Mitochondrial Ceramide Effects on the Retinal Pigment Epithelium in Diabetes

 , , , and
, , , and
Abstract
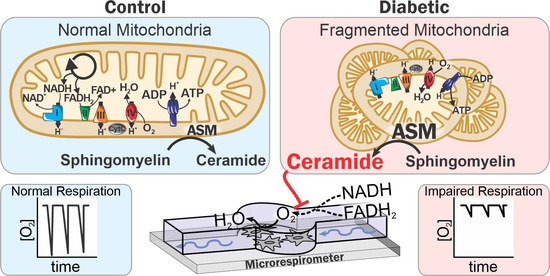
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
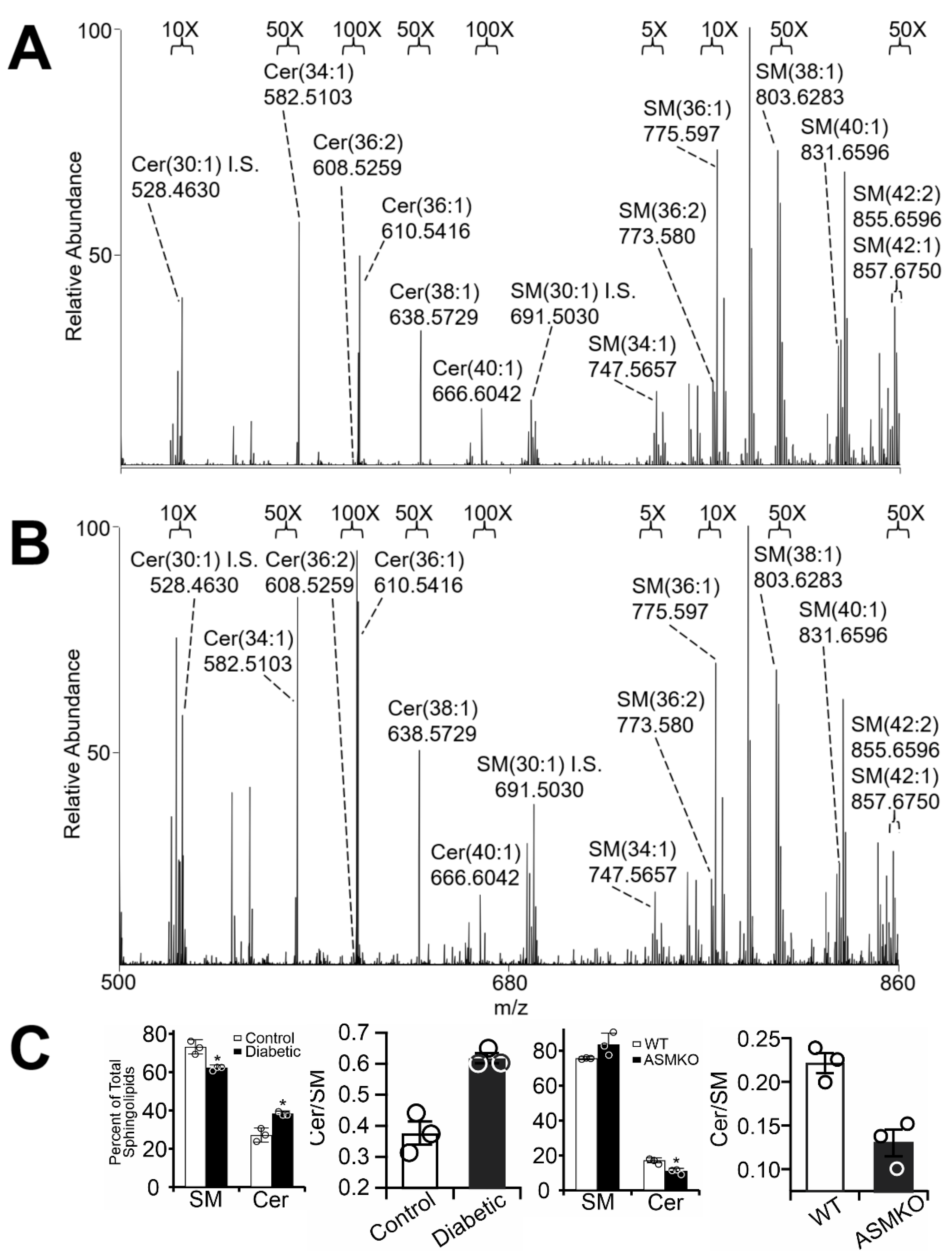
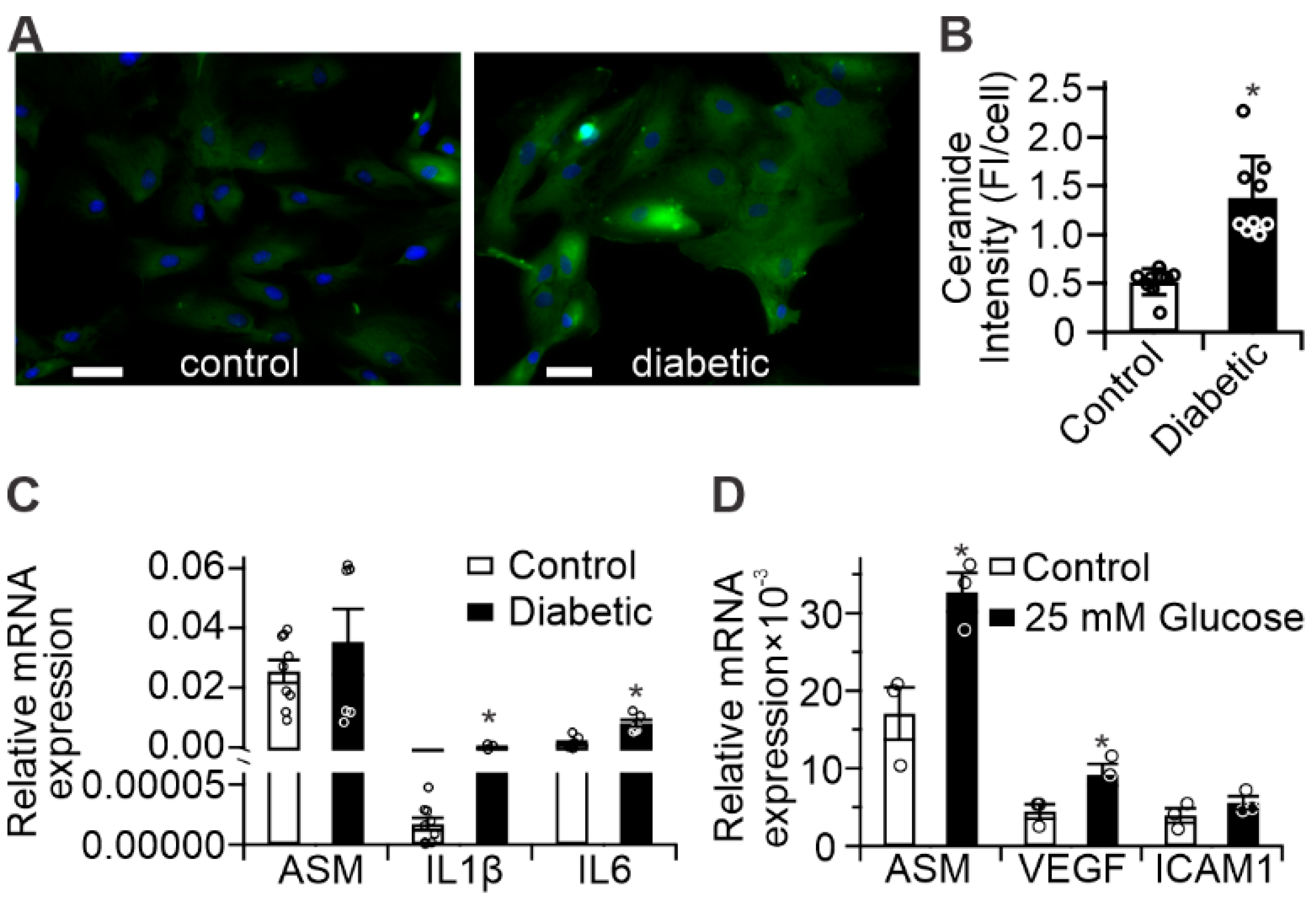
2.1. Diabetes Results in Retinal Mitochondrial Ceramide Accumulation
2.2. Diabetes Results in Pro-Inflammatory Changes in Human Retinal Pigment Epithelium (RPE) Cells
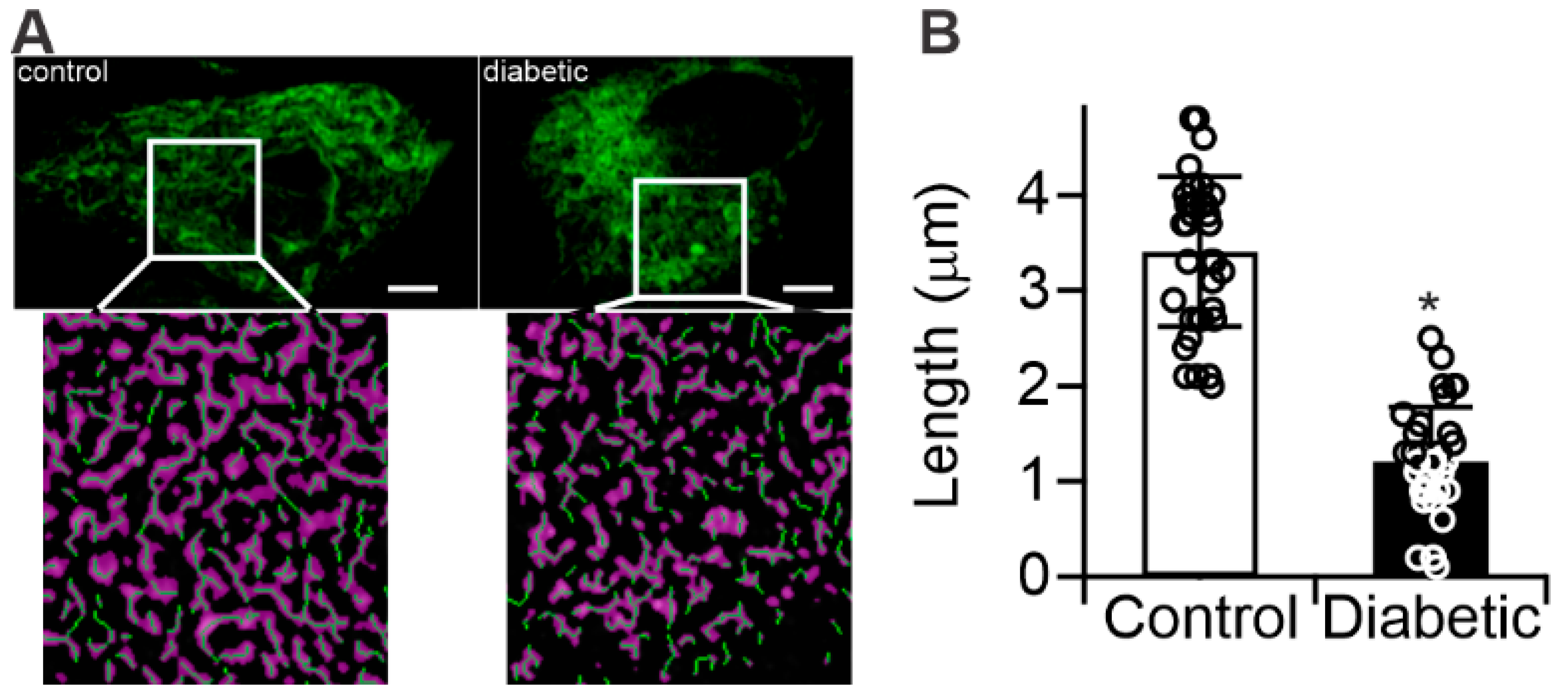
2.3. Diabetes Results in Mitochondrial Fragmentation in Human RPE Cells
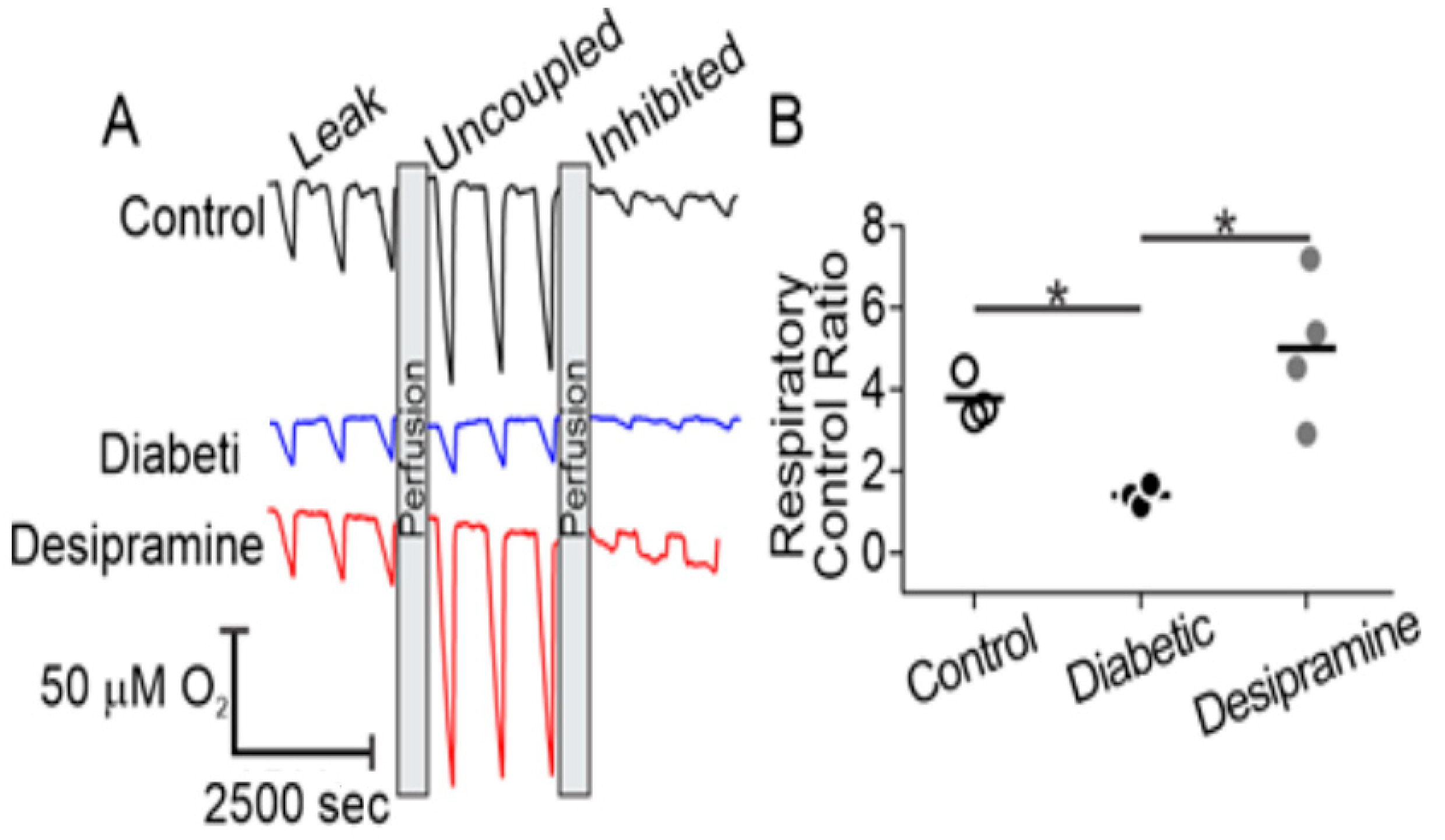
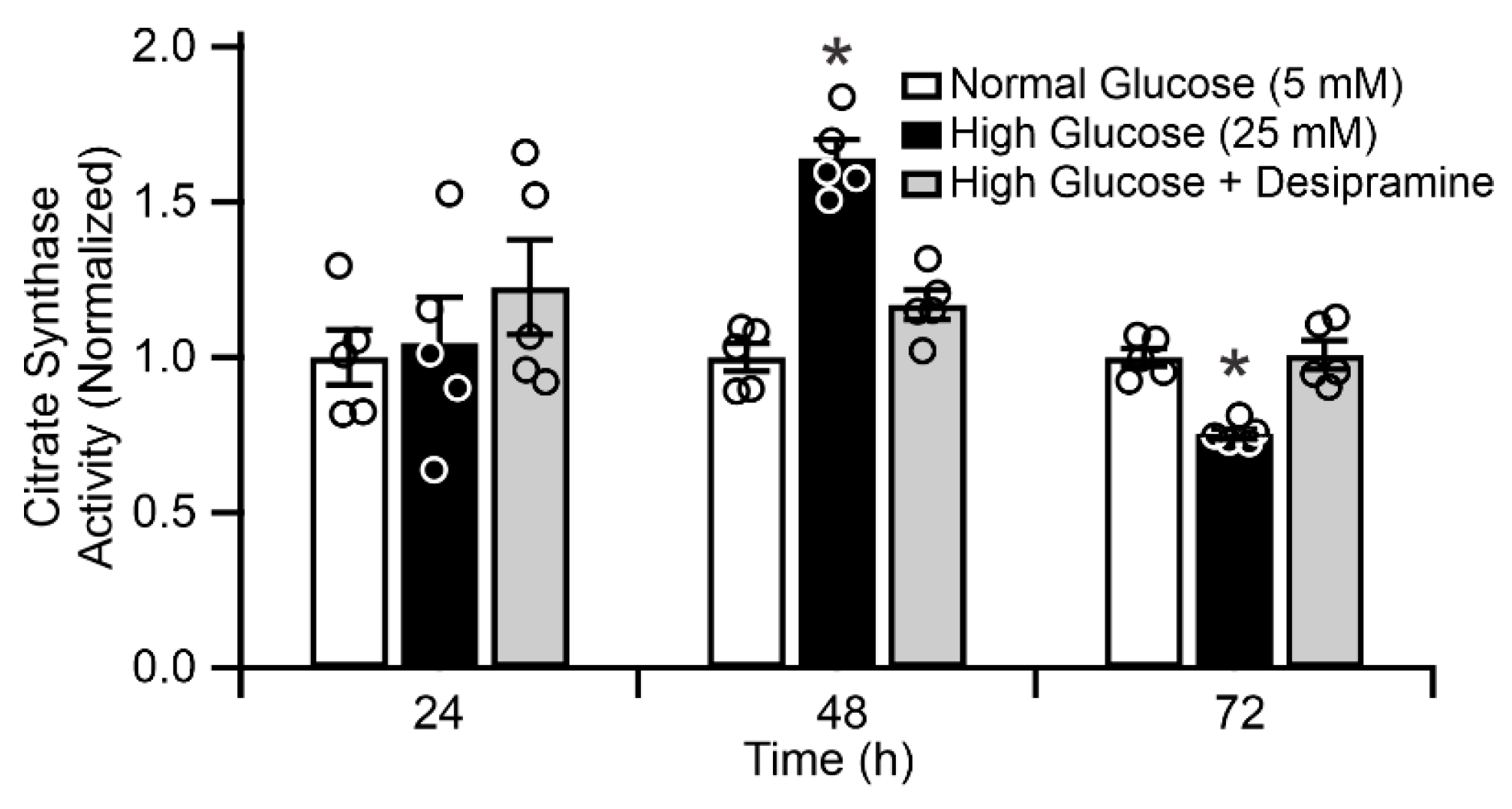
2.4. Diabetes Induces Acid Sphingomyelinase (ASM)-Mediated Changes in Mitochondrial Function of Human RPE Cells
2.5. Mitochondrial ASM Contributes to Impaired Mitochondrial Function In Vitro
3. Discussion
4. Materials and Methods
4.1. Rodents
4.2. Cell Culture
4.3. Mitochondrial Isolation
4.4. Mass Spectrometry
4.5. Immunocytochemistry and Mitochondrial Morphology
4.6. Quantitative Real-Time Polymerase Chain Reaction
4.7. Western Blot Analysis
4.8. Citrate Synthase Activity
4.9. Microrespirometry
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CCCP | Carbonyl cyanide m-chlorophenylhydrazone |
| DR | Diabetic retinopathy |
| ICAM-1 | Intercellular adhesion molecule 1 |
| VDAC | Voltage dependent anion channel |
| LAMP-1 | Lysosome associated membrane protein 1 |
| ASM | Acid sphingomyelinase |
| RCR | Respiratory control ratio |
| RPE | Retinal pigment epithelium |
| ROS | Reactive oxygen species |
| STZ | Streptozotocin |
| VEGF | Vascular endothelial growth factor |
References
- Du, Y.; Miller, C.M.; Kern, T.S. Hyperglycemia increases mitochondrial superoxide in retina and retinal cells. Free Radic. Biol. Med. 2003, 35, 1491–1499. [Google Scholar] [CrossRef] [PubMed]
