Functional Heterologous Expression of Mature Lipase LipA from Pseudomonas aeruginosa PSA01 in Escherichia coli SHuffle and BL21 (DE3): Effect of the Expression Host on Thermal Stability and Solvent Tolerance of the Enzyme Produced

 ,
,
Abstract
1. Introduction
2. Results
2.1. Cloning of LipA and its Foldase
Nucleotide Accession Number
2.2. Monitoring of the Recombinant LipA Expression in E. coli SHuffle and E. coli BL21 (DE3) Strains.
2.2.1. Induction with IPTG
2.2.2. Induction with Autoinducing Medium
2.3. Stability in Methanol and Ethanol of LipA Produced by E. coli SHuffle and E. coli BL21 (DE3)
2.4. Stability of LipA Produced by E. coli SHuffle and E. coli BL21 (DE3) to Temperature
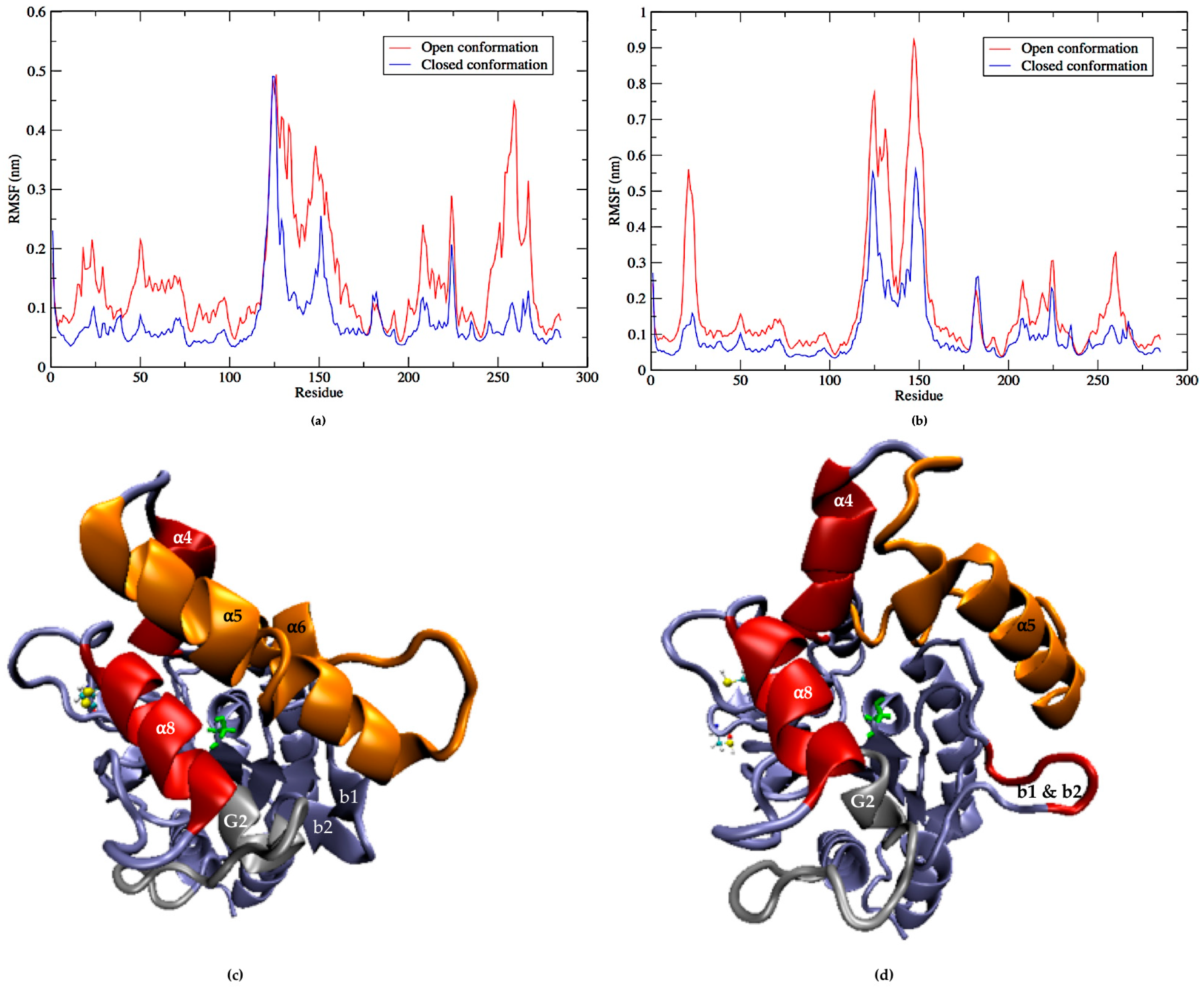
2.5. Flexibility of LipA with and without Disulfide Bonds during the Molecular Dynamics Simulations
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains, Plasmids, and Reagents.
