Time-course Transcriptome of Parageobacillus thermoglucosidasius DSM 6285 Grown in the Presence of Carbon Monoxide and Air



Abstract

1. Introduction
2. Results
2.1. Differential Gene Expression during Aerobic, Anaerobic and Hydrogenogenic Growth Phases in P. thermoglucosidasius
2.2. Metabolic and Functional Shifts in Response to Changing Gas Atmosphere
2.3. Temporal Dynamics of P. thermoglucosidasius DSM 6285 Transcriptome between Aerobic Metabolism to Water Gas SHIFT reaction
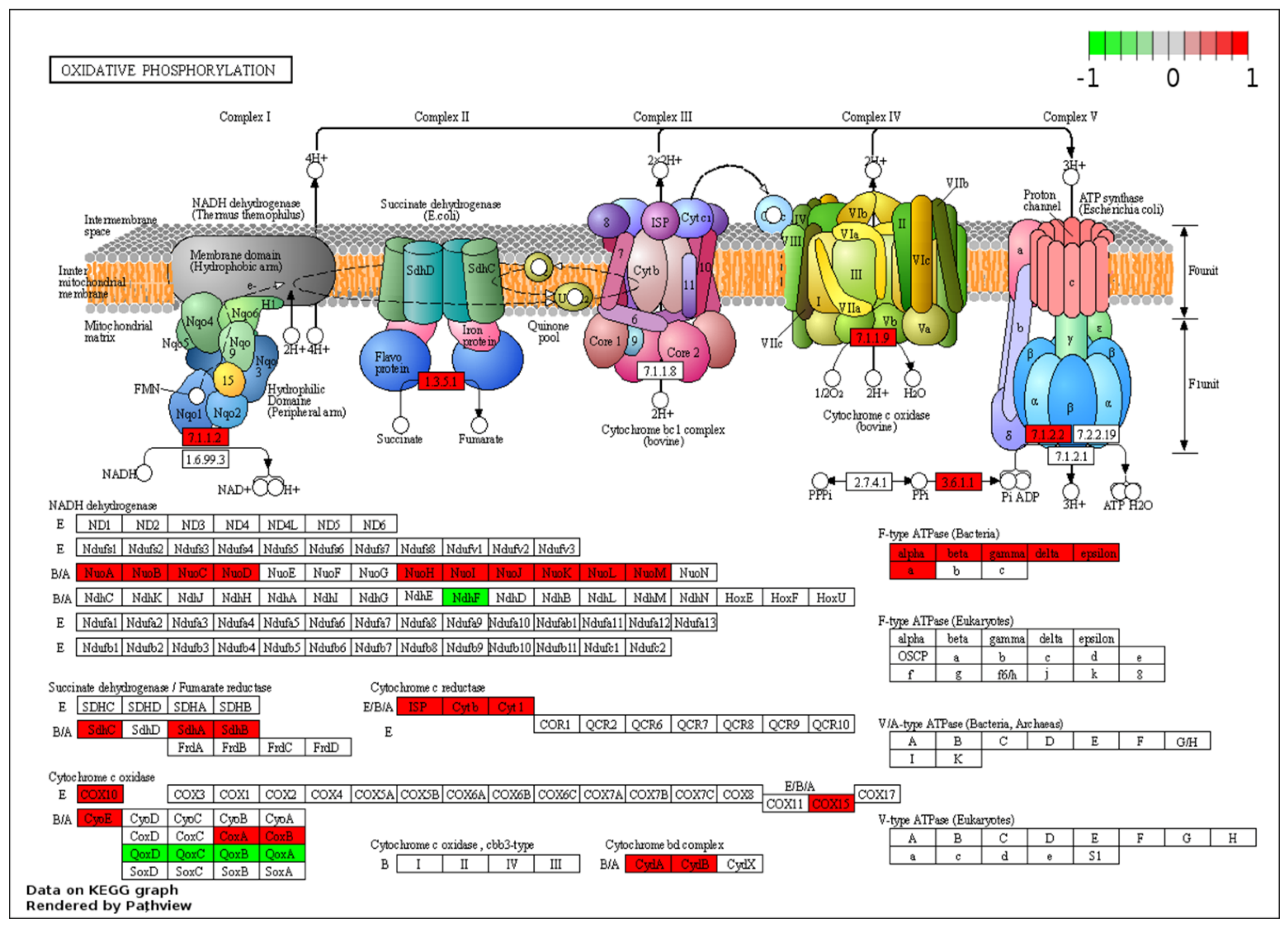
2.4. Transcriptome Pattern of P. thermoglucosidasius DSM 6285 CODH and Hydrogenases
3. Discussion
4. Materials and Methods
4.1. Microorganism and Media
4.2. Experimental Set up and Analytical Methods
4.3. RNA Isolation, Library Preparation, and RNA-Seq
4.4. RNA-Seq Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Midilli, A.; Ay, M.; Dincer, I.; Rosen, M.A. On hydrogen and hydrogen energy strategies: I: Current status and needs. Renew. Sustain. Energy Rev. 2005, 9, 255–271. [Google Scholar] [CrossRef]
- Skinner, J.F. A spectacular demonstration: 2h2 + o2 -> 2h2o. J. Chem. Educ. 1987, 64, 545. [Google Scholar] [CrossRef]
- Nikolaidis, P.; Poullikkas, A. A comparative overview of hydrogen production processes. Renew. Sustain. Energy Rev. 2017, 67, 597–611. [Google Scholar] [CrossRef]
- Ball, M.; Weeda, M. The hydrogen economy—Vision or reality? Int. J. Hydrog. Energy 2015, 40, 7903–7919. [Google Scholar] [CrossRef]
- Kumar, G.R.; Chowdhary, N. Biotechnological and bioinformatics approaches for augmentation of biohydrogen production: A review. Renew. Sustain. Energy Rev. 2016, 56, 1194–1206. [Google Scholar] [CrossRef]
- Barnard, D.; Casanueva, A.; Tuffin, M.; Cowan, D. Extremophiles in biofuel synthesis. Environ. Technol. 2010, 31, 871–888. [Google Scholar] [CrossRef]
- Diender, M.; Stams, A.; Sousa, D. Pathways and bioenergetics of anaerobic carbon monoxide fermentation. Front. Microbiol. 2015, 6, 1275. [Google Scholar] [CrossRef]
- Oelgeschläger, E.; Rother, M. Carbon monoxide-dependent energy metabolism in anaerobic bacteria and archaea. Arch. Microbiol. 2008, 190, 257–269. [Google Scholar] [CrossRef]
- Yoneda, Y.; Yoshida, T.; Kawaichi, S.; Daifuku, T.; Takabe, K.; Sako, Y. Carboxydothermus pertinax sp. Nov., a thermophilic, hydrogenogenic, fe(iii)-reducing, sulfur-reducing carboxydotrophic bacterium from an acidic hot spring. Int. J. Syst. Evol. Microbiol. 2012, 62, 1692–1697. [Google Scholar] [CrossRef]
- Kim, S.M.; Bae, S.S.; Kim, Y.J.; Kim, T.W.; Lim, J.K.; Lee, S.H.; Choi, A.R.; Jeon, J.H.; Lee, J.-H.; Lee, H.S. Co-dependent h2 production by genetically engineered thermococcus onnurineus na1. Appl. Environ. Microbiol. 2013, 79, 2048–2053. [Google Scholar] [CrossRef]