- Trudeau, K.; Molina, A.J.A.; Guo, W.; Roy, S. High Glucose Disrupts Mitochondrial Morphology in Retinal Endothelial Cells: Implications for Diabetic Retinopathy. Am. J. Pathol. 2010, 177, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Opreanu, M.; Lydic, T.A.; Reid, G.E.; McSorley, K.M.; Esselman, W.J.; Busik, J.V. Inhibition of cytokine signaling in human retinal endothelial cells through downregulation of sphingomyelinases by docosahexaenoic acid. Invest. Ophthalmol. Vis. Sci. 2010, 51, 3253–3263. [Google Scholar] [CrossRef] [PubMed]
- Simó, R.; Villarroel, M.; Corraliza, L.; Hernández, C.; Garcia-Ramírez, M. The retinal pigment epithelium: Something more than a constituent of the blood-retinal barrier—Implications for the pathogenesis of diabetic retinopathy. J. Biomed. Biotechnol. 2009, 2010, 15. [Google Scholar] [CrossRef]
- Sparrrow, J.R.; Hicks, D.; Hamel, C.P. The Retinal Pigment Epithelium in Health and Disease. Curr. Mol. Med. 2010, 10, 802–823. [Google Scholar] [CrossRef]
- Romero-Aroca, P.; Baget-Bernaldiz, M.; Pareja-Rios, A.; Lopez-Galvez, M.; Navarro-Gil, R.; Verges, R. Diabetic Macular Edema Pathophysiology: Vasogenic versus Inflammatory. J. Diabetes Res. 2016, 2016. [Google Scholar] [CrossRef] [Green Version]
- Berkowitz, B.A.; Olds, H.K.; Richards, C.; Joy, J.; Rosales, T.; Podolsky, R.H.; Childers, K.L.; Brad Hubbard, W.; Sullivan, P.G.; Gao, S.; et al. Novel imaging biomarkers for mapping the impact of mild mitochondrial uncoupling in the outer retina in vivo. PLoS ONE 2020, 15, e0226840. [Google Scholar] [CrossRef]
- Saint-Geniez, M.; Kurihara, T.; Sekiyama, E.; Maldonado, A.E.; D’Amore, P.A. An essential role for RPE-derived soluble VEGF in the maintenance of the choriocapillaris. Proc. Natl. Acad. Sci. USA 2009, 106, 18751–18756. [Google Scholar] [CrossRef] [Green Version]
- Hammer, S.S.; Busik, J.V. The role of dyslipidemia in diabetic retinopathy. Vision Res. 2017, 139, 228–236. [Google Scholar] [CrossRef]
- Newton, J.; Lima, S.; Maceyka, M.; Spiegel, S. Revisiting the sphingolipid rheostat: Evolving concepts in cancer therapy. Exp. Cell Res. 2015, 333, 195–200. [Google Scholar] [CrossRef] [Green Version]
