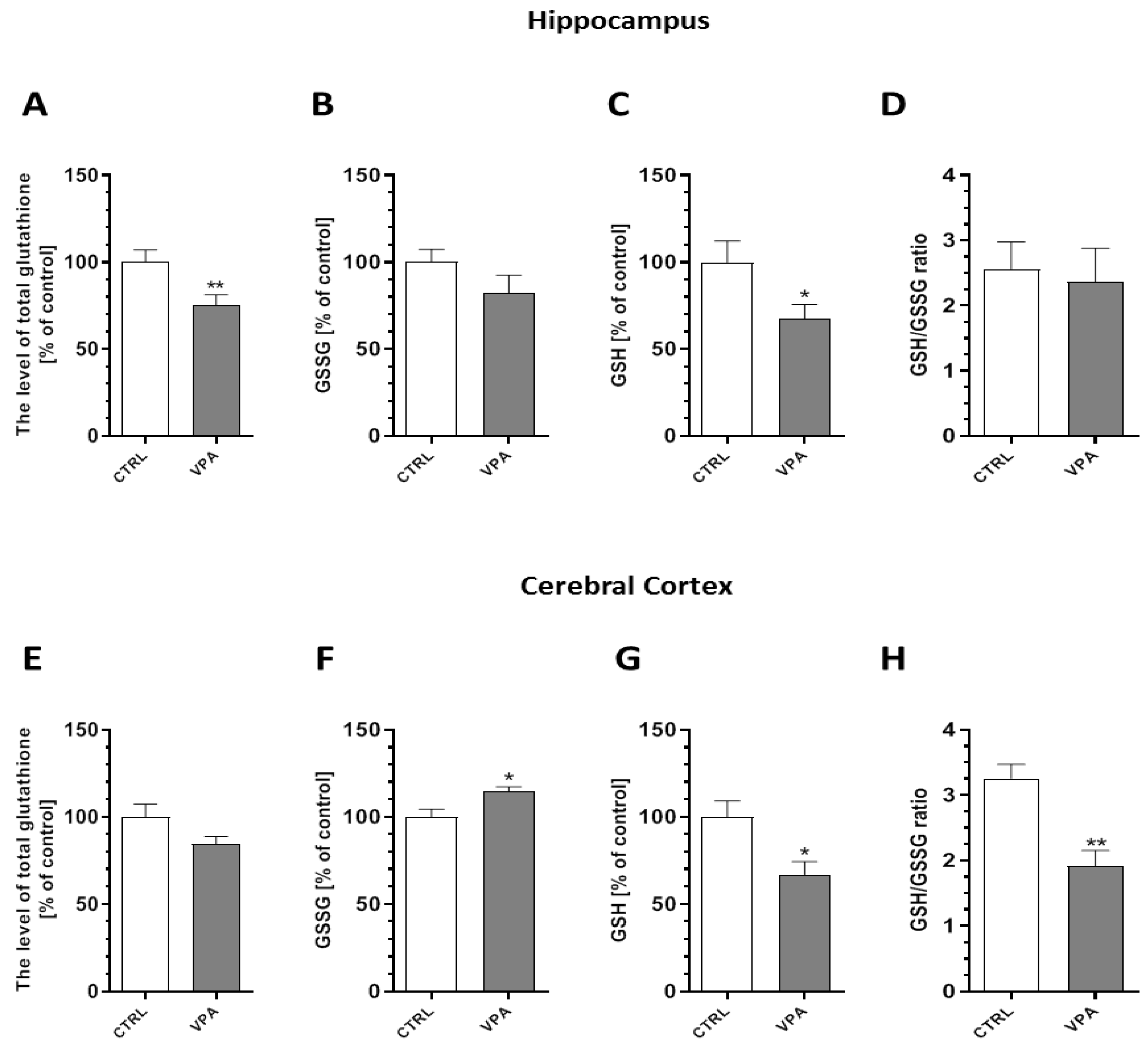
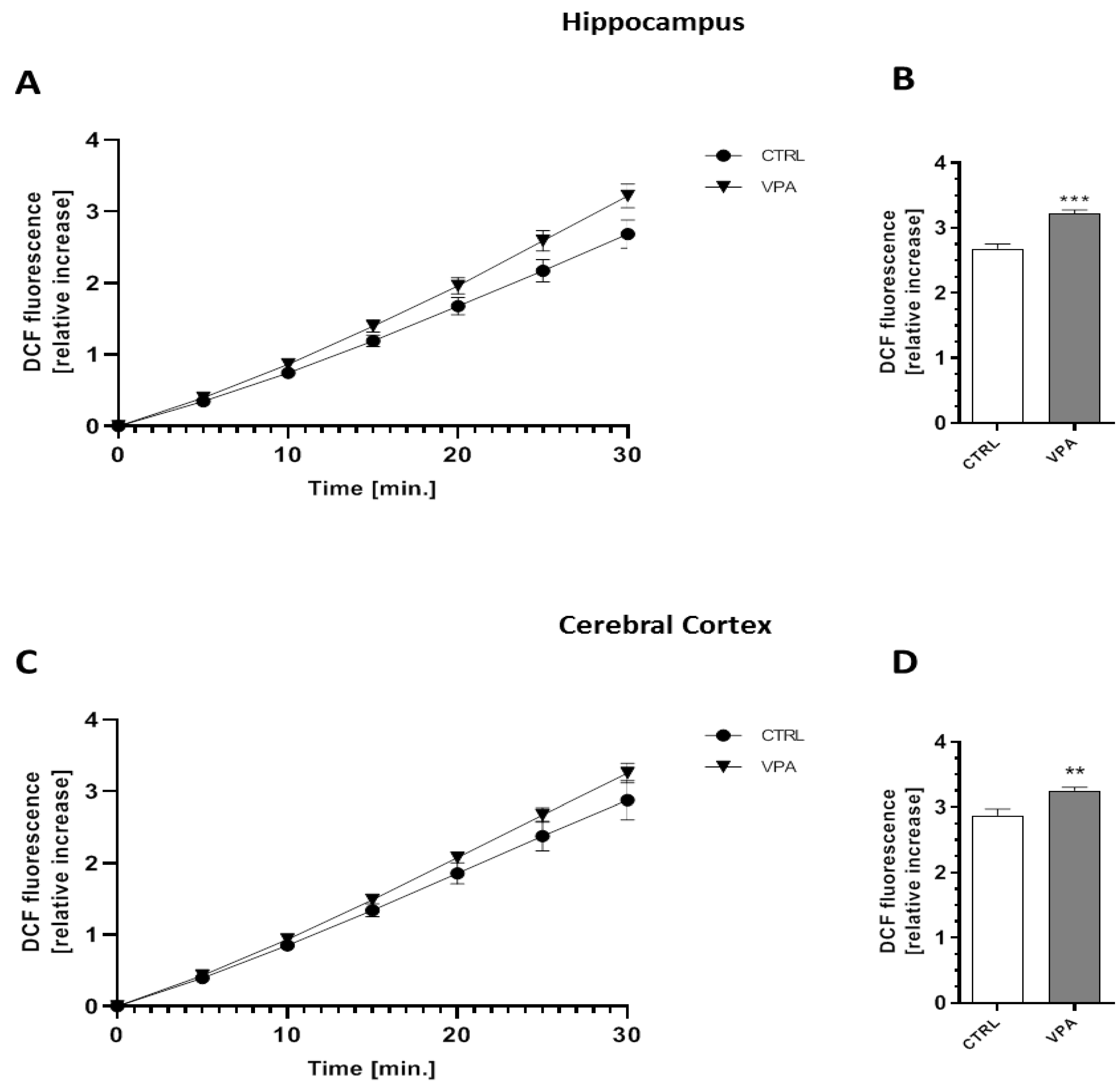
2.5. Prenatal Exposure to VPA Evoked Oxidative Stress and ROS Generation in the Brains of Young-Adult Offspring
2.6. Prenatal Exposure to VPA Affected the Microglia in the Young-Adult Offspring
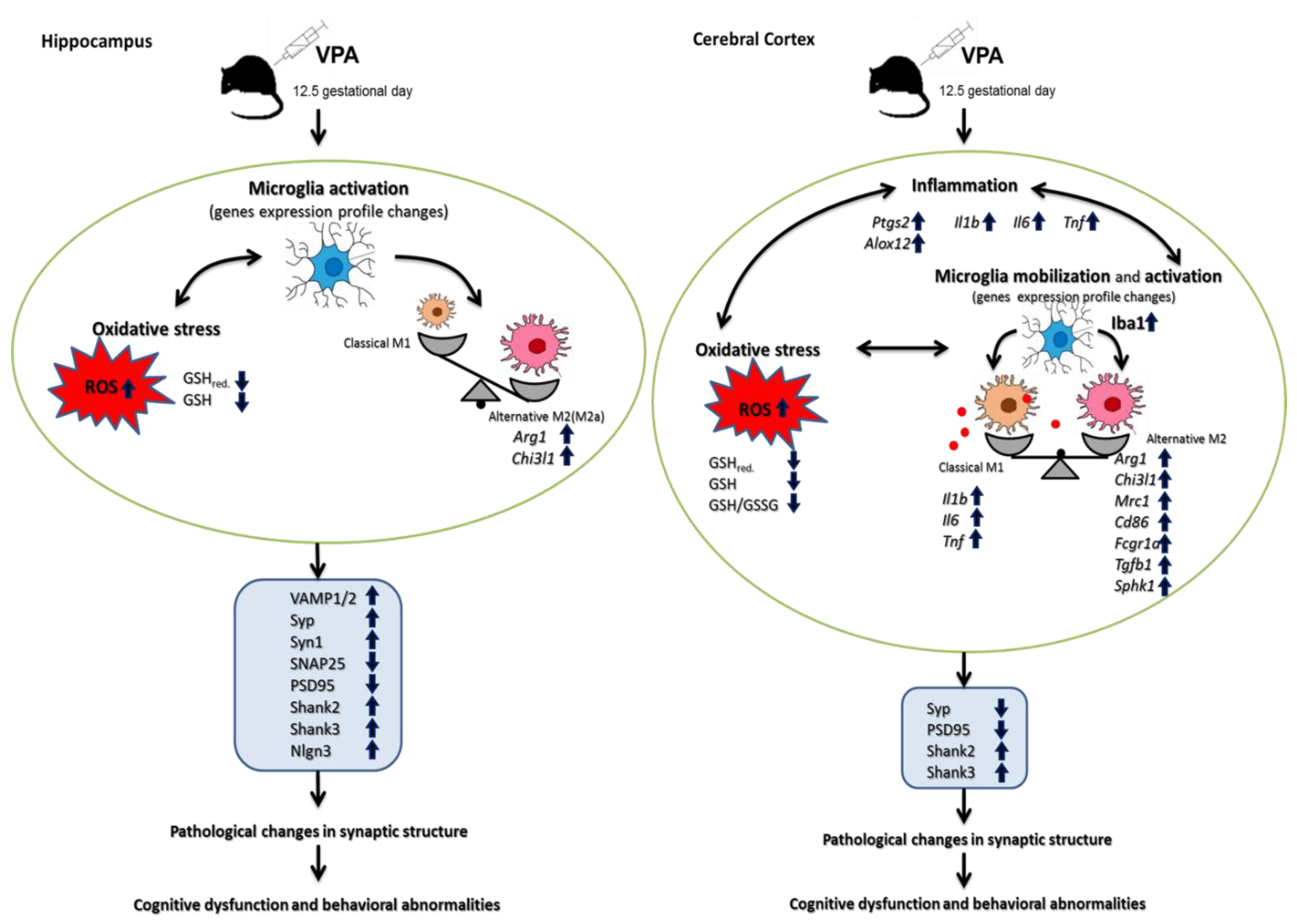
3. Discussion
4. Materials and Methods
4.1. Ethical Statement
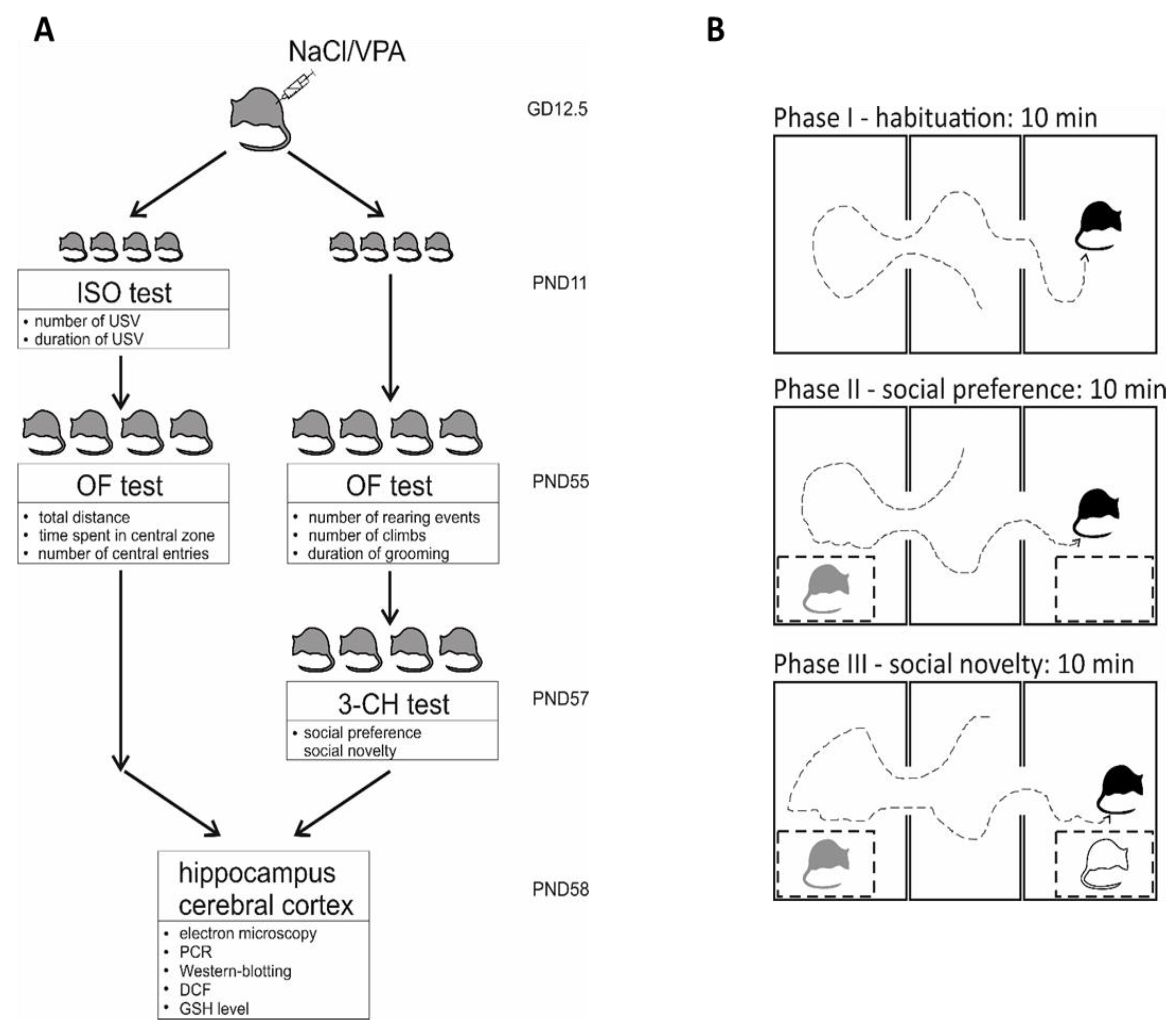
4.2. Animals—In Vivo Model of ASD
4.3. Behavioural Analysis
4.3.1. Juvenile Isolation Test (ISO)
4.3.2. Open Field Test (OF)
4.3.3. Three-Chamber Sociability and Social Novelty Test
4.4. Electron Microscopic Analysis of Brain Samples (TEM Analysis)
4.5. Quantitative Real Time Polymerase Chain Reaction (qRT-PCR)
4.6. Immunochemical Determination of Protein Levels (Western Blot Analysis)
4.7. Determination of Glutathione Levels
4.8. Determination of Reactive Oxygen Species (ROS) Level
4.9. Statistical Analysis of Biochemical and Behavioural Results
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ASD | Autism Spectrum Disorder |
| VPA | Valproic acid |
| Nlgns | Neuroligins |
| PSD95 | Post-synaptic density protein 95 |
| Shank | SH3 and multiple ankyrin repeat domains protein |
| Syn | Synapsin |
| Nrnx | Neurexin |
| SNAP25 | Synaptosomal-associated protein of 25 kDa |
| SNARE | Soluble N-ethylmaleimide-sensitive factor (NSF) attachment protein (SNAP) receptor |
| v-SNARE | Vesicle-associated membrane proteins |
| t-SNARE | Transmembrane-associated proteins |
| VAMP1/2 | Synaptobrevins 1 and 2 |
| Stx1 | Syntaxin1 |
| Syp | Synaptophysin |
| Syt | Synaptotagmin |
| ADHD | Attention deficit hyperactivity disorder |
| BPD | Bipolar disorder |
| SCH | Schizophrenia |
| MDD | Major depressive disorder |
| TEM | Transmission electron microscopy |
| SVs | Synaptic vesicles |
| PSD | Postsynaptic density |
| p-Syn1 | Phospho-synapsin 1(Ser62/Ser67) |
| GSH | Glutathione |
| GSSG | Oxidised glutathione |
| IL-1β | Interleukin-1β |
| IL-6 | Interleukin-6 |
| TNF-α | Tumor necrosis factor α |
| Arg1 | Arginase1 |
| Chi3l1 | Chitinase-3-like protein 1 |
| Mrc1 | Mannose receptor C-type 1 |
| Cd86 | CD86 molecule |
| Fcgr1α | Fc receptor subunits 1 |
| Tgfb1 | Transforming growth factor β |
| Sphk1 | Sphingosine kinase 1 |
| Alox12 | Arachidonate 12-Lipoxygenase |
| LOX12 | 12-Lipoxygenase |
| Ptgs2 | Prostaglandin-endoperoxide synthase 2 |
| COX2 | Cyclooxygenase 2 |
| CNS | Central nervous system |
| NMDA | N-methyl-D-aspartate receptor |
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor |
| AD | Alzheimer’s disease |
| PD | Parkinson’s disease |
| FXS | Fragile X syndrome |
| Iba 1 | Ionised calcium binding adaptor molecule 1 |
| NF-κB | Nuclear factor kappa β |
| BDNF | Brain-derived neurotrophic factor |
| IGF-1 | Insulin like growth factor 1 |
References
- Zoghbi, H.Y.; Bear, M.F. Synaptic dysfunction in neurodevelopmental disorders associated with autism and intellectual disabilities. Cold Spring Harb. Perspect. Biol. 2012, 4, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi-Fakhari, D.; Sahin, M. Autism and the synapse: Emerging mechanisms and mechanism-based therapies. Curr. Opin. Neurol. 2015, 28, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Safari, M.R.; Omrani, M.D.; Noroozi, R.; Sayad, A.; Sarrafzadeh, S.; Komaki, A.; Manjili, F.A.; Mazdeh, M.; Ghaleiha, A.; Taheri, M. Synaptosome-Associated Protein 25 (SNAP25) Gene Association Analysis Revealed Risk Variants for ASD, in Iranian Population. J. Mol. Neurosci. 