- Sokolova, T.; Lebedinsky, A. Thermophilic microbes in environmental and industrial biotechnology. Biotechnol. Thermophiles 2013. [Google Scholar] [CrossRef]
- McMullan, G.; Christie, J.; Rahman, T.; Banat, I.; Ternan, N.; Marchant, R. Habitat, applications and genomics of the aerobic, thermophilic genus Geobacillus. Biochem. Soc. Trans. 2004, 32, 214–217. [Google Scholar] [CrossRef] [PubMed]
- Nazina, T.; Tourova, T.; Poltaraus, A.; Novikova, E.; Grigoryan, A.; Ivanova, A.; Lysenko, A.; Petrunyaka, V.; Osipov, G.; Belyaev, S. Taxonomic study of aerobic thermophilic bacilli: Descriptions of Geobacillus subterraneus gen. Nov., sp. Nov. And Geobacillus uzenensis sp. Nov. From petroleum reservoirs and transfer of Bacillus stearothermophilus, Bacillus thermocatenulatus, Bacillus thermoleovorans, Bacillus kaustophilus, Bacillus thermodenitrificans to Geobacillus as the new combinations G. stearothermophilus, g. Th. Int. J. Syst. Evol. Microbiol. 2001, 51, 433–446. [Google Scholar] [PubMed]
- Zeigler, D.R. The Geobacillus paradox: Why is a thermophilic bacterial genus so prevalent on a mesophilic planet? Microbiology 2014, 160. [Google Scholar] [CrossRef]
- Huang, Z.; Liu, X.; Zhang, S.; Liu, Z. Gh52 xylosidase from Geobacillus stearothermophilus: Characterization and introduction of xylanase activity by site-directed mutagenesis of tyr509. J. Ind. Microbiol. Biotechnol. 2014, 41, 65–74. [Google Scholar]
- Jiang, T.; Cai, M.; Huang, M.; He, H.; Lu, J.; Zhou, X.; Zhang, Y. Characterization of a thermostable raw-starch hydrolyzing α-amylase from deep-sea thermophile Geobacillus sp. Protein Expr. Purif. 2015, 114, 15–22. [Google Scholar] [CrossRef]
- Mok, S.-C.; Teh, A.-H.; Saito, J.A.; Najimudin, N.; Alam, M. Crystal structure of a compact α-amylase from Geobacillus thermoleovorans. Enzym. Microb. Technol. 2013, 53, 46–54. [Google Scholar] [CrossRef]
- Abdel-Fattah, Y.R.; Gaballa, A.A. Identification and over-expression of a thermostable lipase from geobacillus thermoleovorans toshki in Escherichia coli. Microbiol. Res. 2008, 163, 13–20. [Google Scholar] [CrossRef]
- Zhu, W.; Cha, D.; Cheng, G.; Peng, Q.; Shen, P. Purification and characterization of a thermostable protease from a newly isolated Geobacillus sp. Ymtc 1049. Enzym. Microb. Technol. 2007, 40, 1592–1597. [Google Scholar] [CrossRef]
- Christopher, L.P.; Zambare, V.P.; Zambare, A.; Kumar, H.; Malek, L. A thermo-alkaline lipase from a new thermophile Geobacillus thermodenitrificans av-5 with potential application in biodiesel production. J. Chem. Technol. Biotechnol. 2015, 90, 2007–2016. [Google Scholar] [CrossRef]