- Ion, G.; Fajka-Boja, R.; Kovács, F.; Szebeni, G.; Gombos, I.; Czibula, Á.; Matkó, J.; Monostori, É. Acid sphingomyelinase mediated release of ceramide is essential to trigger the mitochondrial pathway of apoptosis by galectin-1. Cell. Signal. 2006, 18, 1887–1896. [Google Scholar] [CrossRef]
- Santana, P.; Peña, L.A.; Haimovitz-Friedman, A.; Martin, S.; Green, D.; McLoughlin, M.; Cordon-Cardo, C.; Schuchman, E.H.; Fuks, Z.; Kolesnick, R. Acid sphingomyelinase-deficient human lymphoblasts and mice are defective in radiation-induced apoptosis. Cell 1996, 86, 189–199. [Google Scholar] [CrossRef] [Green Version]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef]
- Kady, N.M.; Liu, X.; Lydic, T.A.; Syed, M.H.; Navitskaya, S.; Wang, Q.; Hammer, S.S.; O’Reilly, S.; Huang, C.; Seregin, S.S.; et al. ELOVL4-Mediated Production of Very Long-Chain Ceramides Stabilizes Tight Junctions and Prevents Diabetes-Induced Retinal Vascular Permeability. Diabetes 2018, 67, 769–781. [Google Scholar] [CrossRef] [Green Version]
- Opreanu, M.; Tikhonenko, M.; Bozack, S.; Lydic, T.A.; Reid, G.E.; McSorley, K.M.; Sochacki, A.; Perez, G.I.; Esselman, W.J.; Kern, T.; et al. The unconventional role of acid sphingomyelinase in regulation of retinal microangiopathy in diabetic human and animal models. Diabetes 2011, 60, 2370–2378. [Google Scholar] [CrossRef] [Green Version]
- Tikhonenko, M.; Lydic, T.A.; Opreanu, M.; Li Calzi, S.; Bozack, S.; McSorley, K.M.; Sochacki, A.L.; Faber, M.S.; Hazra, S.; Duclos, S.; et al. N-3 polyunsaturated Fatty acids prevent diabetic retinopathy by inhibition of retinal vascular damage and enhanced endothelial progenitor cell reparative function. PLoS ONE 2013, 8, e55177. [Google Scholar] [CrossRef] [Green Version]
- Yu, Z.F.; Nikolova-Karakashian, M.; Zhou, D.; Cheng, G.; Schuchman, E.H.; Mattson, M.P. Pivotal role for acidic sphingomyelinase in cerebral ischemia-induced ceramide and cytokine production, and neuronal apoptosis. J. Mol. Neurosci. 2000, 15, 85–97. [Google Scholar] [CrossRef]