2017, 61, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Bronzuoli, M.R.; Facchinetti, R.; Ingrassia, D.; Sarvadio, M.; Schiavi, S.; Steardo, L.; Verkhratsky, A.; Trezza, V.; Scuderi, C. Neuroglia in the autistic brain: Evidence from a preclinical model. Mol. Autism 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Baio, J.; Wiggins, L.; Christensen, D.L.; Maenner, M.J.; Daniels, J.; Warren, Z.; Kurzius-Spencer, M.; Zahorodny, W.; Robinson Rosenberg, C.; White, T.; et al. Prevalence of Autism Spectrum Disorder Among Children Aged 8 Years -Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2014. MMWR Surveill Summ. 2018, 67, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Deckmann, I.; Schwingel, G.B.; Fontes-Dutra, M.; Bambini-Junior, V.; Gottfried, C. Neuroimmune alterations in autism: A translational analysis focusing on the animal model of autism induced by prenatal exposure to valproic acid. NeuroImmunoModulation 2018, 25, 285–299. [Google Scholar] [CrossRef] [PubMed]
- Chaste, P.; Leboyer, M. Autism risk factors: Genes, environment, and gene-environment interactions. Dialogues Clin. Neurosci. 2012, 14, 281–292. [Google Scholar]
- Guang, S.; Pang, N.; Deng, X.; Yang, L.; He, F.; Wu, L.; Chen, C.; Yin, F.; Peng, J. Synaptopathology involved in autism spectrum disorder. Front. Cell Neurosci. 2018, 12, 1–28. [Google Scholar] [CrossRef]
- Howell, B.W.; Smith, K.M. Synaptic structural protein dysfunction leads to altered excitation inhibition ratios in models of autism spectrum disorder. Pharmacol Res. 2019, 139, 207–214. [Google Scholar] [CrossRef]
- Talebizadeh, Z.; Bittel, D.C.; Veatch, O.J.; Butler, M.G.; Takahashi, T.N.; Miles, J.H.; Wang, C.H. Letter to the editor: Do known mutations in neuroligin genes (NLGN3 and NLGN4) cause autism? J. Autism Dev. Disord. 2004, 34, 735–736. [Google Scholar] [CrossRef]
- Talebizadeh, Z.; Lam, D.Y.; Theodoro, M.F.; Bittel, D.C.; Lushington, G.H.; Butler, M.G. Novel splice isoforms for NLGN3 and NLGN4 with possible implications in autism. J. Med. Genet. 2006, 43, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Oliveira, G.; Coutinho, A.; Yang, C.; Feng, J.; Katz, C.; Sram, J.; Bockholt, A.; Jones, I.R.; Craddock, N.; et al. Analysis of the neuroligin 3 and 4 genes in autism and other neuropsychiatric patients. Mol. Psychiatry 2005, 10, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, J.; Man, H.Y. Fundamental Elements in Autism: From Neurogenesis and Neurite Growth to Synaptic Plasticity. Front. Cell. Neurosci. 2017, 11, 1–44. [Google Scholar]
- Chen, X.; Nelson, C.D.; Li, X.; Winters, C.A.; Azzam, R.; Sousa, A.A.; Leapman, R.D.; Gainer, H.; Sheng, M.; Reese, T.S. PSD-95 Is Required to Sustain the Molecular Organization of the Postsynaptic Density. J. Neurosci. 2011, 31, 6329–6338. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.; Kimura, H.; Wang, C.; Ishizuka, K.; Kushima, I.; Arioka, Y.; Arioka, Y.; Yoshimi, A.; Nakamura, Y.; Shiino, T.; et al. Resequencing and association analysis of Six PSD-95-related genes as possible susceptibility genes for schizophrenia and autism spectrum disorders. Sci. Rep. 2016, 6, 27491. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Bozdagi, O.; Scattoni, M.L.; Wohr, M.; Roullet, F.I.; Katz, A.M.; Abrams, D.N.; Kalikhman, D.; Simon, H.; Woldeyohannes, L.; et al. Reduced Excitatory Neurotransmission and Mild Autism-Relevant Phenotypes in Young-adult Shank3 Null Mutant Mice. J. Neurosci. 2012, 32, 6525–6541. [Google Scholar] [CrossRef]
- Peça, J.; Feliciano, C.; Ting, J.T.; Wang, W.; Wells, M.F.; Venkatraman, T.N.; Lascola, C.D.; Fu, Z.; Feng, G. Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 2011, 472, 437–442. [Google Scholar] [CrossRef]
- Coley, A.A.; Gao, W.J. PSD95: A synaptic protein implicated in schizophrenia or autism? Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2018, 82, 187–194. [Google Scholar] [CrossRef]