- De Maayer, P.; Brumm, P.J.; Mead, D.A.; Cowan, D.A. Comparative analysis of the Geobacillus hemicellulose utilization locus reveals a highly variable target for improved hemicellulolysis. BMC Genom. 2014, 15, 836. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.P.; Rabe, K.S.; Takasumi, J.L.; Kadisch, M.; Arnold, F.H.; Liao, J.C. Isobutanol production at elevated temperatures in thermophilic Geobacillus thermoglucosidasius. Metab. Eng. 2014, 24. [Google Scholar] [CrossRef] [PubMed]
- Peeples, T.L. 10 bioremediation using extremophiles. In Microbial Biodegradation and Bioremediation; Elsevier: Amsterdam, The Netherlands, 2014; pp. 251–268. [Google Scholar]
- Mohr, T.; Aliyu, H.; Küchlin, R.; Polliack, S.; Zwick, M.; Neumann, A.; Cowan, D.; de Maayer, P. Co-dependent hydrogen production by the facultative anaerobe Parageobacillus thermoglucosidasius. Microb. Cell Factories 2018, 17, 108. [Google Scholar] [CrossRef] [PubMed]
- Mohr, T.; Aliyu, H.; Küchlin, R.; Zwick, M.; Cowan, D.; Neumann, A.; de Maayer, P. Comparative genomic analysis of Parageobacillus thermoglucosidasius strains with distinct hydrogenogenic capacities. BMC Genom. 2018, 19, 880. [Google Scholar] [CrossRef]
- Antonopoulou, G.; Ntaikou, I.; Stamatelatou, K.; Lyberatos, G. Biological and fermentative production of hydrogen. In Handbook of Biofuels Production; Elsevier: Amsterdam, The Netherlands, 2011; pp. 305–346. [Google Scholar]
- Mohr, T.; Aliyu, H.; Biebinger, L.; Gödert, R.; Hornberger, A.; Cowan, D.; de Maayer, P.; Neumann, A. Effects of different operating parameters on hydrogen production by Parageobacillus thermoglucosidasius dsm 6285. AMB Express 2019, 9, 207. [Google Scholar] [CrossRef]
- Uniprot. Uniprot: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Morishima, K. Blastkoala and ghostkoala: Kegg tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef]
- Higgins, C.F. Abc transporters: Physiology, structure and mechanism—An overview. Res. Microbiol. 2001, 152, 205–210. [Google Scholar] [CrossRef]
- Murugasu-Oei, B.; Tay, A.; Dick, T. Upregulation of stress response genes and abc transporters in anaerobic stationary-phase Mycobacterium smegmatis. Mol. Gen. Genet. MGG 1999, 262, 677–682. [Google Scholar] [CrossRef]
- Pinhal, S.; Ropers, D.; Geiselmann, J.; de Jong, H. Acetate metabolism and the inhibition of bacterial growth by acetate. J. Bacteriol. 2019, 201, e00147-19. [Google Scholar] [CrossRef]