- Novgorodov, S.A.; Gudz, T.I. Ceramide and mitochondria in ischemic brain injury. Int. J. Biochem. Mol. Biol. 2011, 2, 347–361. [Google Scholar]
- Siskind, L.J.; Kolesnick, R.N.; Colombini, M. Ceramide channels increase the permeability of the mitochondrial outer membrane to small proteins. J. Biol. Chem. 2002, 277, 26796–26803. [Google Scholar] [CrossRef] [Green Version]
- France-Lanord, V.; Brugg, B.; Michel, P.P.; Agid, Y.; Ruberg, M. Mitochondrial free radical signal in ceramide-dependent apoptosis: A putative mechanism for neuronal death in Parkinson’s disease. J. Neurochem. 1997, 69, 1612–1621. [Google Scholar] [CrossRef]
- Gudz, T.I.; Tserng, K.-Y.; Hoppel, C.L. Direct Inhibition of Mitochondrial Respiratory Chain Complex III by Cell-permeable Ceramide. J. Biol. Chem. 1997, 272, 24154–24158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Managò, A.; Becker, K.A.; Carpinteiro, A.; Wilker, B.; Soddemann, M.; Seitz, A.P.; Edwards, M.J.; Grassmé, H.; Szabò, I.; Gulbins, E. Pseudomonas aeruginosa Pyocyanin Induces Neutrophil Death via Mitochondrial Reactive Oxygen Species and Mitochondrial Acid Sphingomyelinase. Antioxid. Redox Signal. 2015, 22, 1097–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novgorodov, S.A.; Szulc, Z.M.; Luberto, C.; Jones, J.A.; Bielawski, J.; Bielawska, A.; Hannun, Y.A.; Obeid, L.M. Positively charged ceramide is a potent inducer of mitochondrial permeabilization. J. Biol. Chem. 2005, 280, 16096–16105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, T.E.; Han, X.; Kelly, S.; Merrill, A.H.; Martin, R.E.; Anderson, R.E.; Gardner, T.W.; Kester, M. Diabetes alters sphingolipid metabolism in the retina: A potential mechanism of cell death in diabetic retinopathy. Diabetes 2006, 55, 3573–3580. [Google Scholar] [CrossRef] [Green Version]