- Monteiro, P.; Feng, G. SHANK proteins: Roles at the synapse and in autism spectrum disorder. Nat. Rev. Neurosci. 2017, 18, 147–157. [Google Scholar] [CrossRef]
- Torres, V.I.; Vallejo, D.; Inestrosa, N.C. Emerging Synaptic Molecules as Candidates in the Etiology of Neurological Disorders. Neural Plast. 2017, 2017, 8081758. [Google Scholar] [CrossRef]
- Chen, J.; Yu, S.; Fu, Y.; Li, X. Synaptic proteins and receptors defects in autism spectrum disorders. Front. Cell. Neurosci. 2014, 8, 276. [Google Scholar] [CrossRef] [PubMed]
- Fassio, A.; Patry, L.; Congia, S.; Onofri, F.; Piton, A.; Gauthier, J.; Pozzi, D.; Messa, M.; Defranchi, E.; Fadda, M.; et al. SYN1 loss-of-function mutations in autism and partial epilepsy cause impaired synaptic function. Hum. Mol. Genet. 2011, 20, 2297–2307. [Google Scholar] [CrossRef] [PubMed]
- Sauter, S.; Von Beust, G.; Burfeind, P.; Weise, A.; Starke, H.; Liehr, T.; Zoll, B. Autistic disorder and chromosomal mosaicism 46,XY[123]/46,XY,del(20)(pter --> p12.2)[10]. Am. J. Med. Genet. A 2003, 20, 533–536. [Google Scholar] [CrossRef] [PubMed]
- Won, H.; Mah, W.; Kim, E. Autism spectrum disorder causes, mechanisms, and treatments: Focus on neuronal synapses. Front. Mol. Neurosci. 2013, 6, 1–19. [Google Scholar] [CrossRef]
- Südhof, T.C. The synaptic vesicle cycle. Annu. Rev. Neurosci. 2004, 27, 509–547. [Google Scholar] [CrossRef]
- Südhof, T.C.; Rothman, J.E. Membrane fusion: Grappling with SNARE and SM proteins. Science 2009, 76, 736–753. [Google Scholar] [CrossRef]
- Südhof, T.C.; Rizo, J. Synaptic vesicle exocytosis. Cold Spring Harb. Perspect. Biol. 2011, 3, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Südhof, T.C. A molecular machine for neurotransmitter release: Synaptotagmin and beyond. Nat. Med. 2013, 19, 1227–1231. [Google Scholar] [CrossRef]
- Valtorta, F.; Pennuto, M.; Bonanomi, D.; Benfenati, F. Synaptophysin: Leading actor or walk-on role in synaptic vesicle exocytosis? BioEssays 2004, 26, 445–453. [Google Scholar] [CrossRef]
- Cupertino, R.B.; Kappel, D.B.; Bandeira, C.E.; Schuch, J.B.; da Silva, B.S.; Müller, D.; Bau, C.H.; Mota, N.R. SNARE complex in developmental psychiatry: Neurotransmitter exocytosis and beyond. J. Neural Transm. 2016, 123, 867–883. [Google Scholar] [CrossRef]
- Schneider, T.; Przewłocki, R. Behavioral alterations in rats prenatally to valproic acid: Animal model of autism. Neuropsychopharmacology 2005, 30, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Wöhr, M.; Schwarting, R.K. Affective communication in rodents: Ultrasonic vocalizations as a tool for research on emotion and motivation. Cell Tissue Res. 2013, 354, 81–97. [Google Scholar] [CrossRef]
- Zieminska, E.; Toczylowska, B.; Diamandakis, D.; Hilgier, W.; Filipkowski, R.K.; Polowy, R.; Orzel, J.; Gorka, M.; Lazarewicz, J.W. Glutamate, Glutamine and GABA Levels in Rat Brain Measured Using MRS, HPLC and NMR Methods in Study of Two Models of Autism. Front. Mol. Neurosci. 2018, 11, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Peralta, F.; Fuentealba, C.; Fiedler, J.; Aliaga, E. Prenatal valproate treatment produces autistic-like behavior and increases metabotropic glutamate receptor 1A-immunoreactivity in the hippocampus of juvenile rats. Mol Med. Rep. 2016, 14, 2807–2814. [Google Scholar] [CrossRef] [PubMed]
- Wellmann, K.A.; Varlinskaya, E.I.; Mooney, S.M. D-Cycloserine ameliorates social alterations that result from prenatal exposure to valproic acid. Brain Res. Bull. 2014, 108, 1–9. [Google Scholar] [CrossRef][Green Version]