- Majewski, R.A.; Domach, M.M. Simple constrained-optimization view of acetate overflow in E. coli. Biotechnol. Bioeng. 1990, 35, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.J.; Sapra, R.; Joyner, D.; Hazen, T.C.; Myers, S.; Reichmuth, D.; Blanch, H.; Keasling, J.D. Analysis of metabolic pathways and fluxes in a newly discovered thermophilic and ethanol-tolerant Geobacillus strain. Biotechnol. Bioeng. 2009, 102, 1377–1386. [Google Scholar] [CrossRef]
- Cypionka, H.; Meyer, O. Carbon monoxide-insensitive respiratory chain of Pseudomonas carboxydovorans. J. Bacteriol. 1983, 156, 1178–1187. [Google Scholar] [CrossRef] [PubMed]
- Wareham, L.K.; Begg, R.; Jesse, H.E.; van Beilen, J.W.A.; Ali, S.; Svistunenko, D.; McLean, S.; Hellingwerf, K.J.; Sanguinetti, G.; Poole, R.K. Carbon monoxide gas is not inert, but global, in its consequences for bacterial gene expression, iron acquisition, and antibiotic resistance. Antioxid. Redox Signal. 2016, 24, 1013–1028. [Google Scholar]
- Fang, X.; Wallqvist, A.; Reifman, J. Modeling phenotypic metabolic adaptations of Mycobacterium tuberculosis h37rv under hypoxia. PLoS Comput. Biol. 2012, 8, e1002688. [Google Scholar] [CrossRef]
- Osterman, I.A.; Dikhtyar, Y.Y.; Bogdanov, A.A.; Dontsova, O.A.; Sergiev, P.V. Regulation of flagellar gene expression in bacteria. Biochemistry (Moscow) 2015, 80, 1447–1456. [Google Scholar] [CrossRef]
- Müller-Herbst, S.; Wüstner, S.; Mühlig, A.; Eder, D.; Fuchs, T.M.; Held, C.; Ehrenreich, A.; Scherer, S. Identification of genes essential for anaerobic growth of Listeria monocytogenes. Microbiology 2014, 160, 752–765. [Google Scholar] [CrossRef]
- Cotter, P.D.; Hill, C. Surviving the acid test: Responses of gram-positive bacteria to low ph. Microbiol. Mol. Biol. Rev. 2003, 67, 429–453. [Google Scholar] [CrossRef]
- Wolfe, A.J. The acetate switch. Microbiol. Mol. Biol. Rev. 2005, 69, 12–50. [Google Scholar] [CrossRef]
- Horlbog, J.A.; Stevens, M.J.A.; Stephan, R.; Guldimann, C. Global transcriptional response of three highly acid-tolerant field strains of listeria monocytogenes to hcl stress. Microorganisms 2019, 7, 455. [Google Scholar] [CrossRef]
- Vignais, P.M.; Billoud, B. Occurrence, classification, and biological function of hydrogenases: An overview. Chem. Rev. 2007, 107, 4206–4272. [Google Scholar] [CrossRef] [PubMed]
- Greening, C.; Biswas, A.; Carere, C.R.; Jackson, C.J.; Taylor, M.C.; Stott, M.B.; Cook, G.M.; Morales, S.E. Genomic and metagenomic surveys of hydrogenase distribution indicate h 2 is a widely utilised energy source for microbial growth and survival. ISME J. 2016, 10, 761–777. [Google Scholar] [CrossRef] [PubMed]
