Oncogenic Potential of Bisphenol A and Common Environmental Contaminants in Human Mammary Epithelial Cells

 , and
, and
Abstract
1. Introduction
2. Results
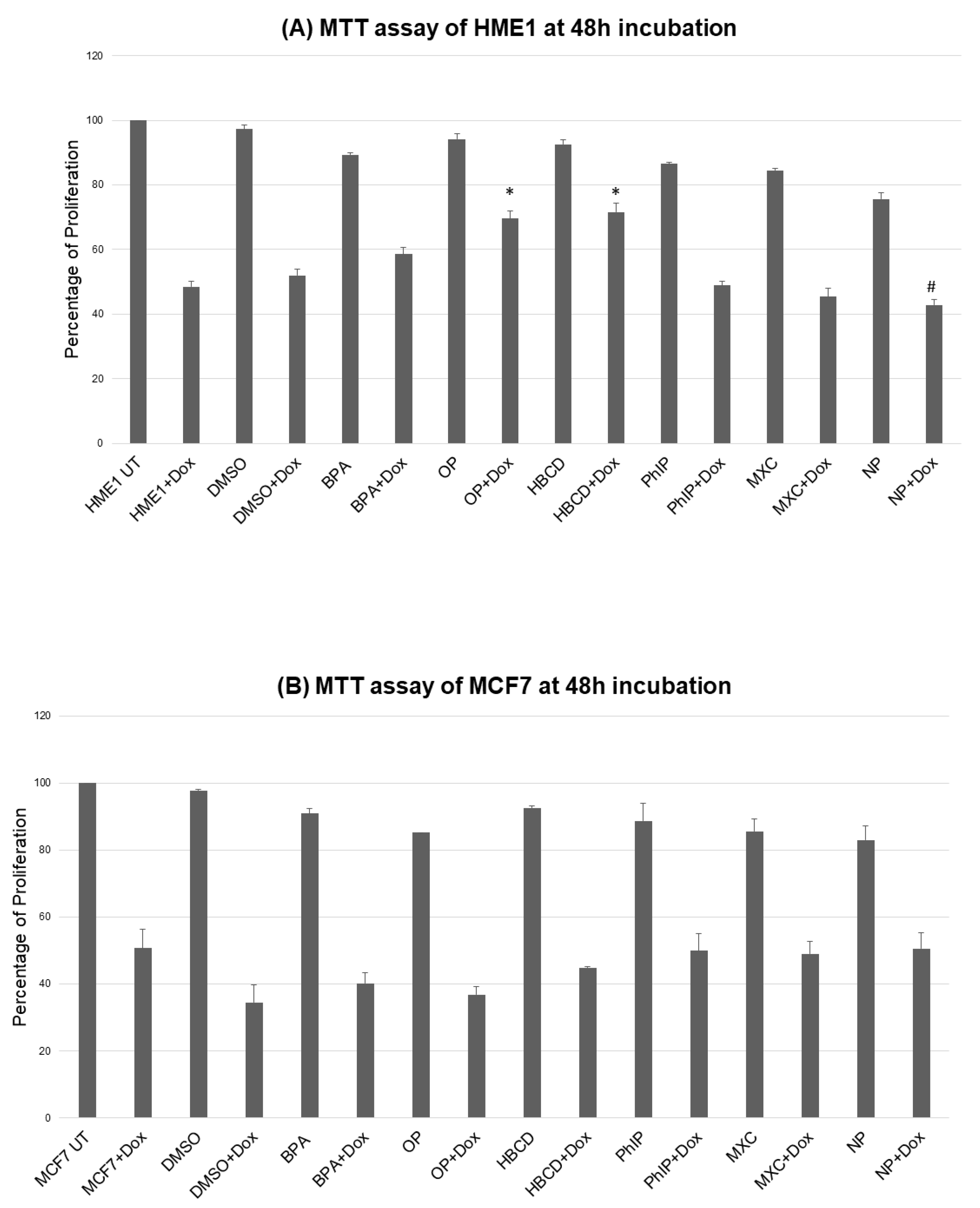
2.1. Environmental Contaminants and Cell Viability
2.2. DNA Damage by Environmental Contaminants
2.3. Disruption of the Cell Cycle by Environmental Contaminants
2.4. Methylation Changes in Tumor Suppressor Genes and LINE1
2.5. Environmental Contaminants and Collagen Invasion
2.6. Colony Formation in Soft Agar
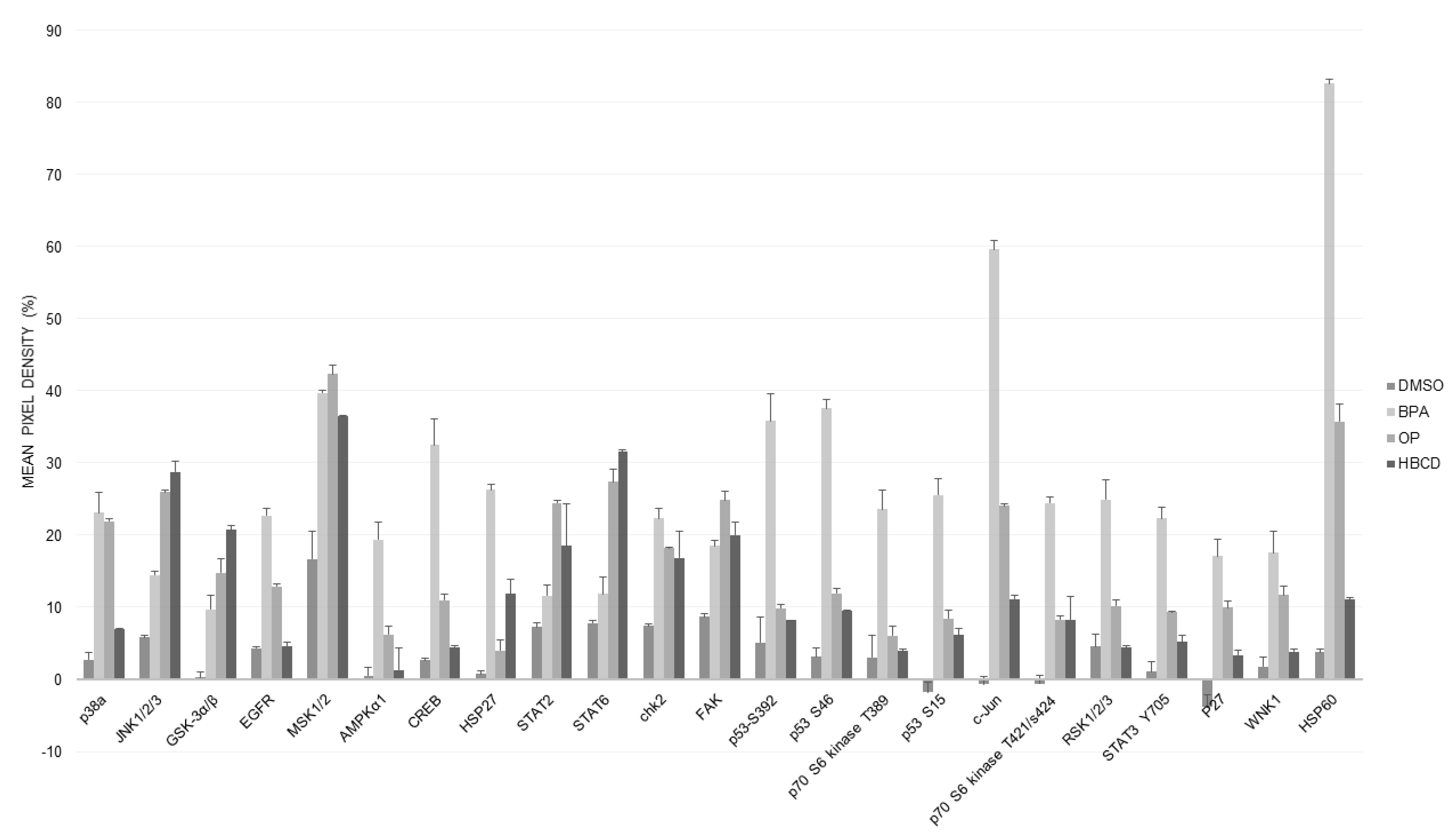
2.7. Proteomic Analysis (Human Phospho Kinase Array)
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Toxic Chemicals and Viability Assays
4.3. Detection of DNA Damage Response by Western Blot Analysis
4.4. Analysis of DNA Content and Cell Cycle Progression
4.5. Methylation Specific Multiplex-Ligation Dependent Probe Amplification (MS-MLPA)
4.6. Invasion Assay
4.7. Colony Formation in Soft Agar
4.8. Proteome Profile/Human Phospho-Kinase Array
4.9. Data Analysis:
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Azubuike, S.O.; Muirhead, C.; Hayes, L.; McNally, R. Rising global burden of breast cancer: The case of sub-Saharan Africa (with emphasis on Nigeria) and implications for regional development: A review. World J. Surg. Oncol. 2018, 16, 63. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Franco, M.M.; Leon-Rodriguez, E. Delays in Breast Cancer Detection and Treatment in Developing Countries. Breast Cancer (Auckl) 2018, 12. [Google Scholar] [CrossRef] [PubMed]
- Dey, S.; Soliman, A.S.; Hablas, A.; Seifeldein, I.A.; Ismail, K.; Ramadan, M.; El-Hamzawy, H.; Wilson, M.L.; Banerjee, M.; Boffetta, P.; et al. Urban-rural differences in breast cancer incidence in Egypt (1999–2006). Breast 2010, 19, 417–423. [Google Scholar] [CrossRef] [PubMed]
- McDonald, J.T.; Farnworth, M.; Liu, Z. Cancer and the healthy immigrant effect: A statistical analysis of cancer diagnosis using a linked Census-cancer registry administrative database. BMC Public Health 2017, 17, 296. [Google Scholar] [CrossRef] [PubMed]
- Peto, J. Cancer epidemiology in the last century and the next decade. Nature 2001, 411, 390–395. [Google Scholar] [CrossRef]
- Abdel-Rahman, W.M.; Moustafa, Y.M.; Ahmed, B.O.; Mostafa, R.M. Endocrine disruptors and breast cancer risk–time to consider the environment. Asian Pac. J. Cancer Prev. 2012, 13, 5937–5946. [Google Scholar] [CrossRef]