4.2. Cloning of lipA and lif
4.3. Expression of lipA in Strains of E. coli SHuffle and E. coli BL21 (DE3)
4.3.1. Induction with IPTG
4.3.2. Inducing with Autoinducing Medium
4.4. SDS-PAGE Analysis and Protein Concentration
4.5. Determination of Lipase Activity
4.6. Evaluation of the Stability of Recombinant LipA at Different Temperatures
4.7. Evaluation of the Stability of Recombinant LipA in Methanol and Ethanol
4.8. Molecular Dynamics Simulations of LipA with and without SS-Bridge
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DMSO | Dimethyl sulfoxide |
| IPTG | Isopropyl -β- d-1-thiogalactopyranoside |
| PCR | Polymerase chain reaction |
| MCS | Multi cloning site |
| CFU | Colony-forming unit |
| OD600 | Optical density at 600 nm |
| KDa | Kilodalton |
| PMSF | Phenylmethyl sulfonyl fluoride |
| Ps | Picoseconds |
| SDS-PAGE | Phenylmethyl sulfonyl fluoride |
| MD | Molecular dynamics |
| K | Kelvin |
| μL | Microliter |
| NVT | Constant number, volume and temperature |
| NPT | Constant number, pressure and temperature |
| fs | Femtoseconds |
Appendix A


References
- Hasan, F.; Shah, A.A.; Hameed, A. Industrial applications of microbial lipases. Enzyme Microb. Technol. 2006, 39, 235–251. [Google Scholar]
- Houde, A.; Kademi, A.; Leblanc, D. Lipases and Their Industrial Applications: An Overview. Appl. Biochem. Biotechnol. 2004, 118, 155–170. [Google Scholar] [PubMed]
- Andualema, B.; Gessesse, A. Microbial lipases and their industrial applications: Review. Biotechnology 2012, 11, 100–118. [Google Scholar] [CrossRef]
- Sarrouh, B.; Santos, T.M.; Miyoshi, A.; Dias, R.; Azevedo, V. Up-To-Date Insight on Industrial Enzymes Applications and Global Market. J. Bioprocess. Biotech. 2012, S4, 1–10. [Google Scholar]
- Jaeger, K.; Liebeton, K.; Zonta, A.; Schimossek, K.; Reetz, M.T. Biotechnological application of Pseudomonas aeruginosa lipase: Efficient kinetic resolution of amines and alcohols. Appl. Microbiol. Biotechnol. 1996, 46, 99–105. [Google Scholar] [CrossRef]
- Kanwar, S.S.; Verma, M.L.; Maheshwari, C.; Chauhan, S.; Chimni, S.S.; Chauhan, G.S. Properties of Poly (AAc-co-HPMA-cl-EGDMA) Hydrogel-Bound Lipase of Pseudomonas aeruginosa MTCC-4713 and Its Use in Synthesis of Methyl Acrylate. J. Appl. Polym. Sci. 2006, 104, 183–191. [Google Scholar] [CrossRef]
- Liebeton, K.; Zacharias, A.; Jaeger, K.E. Disulfide bond in Pseudomonas aeruginosa lipase stabilizes the structure but is not required for interaction with its foldase. J. Bacteriol. 2001, 183, 597–603. [Google Scholar] [CrossRef]
- Urban, A.; Leipelt, M.; Eggert, T.; Jaeger, K.E. DsbA and DsbC affect extracellular enzyme formation in Pseudomonas aeruginosa. J. Bacteriol. 2001, 183, 587–596. [Google Scholar] [CrossRef]
- Jaeger, K.; Eggert, T. Lipases for biotechnology. Curr. Opin. Biotechnol. 2002, 13, 390–397. [Google Scholar]
- Wu, X.; You, P.; Su, E.; Xu, J.; Gao, B.; Wei, D. In vivo functional expression of a screened P. aeruginosa chaperone-dependent lipase in E. coli. BMC Biotechnol. 2012, 12, 1–9. [Google Scholar]
