Cell-Clearing Systems Bridging Repeat Expansion Proteotoxicity and Neuromuscular Junction Alterations in ALS and SBMA

 , ,
, , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
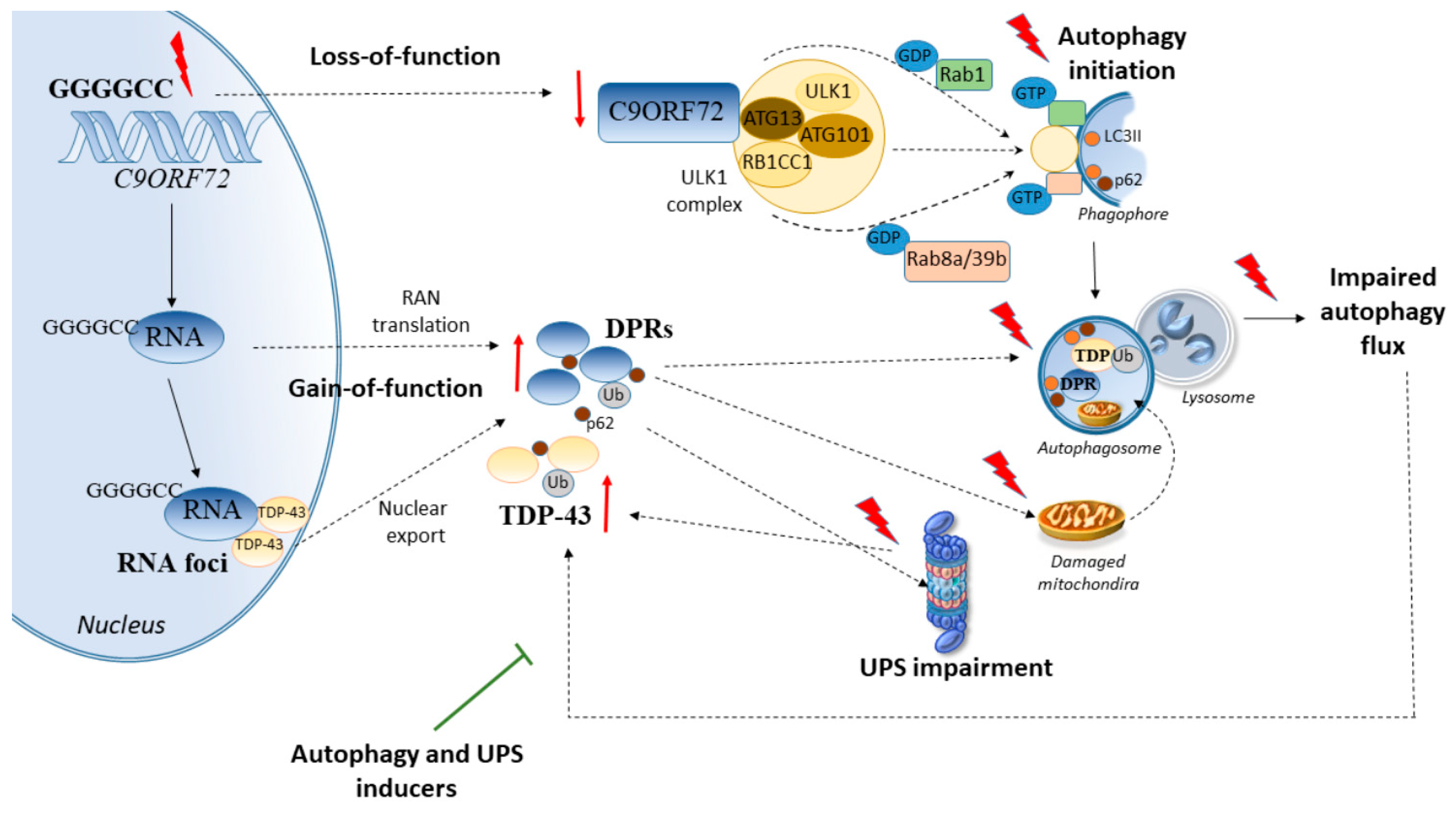
2. Cell-Clearing Systems and C9ORF72 Repeat Expansions in ALS
2.1. C9ORF72 Synergizes with Genetic Disease Modifiers to Alter Cell-Clearing Pathways
2.1.1. ATXN-2
2.1.2. VCP
2.1.3. PGRN
2.1.4. DCTN1
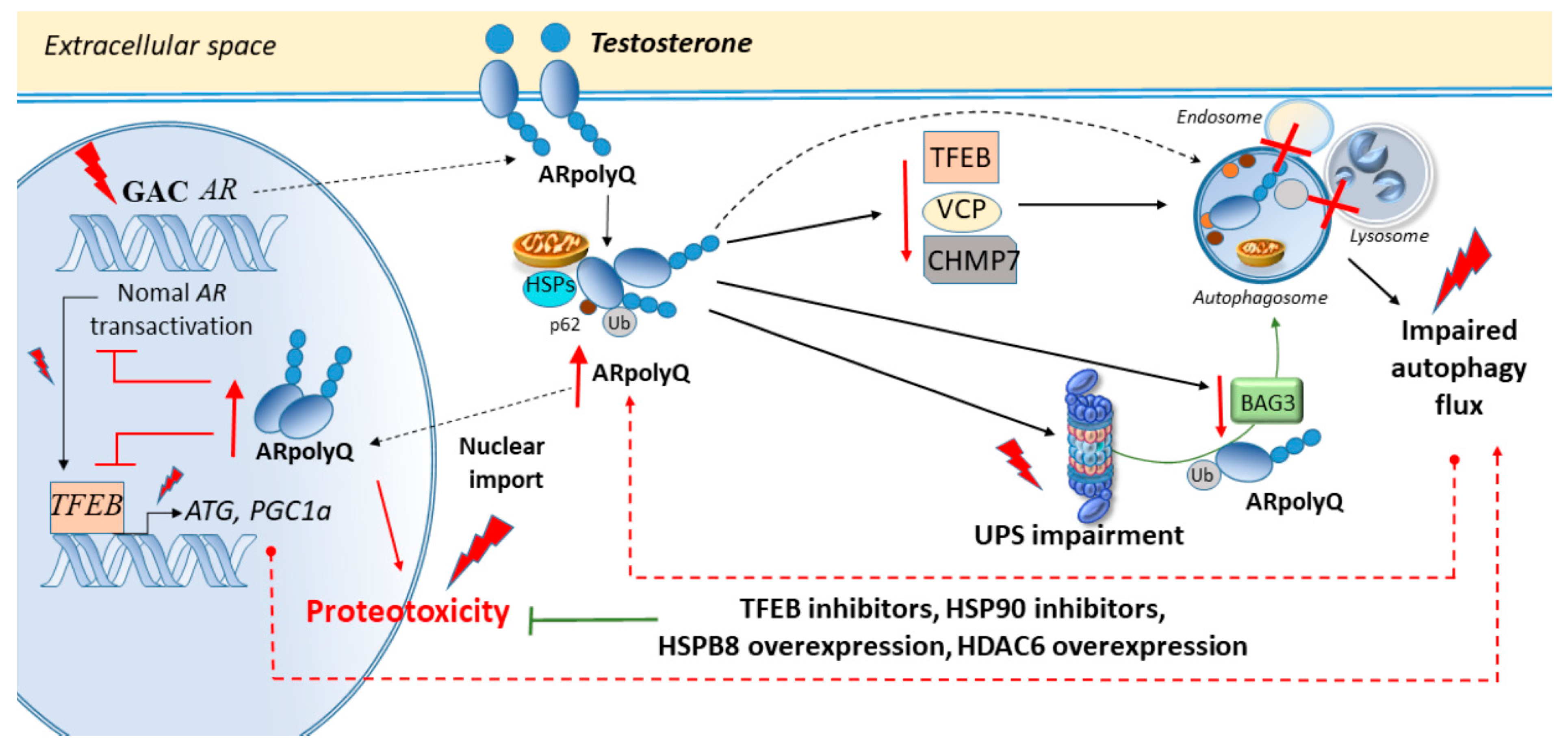
3. Cell-Clearing Systems and AR Nucleotide Repeat Expansions in SBMA
Autophagy and the UPS in SBMA Muscle and Axons
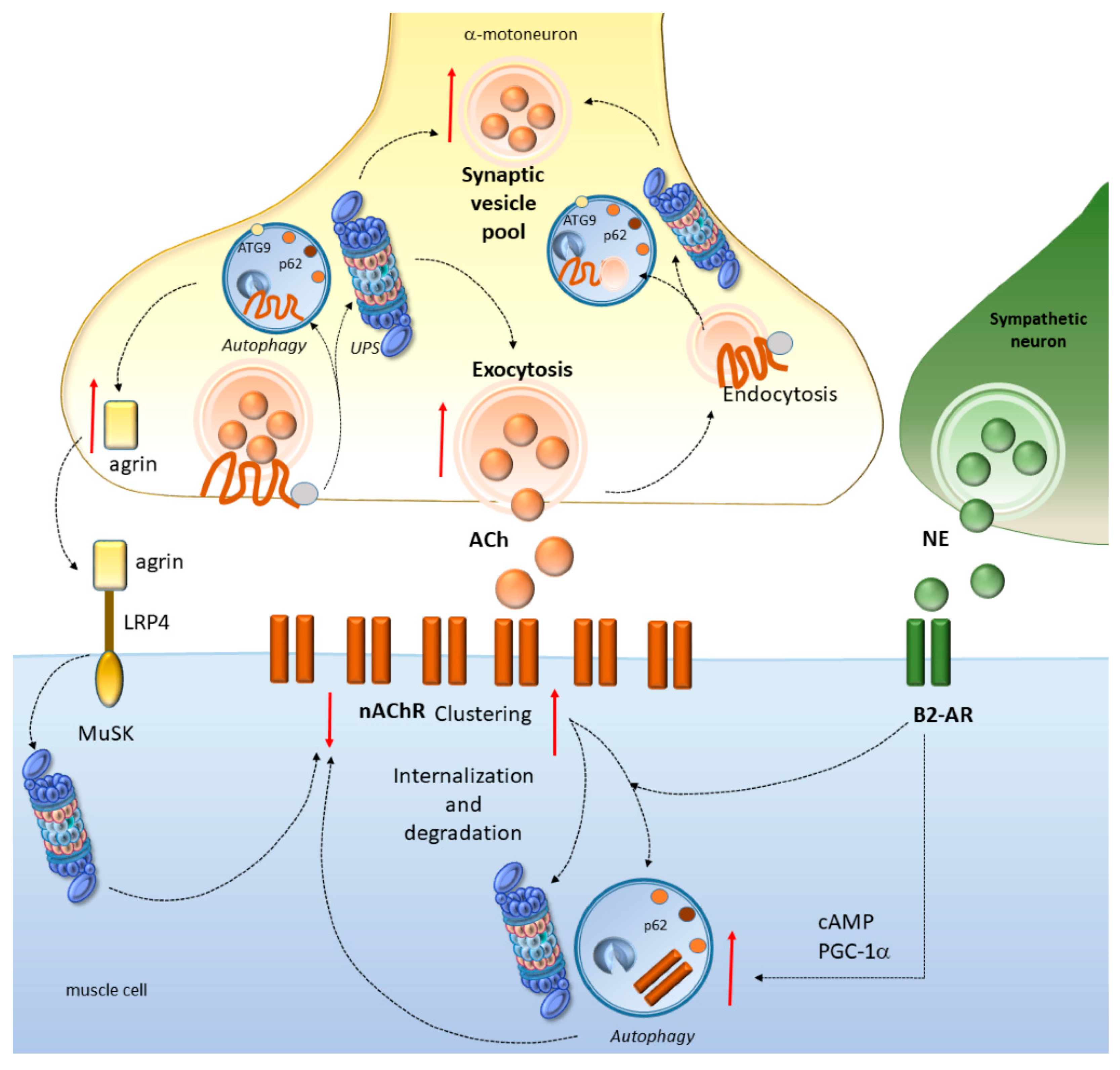
4. Potential Mechanisms Linking Cell-Clearing Systems and Neuromuscular Junction Alterations in ALS and SBMA
4.1. Autophagy and the UPS Regulate NMJ Development and Function
4.2. Autophagy and the UPS Regulate Neurotransmission at the NMJ
4.3. Autophagy and the UPS Regulate nAChR Turnover at the NMJ
4.4. Autophagy Converging with the Sympathetic Innervation of NMJs
5. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| UPS | ubiquitin proteasome system |
| C9ORF72 | chromosome 9 open reading frame 72 |
| AR | androgen receptor |
| ALS | amyotrophic lateral sclerosis |
| SMBA | spinal bulbar muscular atrophy |
| FTD | frontotemporal dementia |
| NMJ | neuromuscular junction |
| ARpolyQ | polyglutamine-expanded AR |
| DPR | dipeptide repeat |
| AChRs | acetylcholine receptors |
| B2-ARs | adrenergic beta2 receptors |
| TDP-43 | TAR DNA-binding protein 43 |
| SOD1 | superoxide dismutase 1 |
| TARDBP | transcription of RNA activating protein/TAR DNA-binding protein |
| FUS | fused in sarcoma |
| OPTN | optineurin |
| UBQLN2 | ubiquilin-2 |
| PGRN | progranulin |
| ATXN-2 | ataxin-2 |
| VCP | valosin-containing protein |
| DCTN1 | dynactin |
| mTORC1 | mammalian target of rapamycin complex 1 |
| SQSTM1/p62 | sequestosome 1/p62 |
| HSPA8 | heat shock protein family A (Hsp70) member 8 |
| Hsc70 | heat shock cognate 71 kDa protein |
| BAG1/3 | Bcl-2-associated athanogene 1/3 |
| TFEB | transcription factor EB |
| GSK3b | glycogen synthase kinase 3beta |
| CHMP2/7 | charged multivesicular body protein 2/7 |
| ESCRT-III | endosomal sorting complexes required for transport III |
| TMEM184b | transmembrane protein 184B |
| MuSK | muscle-specific receptor tyrosine kinase |
| PPARGC1A or PGC-1α | peroxisome proliferator-activated receptor γ-coactivator 1α |
References
- Madeo, F.; Eisenberg, T.; Kroemer, G. Autophagy for the avoidance of neurodegeneration. Genes Dev. 2009, 23, 2253–2259. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A.; Kwon, Y.T. Degradation of misfolded proteins in neurodegenerative diseases: Therapeutic targets and strategies. Exp. Mol. Med. 2015, 47, e147. [Google Scholar] [CrossRef] [PubMed]