- Novgorodov, S.A.; Wu, B.X.; Gudz, T.I.; Bielawski, J.; Ovchinnikova, T.V.; Hannun, Y.A.; Obeid, L.M. Novel pathway of ceramide production in mitochondria: Thioesterase and neutral ceramidase produce ceramide from sphingosine and acyl-CoA. J. Biol. Chem. 2011, 286, 25352–25362. [Google Scholar] [CrossRef] [Green Version]
- Lydic, T.A.; Busik, J.V.; Reid, G.E. A monophasic extraction strategy for the simultaneous lipidome analysis of polar and nonpolar retina lipids. J. Lipid Res. 2014, 55, 1797–1809. [Google Scholar] [CrossRef] [Green Version]
- Kogot-Levin, A.; Saada, A. Ceramide and the mitochondrial respiratory chain. Biochimie 2014, 100, 88–94. [Google Scholar] [CrossRef]
- Levitsky, Y.; Pegouske, D.J.; Hammer, S.S.; Frantz, N.L.; Fisher, K.P.; Muchnik, A.B.; Saripalli, A.R.; Kirschner, P.; Bazil, J.N.; Busik, J.V.; et al. Micro-respirometry of whole cells and isolated mitochondria. RSC Adv. 2019, 9, 33257–33267. [Google Scholar] [CrossRef]
- Pesta, D.; Gnaiger, E. High-Resolution Respirometry: OXPHOS Protocols for Human Cells and Permeabilized Fibers from Small Biopsies of Human Muscle. In Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2012; pp. 25–58. [Google Scholar]
- Brand, M.D.; Nicholls, D.G. Assessing mitochondrial dysfunction in cells. Biochem. J. 2011, 435, 297–312. [Google Scholar] [CrossRef] [Green Version]
- Erdreich-Epstein, A.; Tran, L.B.; Bowman, N.N.; Wang, H.; Cabot, M.C.; Durden, D.L.; Vlckova, J.; Reynolds, C.P.; Stins, M.F.; Groshen, S.; et al. Ceramide signaling in fenretinide-induced endothelial cell apoptosis. J. Biol. Chem. 2002, 277, 49531–49537. [Google Scholar] [CrossRef] [Green Version]
- Perry, D.M.; Newcomb, B.; Adada, M.; Wu, B.X.; Roddy, P.; Kitatani, K.; Siskind, L.; Obeid, L.M.; Hannun, Y.A. Defining a Role for Acid Sphingomyelinase in the p38/Interleukin-6 Pathway. J. Biol. Chem. 2014, 289, 22401–22412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novgorodov, S.A.; Riley, C.L.; Yu, J.; Keffler, J.A.; Clarke, C.J.; Van Laer, A.O.; Baicu, C.F.; Zile, M.R.; Gudz, T.I. Lactosylceramide contributes to mitochondrial dysfunction in diabetes. J. Lipid Res. 2016, 57, 546–562. [Google Scholar] [CrossRef] [Green Version]