- Madan, B.; Mishra, P. Co-expression of the lipase and foldase of Pseudomonas aeruginosa to a functional lipase in Escherichia coli. Appl. Microbiol. Biotechnol. 2010, 85, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Ogino, H.; Katou, Y.; Akagi, R.; Mimitsuka, T.; Hiroshima, S.; Gemba, Y.; Doukyu, N.; Yasuda, M.; Ishimi, K.; Ishikawa, H. Cloning and expression of gene, and activation of an organic solvent-stable lipase from Pseudomonas aeruginosa LST-03. Extremophiles 2007, 11, 809–817. [Google Scholar] [PubMed]
- Xu, Y.; Yasin, A.; Tang, R.; Scharer, J.M.; Moo-Young, M.; Chou, C.P. Heterologous expression of lipase in Escherichia coli is limited by folding and disulfide bond formation. Appl. Microbiol. Biotechnol. 2008, 81, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Rosano, G.L.; Ceccarelli, E.A. Recombinant protein expression in Escherichia coli: Advances and challenges. Front. Microbiol. 2014, 5, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Rosenau, F.; Jaeger, K. Bacterial lipases from Pseudomonas: regulation of gene expression and mechanisms of secretion. Biochimie 2000, 82, 1023–1032. [Google Scholar]
- Hobson, A.H.; Buckley, C.M.; Aamand, J.L.; Jørgensen, S.T.; Diderichsen, B.; McConnell, D.J. Activation of a bacterial lipase by its chaperone. Proc. Natl. Acad. Sci. USA 1993, 90, 5682–5686. [Google Scholar] [CrossRef]
- Peng, R.; Lin, J.; Wei, D. Co-expression of an organic solvent-tolerant lipase and its cognate foldase of Pseudomonas aeruginosa CS-2 and the application of the immobilized recombinant lipase. Appl. Biochem. Biotechnol. 2011, 165, 926–937. [Google Scholar] [CrossRef]
- Akbari, N.; Khajeh, K.; Ghaemi, N.; Salemi, Z. Efficient refolding of recombinant lipase from Escherichia coli inclusion bodies by response surface methodology. Protein Expr. Purif. 2010, 70, 254–259. [Google Scholar] [CrossRef]
- Quyen, D.T.; Schmidt-Dannert, C.; Schmid, R.D. High-level formation of active Pseudomonas cepacia lipase after heterologous expression of the encoding gene and its modified chaperone in Escherichia coli and rapid in vitro refolding. Appl. Environ. Microbiol. 1999, 65, 787–794. [Google Scholar] [CrossRef]
- Kadokura, H.; Katzen, F.; Beckwith, J. Protein Disulfide Bond Formation in Prokaryotes. Annu. Rev. Biochem. 2003, 72, 111–135. [Google Scholar] [CrossRef]
- Lobstein, J.; Emrich, C.A.; Jeans, C.; Faulkner, M.; Riggs, P.; Berkmen, M. SHuffle, a novel Escherichia coli protein expression strain capable of correctly folding disulfide bonded proteins in its cytoplasm. Microb. Cell Fact. 2012, 11, 753. [Google Scholar] [CrossRef] [PubMed]
- Ren, G.; Ke, N.; Berkmen, M. Use of the SHuffle Strains in Production of Proteins. In Current Protocols in Protein Science; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016; pp. 5.26.1–5.26.21. ISBN 1934-3663. [Google Scholar]
- Ogino, H.; Ishikawa, H. Enzymes which are stable in the presence of organic solvents. J. Biosci. Bioeng. 2001, 91, 109–116. [Google Scholar] [CrossRef]
- Ogino, H.; Uchiho, T.; Yokoo, J.; Kobayashi, R.; Ichise, R.; Ishikawa, H. Role of intermolecular disulfide bonds of the organic solvent-stable PST-01 protease in its organic solvent stability. Appl. Environ. Microbiol. 2001, 67, 942–947. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Dhar, K.; Kanwar, S.S.; Arora, P.K. Lipase catalysis in organic solvents: Advantages and applications. Biol. Proced. Online 2016, 18, 2. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.L.; Han, S.Y.; Zheng, S.P.; Lin, Y. Enhancing thermostability of a Rhizomucor miehei lipase by engineering a disulfide bond and displaying on the yeast cell surface. Appl. Microbiol. Biotechnol. 2009, 85, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.W.; Tan, N.J.; Xiao, R.; Xu, Y. Engineering a Disulfide Bond in the Lid Hinge Region of Rhizopus chinensis Lipase: Increased Thermostability and Altered Acyl Chain Length Specificity. PLoS One 2012, 7, e46388. [Google Scholar] [CrossRef]