- Ryskalin, L.; Busceti, C.L.; Limanaqi, F.; Biagioni, F.; Gambardella, S.; Fornai, F. A Focus on the Beneficial Effects of Alpha Synuclein and a Re-Appraisal of Synucleinopathies. Curr. Protein Pept. Sci. 2018, 19, 598–611. [Google Scholar] [CrossRef] [PubMed]
- Limanaqi, F.; Biagioni, F.; Gambardella, S.; Familiari, P.; Frati, A.; Fornai, F. Promiscuous Roles of Autophagy and Proteasome in Neurodegenerative Proteinopathies. Int. J. Mol. Sci. 2020, 21, 3028. [Google Scholar] [CrossRef]
- Jimenez-Sanchez, M.; Thomson, F.; Zavodszky, E.; Rubinsztein, D.C. Autophagy and polyglutamine diseases. Prog. Neurobiol. 2012, 97, 67–82. [Google Scholar] [CrossRef]
- Pearce, M.M.P.; Kopito, R.R. Prion-Like Characteristics of Polyglutamine-Containing Proteins. Cold Spring Harb. Perspect. Med. 2018, 8, a024257. [Google Scholar] [CrossRef]
- Babić Leko, M.; Župunski, V.; Kirincich, J.; Smilović, D.; Hortobágyi, T.; Hof, P.R.; Šimić, G. Molecular Mechanisms of Neurodegeneration Related to C9orf72 Hexanucleotide Repeat Expansion. Behav. Neurol. 2019, 2019, 2909168. [Google Scholar] [CrossRef]
- Davies, J.E.; Sarkar, S.; Rubinsztein, D.C. The ubiquitin proteasome system in Huntington’s disease and the spinocerebellar ataxias. BMC Biochem. 2007, 8 (Suppl. 1), S2. [Google Scholar] [CrossRef]
- Abel, A.; Walcott, J.; Woods, J.; Duda, J.; Merry, D.E. Expression of expanded repeat androgen receptor produces neurologic disease in transgenic mice. Hum. Mol. Genet. 2001, 10, 107–116. [Google Scholar] [CrossRef][Green Version]
- Cicardi, M.E.; Cristofani, R.; Crippa, V.; Ferrari, V.; Tedesco, B.; Casarotto, E.; Chierichetti, M.; Galbiati, M.; Piccolella, M.; Messi, E.; et al. Autophagic and Proteasomal Mediated Removal of Mutant Androgen Receptor in Muscle Models of Spinal and Bulbar Muscular Atrophy. Front. Endocrinol. (Lausanne) 2019, 10, 569. [Google Scholar] [CrossRef]
- Van Blitterswijk, M.; DeJesus-Hernandez, M.; Rademakers, R. How do C9ORF72 repeat expansions cause amyotrophic lateral sclerosis and frontotemporal dementia: Can we learn from other noncoding repeat expansion disorders? Curr. Opin. Neurol. 2012, 25, 689–700. [Google Scholar] [CrossRef] [PubMed]
- La Spada, A.R.; Wilson, E.M.; Lubahn, D.B.; Harding, A.E.; Fischbeck, K.H. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 1991, 352, 77–79. [Google Scholar] [CrossRef] [PubMed]
- Parboosingh, J.S.; Figlewicz, D.A.; Krizus, A.; Meininger, V.; Azad, N.A.; Newman, D.S.; Rouleau, G.A. Spinobulbar muscular atrophy can mimic ALS: The importance of genetic testing in male patients with atypical ALS. Neurology 1997, 49, 568–572. [Google Scholar] [CrossRef] [PubMed]