- Merrouch, M.; Hadj-Saïd, J.; Domnik, L.; Dobbek, H.; Léger, C.; Dementin, S.; Fourmond, V. O2 inhibition of ni-containing co dehydrogenase is partly reversible. Chem. A Eur. J. 2015, 21, 18934–18938. [Google Scholar] [CrossRef] [PubMed]
- Ainala, S.K.; Seol, E.; Sekar, B.S.; Park, S. Improvement of carbon monoxide-dependent hydrogen production activity in Citrobacter amalonaticus y19 by over-expressing the co-sensing transcriptional activator, cooa. Int. J. Hydrog. Energy 2014, 39, 10417–10425. [Google Scholar] [CrossRef]
- Dutta, D.; De, D.; Chaudhuri, S.; Bhattacharya, S.K. Hydrogen production by cyanobacteria. Microb. Cell Factories 2005, 4, 36. [Google Scholar] [CrossRef] [PubMed]
- Casalot, L.; Rousset, M. Maturation of the [nife] hydrogenases. Trends Microbiol. 2001, 9, 228–237. [Google Scholar] [CrossRef]
- Jabłońska, J.; Tawfik, D.S. The number and type of oxygen-utilizing enzymes indicates aerobic vs. Anaerobic phenotype. Free Radic. Biol. Med. 2019, 140, 84–92. [Google Scholar] [CrossRef]
- Xu, J.; Ding, Z.; Liu, B.; Sophia, M.Y.; Li, J.; Zhang, Z.; Liu, Y.; Li, J.; Liu, L.; Zhou, A. Structure of the cytochrome aa3-600 heme-copper menaquinol oxidase bound to inhibitor hqno shows tm0 is part of the quinol binding site. Proc. Natl. Acad. Sci. USA 2020, 117, 872–876. [Google Scholar] [CrossRef]
- Castresana, J.; Lübben, M.; Saraste, M.; Higgins, D.G. Evolution of cytochrome oxidase, an enzyme older than atmospheric oxygen. EMBO J. 1994, 13, 2516–2525. [Google Scholar] [CrossRef]
- Winstedt, L.; von Wachenfeldt, C. Terminal Oxidases of Bacillus subtilis Strain 168: One Quinol Oxidase, Cytochromeaa 3 or Cytochrome bd, Is Required for Aerobic Growth. J. Bacteriol. 2000, 182, 6557. [Google Scholar] [CrossRef]
- Zamboni, N.; Sauer, U. Knockout of the high-coupling cytochrome aa3 oxidase reduces tca cycle fluxes in Bacillus subtilis. FEMS Microbiol. Lett. 2003, 226, 121–126. [Google Scholar] [CrossRef][Green Version]
- Pesavento, C.; Becker, G.; Sommerfeldt, N.; Possling, A.; Tschowri, N.; Mehlis, A.; Hengge, R. Inverse regulatory coordination of motility and curli-mediated adhesion in escherichia coli. Genes Dev. 2008, 22, 2434–2446. [Google Scholar]
- Belas, R. Biofilms, flagella, and mechanosensing of surfaces by bacteria. Trends Microbiol. 2014, 22, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Ragsdale, S.W. Life with carbon monoxide. Crit. Rev. Biochem. Mol. Biol. 2004, 39, 165–195. [Google Scholar] [CrossRef]
- Kim, J.-R.; Oh, Y.-K.; Yoon, Y.-J.; Lee, E.-Y.; Park, S. Oxygen sensitivity of carbon monoxide-dependent hydrogen production activity in citrobacter sp. J. Microbiol. Biotechnol. 2003, 13, 717–724. [Google Scholar]