- Le, Q.A.T.; Joo, J.C.; Yoo, Y.J.; Kim, Y.H. Development of thermostable Candida antarctica lipase B through novel in silico design of disulfide bridge. Biotechnol. Bioeng. 2012, 109, 867–876. [Google Scholar] [CrossRef] [PubMed]
- Godoy, C.A.; Klett, J.; Di Geronimo, B.; Hermoso, J.A.; Guisán, J.M.; Carrasco-López, C. Disulfide engineered lipase to enhance the catalytic activity: A structure-based approach on btl2. Int. J. Mol. Sci. 2019, 20, 5245. [Google Scholar] [CrossRef]
- Sørensen, H.P.; Mortensen, K.K. Advanced genetic strategies for recombinant protein expression in Escherichia coli. J. Biotechnol. 2005, 115, 113–128. [Google Scholar] [CrossRef]
- Terpe, K. Overview of bacterial expression system for heterologous protein production: From molecular and biochemical fundamentals to commercial systems. Appl. Microbiol. Biotechnol. 2006, 72, 211–222. [Google Scholar] [CrossRef]
- Studier, F.W. Protein production by auto-induction in high-density shaking cultures. Protein Expr. Purif. 2005, 41, 207–234. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Kuipers, G.; Niemiec, Ł.; Baumgarten, T.; Slotboom, D.J.; De Gier, J.; Hjelm, A. High-level production of membrane proteins in E. coli BL21(DE3) by omitting the inducer IPTG. Microb. Cell Fact. 2015, 14, 142. [Google Scholar] [PubMed]
- Ogino, H.; Miyamoto, K.; Yasuda, M.; Ishimi, K.; Ishikawa, H. Growth of organic solvent-tolerant Pseudomonas aeruginosa LST-03 in the presence of various organic solvents and production of lipolytic enzyme in the presence of cyclohexane. Biochem. Eng. J. 1999, 4, 1–6. [Google Scholar] [CrossRef]
- Nardini, M.; Lang, D.A.; Liebeton, K.; Jaeger, K.E.; Dijkstra, B.W. Crystal Structure of Pseudomonas aeruginosa Lipase in the open conformation. The Prototype for Family I.1 of Bacterial Lipases. J. Biol. Chem. 2000, 275, 31219–31225. [Google Scholar] [CrossRef] [PubMed]
- Thiruvengadam, K.; Baskaran, S.K.; Pennathur, G. Understanding domain movements and interactions of Pseudomonas aeruginosa lipase with lipid molecule tristearoyl glycerol: A molecular dynamics approach. J. Mol. Graph. Model. 2018, 85, 190–197. [Google Scholar]
- Kawata, T.; Ogino, H. Amino acid residues involved in organic solvent-stability of the LST-03 lipase. Biochem. Biophys. Res. Commun. 2010, 400, 384–388. [Google Scholar]
- Liebeton, K.; Zonta, A.; Schimossek, K.; Nardini, M.; Lang, D.; Dijkstra, B.W.; Reetz, M.T.; Jaeger, K.E. Directed evolution of an enantioselective lipase. Chem. Biol. 2000, 7, 709–718. [Google Scholar]
- Smith, R.S.; Iglewski, B.H.P. P. aeruginosa quorum-sensing systems and virulence. Curr. Opin. Microbiol. 2003, 6, 56–60. [Google Scholar] [CrossRef]
- Ogino, H.; Inoue, S.; Yasuda, M.; Doukyu, N. Hyper-activation of foldase-dependent lipase with lipase-specific foldase. J. Biotechnol. 2013, 166, 20–24. [Google Scholar] [CrossRef]
