Genomic Analysis of Vavilov’s Historic Chickpea Landraces Reveals Footprints of Environmental and Human Selection

 , , ,
, , ,  ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Germplasm Resources and Phenotyping
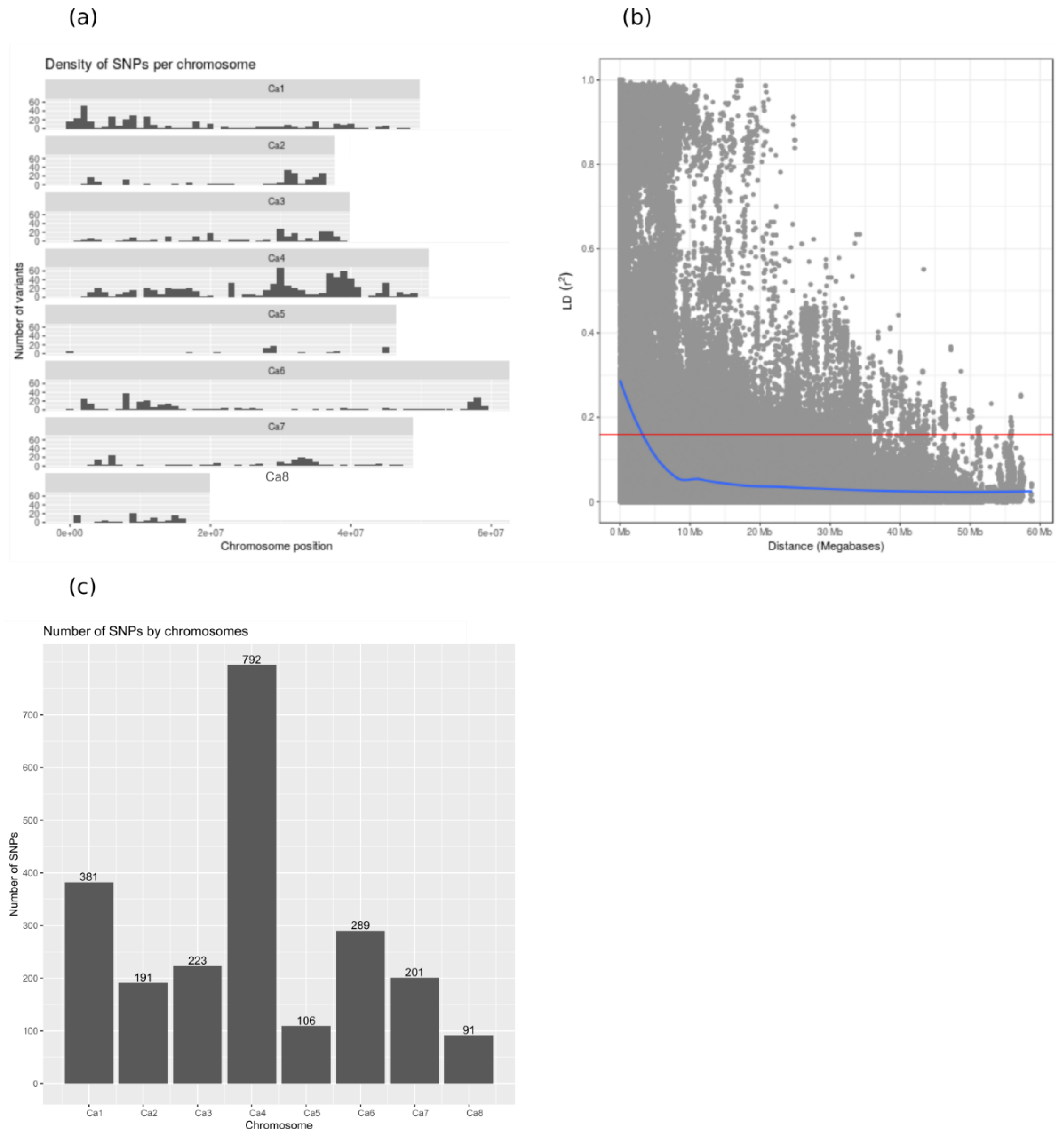
2.2. Marker Polymorphism Analysis
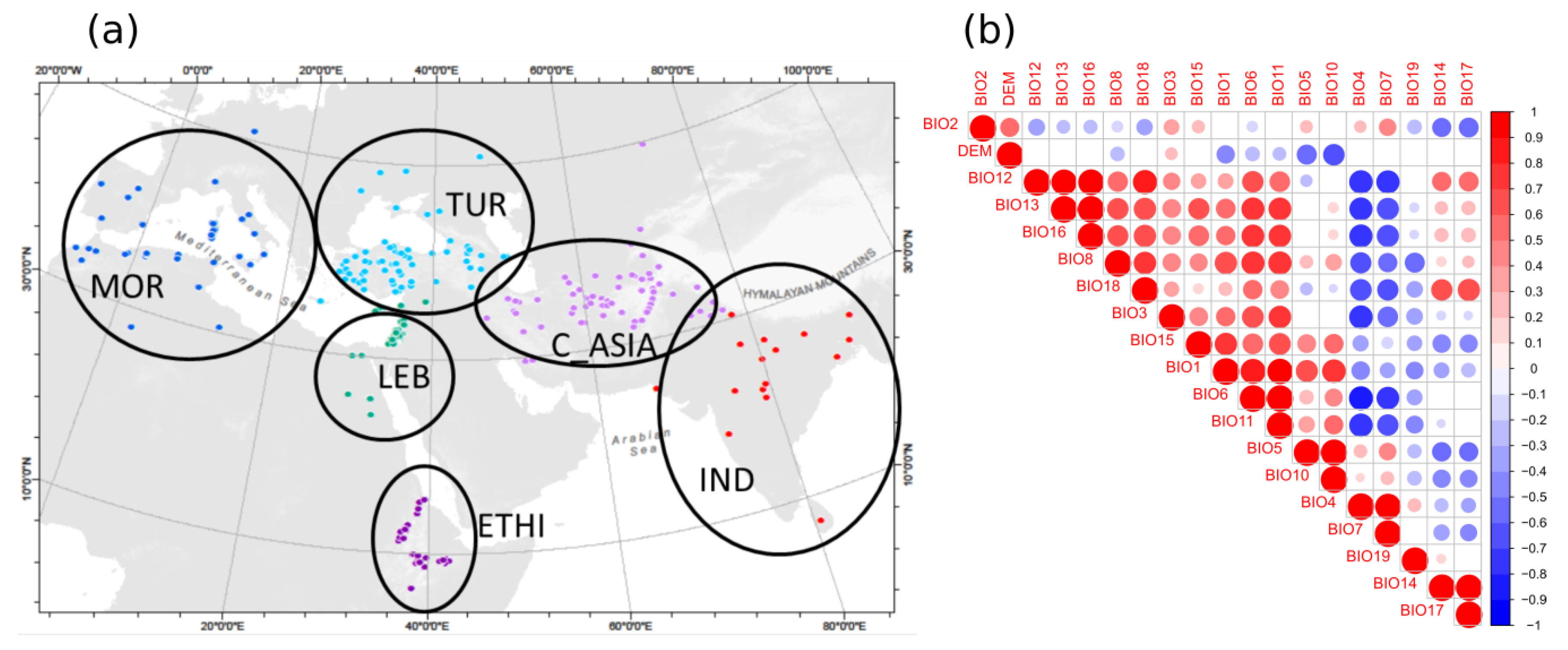
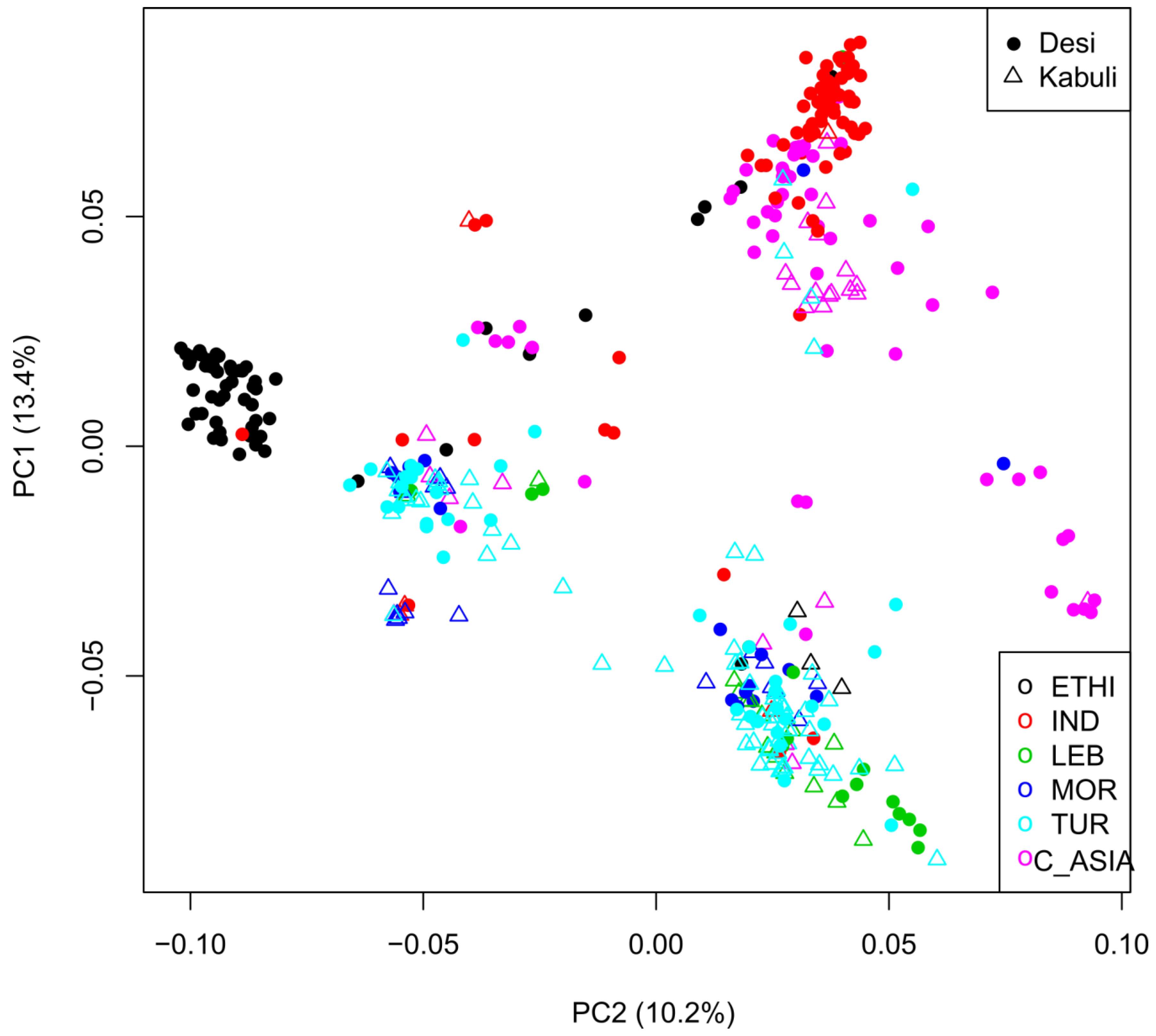
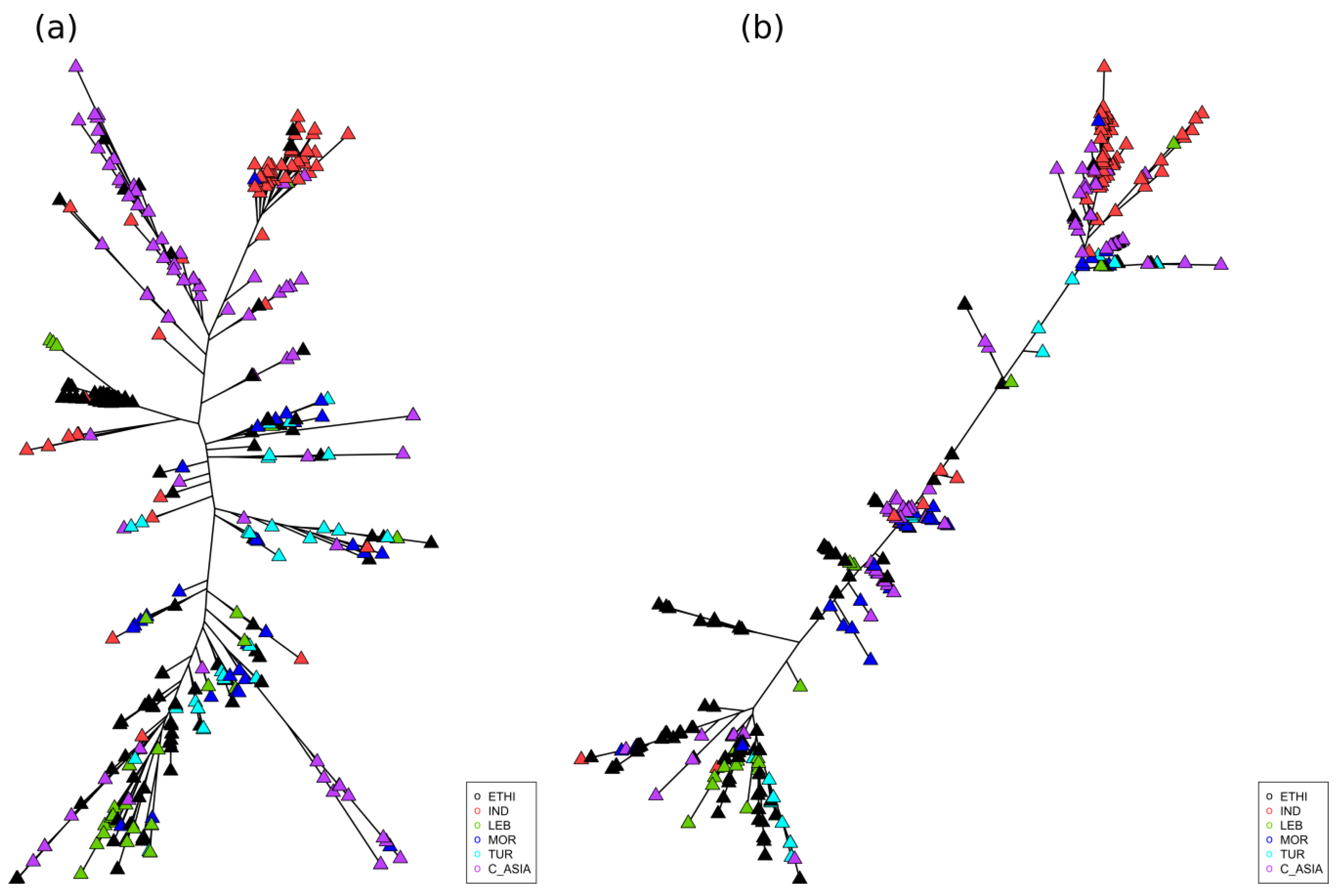
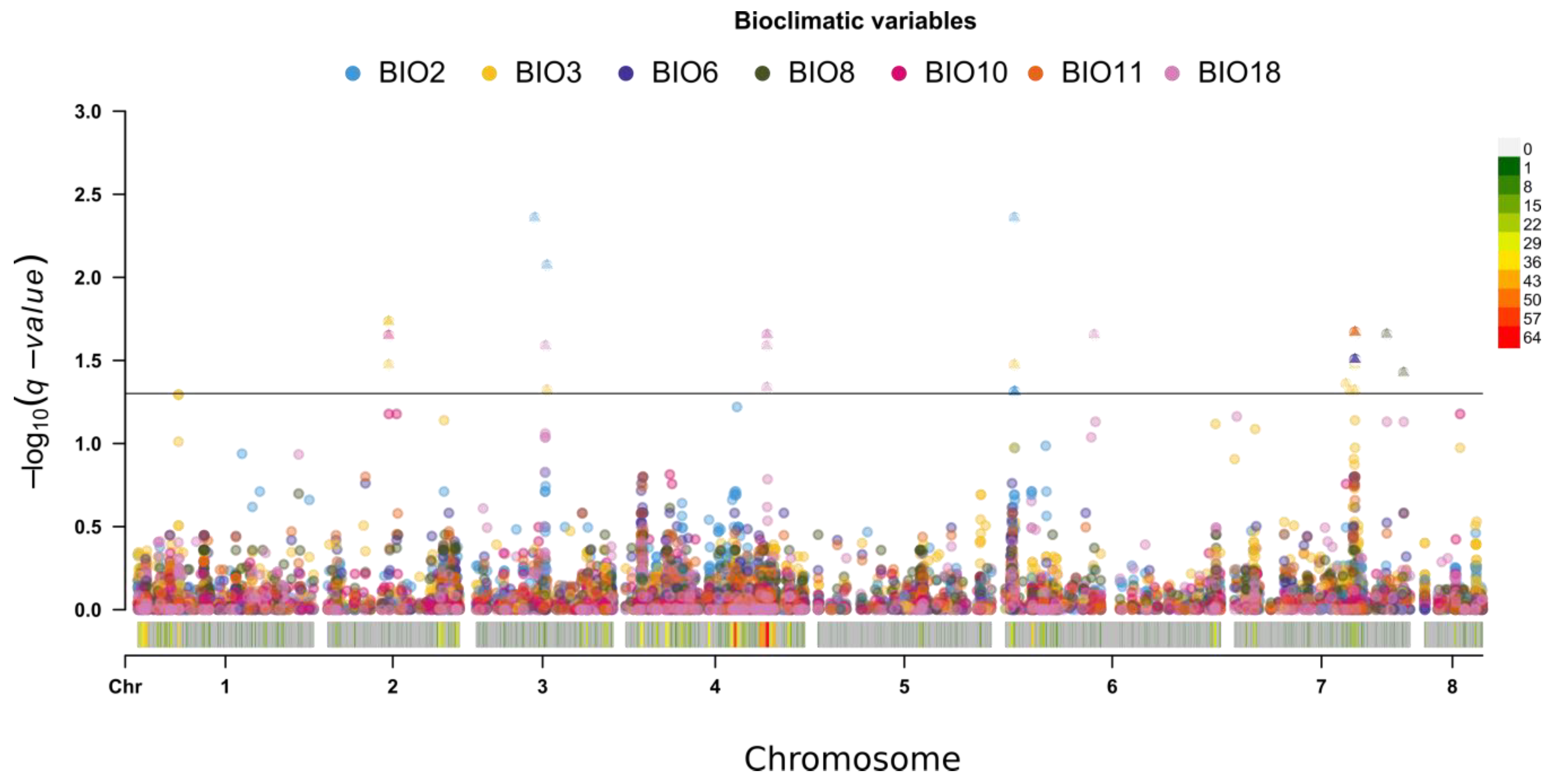
2.3. Geographic Analyses
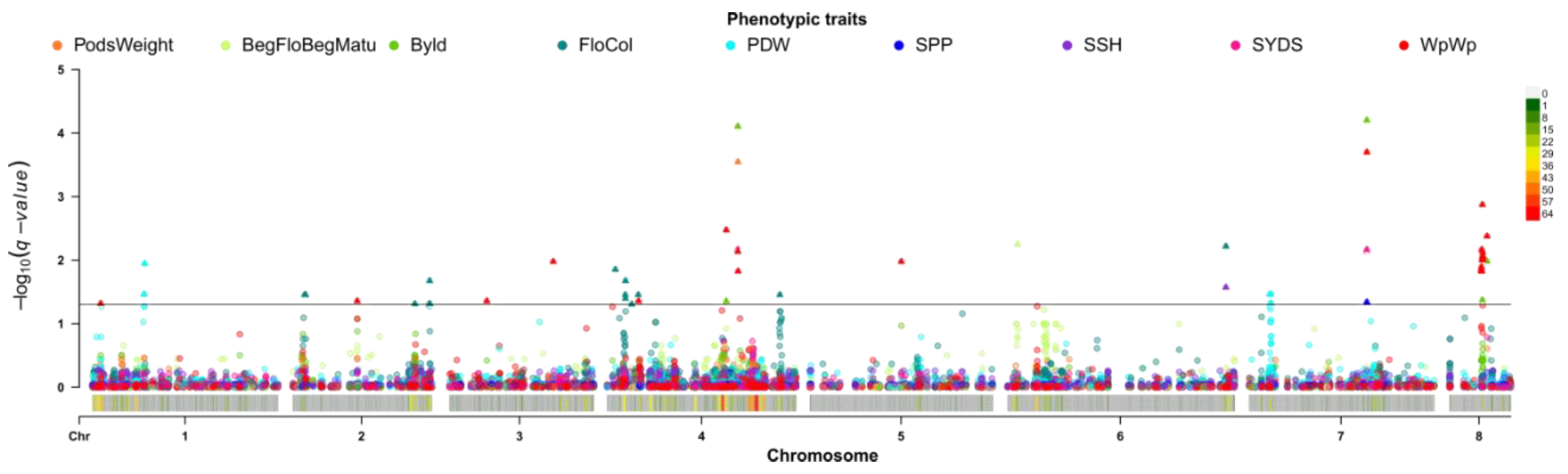
2.4. Single Trait Associations
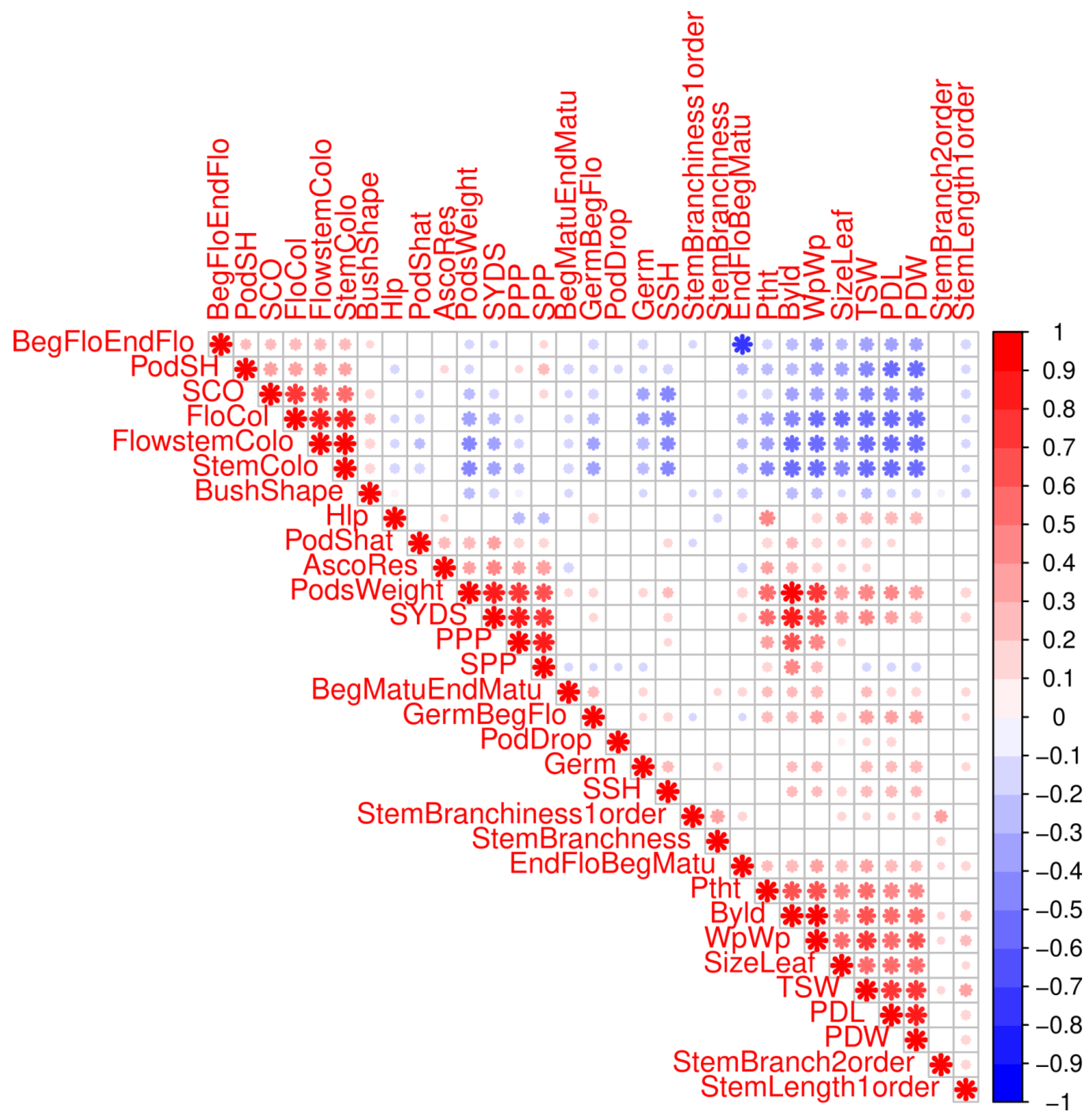
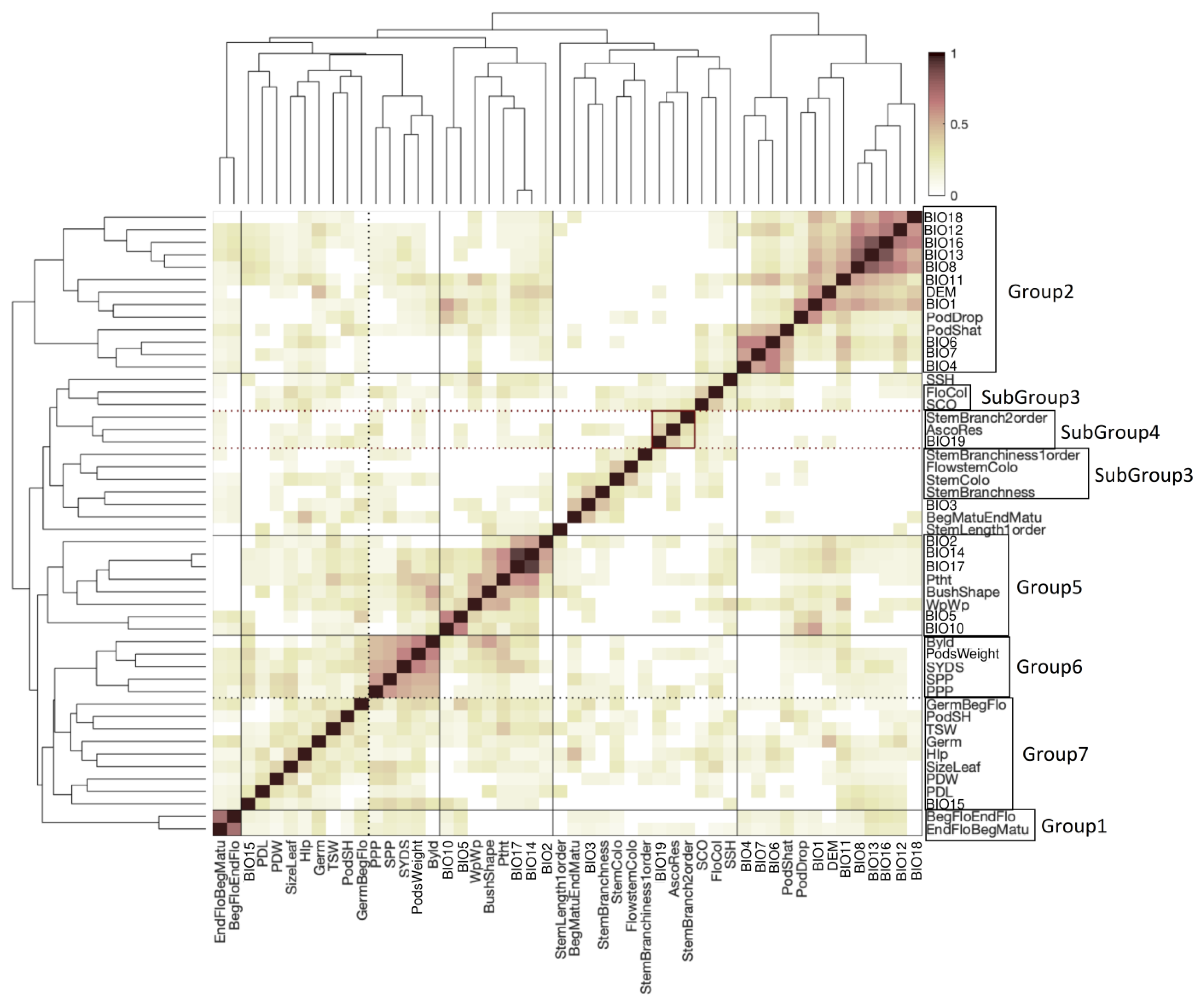
2.5. Clustering of Phenotypes and Variables Sharing Enriched Haploblocks
3. Discussion
4. Materials and Methods
4.1. Germplasm Resources and Phenotyping
4.2. Genotyping by Sequencing (GBS) and SNP Calling
4.3. Genetic Data Analyses
4.4. Bioclimatic Analysis
4.5. Mapping Approaches
4.6. Biogeographic Analyses
4.7. Haploblock Enrichment Analysis and Clustering of Enriched Haploblocks
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fairchild, D. The World was My Garden: Travels of Plant Explorer; LWW: New York, NY, USA, 1939; 495p. [Google Scholar]