- Kappler, L.; Li, J.; Häring, H.U.; Weigert, C.; Lehmann, R.; Xu, G.; Hoene, M. Purity matters: A workflow for the valid high-resolution lipid profiling of mitochondria from cell culture samples. Sci. Rep. 2016, 6, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Franko, A.; Baris, O.R.; Bergschneider, E.; Von Toerne, C.; Hauck, S.M.; Aichler, M.; Walch, A.K.; Wurst, W.; Wiesner, R.J.; Johnston, I.C.D.; et al. Efficient isolation of pure and functional mitochondria from mouse tissues using automated tissue disruption and enrichment with anti-TOM22 magnetic beads. PLoS ONE 2013, 8, e82392. [Google Scholar] [CrossRef]
- Clayton, D.A.; Shadel, G.S. Isolation of mitochondria from cells and tissues. Cold Spring Harb. Protoc. 2014, 2014, 1040–1041. [Google Scholar] [CrossRef] [Green Version]
- Larsen, S.; Nielsen, J.; Hansen, C.N.; Nielsen, L.B.; Wibrand, F.; Stride, N.; Schroder, H.D.; Boushel, R.; Helge, J.W.; Dela, F.; et al. Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J. Physiol. 2012, 590, 3349–3360. [Google Scholar] [CrossRef]
- Kern, T.S.; Antonetti, D.A.; Smith, L.E.H. Pathophysiology of Diabetic Retinopathy: Contribution and Limitations of Laboratory Research. Ophthalmic Res. 2019, 62, 196–202. [Google Scholar] [CrossRef]
- Babiychuk, E.B.; Atanassoff, A.P.; Monastyrskaya, K.; Brandenberger, C.; Studer, D.; Allemann, C.; Draeger, A. The targeting of plasmalemmal ceramide to mitochondria during apoptosis. PLoS ONE 2011, 6, e23706. [Google Scholar] [CrossRef] [Green Version]
- Kowluru, R.A. Diabetic retinopathy, metabolic memory and epigenetic modifications. Vision Res. 2017, 139, 30–38. [Google Scholar] [CrossRef]
- Alivand, M.R.; Soheili, Z.S.; Pornour, M.; Solali, S.; Sabouni, F. Novel Epigenetic Controlling of Hypoxia Pathway Related to Overexpression and Promoter Hypomethylation of TET1 and TET2 in RPE Cells. J. Cell. Biochem. 2017, 118, 3193–3204. [Google Scholar] [CrossRef]