- Quyen, T.D.; Vu, C.H.; Thu Le, G.T. Enhancing functional production of a chaperone-dependent lipase in Escherichia coli using the dual expression cassette plasmid. Microb. Cell Fact. 2012, 11, 29. [Google Scholar] [CrossRef]
- West, S.E.; Iglewski, B.H. Codon usage in Pseudomonas aeruginosa. Nucleic Acids Res. 1988, 16, 9323–9335. [Google Scholar] [CrossRef] [PubMed]
- Briand, L.; Marcion, G.; Kriznik, A.; Heydel, J.M.; Artur, Y.; Garrido, C.; Seigneuric, R.; Neiers, F. A self-inducible heterologous protein expression system in Escherichia coli. Sci. Rep. 2016, 6, 33037. [Google Scholar] [CrossRef] [PubMed]
- Nair, R.; Salvi, P.; Banerjee, S.; Raiker, V.A.; Bandyopadhyay, S.; Soorapaneni, S.; Kotwal, P.; Padmanabhan, S. Yeast extract mediated autoinduction of lacUV5 promoter: An insight. N. Biotechnol. 2009, 26, 282–288. [Google Scholar] [PubMed]
- Mercatelli, E.; Barbieri, L.; Luchinat, E.; Banci, L. Direct structural evidence of protein redox regulation obtained by in-cell NMR. Biochim. Biophys. Acta - Mol. Cell Res. 2016, 1863, 198–204. [Google Scholar]
- de Marco, A. Strategies for successful recombinant expression of disulfide bond-dependent proteins in Escherichia coli. Microb. Cell Fact. 2009, 8, 26. [Google Scholar] [CrossRef]
- Calce, E.; Vitale, R.M.; Scaloni, A.; Amodeo, P.; De Luca, S. Air oxidation method employed for the disulfide bond formation of natural and synthetic peptides. Amino Acids 2015, 47, 1507–1515. [Google Scholar] [CrossRef]
- Haworth, N.L.; Gready, J.E.; George, R.A.; Wouters, M.A. Evaluating the stability of disulfide bridges in proteins: A torsional potential energy surface for diethyl disulfide. Mol. Simul. 2007, 33, 475–485. [Google Scholar] [CrossRef]
- Zhang, Y.; Schulten, K.; Gruebele, M.; Bansal, P.S.; Wilson, D.; Daly, N.L. Disulfide bridges: Bringing together frustrated structure in a bioactive peptide. Biophys. J. 2016, 110, 1744–1752. [Google Scholar]
- Dror, A.; Shemesh, E.; Dayan, N.; Fishman, A. Protein Engineering by Random Mutagenesis and Structure-Guided Consensus of Geobacillus stearothermophilus Lipase T6 for Enhanced Stability in Methanol. Appl. Environ. Microbiol. 2014, 80, 1515–1527. [Google Scholar] [CrossRef]
- Sambrook, J.; Russell, D.W. Molecular Cloning A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2012; ISBN 978-087969577-3. [Google Scholar]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar]
- Ernst, O.; Zor, T. Linearization of the Bradford Protein Assay. J. Vis. Exp. 2010, 38, e1918. [Google Scholar]
- Selvin, J.; Kennedy, J.; Lejon, D.P.H.; Kiran, G.S.; Dobson, A.D.W. Isolation identification and biochemical characterization of a novel halo-tolerant lipase from the metagenome of the marine sponge Haliclona simulans. Microb. Cell Fact. 2012, 11, 72. [Google Scholar] [PubMed]
- Xie, Y.; An, J.; Yang, G.; Wu, G.; Zhang, Y.; Cui, L.; Feng, Y. Enhanced Enzyme Kinetic Stability by Increasing Rigidity within the Active Site. J. Biol. Chem. 2014, 289, 7994–8006. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar]