- Hughes, E.M.; Calcagno, P.; Clarke, M.; Sanchez, C.; Smith, K.; Kelly, J.P.; Finn, D.P.; Roche, M. Prenatal exposure to valproic acid reduces social responses and alters mRNA levels of opioid receptor and pre-pro-peptide in discrete brain regions of young-adult and adult male rats. Brain Res. 2020, 1732, 146675. [Google Scholar] [CrossRef]
- Schiavi, S.; Iezzi, D.; Manduca, A.; Leone, S.; Melancia, F.; Carbone, C.; Petrella, M.; Mannaioni, G.; Masi, A.; Trezza, V. Reward-Related Behavioral, Neurochemical and Electrophysiological Changes in a Rat Model of Autism Based on Prenatal Exposure to Valproic Acid. Front. Cell Neurosci. 2019, 13, 479. [Google Scholar] [CrossRef]
- Kumar, H.; Sharma, B.M.; Sharma, B. Benefits of agomelatine in behavioral, neurochemical and blood brain barrier alterations in prenatal valproic acid induced autism spectrum disorder. Neurochem Int. 2015, 91, 34–45. [Google Scholar] [CrossRef]
- Cohen, O.S.; Varlinskaya, E.I.; Wilson, C.A.; Glatt, S.J.; Mooney, S.M. Acute prenatal exposure to a moderate dose of valproic acid increases social behaviour and alters gene expression in rats. Int. J. Dev. Neurosci. 2013, 31, 740–750. [Google Scholar] [CrossRef]
- Stefanik, P.; Olexová, L.; Kršková, L. Increased sociability and gene expression of oxytocin and its receptor in the brains of rats affected prenatally by valproic acid. Pharmacol. Biochem. Behav. 2015, 131, 42–50. [Google Scholar] [CrossRef]
- Zhan, Y.; Kong, X. Synaptic Dysfunction Attributes to Autism Spectrum Disorder. N. Am. J. Med. Sci. 2011, 4, 112–115. [Google Scholar] [CrossRef]
- Gordon, S.L.; Harper, C.B.; Smillie, K.J.; Cousin, M.A. A fine balance of synaptophysin levels underlies efficient retrieval of synaptobrevin II to synaptic vesicles. PLoS ONE 2016, 11, e0149457. [Google Scholar] [CrossRef] [PubMed]
- Corradi, A.; Fadda, M.; Piton, A.; Patry, L.; Marte, A.; Rossi, P.; Cadieux-Dion, M.; Gauthier, J.; Lapointe, L.; Mottron, L.; et al. SYN2 is an autism predisposing gene: Loss-of-function mutations alter synaptic vesicle cycling and axon outgrowth. Hum. Mol. Genet. 2014, 23, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Guerini, F.R.; Bolognesi, E.; Chiappedi, M.; Manca, S.; Ghezzo, A.; Agliardi, C.; Sotgiu, S.; Usai, S.; Matteoli, M.; Clerici, M. SNAP-25 single nucleotide polymorphisms are associated with hyperactivity in autism spectrum disorders. Pharmacol. Res. 2011, 64, 283–288. [Google Scholar] [CrossRef]
- Braida, D.; Guerini, F.R.; Ponzoni, L.; Corradini, I.; De Astis, S.; Pattini, L.; Bolognesi, E.; Benfante, R.; Fornasari, D.; Chiappedi, M.; et al. Association between SNAP-25 gene polymorphisms and cognition in autism: Functional consequences and potential therapeutic strategies. Transl. Psychiatry 2015, 5, 1–11. [Google Scholar] [CrossRef]
- Joshi, H.; Sharma, R.; Prashar, S.; Ho, J.; Thomson, S.; Mishra, R. Differential expression of synapsin I and II upon treatment by lithium and valproic acid in various brain regions. Int. J. Neuropsychopharmacol. 2018, 21, 616–622. [Google Scholar] [CrossRef]
- Béïque, J.C.; Andrade, R. PSD-95 regulates synaptic transmission and plasticity in rat cerebral cortex. J. Physiol. 2003, 546, 859–867. [Google Scholar] [CrossRef]
- El-Husseini, A.E.; Schnell, E.; Chetkovich, D.M.; Nicoll, R.A.; Bredt, D.S. PSD-95 involvement in maturation of excitatory synapses. Science 2000, 290, 1364–1368. [Google Scholar]
- Kim, M.J.; Futai, K.; Jo, J.; Hayashi, Y.; Cho, K.; Sheng, M. Synaptic Accumulation of PSD-95 and Synaptic Function Regulated by Phosphorylation of Serine-295 of PSD-95. Neuron 2007, 56, 488–502. [Google Scholar] [CrossRef]