- Maxted, N.; Dulloo, M.E.; Ford-Lloyd, B.V. Enhancing Crop Genepool Use: Capturing Wild Relative and Landrace Diversity for Crop Improvement; CABI: Oxfordshire, UK, 2016; 469p. [Google Scholar]
- Vishnyakova, M.A.; Burlyaeva, M.O.; Bulyntsev, S.V.; Seferova, I.V.; Plekhanova, E.S.; Nuzhdin, S.V. Chickpea landraces from centers of the crop origin: Diversity and differences. Sel’skokhozyaistvennaya biologiya. Agric. Biol. 2017, 52, 976–985. [Google Scholar]
- Plekhanova, E.; Vishnyakova, M.A.; Bulyntsev, S.; Chang, P.L.; Carrasquilla-Garcia, N.; Negash, K.; Nuzhdin, S.V. Genomic and phenotypic analysis of Vavilov’s historic landraces reveals the impact of environment and genomic islands of agronomic traits. Sci. Rep. 2017, 7, 4816. [Google Scholar] [CrossRef] [PubMed]
- Redden, R.J.; Berger, J.D. History and origin of Chickpea. In Chickpea Breeding & Management; Yadav, S.S., Redden, R., Chen, W., Sharma, B., Eds.; CABI: Wallingford, UK, 2007; pp. 1–13. [Google Scholar]