- Ho, V.; Brunetti, V.; Peacock, S.; Massey, T.E.; Godschalk, R.W.L.; van Schooten, F.J.; Ashbury, J.E.; Vanner, S.J.; King, W.D. Exposure to meat-derived carcinogens and bulky DNA adduct levels in normal-appearing colon mucosa. Mutat. Res. 2017, 821, 5–12. [Google Scholar] [CrossRef]
- Chiavarini, M.; Bertarelli, G.; Minelli, L.; Fabiani, R. Dietary Intake of Meat Cooking-Related Mutagens (HCAs) and Risk of Colorectal Adenoma and Cancer: A Systematic Review and Meta-Analysis. Nutrients 2017, 9, 514. [Google Scholar] [CrossRef]
- Chen, J.X.; Wang, H.; Liu, A.; Zhang, L.; Reuhl, K.; Yang, C.S. From the Cover: PhIP/DSS-Induced Colon Carcinogenesis in CYP1A-Humanized Mice and the Possible Role of Lgr5+ Stem Cells. Toxicol. Sci. 2017, 155, 224–233. [Google Scholar] [CrossRef]
- Gould, J.C.; Leonard, L.S.; Maness, S.C.; Wagner, B.L.; Conner, K.; Zacharewski, T.; Safe, S.; McDonnell, D.P.; Gaido, K.W. Bisphenol A interacts with the estrogen receptor alpha in a distinct manner from estradiol. Mol. Cell. Endocrinol. 1998, 142, 203–214. [Google Scholar] [CrossRef]
- Kim, H.S.; Han, S.Y.; Yoo, S.D.; Lee, B.M.; Park, K.L. Potential estrogenic effects of bisphenol-A estimated by in vitro and in vivo combination assays. J. Toxicol. Sci. 2001, 26, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Terasaka, S.; Kiyama, R. Bisphenol A induces a rapid activation of Erk1/2 through GPR30 in human breast cancer cells. Environ. Pollut. 2011, 159, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Quesada, I.; Fuentes, E.; Viso-Leon, M.C.; Soria, B.; Ripoll, C.; Nadal, A. Low doses of the endocrine disruptor bisphenol-A and the native hormone 17beta-estradiol rapidly activate transcription factor CREB. FASEB J. 2002, 16, 1671–1673. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, H.; Liu, S. Low-Dose Bisphenol A Exposure: A Seemingly Instigating Carcinogenic Effect on Breast Cancer. Adv. Sci. (Weinh) 2016, 4, 1600248. [Google Scholar] [CrossRef]
- Hafezi, S.A.; Abdel-Rahman, W.M. The Endocrine Disruptor Bisphenol A (BPA) Exerts a Wide Range of Effects in Carcinogenesis and Response to Therapy. Curr. Mol. Pharm. 2019, 12, 230–238. [Google Scholar] [CrossRef]
- Kim, H.; Kim, H.S.; Moon, W.K. Comparison of transcriptome expression alterations by chronic exposure to low-dose bisphenol A in different subtypes of breast cancer cells. Toxicol. Appl. Pharmacol. 2019, 385, 114814. [Google Scholar] [CrossRef]