- Shao, C.Y.; Mirra, S.S.; Sait, H.B.R.; Sacktor, T.C.; Sigurdsson, E.M. Postsynaptic degeneration as revealed by PSD-95 reduction occurs after advanced Aβ and tau pathology in transgenic mouse models of Alzheimer’s disease. Acta Neuropathol. 2011, 122, 285–292. [Google Scholar] [CrossRef]
- Sultana, R.; Banks, W.A.; Butterfield, D.A. Decreased levels of PSD95 and two associated proteins and increased levels of BCl2 and caspase 3 in hippocampus from subjects with amnestic mild cognitive impairment: Insights into their potential roles for loss of synapses and memory, accumulation of Aβ. J. Neurosci. Res. 2010, 88, 469–477. [Google Scholar] [PubMed]
- Tu, S.; Okamoto, S.; Lipton, S.A.; Xu, H. Oligomeric Aβ-induced synaptic dysfunction in Alzheimer’s disease. Mol. Neurodegener. 2014, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Nash, J.E.; Johnston, T.H.; Collingridge, G.L.; Garner, C.C.; Brotchie, J.M. Subcellular redistribution of the synapse-associated proteins PSD-95 and SAP97 in animal models of Parkinson’s disease and L-DOPA-induced dyskinesia. FASEB J. 2005, 19, 583–585. [Google Scholar] [CrossRef] [PubMed]
- Matosin, N.; Fernandez-Enright, F.; Lum, J.S.; Engel, M.; Andrews, J.L.; Gassen, N.C.; Wagner, K.V.; Schmidt, M.V.; Newell, K.A. Molecular evidence of synaptic pathology in the CA1 region in schizophrenia. Npj Schizophr. 2016, 2, 1–8. [Google Scholar] [CrossRef]
- Catts, V.S.; Derminio, D.S.; Hahn, C.G.; Weickert, C.S. Postsynaptic density levels of the NMDA receptor NR1 subunit and PSD-95 protein in prefrontal cortex from people with schizophrenia. Npj Schizophr. 2015, 1, 1–8. [Google Scholar] [CrossRef]
- Lelieveld, S.H.; Reijnders, M.R.F.; Pfundt, R.; Yntema, H.G.; Kamsteeg, E.J.; De Vries, P.; Willemsen, M.H.; Kleefstra, T.; Löhner, K.; Vreeburg, M.; et al. Meta-analysis of 2,104 trios provides support for 10 new genes for intellectual disability. Nat. Neurosci. 2016, 19, 1194–1196. [Google Scholar] [CrossRef]
- Feyder, M.; Karlsson, R.M.; Mathur, P.; Lyman, M.; Bock, R.; Momenan, R.; Munasinghe, J.; Scattoni, M.L.; Ihne, J.; Camp, M.; et al. Association of mouse Dlg4 (PSD-95) gene deletion and human DLG4 gene variation with phenotypes relevant to autism spectrum disorders and Williams’ syndrome. Am. J. Psychiatry 2010, 167, 1508–1517. [Google Scholar] [CrossRef]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Cicek, A.E.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef]
- Amal, H.; Barak, B.; Bhat, V.; Gong, G.; Joughin, B.A.; Wishnok, J.S.; Feng, G.; Tannenbaum, S.R. Shank3 mutation in a mouse model of autism leads to changes in the S-nitroso-proteome and affects key proteins involved in vesicle release and synaptic function. Mol. Psychiatry 2018, 10, 1–25. [Google Scholar] [CrossRef]
- Sala, C.; Vicidomini, C.; Bigi, I.; Mossa, A.; Verpelli, C. Shank synaptic scaffold proteins: Keys to understanding the pathogenesis of autism and other synaptic disorders. J. Neurochem. 2015, 135, 849–858. [Google Scholar] [CrossRef]
- Yoo, J.; Bakes, J.; Bradley, C.; Collingridge, G.L.; Kaang, B.K. Shank mutant mice as an animal model of autism. Philos. Trans. R. Soc. B: Biol. Sci. 2014, 369, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Betancur, C.; Sakurai, T.; Buxbaum, J.D. The emerging role of synaptic cell-adhesion pathways in the pathogenesis of autism spectrum disorders. Trends Neurosci. 2009, 32, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Bozdagi, O.; Sakurai, T.; Papapetrou, D.; Wang, X.; Dickstein, D.L.; Takahashi, N.; Kajiwara, Y.; Yang, M.; Katz, A.M.; Scattoni, M.L.; et al. Haploinsufficiency of the autism-associated Shank3 gene leads to deficits in synaptic function, social interaction, and social communication. Mol. Autism 2010, 1, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Harris, K.P.; Akbergenova, Y.; Cho, R.W.; Baas-Thomas, M.S.; Littleton, J.T. Shank Modulates Postsynaptic Wnt Signaling to Regulate Synaptic Development. J. Neurosci. 2016, 36, 5820–5832. [Google Scholar] [CrossRef] [PubMed]