- Von Wettberg, E.J.; Chang, P.L.; Başdemir, F.; Carrasquila-Garcia, N.; Korbu, L.B.; Moenga, S.M.; Cordeiro, M.A. Ecology and community genomics of an important crop wild relative as a prelude to agricultural innovation. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Varma Penmetsa, R.; Carrasquilla-Garcia, N.; Bergmann, E.M.; Vance, L.; Castro, B.; Kassa, M.T.; Coyne, C.J. Multiple post-domestication origins of kabuli chickpea through allelic variation in a diversification-associated transcription factor. New Phytol. 2016, 211, 1440–1451. [Google Scholar] [CrossRef] [PubMed]
- Vavilov, N.I. The origin, variation, immunity and breeding of cultivated plants (Translated by S.K. Chestitee). Chron. Botonica 1951, 13, 1–366. [Google Scholar]
- Kale, S.M.; Jaganathan, D.; Ruperao, P.; Chen, C.; Punna, R.; Kudapa, H.; Garg, V. Prioritization of candidate genes in ‘QTL-hotspot’ region for drought tolerance in chickpea (Cicer arietinum L.). Sci. Rep. 2015, 5, 15296. [Google Scholar] [CrossRef]
- Thudi, M.; Khan, A.W.; Kumar, V.; Gaur, P.M.; Katta, K.; Garg, V.; Varshney, R.K. Whole genome re-sequencing reveals genome-wide variations among parental lines of 16 mapping populations in chickpea (Cicer arietinum L.). BMC Plant Biol. 2016, 16, 10. [Google Scholar] [CrossRef]