- Pradat, P.F. SBMA: A rare disease but a classic ALS mimic syndrome. Presse Med. 2014, 43, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Carnio, S.; LoVerso, F.; Baraibar, M.A.; Longa, E.; Khan, M.M.; Maffei, M.; Reischl, M.; Canepari, M.; Loefler, S.; Kern, H.; et al. Autophagy impairment in muscle induces neuromuscular junction degeneration and precocious aging. Cell Rep. 2014, 8, 1509–1521. [Google Scholar] [CrossRef] [PubMed]
- Malik, B.; Devine, H.; Patani, R.; La Spada, A.R.; Hanna, M.G.; Greensmith, L. Gene expression analysis reveals early dysregulation of disease pathways and links Chmp7 to pathogenesis of spinal and bulbar muscular atrophy. Sci. Rep. 2019, 9, 3539. [Google Scholar] [CrossRef]
- Rudnick, N.D.; Griffey, C.J.; Guarnieri, P.; Gerbino, V.; Wang, X.; Piersaint, J.A.; Tapia, J.C.; Rich, M.M.; Maniatis, T. Distinct roles for motor neuron autophagy early and late in the SOD1G93A mouse model of ALS. Proc. Natl. Acad. Sci. USA 2017, 114, E8294–E8303. [Google Scholar] [CrossRef]
- Zhu, Q.; Jiang, J.; Gendron, T.F.; McAlonis-Downes, M.; Jiang, L.; Taylor, A.; Diaz Garcia, S.; Ghosh Dastidar, S.; Rodriguez, M.J.; King, P.; et al. Reduced C9ORF72 function exacerbates gain of toxicity from ALS/FTD-causing repeat expansion in C9orf72. Nat. Neurosci. 2020. [Google Scholar] [CrossRef]
- Khosravi, B.; LaClair, K.D.; Riemenschneider, H.; Zhou, Q.; Frottin, F.; Mareljic, N.; Czuppa, M.; Farny, D.; Hartmann, H.; Michaelsen, M.; et al. Cell-to-cell transmission of C9orf72 poly-(Gly-Ala) triggers key features of ALS/FTD. EMBO J. 2020, 39, e102811. [Google Scholar] [CrossRef]
- Boivin, M.; Pfister, V.; Gaucherot, A.; Ruffenach, F.; Negroni, L.; Sellier, C.; Charlet-Berguerand, N. Reduced autophagy upon C9ORF72 loss synergizes with dipeptide repeat protein toxicity in G4C2 repeat expansion disorders. EMBO J. 2020, 39, e100574. [Google Scholar] [CrossRef]
- Cristofani, R.; Crippa, V.; Rusmini, P.; Cicardi, M.E.; Meroni, M.; Licata, N.V.; Sala, G.; Giorgetti, E.; Grunseich, C.; Galbiati, M.; et al. Inhibition of retrograde transport modulates misfolded protein accumulation and clearance in motoneuron diseases. Autophagy 2017, 13, 1280–1303. [Google Scholar] [CrossRef] [PubMed]
- Webster, C.P.; Smith, E.F.; Bauer, C.S.; Moller, A.; Hautbergue, G.M.; Ferraiuolo, L.; Myszczynska, M.A.; Higginbottom, A.; Walsh, M.J.; Whitworth, A.J.; et al. The C9orf72 protein interacts with Rab1a and the ULK1 complex to regulate initiation of autophagy. EMBO J. 2016, 35, 1656–1676. [Google Scholar] [CrossRef] [PubMed]