- Burley, S.K.; Berman, H.M.; Kleywegt, G.J.; Markley, J.L.; Nakamura, H.; Velankar, S. Protein Data Bank (PDB): The Single Global Macromolecular Structure Archive. Methods Mol. Biol. 2017, 1607, 627–641. [Google Scholar]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple AMBER force fields and development of improved protein backbone parameters. Proteins 2006, 65, 712–725. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| E. coli SHuffle | E. coli BL21 (DE3) | |||
|---|---|---|---|---|
| Time (h) | Specific Activity U/mg with Lactose | Specific Activity U/mg without Lactose | Specific Activity U/mg with Lactose | Specific Activity U/mg without Lactose |
| 9 | 178 ± 0.2 | 112 ± 10 | 205 ± 21 | 143 ± 28 |
| 13 | 282 ± 1 | 197 ± 1 | 252 ± 21 | 338 ± 36 |
| 18 | 256 ± 26 | 202 ± 6 | 246 ± 20 | 267 ± 13 |
| 30 | 365 ± 20 | 463 ± 15 | 283 ± 17 | 354 ± 41 |
| 36 | 543 ± 29 | 465 ± 26 | 243 ± 5 | 349 ± 22 |
| Primer | Sequence | Feature |
|---|---|---|
| PafoR | 5′-ATGAAGAAGAAGTCTCTGCTCC-3′ | Amplification of the whole sequence of lipA. |
| Parev | 5′-CTACAGGCTGGCGTTCTTCAG-3′ | Amplification of the whole sequence of lipA. |
| Folfor | 5′-ATGGTGCCGGCCCCCCAGGTCATG-3′ | Amplification of the whole sequence of lif. |
| Perm | 5′-TCAGCGCTGCTCGGCCTGGCGCAT-3′ | Amplification of the whole sequence of lif. [17]; |
| NdelipF | 5′-GGAATTCCATATGAGCACCTACACCCAGACC-3′ | Amplification of lipA without signal peptide; recognition site for NdeI. |
| XholipR | 5′-CCGCTCGAGCTACAGGCTGGCGTTCTTCAG-3′ | Amplification of lipA without signal peptide; recognition site for XhoI; |
| Ncofol | 5′-CATGCCATGGTGCCGGCCCCCCAGGTCATG-3′ | Amplification of lif without inner membrane anchor; recognition site for NcoI. |
| PBR | 5′-CGATAAGCTTTCAGCGCTGCTCGGCCTGG-3′ | Amplification of lif; recognition site for HindIII; |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pulido, I.Y.; Prieto, E.; Pieffet, G.P.; Méndez, L.; Jiménez-Junca, C.A. Functional Heterologous Expression of Mature Lipase LipA from Pseudomonas aeruginosa PSA01 in Escherichia coli SHuffle and BL21 (DE3): Effect of the Expression Host on Thermal Stability and Solvent Tolerance of the Enzyme Produced. Int. J. Mol. Sci. 2020, 21, 3925. https://doi.org/10.3390/ijms21113925
Pulido IY, Prieto E, Pieffet GP, Méndez L, Jiménez-Junca CA. Functional Heterologous Expression of Mature Lipase LipA from Pseudomonas aeruginosa PSA01 in Escherichia coli SHuffle and BL21 (DE3): Effect of the Expression Host on Thermal Stability and Solvent Tolerance of the Enzyme Produced. International Journal of Molecular Sciences. 2020; 21(11):3925. https://doi.org/10.3390/ijms21113925
Chicago/Turabian StylePulido, Ingrid Yamile, Erlide Prieto, Gilles Paul Pieffet, Lina Méndez, and Carlos A. Jiménez-Junca. 2020. "Functional Heterologous Expression of Mature Lipase LipA from Pseudomonas aeruginosa PSA01 in Escherichia coli SHuffle and BL21 (DE3): Effect of the Expression Host on Thermal Stability and Solvent Tolerance of the Enzyme Produced" International Journal of Molecular Sciences 21, no. 11: 3925. https://doi.org/10.3390/ijms21113925
APA StylePulido, I. Y., Prieto, E., Pieffet, G. P., Méndez, L., & Jiménez-Junca, C. A. (2020). Functional Heterologous Expression of Mature Lipase LipA from Pseudomonas aeruginosa PSA01 in Escherichia coli SHuffle and BL21 (DE3): Effect of the Expression Host on Thermal Stability and Solvent Tolerance of the Enzyme Produced. International Journal of Molecular Sciences, 21(11), 3925. https://doi.org/10.3390/ijms21113925