- Chollet, R.; Vidal, J.; O’Leary, M.H. PHOSPHOENOLPYRUVATE CARBOXYLASE: A ubiquitous, highly regulated enzyme in plants. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1996, 47, 273–298. [Google Scholar] [CrossRef]
- Minic, Z.; Rihouey, C.; Do, C.T.; Lerouge, P.; Jouanin, L. Purification and characterization of enzymes exhibiting beta-D-xylosidase activities in stem tissues of Arabidopsis. Plant Physiol. 2004, 135, 867–878. [Google Scholar] [CrossRef]
- Chen, X.; Yang, B.; Huang, W.; Wang, T.; Li, Y.; Zhong, Z.; Yang, L.; Li, S.; Tian, J. Comparative proteomic analysis reveals elevated capacity for photosynthesis in polyphenol oxidase expression-silenced Clematis terniflora DC. Leaves. Int. J. Mol. Sci. 2018, 19, 3897. [Google Scholar] [CrossRef]
- Ryšlavá, H.; Valenta, R.; Hýsková, V.; Křížek, T.; Liberda, J.; Coufal, P. Purification and enzymatic characterization of tobacco leaf β-N-acetylhexosaminidase. Biochimie 2014, 107 Pt B, 263–269. [Google Scholar] [CrossRef]