- Lloyd, V.; Morse, M.; Purakal, B.; Parker, J.; Benard, P.; Crone, M.; Pfiffner, S.; Szmyd, M.; Dinda, S. Hormone-Like Effects of Bisphenol A on p53 and Estrogen Receptor Alpha in Breast Cancer Cells. BioRes. Open Access 2019, 8, 169–184. [Google Scholar] [CrossRef]
- LaPensee, E.W.; LaPensee, C.R.; Fox, S.; Schwemberger, S.; Afton, S.; Ben-Jonathan, N. Bisphenol A and estradiol are equipotent in antagonizing cisplatin-induced cytotoxicity in breast cancer cells. Cancer Lett. 2010, 290, 167–173. [Google Scholar] [CrossRef]
- Lapensee, E.W.; Tuttle, T.R.; Fox, S.R.; Ben-Jonathan, N. Bisphenol A at low nanomolar doses confers chemoresistance in estrogen receptor-alpha-positive and -negative breast cancer cells. Environ. Health Perspect. 2009, 117, 175–180. [Google Scholar] [CrossRef]
- Chamard-Jovenin, C.; Thiebaut, C.; Chesnel, A.; Bresso, E.; Morel, C.; Smail-Tabbone, M.; Devignes, M.D.; Boukhobza, T.; Dumond, H. Low-Dose Alkylphenol Exposure Promotes Mammary Epithelium Alterations and Transgenerational Developmental Defects, But Does Not Enhance Tumorigenic Behavior of Breast Cancer Cells. Front. Endocrinol. (Lausanne) 2017, 8, 272. [Google Scholar] [CrossRef] [PubMed]
- Ajj, H.; Chesnel, A.; Pinel, S.; Plenat, F.; Flament, S.; Dumond, H. An alkylphenol mix promotes seminoma derived cell proliferation through an ERalpha36-mediated mechanism. PLoS ONE 2013, 8, e61758. [Google Scholar] [CrossRef] [PubMed]
- Park, M.A.; Hwang, K.A.; Lee, H.R.; Yi, B.R.; Choi, K.C. Cell Growth of BG-1 Ovarian Cancer Cells was Promoted by 4-Tert-octylphenol and 4-Nonylphenol via Downregulation of TGF-beta Receptor 2 and Upregulation of c-myc. Toxicol. Res. 2011, 27, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Yi, B.R.; Go, R.E.; Hwang, K.A.; Nam, K.H.; Choi, K.C. Methoxychlor and triclosan stimulates ovarian cancer growth by regulating cell cycle- and apoptosis-related genes via an estrogen receptor-dependent pathway. Environ. Toxicol. Pharm. 2014, 37, 1264–1274. [Google Scholar] [CrossRef]
- Eldakroory, S.A.; Morsi, D.E.; Abdel-Rahman, R.H.; Roshdy, S.; Gouida, M.S.; Khashaba, E.O. Correlation between toxic organochlorine pesticides and breast cancer. Hum. Exp. Toxicol. 2017, 36, 1326–1334. [Google Scholar] [CrossRef]
- Li, R.J.; Gao, H.; Na, G.S.; Lu, Z.H.; Yao, Y.; Yang, F. Hexabromocyclododecane-induced Genotoxicity in Cultured Human Breast Cells through DNA Damage. Biomed. Environ. Sci. 2017, 30, 296–300. [Google Scholar]
- Kim, S.H.; Nam, K.H.; Hwang, K.A.; Choi, K.C. Influence of hexabromocyclododecane and 4-nonylphenol on the regulation of cell growth, apoptosis and migration in prostatic cancer cells. Toxicol. In Vitro 2016, 32, 240–247. [Google Scholar] [CrossRef]