- Desjardins, D.; Liu, Y.; Crosson, C.E.; Ablonczy, Z. Histone Deacetylase Inhibition Restores Retinal Pigment Epithelium Function in Hyperglycemia. PLoS ONE 2016, 11, e0162596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolinko, A.H.; Chwa, M.; Atilano, S.R.; Kenney, M.C. African and Asian Mitochondrial DNA Haplogroups Confer Resistance Against Diabetic Stresses on Retinal Pigment Epithelial Cybrid Cells In Vitro. Mol. Neurobiol. 2020, 57, 1636–1655. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.H.; Tong, P.; Gu, L.M.; Li, W.J. Astragalus polysaccharide attenuates metabolic memory-triggered ER stress and apoptosis via regulation of miR-204/SIRT1 axis in retinal pigment epithelial cells. Biosci. Rep. 2020, 40, 1–15. [Google Scholar] [CrossRef]
- Roy, S.; Sala, R.; Cagliero, E.; Lorenzi, M. Overexpression of fibronectin induced by diabetes or high glucose: Phenomenon with a memory. Proc. Natl. Acad. Sci. USA 1990, 87, 404–408. [Google Scholar] [CrossRef] [Green Version]
- Duraisamy, A.J.; Mohammad, G.; Kowluru, R.A. Mitochondrial fusion and maintenance of mitochondrial homeostasis in diabetic retinopathy. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 1617–1626. [Google Scholar] [CrossRef]
- Devi, T.S.; Yumnamcha, T.; Yao, F.; Somayajulu, M.; Kowluru, R.A.; Singh, L.P. TXNIP mediates high glucose-induced mitophagic flux and lysosome enlargement in human retinal pigment epithelial cells. Biol. Open 2019, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Qi, X.; Mitter, S.K.; Yan, Y.; Busik, J.V.; Grant, M.B.; Boulton, M.E. Diurnal Rhythmicity of Autophagy Is Impaired in the Diabetic Retina. Cells 2020, 9, 905. [Google Scholar] [CrossRef] [Green Version]
- Weinbach, E.C.; Costa, J.L.; Nelson, B.D.; Claggett, C.E.; Hundal, T.; Bradley, D.; Morris, S.J. Effects of tricyclic antidepressant drugs on energy-linked reactions in mitochondria. Biochem. Pharmacol. 1986, 35, 1445–1451. [Google Scholar] [CrossRef]