- Hellens, R.P.; Moreau, C.; Lin-Wang, K.; Schwinn, K.E.; Thomson, S.J.; Fiers, M.W.; Davies, K.M. Identification of mendel’s white flower character. PLoS ONE 2010, 5, e13230. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: Analysis and visualization of, L.D. and haplotype maps. Bioinformatics 2005, 21, 263–265. [Google Scholar] [CrossRef] [PubMed]
- Sergushichev, A. An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. BioRxiv 2016, 060012. [Google Scholar] [CrossRef]
- Olsen, K.M.; Wendel, J.F. A bountiful harvest: Genomic insights into crop domestication phenotypes. Annu. Rev. Plant Biol. 2013, 64, 47–70. [Google Scholar] [CrossRef]
- Upadhyaya, H.D.; Bajaj, D.; Das, S.; Saxena, M.S.; Badoni, S.; Kumar, V.; Parida, S.K. A genome-scale integrated approach aids in genetic dissection of complex flowering time trait in chickpea. Plant Mol. Biol. 2015, 89, 403–420. [Google Scholar] [CrossRef]
- Bajaj, D.; Upadhyaya, H.D.; Das, S.; Kumar, V.; Gowda, C.L.L.; Sharma, S.; Parida, S.K. Identification of candidate genes for dissecting complex branchnumber trait in chickpea. Plant Sci. 2016, 245, 61–70. [Google Scholar] [CrossRef]