- Cortes, C.J.; Miranda, H.C.; Frankowski, H.; Batlevi, Y.; Young, J.E.; Le, A.; Ivanov, N.; Sopher, B.L.; Carromeu, C.; Muotri, A.R.; et al. Polyglutamine-expanded androgen receptor interferes with TFEB to elicit autophagy defects in SBMA. Nat. Neurosci. 2014, 17, 1180–1189. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Lan, M.; Mojsilovic-Petrovic, J.; Choi, W.H.; Safren, N.; Barmada, S.; Lee, M.J.; Kalb, R. The Proline/Arginine Dipeptide from Hexanucleotide Repeat Expanded C9ORF72 Inhibits the Proteasome. eNeuro 2017, 4. [Google Scholar] [CrossRef] [PubMed]
- Thibaudeau, T.A.; Anderson, R.T.; Smith, D.M. A common mechanism of proteasome impairment by neurodegenerative disease-associated oligomers. Nat. Commun. 2018, 9, 1097. [Google Scholar] [CrossRef] [PubMed]
- Abo-Rady, M.; Kalmbach, N.; Pal, A.; Schludi, C.; Janosch, A.; Richter, T.; Freitag, P.; Bickle, M.; Kahlert, A.K.; Petri, S.; et al. Knocking out C9ORF72 Exacerbates Axonal Trafficking Defects Associated with Hexanucleotide Repeat Expansion and Reduces Levels of Heat Shock Proteins. Stem Cell Rep. 2020, 14, 390–405. [Google Scholar] [CrossRef]
- Thomas, P.S., Jr.; Fraley, G.S.; Damian, V.; Woodke, L.B.; Zapata, F.; Sopher, B.L.; Plymate, S.R.; La Spada, A.R. Loss of endogenous androgen receptor protein accelerates motor neuron degeneration and accentuates androgen insensitivity in a mouse model of X-linked spinal and bulbar muscular atrophy. Hum. Mol. Genet. 2006, 15, 2225–2238. [Google Scholar] [CrossRef]
- Blessing, A.M.; Rajapakshe, K.; Reddy Bollu, L.; Shi, Y.; White, M.A.; Pham, A.H.; Lin, C.; Jonsson, P.; Cortes, C.J.; Cheung, E.; et al. Transcriptional regulation of core autophagy and lysosomal genes by the androgen receptor promotes prostate cancer progression. Autophagy 2017, 13, 506–521. [Google Scholar] [CrossRef]
- Ciura, S.; Sellier, C.; Campanari, M.L.; Charlet-Berguerand, N.; Kabashi, E. The most prevalent genetic cause of ALS-FTD, C9orf72 synergizes the toxicity of ATXN2 intermediate polyglutamine repeats through the autophagy pathway. Autophagy 2016, 12, 1406–1408. [Google Scholar] [CrossRef]
- Pasquali, L.; Ruffoli, R.; Fulceri, F.; Pietracupa, S.; Siciliano, G.; Paparelli, A.; Fornai, F. The role of autophagy: What can be learned from the genetic forms of amyotrophic lateral sclerosis. CNS Neurol. Disord. Drug Targets 2010, 9, 268–278. [Google Scholar] [CrossRef]
- Ferrucci, M.; Fulceri, F.; Toti, L.; Soldani, P.; Siciliano, G.; Paparelli, A.; Fornai, F. Protein clearing pathways in ALS. Arch. Ital. Biol. 2011, 149, 121–149. [Google Scholar] [CrossRef] [PubMed]
- Maurel, C.; Dangoumau, A.; Marouillat, S.; Brulard, C.; Chami, A.; Hergesheimer, R.; Corcia, P.; Blasco, H.; Andres, C.R.; Vourc’h, P. Causative Genes in Amyotrophic Lateral Sclerosis and Protein Degradation Pathways: A Link to Neurodegeneration. Mol. Neurobiol. 2018, 55, 6480–6499. [Google Scholar] [CrossRef] [PubMed]