- Park, M.A.; Hwang, K.A.; Lee, H.R.; Yi, B.R.; Jeung, E.B.; Choi, K.C. Cell growth of BG-1 ovarian cancer cells is promoted by di-n-butyl phthalate and hexabromocyclododecane via upregulation of the cyclin D and cyclin-dependent kinase-4 genes. Mol. Med. Rep. 2012, 5, 761–766. [Google Scholar]
- An, J.; Wang, X.; Guo, P.; Zhong, Y.; Zhang, X.; Yu, Z. Hexabromocyclododecane and polychlorinated biphenyls increase resistance of hepatocellular carcinoma cells to cisplatin through the phosphatidylinositol 3-kinase/protein kinase B pathway. Toxicol. Lett. 2014, 229, 265–272. [Google Scholar] [CrossRef]
- Choudhary, S.; Sood, S.; Donnell, R.L.; Wang, H.C. Intervention of human breast cell carcinogenesis chronically induced by 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine. Carcinogenesis 2012, 33, 876–885. [Google Scholar] [CrossRef]
- Papaioannou, M.D.; Koufaris, C.; Gooderham, N.J. The cooked meat-derived mammary carcinogen 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) elicits estrogenic-like microRNA responses in breast cancer cells. Toxicol. Lett. 2014, 229, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Gu, D.; Turesky, R.J.; Tao, Y.; Langouet, S.A.; Nauwelaers, G.C.; Yuan, J.M.; Yee, D.; Yu, M.C. DNA adducts of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine and 4-aminobiphenyl are infrequently detected in human mammary tissue by liquid chromatography/tandem mass spectrometry. Carcinogenesis 2012, 33, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Mlynarcikova, A.; Macho, L.; Fickova, M. Bisphenol A alone or in combination with estradiol modulates cell cycle- and apoptosis-related proteins and genes in MCF7 cells. Endocr. Regul. 2013, 47, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Delgado, M.; Ribeiro-Varandas, E. Bisphenol A at the reference level counteracts doxorubicin transcriptional effects on cancer related genes in HT29 cells. Toxicol. In Vitro 2015, 29, 2009–2014. [Google Scholar] [CrossRef]
- Dorosh, A.; Ded, L.; Elzeinova, F.; Peknicova, J. Assessing oestrogenic effects of brominated flame retardants hexabromocyclododecane and tetrabromobisphenol A on MCF-7 cells. Folia Biol. (Praha) 2011, 57, 35–39. [Google Scholar]
- Abdel-Rahman, W.M. Genomic instability and carcinogenesis: An update. Curr. Genom. 2008, 9, 535–541. [Google Scholar] [CrossRef]