- Kujur, A.; Upadhyaya, H.D.; Bajaj, D.; Gowda, C.L.L.; Sharma, S.; Tyagi, A.K.; Parida, S.K. Identification of candidate genes and natural allelic variants for QTLs governing plant height in chickpea. Sci. Rep. 2016, 6, 27968. [Google Scholar] [CrossRef]
- Das, S.; Upadhyaya, H.D.; Srivastava, R.; Bajaj, D.; Gowda, C.L.; Sharma, S.; Singh, S.; Tyagi, A.K.; Parida, S.K. Genome-wide insertion-deletion (InDel) marker discovery and genotyping for genomics-assisted breeding applications in chickpea. DNA Res. 2015, 22, 377–386. [Google Scholar] [CrossRef]
- Ortega, R.; Hecht, V.F.G.; Freeman, J.S.; Rubio, J.; Carrasquilla-Garcia, N.; Mir, R.R.; Penmetsa, R.V.; Cook, D.R.; Millan, T.; Weller, J.L. Altered Expression of an, F.T. Cluster underlies a major locus controlling domestication-related changes to chickpea phenology and growth habit. Front. Plant Sci. 2019, 10, 824. [Google Scholar] [CrossRef]
- Varshney, R.K.; Thudi, M.; Roorkiwal, M.; He, W.; Upadhyaya, H.D.; Yang, W.; Doddamani, D. Resequencing of 429 chickpea accessions from 45 countries provides insights into genome diversity, domestication and agronomic traits. Nat Genet. 2019, 51, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Sokolkova, A.B.; Chang, P.L.; Carrasquila-Garcia, N.; Noujdina, N.V.; Cook, D.R.; Nuzhdin, S.V.; Samsonova, M.G. The signatures of ecological adaptation in the genomes of chickpea landraces. Biophysics 2020, 65, 237–240. [Google Scholar] [CrossRef]
- Van-Oss, R.P.; Gopher, A.; Kerem, Z.; Peleg, Z.; Lev-Yadun, S.; Sherman, A.; Abbo, S. Independent selection for seed free tryptophan content and vernalization response in chickpea domestication. Plant Breed. 2018, 137, 290–300. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; DePristo, M.A. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; McVean, G. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Zheng, X.; Levine, D.; Shen, J.; Gogarten, S.; Laurie, C.; Weir, B. A High-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 2012, 28, 3326–3328. [Google Scholar] [CrossRef]
- Python Software Foundation. Python Language Reference, Version 2.7. Available online: http://www.python.org (accessed on 20 June 2018).