- Xavier, J.C.; Preiner, M.; Martin, W.F. Something special about co-dependent co2 fixation. FEBS J. 2018, 285, 4181–4195. [Google Scholar] [CrossRef]
- Wu, J.; Zhan, M.; Chang, Y.; Gao, H.; Ye, J.; Yu, R.; Ding, Z. Mechanistic insights into stress response and metabolic activity resilience of Nitrosomonas europaea cultures to long-term ceo 2 nanoparticle exposure. Environ. Sci. Nano 2019, 6, 2215–2227. [Google Scholar] [CrossRef]
- Rossoni, A.W.; Schönknecht, G.; Lee, H.J.; Rupp, R.L.; Flachbart, S.; Mettler-Altmann, T.; Weber, A.P.M.; Eisenhut, M. Cold acclimation of the thermoacidophilic red alga Galdieria sulphuraria: Changes in gene expression and involvement of horizontally acquired genes. Plant Cell Physiol. 2018, 60, 702–712. [Google Scholar] [CrossRef]
- Jones, P.G.; Inouye, M. The cold-shock response—A hot topic. Mol. Microbiol. 1994, 11, 811–818. [Google Scholar] [CrossRef]
- Tai, S.L.; Daran-Lapujade, P.; Walsh, M.C.; Pronk, J.T.; Daran, J.-M. Acclimation of Saccharomyces cerevisiae to low temperature: A chemostat-based transcriptome analysis. Mol. Biol. Cell 2007, 18, 5100–5112. [Google Scholar] [CrossRef]
- Singleton, C.; Gilman, J.; Rollit, J.; Zhang, K.; Parker, D.A.; Love, J. A design of experiments approach for the rapid formulation of a chemically defined medium for metabolic profiling of industrially important microbes. PLoS ONE 2019, 14, e0218208. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.K.; Scholz, M.B.; Teal, T.K.; Abramovitch, R.B. Sparta: Simple program for automated reference-based bacterial rna-seq transcriptome analysis. BMC Bioinform. 2016, 17, 66. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Andrews, S. Fastqc: A Quality Control Tool for High Throughput Sequence Data; Babraham Bioinformatics, Babraham Institute: Cambridge, UK, 2010. [Google Scholar]
- Langmead, B. Aligning short sequencing reads with bowtie. Curr. Protoc. Bioinform. 2010, 32, 11.7.1–11.7.14. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. Htseq—A python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. Edger: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- McDowell, I.C.; Manandhar, D.; Vockley, C.M.; Schmid, A.K.; Reddy, T.E.; Engelhardt, B.E. Clustering gene expression time series data using an infinite gaussian process mixture model. PLoS Comput. Biol. 2018, 14, e1005896. [Google Scholar] [CrossRef]
- Zheng, Q.; Wang, X.-J. Goeast: A web-based software toolkit for gene ontology enrichment analysis. Nucleic Acids Res. 2008, 36, W358–W363. [Google Scholar] [CrossRef]