- Bromer, J.G.; Zhou, Y.; Taylor, M.B.; Doherty, L.; Taylor, H.S. Bisphenol-A exposure in utero leads to epigenetic alterations in the developmental programming of uterine estrogen response. FASEB J. 2010, 24, 2273–2280. [Google Scholar] [CrossRef]
- Laing, L.V.; Viana, J.; Dempster, E.L.; Trznadel, M.; Trunkfield, L.A.; Uren Webster, T.M.; van Aerle, R.; Paull, G.C.; Wilson, R.J.; Mill, J.; et al. Bisphenol A causes reproductive toxicity, decreases dnmt1 transcription, and reduces global DNA methylation in breeding zebrafish (Danio rerio). Epigenetics 2016, 11, 526–538. [Google Scholar] [CrossRef]
- Fernandez, S.V.; Huang, Y.; Snider, K.E.; Zhou, Y.; Pogash, T.J.; Russo, J. Expression and DNA methylation changes in human breast epithelial cells after bisphenol A exposure. Int. J. Oncol. 2012, 41, 369–377. [Google Scholar] [CrossRef]
- Lotsari, J.E.; Gylling, A.; Abdel-Rahman, W.M.; Nieminen, T.T.; Aittomaki, K.; Friman, M.; Pitkanen, R.; Aarnio, M.; Jarvinen, H.J.; Mecklin, J.P.; et al. Breast carcinoma and Lynch syndrome: Molecular analysis of tumors arising in mutation carriers, non-carriers, and sporadic cases. Breast Cancer Res. 2012, 14, R90. [Google Scholar] [CrossRef]
- Goodson, W.H., 3rd; Luciani, M.G.; Sayeed, S.A.; Jaffee, I.M.; Moore, D.H., 2nd; Dairkee, S.H. Activation of the mTOR pathway by low levels of xenoestrogens in breast epithelial cells from high-risk women. Carcinogenesis 2011, 32, 1724–1733. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Yang, B.J.; Li, N.; Feng, L.M.; Shi, X.Y.; Zhao, W.H.; Liu, S.J. Bisphenol A and hormone-associated cancers: Current progress and perspectives. Medicine 2015, 94, e211. [Google Scholar] [CrossRef] [PubMed]
- Atlas, E.; Dimitrova, V. Bisphenol S and Bisphenol A disrupt morphogenesis of MCF-12A human mammary epithelial cells. Sci. Rep. 2019, 9, 16005. [Google Scholar] [CrossRef] [PubMed]
- Mao, W.; Song, Y.; Sui, H.; Cao, P.; Liu, Z. Analysis of individual and combined estrogenic effects of bisphenol, nonylphenol and diethylstilbestrol in immature rats with mathematical models. Environ. Health Prev. Med. 2019, 24, 32. [Google Scholar] [CrossRef] [PubMed]
- Dairkee, S.H.; Seok, J.; Champion, S.; Sayeed, A.; Mindrinos, M.; Xiao, W.; Davis, R.W.; Goodson, W.H. Bisphenol A induces a profile of tumor aggressiveness in high-risk cells from breast cancer patients. Cancer Res. 2008, 68, 2076–2080. [Google Scholar] [CrossRef] [PubMed]