- Manzano, R.; Sorarú, G.; Grunseich, C.; Fratta, P.; Zuccaro, E.; Pennuto, M.; Rinaldi, C. Beyond motor neurons: Expanding the clinical spectrum in Kennedy’s disease. J. Neurol. Neurosurg. Psychiatry 2018, 89, 808–812. [Google Scholar] [CrossRef] [PubMed]
- Starr, A.; Sattler, R. Synaptic dysfunction and altered excitability in C9ORF72 ALS/FTD. Brain Res. 2018, 1693, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Halievski, K.; Henley, C.; Atchison, W.D.; Katsuno, M.; Adachi, H.; Sobue, G.; Breedlove, S.M.; Jordan, C.L. Defects in Neuromuscular Transmission May Underlie Motor Dysfunction in Spinal and Bulbar Muscular Atrophy. J. Neurosci. 2016, 36, 5094–5106. [Google Scholar] [CrossRef]
- Dupuis, L.; Echaniz-Laguna, A. Skeletal muscle in motor neuron diseases: Therapeutic target and delivery route for potential treatments. Curr. Drug Targets 2010, 11, 1250–1261. [Google Scholar] [CrossRef]
- Clark, J.A.; Southam, K.A.; Blizzard, C.A.; King, A.E.; Dickson, T.C. Axonal degeneration, distal collateral branching and neuromuscular junction architecture alterations occur prior to symptom onset in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. J. Chem. Neuroanat. 2016, 76, 35–47. [Google Scholar] [CrossRef]
- Onodera, K.; Shimojo, D.; Ishihara, Y.; Yano, M.; Miya, F.; Banno, H.; Kuzumaki, N.; Ito, T.; Okada, R.; de Araújo Herculano, B.; et al. Unveiling synapse pathology in spinal bulbar muscular atrophy by genome-wide transcriptome analysis of purified motor neurons derived from disease specific iPSCs. Mol. Brain 2020, 13, 18. [Google Scholar] [CrossRef]
- Shen, D.N.; Zhang, L.H.; Wei, E.Q.; Yang, Y. Autophagy in synaptic development, function, and pathology. Neurosci. Bull. 2015, 31, 416–426. [Google Scholar] [CrossRef]
- Uytterhoeven, V.; Lauwers, E.; Maes, I.; Miskiewicz, K.; Melo, M.N.; Swerts, J.; Kuenen, S.; Wittocx, R.; Corthout, N.; Marrink, S.J.; et al. Hsc70-4 Deforms Membranes to Promote Synaptic Protein Turnover by Endosomal Microautophagy. Neuron 2015, 88, 735–748. [Google Scholar] [CrossRef]
- Ding, M.; Shen, K. The role of the ubiquitin proteasome system in synapse remodeling and neurodegenerative diseases. Bioessays 2008, 30, 1075–1083. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, B.J.; Wilson, S.M.; Jung, H.; Miller, R.J. Altered neurotransmitter release machinery in mice deficient for the deubiquitinating enzyme Usp14. Am. J. Physiol. Cell. Physiol. 2012, 302, C698–C708. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.J.; Li, X.X.; Yu, Y.; Chiu, A.P.; Lo, L.H.; To, J.C.; Rowlands, D.K.; Keng, V.W. Schwann cell-specific PTEN and EGFR dysfunctions affect neuromuscular junction development by impairing Agrin signaling and autophagy. Biochem. Biophys. Res. Commun. 2019, 515, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Rudolf, R.; Straka, T. Nicotinic acetylcholine receptor at vertebrate motor endplates: Endocytosis, recycling, and degradation. Neurosci. Lett. 2019, 711, 134434. [Google Scholar] [CrossRef]