- Han, K.; Holder, J.L.; Schaaf, C.P.; Lu, H.; Chen, H.; Kang, H.; Tang, J.; Wu, Z.; Hao, S.; Cheung, S.W.; et al. SHANK3 overexpression causes manic-like behaviour with unique pharmacogenetic properties. Nature 2013, 503, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Arons, M.H.; Thynne, C.J.; Grabrucker, A.M.; Li, D.; Schoen, M.; Cheyne, J.E.; Boeckers, T.M.; Montgomery, J.M.; Garner, C.C. Autism-associated mutations in ProSAP2/Shank3 impair synaptic transmission and neurexin-neuroligin-mediated transsynaptic signaling. J. Neurosci. 2012, 32, 14966–14978. [Google Scholar] [CrossRef]
- Chmielewska, J.J.; Kuzniewska, B.; Milek, J.; Urbanska, K.; Dziembowska, M. Neuroligin 1, 2, and 3 Regulation at the Synapse: FMRP-Dependent Translation and Activity-Induced Proteolytic Cleavage. Mol. Neurobiol. 2019, 56, 2741–2759. [Google Scholar] [CrossRef]
- Tabuchi, K.; Blundell, J.; Etherton, M.R.; Hammer, R.E.; Liu, X.; Powell, C.M.; Südhof, T.C. A neuroligin-3 mutation implicated in autism increases inhibitory synaptic transmission in mice. Science 2007, 318, 71–76. [Google Scholar] [CrossRef]
- Kolozsi, E.; Mackenzie, R.N.; Roullet, F.I.; Decatanzaro, D.; Foster, J.A. Prenatal exposure to valproic acid leads to reduced expression of synaptic adhesion molecule neuroligin 3 in mice. Neuroscience 2009, 163, 1201–1210. [Google Scholar] [CrossRef]
- Rossignol, D.A.; Frye, R.E. Evidence linking oxidative stress, mitochondrial dysfunction, and inflammation in the brain of individuals with autism. Front. Physiol. 2014, 5, 1–15. [Google Scholar] [CrossRef]
- Chauhan, A.; Audhya, T.; Chauhan, V. Brain region-specific glutathione redox imbalance in autism. Neurochem. Res. 2012, 37, 1681–1689. [Google Scholar] [CrossRef] [PubMed]
- Frustaci, A.; Neri, M.; Cesario, A.; Adams, J.B.; Domenici, E.; Dalla Bernardina, B.; Bonassi, S. Oxidative stress-related biomarkers in autism: Systematic review and meta-analyses. Free Radic. Biol. Med. 2012, 52, 2128–2141. [Google Scholar] [CrossRef] [PubMed]
- Rose, S.; Melnyk, S.; Pavliv, O.; Bai, S.; Nick, T.G.; Frye, R.E.; James, S.J. Evidence of oxidative damage and inflammation associated with low glutathione redox status in the autism brain. Transl. Psychiatry 2012, 2, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxidative Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Kern, J.K.; Jones, A.M. Evidence of toxicity, oxidative stress, and neuronal insult in autism. J. Toxicol. Environ. Health-Part B: Crit. Rev. 2006, 9, 485–499. [Google Scholar] [CrossRef]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, 1–10. [Google Scholar] [CrossRef]
- Shih, R.H.; Wang, C.Y.; Yang, C.M. NF-kappaB Signaling Pathways in Neurological Inflammation: A Mini Review. Front. Mol. Neurosci. 2015, 8, 1–8. [Google Scholar] [CrossRef]
- Minghetti, L. Cyclooxygenase-2 (COX-2) in inflammatory and degenerative brain diseases. J. Neuropathol. Exp. Neurol. 2004, 63, 901–910. [Google Scholar] [CrossRef]
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M.K. Are “resting” microglia more “M2”? Front. Immunol. 2014, 5, 1–4. [Google Scholar] [CrossRef]