- Duncan, E.A.; Anest, V.; Cogswell, P.; Baldwin, A.S. The kinases MSK1 and MSK2 are required for epidermal growth factor-induced, but not tumor necrosis factor-induced, histone H3 Ser10 phosphorylation. J. Biol. Chem. 2006, 281, 12521–12525. [Google Scholar] [CrossRef] [PubMed]
- Menard, S.; Casalini, P.; Campiglio, M.; Pupa, S.M.; Tagliabue, E. Role of HER2/neu in tumor progression and therapy. Cell. Mol. Life Sci. 2004, 61, 2965–2978. [Google Scholar]
- Kyriakopoulou, K.; Kefali, E.; Piperigkou, Z.; Bassiony, H.; Karamanos, N.K. Advances in targeting epidermal growth factor receptor signaling pathway in mammary cancer. Cell. Signal. 2018, 51, 99–109. [Google Scholar] [CrossRef]
- Voudouri, K.; Nikitovic, D.; Berdiaki, A.; Kletsas, D.; Karamanos, N.K.; Tzanakakis, G.N. IGF-I/EGF and E2 signaling crosstalk through IGF-IR conduit point affects breast cancer cell adhesion. Matrix Biol. 2016, 56, 95–113. [Google Scholar] [CrossRef]
- Gao, S.; Ge, A.; Xu, S.; You, Z.; Ning, S.; Zhao, Y.; Pang, D. PSAT1 is regulated by ATF4 and enhances cell proliferation via the GSK3beta/beta-catenin/cyclin D1 signaling pathway in ER-negative breast cancer. J. Exp. Clin. Cancer Res. 2017, 36, 179. [Google Scholar] [CrossRef]
- Song, N.; Zhong, J.; Hu, Q.; Gu, T.; Yang, B.; Zhang, J.; Yu, J.; Ma, X.; Chen, Q.; Qi, J.; et al. FGF18 Enhances Migration and the Epithelial-Mesenchymal Transition in Breast Cancer by Regulating Akt/GSK3beta/Beta-Catenin Signaling. Cell. Physiol. Biochem. 2018, 49, 1019–1032. [Google Scholar] [CrossRef] [PubMed]
- Halacli, S.O.; Dogan, A.L. FOXP1 regulation via the PI3K/Akt/p70S6K signaling pathway in breast cancer cells. Oncol. Lett. 2015, 9, 1482–1488. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.D.; Chen, L.; Zhang, M.; Qi, H.J.; Chen, L.; Chen, H.F.; Zhong, M.K.; Shi, X.J.; Li, Q.Y. Downregulation of ERRalpha inhibits angiogenesis in human umbilical vein endothelial cells through regulating VEGF production and PI3K/Akt/STAT3 signaling pathway. Eur. J. Pharm. 2015, 769, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Houles, T.; Roux, P.P. Defining the role of the RSK isoforms in cancer. Semin. Cancer Biol. 2018, 48, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Crawley, S.; Hokari, R.; Kwon, S.; Kim, Y.S. Bile acid regulates MUC2 transcription in colon cancer cells via positive EGFR/PKC/Ras/ERK/CREB, PI3K/Akt/IkappaB/NF-kappaB and p38/MSK1/CREB pathways and negative JNK/c-Jun/AP-1 pathway. Int. J. Oncol. 2010, 36, 941–953. [Google Scholar]