- Rudolf, R.; Deschenes, M.R.; Sandri, M. Neuromuscular junction degeneration in muscle wasting. Curr. Opin. Clin. Nutr. Metab. Care 2016, 19, 177–181. [Google Scholar] [CrossRef]
- Aránguiz-Urroz, P.; Canales, J.; Copaja, M.; Troncoso, R.; Vicencio, J.M.; Carrillo, C.; Lara, H.; Lavandero, S.; Díaz-Araya, G. Beta(2)-adrenergic receptor regulates cardiac fibroblast autophagy and collagen degradation. Biochim. Biophys. Acta 2011, 1812, 23–31. [Google Scholar] [CrossRef]
- Lynch, E.; Semrad, T.; Belsito, V.S.; FitzGibbons, C.; Reilly, M.; Hayakawa, K.; Suzuki, M. C9ORF72-related cellular pathology in skeletal myocytes derived from ALS-patient induced pluripotent stem cells. Dis. Model Mech. 2019, 12, dmm039552. [Google Scholar] [CrossRef]
- Cykowski, M.D.; Powell, S.Z.; Appel, J.W.; Arumanayagam, A.S.; Rivera, A.L.; Appel, S.H. Phosphorylated TDP-43 (pTDP-43) aggregates in the axial skeletal muscle of patients with sporadic and familial amyotrophic lateral sclerosis. Acta Neuropathol. Commun. 2018, 6, 28. [Google Scholar] [CrossRef]
- Ho, W.Y.; Tai, Y.K.; Chang, J.C.; Liang, J.; Tyan, S.H.; Chen, S.; Guan, J.L.; Zhou, H.; Shen, H.M.; Koo, E.; et al. The ALS-FTD-linked gene product, C9orf72, regulates neuronal morphogenesis via autophagy. Autophagy 2019, 15, 827–842. [Google Scholar] [CrossRef]
- Guo, Q.; Lehmer, C.; Martínez-Sánchez, A.; Rudack, T.; Beck, F.; Hartmann, H.; Pérez-Berlanga, M.; Frottin, F.; Hipp, M.S.; Hartl, F.U.; et al. In Situ Structure of Neuronal C9orf72 Poly-GA Aggregates Reveals Proteasome Recruitment. Cell 2018, 172, 696–705.e12. [Google Scholar] [CrossRef]
- Yamakawa, M.; Ito, D.; Honda, T.; Kubo, K.; Noda, M.; Nakajima, K.; Suzuki, N. Characterization of the dipeptide repeat protein in the molecular pathogenesis of c9FTD/ALS. Hum. Mol. Genet. 2015, 24, 1630–1645. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; Gendron, T.F.; Grima, J.C.; Sasaguri, H.; Jansen-West, K.; Xu, Y.F.; Katzman, R.B.; Gass, J.; Murray, M.E.; Shinohara, M.; et al. C9ORF72 poly(GA) aggregates sequester and impair HR23 andnucleocytoplasmic transport proteins. Nat. Neurosci. 2016, 19, 668–677. [Google Scholar] [CrossRef] [PubMed]
- Boeve, B.F.; Boylan, K.B.; Graff-Radford, N.R.; DeJesus-Hernandez, M.; Knopman, D.S.; Pedraza, O.; Vemuri, P.; Jones, D.; Lowe, V.; Murray, M.E.; et al. Characterization of frontotemporal dementia and/or amyotrophic lateral sclerosis associated with the GGGGCC repeat expansion in C9ORF72. Brain 2012, 135, 765–783. [Google Scholar] [CrossRef] [PubMed]