- Zhao, Q.; Xie, X.; Fan, Y.; Zhang, J.; Jiang, W.; Wu, X.; Yan, S.; Chen, Y.; Peng, C.; You, Z. Phenotypic dysregulation of microglial activation in young offspring rats with maternal sleep deprivation-induced cognitive impairment. Sci. Rep. 2015, 5, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kaur, C.; Rathnasamy, G.; Ling, E.A. Biology of microglia in the developing brain. J. Neuropathol. Exp. Neurol. 2017, 76, 736–753. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, J.; You, Z. Switching of the Microglial Activation Phenotype Is a Possible Treatment for Depression Disorder. Front. Cell. Neurosci. 2018, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sudduth, T.L.; Schmitt, F.A.; Nelson, P.T.; Wilcock, D.M. Neuroinflammatory phenotype in early Alzheimer’s disease. Neurobiol. Aging 2013, 34, 1051–1059. [Google Scholar] [CrossRef]
- Hopperton, K.E.; Mohammad, D.; Trépanier, M.O.; Giuliano, V.; Bazinet, R.P. Markers of microglia in post-mortem brain samples from patients with Alzheimer’s disease: A systematic review. Mol. Psychiatry 2018, 23, 177–198. [Google Scholar] [CrossRef]
- Petrelli, F.; Pucci, L.; Bezzi, P. Astrocytes and Microglia and Their Potential Link with Autism Spectrum Disorders. Front. Cell. Neurosci. 2016, 10, 1–8. [Google Scholar]
- Li, X.; Chauhan, A.; Sheikh, A.M.; Patil, S.; Chauhan, V.; Li, X.M.; Brown, T.; Malik, M. Elevated immune response in the brain of autistic patients. J. Neuroimmunol. 2009, 207, 1–13. [Google Scholar] [CrossRef]
- Wu, H.; Wang, X.; Gao, J.; Liang, S.; Hao, Y.; Sun, C.; Xia, W.; Cao, Y.; Wu, L. Fingolimod (FTY720) attenuates social deficits, learning and memory impairments, neuronal loss and neuroinflammation in the rat model of autism. Life Sci. 2017, 173, 43–54. [Google Scholar] [CrossRef]
- Hegazy, H.G.; Ali, E.H.; Elgoly, A.H. Interplay between pro-inflammatory cytokines and brain oxidative stress biomarkers: Evidence of parallels between butyl paraben intoxication and the valproic acid brain physiopathology in autism rat model. Cytokine 2015, 71, 173–180. [Google Scholar] [CrossRef]
- Uchida, K.; Nakamura, M.; Ozawa, H.; Katoh, S.; Toyama, Y. Neuroprotection and Regeneration of the Spinal Cord; Chapter: Roles of Microglia in Spinal Cord Injury; Springer: Tokyo, Japan, 2014; pp. 43–57. [Google Scholar]
- Parkhurst, C.N.; Yang, G.; Ninan, I.; Savas, J.N.; Yates, J.R.; Lafaille, J.J.; Hempstead, B.L.; Littman, D.R.; Gan, W.B. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 2013, 155, 1596–1609. [Google Scholar] [CrossRef]
- Moseley, R.L.; Hitchiner, R.; Kirkby, J.A. Self-reported sex differences in high-functioning adults with autism: A meta-analysis. Molecular autism 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Grove, R.; Hoekstra, R.A.; Wierda, M.; Begeer, S. Exploring sex differences in autistic traits: A factor analytic study of adults with autism. Autism: Int. J. Res. Pract. 2017, 21, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.C.; Kim, P.; Go, H.S.; Choi, C.S.; Park, J.H.; Kim, H.J.; Jeon, S.J.; Dela Pena, I.C.; Han, S.H.; Cheong, J.H.; et al. Male-specific alteration in excitatory post-synaptic development and social interaction in pre-natal valproic acid exposure model of autism spectrum disorder. J. Neurochem. 2013, 124, 832–843. [Google Scholar] [CrossRef] [PubMed]
- Mowery, T.M.; Wilson, S.M.; Kostylev, P.V.; Dina, B.; Buchholz, J.B.; Prieto, A.L.; Garraghty, P.E. Embryological exposure to valproic acid disrupts morphology of the deep cerebellar nuclei in a sexually dimorphic way. Int. J. Dev. Neurosci. 2015, 40, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Kazlauskas, N.; Seiffe, A.; Campolongo, M.; Zappala, C.; Depino, A.M. Sex-specific effects of prenatal valproic acid exposure on sociability and neuroinflammation: Relevance for susceptibility and resilience in autism. Psychoneuroendocrinology 2019, 110, 104441. [Google Scholar] [CrossRef]