- R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. 2018. Available online: https://www.R-project.org/ (accessed on 20 June 2018).
- Lee, T.H.; Guo, H.; Wang, X.; Kim, C.; Paterson, A.H. SNPhylo: A pipeline to construct a phylogenetic tree from huge SNP data. BMC Genom. 2014, 15, 162. [Google Scholar] [CrossRef]
- Revell, L.J. phytools: An, R. package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 2012, 3, 217–223. [Google Scholar] [CrossRef]
- Paradis, E.; Schliep, K. Ape 5.0: An Environment for Modern Phylogenetics and Evolutionary Analyses in R. Bioinformatics. 2018. Available online: https://doi.org/10.1093/bioinformatics/bty633 (accessed on 15 June 2018).
- Mann, H.B.; Whitney, D.R. On a test of whether one of two random variables is stochastically larger than the other. Ann. Math. Stat. 1947, 18, 50–60. [Google Scholar] [CrossRef]
- Yang, J.; Lee, S.H.; Goddard, M.E.; Visscher, P.M. GCTA: A tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 2011, 88, 76–82. [Google Scholar] [CrossRef]
- Shapiro, S.S.; Wilk, M.B. An analysis of variance test for normality (complete samples). Biometrika 1965, 52, 591–611. [Google Scholar] [CrossRef]
- Harrell, F.E., Jr. Hmisc: Harrell Miscellaneous. R Package Version 4.1-1. 2018. Available online: https://CRAN.R-project.org/package=Hmisc (accessed on 15 June 2018).
- Lippert, C.; Listgarten, J.; Liu, Y.; Kadie, C.M.; Davidson, R.I.; Heckerman, D. FaST linear mixed models for genome-wide association studies. Nat. Methods 2011, 8, 833–835. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.W. Labdsv: Ordination and Multivariate Analysis for Ecology. R Package Version 1.8-0. 2016. Available online: http://CRAN.R-project.org/package=labdsv (accessed on 15 June 2018).
- Storey, J.D. The positive false discovery rate: A Bayesian interpretation and the q-Value. Source Ann. Stat. Ann. Stat. 2003, 31, 2013–2035. [Google Scholar] [CrossRef]
- CMplot: Circle Manhattan Plot. Available online: https://github.com/YinLiLin/R-CMplot (accessed on 20 June 2018).
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly Austin 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Dash, S.; Campbell, J.D.; Cannon, E.K.; Cleary, A.M.; Huang, W.; Kalberer, S.R.; Weeks, N.T. Legume information system (LegumeInfo. org): A key component of a set of federated data resources for the legume family. Nucl. Acids Res. 2016, 44, D1181–D1188. [Google Scholar] [CrossRef]
- Li, J.; Dai, X.; Liu, T.; Zhao, P.X. LegumeIP: An integrative database for comparative genomics and transcriptomics of model legumes. Nucleic Acids Res. 2012, 40, 1221–1229. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sokolkova, A.; Bulyntsev, S.V.; Chang, P.L.; Carrasquilla-Garcia, N.; Igolkina, A.A.; Noujdina, N.V.; von Wettberg, E.; Vishnyakova, M.A.; Cook, D.R.; Nuzhdin, S.V.; et al. Genomic Analysis of Vavilov’s Historic Chickpea Landraces Reveals Footprints of Environmental and Human Selection. Int. J. Mol. Sci. 2020, 21, 3952. https://doi.org/10.3390/ijms21113952
Sokolkova A, Bulyntsev SV, Chang PL, Carrasquilla-Garcia N, Igolkina AA, Noujdina NV, von Wettberg E, Vishnyakova MA, Cook DR, Nuzhdin SV, et al. Genomic Analysis of Vavilov’s Historic Chickpea Landraces Reveals Footprints of Environmental and Human Selection. International Journal of Molecular Sciences. 2020; 21(11):3952. https://doi.org/10.3390/ijms21113952
Chicago/Turabian StyleSokolkova, Alena, Sergey V. Bulyntsev, Peter L. Chang, Noelia Carrasquilla-Garcia, Anna A. Igolkina, Nina V. Noujdina, Eric von Wettberg, Margarita A. Vishnyakova, Douglas R. Cook, Sergey V. Nuzhdin, and et al. 2020. "Genomic Analysis of Vavilov’s Historic Chickpea Landraces Reveals Footprints of Environmental and Human Selection" International Journal of Molecular Sciences 21, no. 11: 3952. https://doi.org/10.3390/ijms21113952
APA StyleSokolkova, A., Bulyntsev, S. V., Chang, P. L., Carrasquilla-Garcia, N., Igolkina, A. A., Noujdina, N. V., von Wettberg, E., Vishnyakova, M. A., Cook, D. R., Nuzhdin, S. V., & Samsonova, M. G. (2020). Genomic Analysis of Vavilov’s Historic Chickpea Landraces Reveals Footprints of Environmental and Human Selection. International Journal of Molecular Sciences, 21(11), 3952. https://doi.org/10.3390/ijms21113952