- Cooper-Knock, J.; Hewitt, C.; Highley, J.R.; Brockington, A.; Milano, A.; Man, S.; Martindale, J.; Hartley, J.; Walsh, T.; Gelsthorpe, C.; et al. Clinico-pathological features in amyotrophic lateral sclerosis with expansions in C9ORF72. Brain 2012, 135, 751–764. [Google Scholar] [CrossRef]
- Al-Sarraj, S.; King, A.; Troakes, C.; Smith, B.; Maekawa, S.; Bodi, I.; Rogelj, B.; Al-Chalabi, A.; Hortobágyi, T.; Shaw, C.E. p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol. 2011, 122, 691–702. [Google Scholar] [CrossRef]
- Sellier, C.; Campanari, M.L.; Julie Corbier, C.; Gaucherot, A.; Kolb-Cheynel, I.; Oulad-Abdelghani, M.; Ruffenach, F.; Page, A.; Ciura, S.; Kabashi, E.; et al. Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J. 2016, 35, 1276–1297. [Google Scholar] [CrossRef]
- van Blitterswijk, M.; van Es, M.A.; Hennekam, E.A.; Dooijes, D.; van Rheenen, W.; Medic, J.; Bourque, P.R.; Schelhaas, H.J.; van der Kooi, A.J.; de Visser, M.; et al. Evidence for an oligogenic basis of amyotrophic lateral sclerosis. Hum. Mol. Genet. 2012, 21, 3776–3784. [Google Scholar] [CrossRef]
- Ratti, A.; Corrado, L.; Castellotti, B.; Del Bo, R.; Fogh, I.; Cereda, C.; Tiloca, C.; D’Ascenzo, C.; Bagarotti, A.; Pensato, V.; et al. C9ORF72 repeat expansion in a large Italian ALS cohort: Evidence of a founder effect. Neurobiol. Aging 2012, 33, e7–e14. [Google Scholar] [CrossRef]
- Millecamps, S.; Boillée, S.; Le Ber, I.; Seilhean, D.; Teyssou, E.; Giraudeau, M.; Moigneu, C.; Vandenberghe, N.; Danel-Brunaud, V.; Corcia, P.; et al. Phenotype difference between ALS patients with expanded repeats in C9ORF72 and patients with mutations in other ALS-related genes. J. Med. Genet. 2012, 49, 258–263. [Google Scholar] [CrossRef]
- Ferrari, R.; Mok, K.; Moreno, J.H.; Cosentino, S.; Goldman, J.; Pietrini, P.; Mayeux, R.; Tierney, M.C.; Kapogiannis, D.; Jicha, G.A.; et al. Screening for C9ORF72 repeat expansion in FTLD. Neurobiol. Aging 2012, 33, e1–e11. [Google Scholar] [CrossRef]
- Van Blitterswijk, M.; Mullen, B.; Heckman, M.G.; Baker, M.C.; DeJesus-Hernandez, M.; Brown, P.H.; Murray, M.E.; Hsiung, G.Y.; Stewart, H.; Karydas, A.M.; et al. Ataxin-2 as potential disease modifier in C9ORF72 expansion carriers. Neurobiol. Aging 2014, 35, e13–e17. [Google Scholar] [CrossRef] [PubMed]
- Kwok, C.T.; Wang, H.Y.; Morris, A.G.; Smith, B.; Shaw, C.; de Belleroche, J. VCP mutations are not a major cause of familial amyotrophic lateral sclerosis in the UK. J. Neurol. Sci. 2015, 349, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Münch, C.; Rosenbohm, A.; Sperfeld, A.D.; Uttner, I.; Reske, S.; Krause, B.J.; Sedlmeier, R.; Meyer, T.; Hanemann, C.O.; Stumm, G.; et al. Heterozygous R1101K mutation of the DCTN1 gene in a family with ALS and FTD. Ann. Neurol. 2005, 58, 777–780. [Google Scholar] [CrossRef] [PubMed]