- Luo, W.; Pant, G.; Bhavnasi, Y.K.; Blanchard, S.G., Jr.; Brouwer, C. Pathview web: User friendly pathway visualization and data integration. Nucleic Acids Res. 2017, 45, W501–W508. [Google Scholar] [CrossRef]
- Hammer-Muntz, O.; Harper, D.; Ryan, P. Past: Paleontological statistics software package for education and data analysis version 3.26. 2001. Available online: https://palaeo-electronica.org/2001_1/past/issue1_01.htm (accessed on 20 May 2020).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Set | Kegg Pathway | Definition | Stat. Mean | Set. Size | q |
|---|---|---|---|---|---|
| P1 | ko01100 | Metabolic pathways | −4.35 | 273 | 6.31 × 10−4 |
| ko00020 | Citrate cycle (TCA cycle) | −5.11 | 19 | 7.18 × 10−4 | |
| ko01130 | Biosynthesis of antibiotics | −4.09 | 90 | 7.18 × 10−4 | |
| ko02040 | Flagellar assembly | −5.24 | 20 | 7.18 × 10−4 | |
| ko00860 | Porphyrin and chlorophyll metabolism | −4.36 | 20 | 1.37 × 10−3 | |
| ko01110 | Biosynthesis of secondary metabolites | −3.37 | 118 | 5.51 × 10−3 | |
| ko01230 | Biosynthesis of amino acids | −3.40 | 50 | 5.51 × 10−3 | |
| ko00010 | Glycolysis / Gluconeogenesis | −3.37 | 28 | 6.31 × 10−3 | |
| ko00190 | Oxidative phosphorylation | −3.37 | 28 | 6.31 × 10−3 | |
| ko03010 | Ribosome | −3.38 | 14 | 1.16 × 10−2 | |
| ko01200 | Carbon metabolism | −2.82 | 56 | 2.01 × 10−2 | |
| ko00620 | Pyruvate metabolism | −2.57 | 25 | 3.71 × 10−2 | |
| ko00640 | Propanoate metabolism | −2.63 | 17 | 3.71 × 10−2 | |
| ko00650 | Butanoate metabolism | −2.46 | 16 | 5.02 × 10−2 | |
| ko01210 | 2-Oxocarboxylic acid metabolism | −2.58 | 10 | 5.02 × 10−2 | |
| ko00720 | Carbon fixation pathways in prokaryotes | −2.37 | 21 | 5.24 × 10−2 | |
| ko01120 | Microbial metabolism in diverse environments | −2.27 | 110 | 5.26 × 10−2 | |
| ko00280 | Valine, leucine and isoleucine degradation | −1.84 | 10 | 1.68 × 10−01 | |
| ko02010 | ABC transporters | 2.71 | 67 | 2.51 × 10−2 | |
| P2 | ko03010 | Ribosome | −5.38 | 49 | 9.20 × 10−05 |
| ko01110 | Biosynthesis of secondary metabolites | −4.59 | 133 | 1.49 × 10−4 | |
| ko01100 | Metabolic pathways | −4.10 | 282 | 5.93 × 10−4 | |
| ko01230 | Biosynthesis of amino acids | −4.27 | 53 | 5.93 × 10−4 | |
| ko01130 | Biosynthesis of antibiotics | −3.53 | 97 | 4.42 × 10−3 | |
| ko01200 | Carbon metabolism | −3.52 | 62 | 4.54 × 10−3 | |
| ko00860 | Porphyrin and chlorophyll metabolism | −3.60 | 22 | 1.00 × 10−2 | |
| ko00020 | Citrate cycle (TCA cycle) | −3.35 | 19 | 1.66 × 10−2 | |
| ko00190 | Oxidative phosphorylation | −2.37 | 32 | 8.68 × 10−2 | |
| ko00620 | Pyruvate metabolism | −2.33 | 27 | 8.68 × 10−2 | |
| ko00720 | Carbon fixation pathways in prokaryotes | −2.33 | 22 | 8.68 × 10−2 | |