- Calderwood, S.K. Heat shock proteins and cancer: Intracellular chaperones or extracellular signalling ligands? Philos. Trans. R. Soc. B Biol. Sci. 2018, 373. [Google Scholar] [CrossRef]
- Rappa, F.; Pitruzzella, A.; Marino Gammazza, A.; Barone, R.; Mocciaro, E.; Tomasello, G.; Carini, F.; Farina, F.; Zummo, G.; Conway de Macario, E.; et al. Quantitative patterns of Hsps in tubular adenoma compared with normal and tumor tissues reveal the value of Hsp10 and Hsp60 in early diagnosis of large bowel cancer. Cell Stress Chaperones 2016, 21, 927–933. [Google Scholar] [CrossRef]
- Zhang, L.; Koivisto, L.; Heino, J.; Uitto, V.J. Bacterial heat shock protein 60 may increase epithelial cell migration through activation of MAP kinases and inhibition of alpha6beta4 integrin expression. Biochem. Biophys. Res. Commun. 2004, 319, 1088–1095. [Google Scholar] [CrossRef]
- Zhang, L.; Pelech, S.L.; Mayrand, D.; Grenier, D.; Heino, J.; Uitto, V.J. Bacterial heat shock protein-60 increases epithelial cell proliferation through the ERK1/2 MAP kinases. Exp. Cell Res. 2001, 266, 11–20. [Google Scholar] [CrossRef]
- Kamath, A.; Joseph, A.M.; Gupta, K.; Behera, D.; Jaiswal, A.; Dewan, R.; Rajala, M.S. Proteomic analysis of HEK293 cells expressing non small cell lung carcinoma associated epidermal growth factor receptor variants reveals induction of heat shock response. Exp. Hematol. Oncol. 2015, 4, 16. [Google Scholar] [CrossRef]
- Tsai, Y.P.; Yang, M.H.; Huang, C.H.; Chang, S.Y.; Chen, P.M.; Liu, C.J.; Teng, S.C.; Wu, K.J. Interaction between HSP60 and beta-catenin promotes metastasis. Carcinogenesis 2009, 30, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Chow, M.T.; Moller, A.; Smyth, M.J. Inflammation and immune surveillance in cancer. Semin. Cancer Biol. 2012, 22, 23–32. [Google Scholar] [CrossRef]
- Saini, J.; Sharma, P.K. Clinical, Prognostic and Therapeutic Significance of Heat Shock Proteins in Cancer. Curr. Drug Targets 2018, 19, 1478–1490. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.E.; Lemieux, G.A.; Hassis, M.E.; Olshen, A.B.; Fisher, S.J.; Werb, Z. Quantitative proteomic analyses of mammary organoids reveals distinct signatures after exposure to environmental chemicals. Proc. Natl. Acad. Sci. USA 2016, 113, E1343–E1351. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Jin, X.; Zhao, N.; Ye, X.; Ying, C. Bisphenol A promotes X-linked inhibitor of apoptosis protein-dependent angiogenesis via G protein-coupled estrogen receptor pathway. J. Appl. Toxicol. 2015, 35, 1309–1317. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Rahman, W.M.; Al-Khayyal, N.A.; Nair, V.A.; Aravind, S.R.; Saber-Ayad, M. Role of AXL in invasion and drug resistance of colon and breast cancer cells and its association with p53 alterations. World J. Gastroenterol. 2017, 23, 3440–3448. [Google Scholar] [CrossRef] [PubMed]