- Milewski, K.; Hilgier, W.; Fręśko, I.; Polowy, R.; Podsiadłowska, A.; Zołocińska, E.; Grymanowska, A.W.; Filipkowski, R.K.; Albrecht, J.; Zielińska, M. Carnosine reduces oxidative stress and reverses attenuation of righting and postural reflexes in rats with thioacetamide- induced liver failure. Neurochem. Res. 2016, 41, 376–384. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary Antibody | Brand/Cat # | Dilution |
| Mouse anti-VAMP1/2 | Santa Cruz Biotechnology sc-20039 | 1:500 5% milk in TBS-T 0.1% |
| Mouse anti-Syp | Santa Cruz Biotechnology sc-55507 | 1:750 TBS-T 0.1% |
| Mouse anti-Syn1 | Santa Cruz Biotechnology sc-390867 | 1:500 TBS-T 0.1% |
| Rabbit anti-p-Syn1(Ser62/Ser67) | Santa Cruz Biotechnology sc-135709 | 1:500 TBS-T 0.1% |
| Rabbit anti-Syt1 | Cell Signalling #3347 | 1:1000 5% BSA in TBS-T 0.1% |
| Rabbit anti-SNAP25 | Cell Signalling #5309 | 1:1000 5% milk in TBS-T 0.1% |
| Mouse anti-Stx1 | Santa Cruz Biotechnology sc-12736 | 1:1000 5% milk in TBS-T 0.1% |
| Mouse anti-PSD95 | Santa Cruz Biotechnology sc-71935 | 1:1000 1% BSA in TBS-T 0.1% |
| Rabbit anti-Shank2 | Cell Signalling #12218S | 1:750 5% milk in TBS-T 0.1% |
| Mouse anti-Shank3 | Abcam ab93607 | 1:1000 5% milk in TBS-T 0.1% |
| Mouse anti-Nlgn1 | Santa Cruz Biotechnology sc-365110 | 1:200 5% milk in TBS-T 0.1% |
| Mouse anti-Nlgn3 | Abcam ab186307 | 1:500 5% milk in TBS-T 0.1% |
| Rabbit anti-Iba1 | Cell Signalling #17198 | 1:1000 TBS-T 0.1% |
| Rabbit anti-GAPDH | Sigma-Aldrich G9545-200UL | 1:50,000 5% milk in TBS-T 0.1% |
| Rabbit anti-vinculin | Cell Signalling #13901 | 1:1000 5% milk in TBS-T 0.1% |
| Secondary Antibody | Brand/Cat # | Dilution |
| anti-mouse IgG | GE Healthcare VXA931V | 1:4000 5% milk in TBS-T 0.1% |
| anti-rabbit IgG | Sigma-Aldrich A0545-1ML | 1:8000 5% milk in TBS-T 0.1% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gąssowska-Dobrowolska, M.; Cieślik, M.; Czapski, G.A.; Jęśko, H.; Frontczak-Baniewicz, M.; Gewartowska, M.; Dominiak, A.; Polowy, R.; Filipkowski, R.K.; Babiec, L.; et al. Prenatal Exposure to Valproic Acid Affects Microglia and Synaptic Ultrastructure in a Brain-Region-Specific Manner in Young-Adult Male Rats: Relevance to Autism Spectrum Disorders. Int. J. Mol. Sci. 2020, 21, 3576. https://doi.org/10.3390/ijms21103576
Gąssowska-Dobrowolska M, Cieślik M, Czapski GA, Jęśko H, Frontczak-Baniewicz M, Gewartowska M, Dominiak A, Polowy R, Filipkowski RK, Babiec L, et al. Prenatal Exposure to Valproic Acid Affects Microglia and Synaptic Ultrastructure in a Brain-Region-Specific Manner in Young-Adult Male Rats: Relevance to Autism Spectrum Disorders. International Journal of Molecular Sciences. 2020; 21(10):3576. https://doi.org/10.3390/ijms21103576
Chicago/Turabian StyleGąssowska-Dobrowolska, Magdalena, Magdalena Cieślik, Grzegorz Arkadiusz Czapski, Henryk Jęśko, Małgorzata Frontczak-Baniewicz, Magdalena Gewartowska, Agnieszka Dominiak, Rafał Polowy, Robert Kuba Filipkowski, Lidia Babiec, and et al. 2020. "Prenatal Exposure to Valproic Acid Affects Microglia and Synaptic Ultrastructure in a Brain-Region-Specific Manner in Young-Adult Male Rats: Relevance to Autism Spectrum Disorders" International Journal of Molecular Sciences 21, no. 10: 3576. https://doi.org/10.3390/ijms21103576
APA StyleGąssowska-Dobrowolska, M., Cieślik, M., Czapski, G. A., Jęśko, H., Frontczak-Baniewicz, M., Gewartowska, M., Dominiak, A., Polowy, R., Filipkowski, R. K., Babiec, L., & Adamczyk, A. (2020). Prenatal Exposure to Valproic Acid Affects Microglia and Synaptic Ultrastructure in a Brain-Region-Specific Manner in Young-Adult Male Rats: Relevance to Autism Spectrum Disorders. International Journal of Molecular Sciences, 21(10), 3576. https://doi.org/10.3390/ijms21103576