| ko01210 | 2-Oxocarboxylic acid metabolism | −2.55 | 11 | 8.68 × 10−2 | |
| ko01212 | Fatty acid metabolism | −2.55 | 11 | 8.68 × 10−2 | |
| ko00630 | Glyoxylate and dicarboxylate metabolism | −2.22 | 21 | 9.25 × 10−2 | |
| ko00970 | Aminoacyl-tRNA biosynthesis | −2.43 | 10 | 9.38 × 10−2 | |
| ko03018 | RNA degradation | −2.43 | 10 | 9.38 × 10−2 | |
| ko00680 | Methane metabolism | −1.91 | 13 | 1.68 × 10−1 | |
| ko02010 | ABC transporters | 3.15 | 64 | 1.07 × 10−2 | |
| P3 | ko03010 | Ribosome | 5.71 | 29 | 2.40 × 10−5 |
| KEGG Pathway | Definition | Cluster Set | Set. Size | p | q |
|---|---|---|---|---|---|
| ko02010 | ABC transporters | 1 | 67 | 1.35 × 10−3 | 7.72 × 10−3 |
| ko00020 | Citrate cycle (TCA cycle) | 2 | 22 | 3.89 × 10−4 | 2.59 × 10−3 |
| ko00190 | Oxidative phosphorylation | 2 | 34 | 5.99 × 10−3 | 2.42 × 10−2 |
| ko00620 | Pyruvate metabolism | 2 | 24 | 3.58 × 10−3 | 1.79 × 10−2 |
| ko00640 | Propanoate metabolism | 2 | 22 | 6.66 × 10−3 | 2.42 × 10−2 |
| ko00860 | Porphyrin and chlorophyll metabolism | 2 | 25 | 1.51 × 10−4 | 1.51 × 10−3 |
| ko01100 | Metabolic pathways | 2 | 304 | 1.59 × 10−5 | 5.25 × 10−4 |
| ko01110 | Biosynthesis of secondary metabolites | 2 | 134 | 2.62 × 10−5 | 5.25 × 10−4 |
| ko01130 | Biosynthesis of antibiotics | 2 | 100 | 7.54 × 10−5 | 1.01 × 10−3 |
| ko01200 | Carbon metabolism | 2 | 52 | 3.43 × 10−4 | 2.59 × 10−3 |
| ko03010 | Ribosome | 2 | 12 | 9.88 × 10−3 | 3.29 × 10−2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aliyu, H.; Mohr, T.; Cowan, D.; de Maayer, P.; Neumann, A. Time-course Transcriptome of Parageobacillus thermoglucosidasius DSM 6285 Grown in the Presence of Carbon Monoxide and Air. Int. J. Mol. Sci. 2020, 21, 3870. https://doi.org/10.3390/ijms21113870
Aliyu H, Mohr T, Cowan D, de Maayer P, Neumann A. Time-course Transcriptome of Parageobacillus thermoglucosidasius DSM 6285 Grown in the Presence of Carbon Monoxide and Air. International Journal of Molecular Sciences. 2020; 21(11):3870. https://doi.org/10.3390/ijms21113870
Chicago/Turabian StyleAliyu, Habibu, Teresa Mohr, Don Cowan, Pieter de Maayer, and Anke Neumann. 2020. "Time-course Transcriptome of Parageobacillus thermoglucosidasius DSM 6285 Grown in the Presence of Carbon Monoxide and Air" International Journal of Molecular Sciences 21, no. 11: 3870. https://doi.org/10.3390/ijms21113870
APA StyleAliyu, H., Mohr, T., Cowan, D., de Maayer, P., & Neumann, A. (2020). Time-course Transcriptome of Parageobacillus thermoglucosidasius DSM 6285 Grown in the Presence of Carbon Monoxide and Air. International Journal of Molecular Sciences, 21(11), 3870. https://doi.org/10.3390/ijms21113870