- Murphy, M.P. How understanding the control of energy metabolism can help investigation of mitochondrial dysfunction, regulation and pharmacology. Biochim. Biophys. Acta Bioenerg. 2001, 1504, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Jaffe, G.J.; Earnest, K.; Fulcher, S.; Lui, M.; Houston, L.L. Antitransferrin Receptor Immunotoxin Inhibits Proliferating Human Retinal Pigment Epithelial Cells. Arch. Ophthalmol. 1990, 108, 1163–1168. [Google Scholar] [CrossRef]
- Clayton, D.A.; Shadel, G.S. Purification of mitochondria by sucrose step density gradient centrifugation. Cold Spring Harb. Protoc. 2014, 2014, 1115–1117. [Google Scholar] [CrossRef]
- Byeon, S.K.; Lee, J.Y.; Lee, J.S.; Moon, M.H. Lipidomic profiling of plasma and urine from patients with Gaucher disease during enzyme replacement therapy by nanoflow liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2015, 1381, 132–139. [Google Scholar] [CrossRef]
- Haimi, P.; Uphoff, A.; Hermansson, M.; Somerharju, P. Software tools for analysis of mass spectrometric lipidome data. Anal. Chem. 2006, 78, 8324–8331. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Levitsky, Y.; Hammer, S.S.; Fisher, K.P.; Huang, C.; Gentles, T.L.; Pegouske, D.J.; Xi, C.; Lydic, T.A.; Busik, J.V.; Proshlyakov, D.A. Mitochondrial Ceramide Effects on the Retinal Pigment Epithelium in Diabetes. Int. J. Mol. Sci. 2020, 21, 3830. https://doi.org/10.3390/ijms21113830
Levitsky Y, Hammer SS, Fisher KP, Huang C, Gentles TL, Pegouske DJ, Xi C, Lydic TA, Busik JV, Proshlyakov DA. Mitochondrial Ceramide Effects on the Retinal Pigment Epithelium in Diabetes. International Journal of Molecular Sciences. 2020; 21(11):3830. https://doi.org/10.3390/ijms21113830
Chicago/Turabian StyleLevitsky, Yan, Sandra S. Hammer, Kiera P. Fisher, Chao Huang, Travan L. Gentles, David J. Pegouske, Caimin Xi, Todd A. Lydic, Julia V. Busik, and Denis A. Proshlyakov. 2020. "Mitochondrial Ceramide Effects on the Retinal Pigment Epithelium in Diabetes" International Journal of Molecular Sciences 21, no. 11: 3830. https://doi.org/10.3390/ijms21113830