- Joensuu, E.I.; Abdel-Rahman, W.M.; Ollikainen, M.; Ruosaari, S.; Knuutila, S.; Peltomaki, P. Epigenetic signatures of familial cancer are characteristic of tumor type and family category. Cancer Res. 2008, 68, 4597–4605. [Google Scholar] [CrossRef]
- Pavicic, W.; Joensuu, E.I.; Nieminen, T.; Peltomaki, P. LINE-1 hypomethylation in familial and sporadic cancer. J. Mol. Med. (Berl. Ger.) 2012, 90, 827–835. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| p-Chk1 | p-Chk2 | p-Histone H2A.X | p-p53 | Total Positive Markers | |||||
|---|---|---|---|---|---|---|---|---|---|
| HME1 | MCF7 | HME1 | MCF7 | HME1 | MCF7 | HME1 | MCF7 | ||
| BPA | ++ | ++ | ++ | ++ | ++ | ++ | ++ | ++ | 8 |
| MXC | - | - | - | + | + | - | - | + | 3 |
| HBCD | + | + | + | ++ | ++ | - | + | + | 7 |
| NP | - | ++ | - | ++ | + | - | - | ++ | 4 |
| OP | + | ++ | ++ | ++ | ++ | + | + | + | 8 |
| PhIP | + | ++ | ++ | ++ | ++ | - | + | + | 7 |
| Total | 9 | 10 | 8 | 10 | |||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nair, V.A.; Valo, S.; Peltomäki, P.; Bajbouj, K.; Abdel-Rahman, W.M. Oncogenic Potential of Bisphenol A and Common Environmental Contaminants in Human Mammary Epithelial Cells. Int. J. Mol. Sci. 2020, 21, 3735. https://doi.org/10.3390/ijms21103735
Nair VA, Valo S, Peltomäki P, Bajbouj K, Abdel-Rahman WM. Oncogenic Potential of Bisphenol A and Common Environmental Contaminants in Human Mammary Epithelial Cells. International Journal of Molecular Sciences. 2020; 21(10):3735. https://doi.org/10.3390/ijms21103735
Chicago/Turabian StyleNair, Vidhya A, Satu Valo, Päivi Peltomäki, Khuloud Bajbouj, and Wael M. Abdel-Rahman. 2020. "Oncogenic Potential of Bisphenol A and Common Environmental Contaminants in Human Mammary Epithelial Cells" International Journal of Molecular Sciences 21, no. 10: 3735. https://doi.org/10.3390/ijms21103735
APA StyleNair, V. A., Valo, S., Peltomäki, P., Bajbouj, K., & Abdel-Rahman, W. M. (2020). Oncogenic Potential of Bisphenol A and Common Environmental Contaminants in Human Mammary Epithelial Cells. International Journal of Molecular Sciences, 21(10), 3735. https://doi.org/10.3390/ijms21103735