- Münch, C.; Sedlmeier, R.; Meyer, T.; Homberg, V.; Sperfeld, A.D.; Kurt, A.; Prudlo, J.; Peraus, G.; Hanemann, C.O.; Stumm, G.; et al. Point mutations of the p150 subunit of dynactin (DCTN1) gene in ALS. Neurology 2004, 63, 724–726. [Google Scholar] [CrossRef]
- Renaud, L.; Picher-Martel, V.; Codron, P.; Julien, J.P. Key role of UBQLN2 in pathogenesis of amyotrophic lateral sclerosis and frontotemporal dementia. Acta Neuropathol. Commun. 2019, 7, 103. [Google Scholar] [CrossRef]
- Crippa, V.; Sau, D.; Rusmini, P.; Boncoraglio, A.; Onesto, E.; Bolzoni, E.; Galbiati, M.; Fontana, E.; Marino, M.; Carra, S.; et al. The small heat shock protein B8 (HspB8) promotes autophagic removal of misfolded proteins involved in amyotrophic lateral sclerosis (ALS). Hum. Mol. Genet. 2010, 19, 3440–3456. [Google Scholar] [CrossRef]
- Crippa, V.; Galbiati, M.; Boncoraglio, A.; Rusmini, P.; Onesto, E.; Giorgetti, E.; Cristofani, R.; Zito, A.; Poletti, A. Motoneuronal and muscle-selective removal of ALS-related misfolded proteins. Biochem. Soc. Trans. 2013, 41, 1598–1604. [Google Scholar] [CrossRef]
- Wang, X.; Fan, H.; Ying, Z.; Li, B.; Wang, H.; Wang, G. Degradation of TDP-43 and its pathogenic form by autophagy and the ubiquitin-proteasome system. Neurosci. Lett. 2010, 469, 112–116. [Google Scholar] [CrossRef]
- Scotter, E.L.; Vance, C.; Nishimura, A.L.; Lee, Y.B.; Chen, H.J.; Urwin, H.; Sardone, V.; Mitchell, J.C.; Rogelj, B.; Rubinsztein, D.C.; et al. Differential roles of the ubiquitin proteasome system and autophagy in the clearance of soluble and aggregated TDP-43 species. J. Cell Sci. 2014, 127, 1263–1278. [Google Scholar] [CrossRef]
- Cicardi, M.E.; Cristofani, R.; Rusmini, P.; Meroni, M.; Ferrari, V.; Vezzoli, G.; Tedesco, B.; Piccolella, M.; Messi, E.; Galbiati, M.; et al. Tdp-25 Routing to Autophagy and Proteasome Ameliorates its Aggregation in Amyotrophic Lateral Sclerosis Target Cells. Sci. Rep. 2018, 8, 12390. [Google Scholar] [CrossRef]
- Watabe, K.; Akiyama, K.; Kawakami, E.; Ishii, T.; Endo, K.; Yanagisawa, H.; Sango, K.; Tsukamoto, M. Adenoviral expression of TDP-43 and FUS genes and shRNAs for protein degradation pathways in rodent motoneurons in vitro and in vivo. Neuropathology 2014, 34, 83–98. [Google Scholar] [CrossRef] [PubMed]
- Marrone, L.; Drexler, H.C.A.; Wang, J.; Tripathi, P.; Distler, T.; Heisterkamp, P.; Anderson, E.N.; Kour, S.; Moraiti, A.; Maharana, S.; et al. FUS pathology in ALS is linked to alterations in multiple ALS-associated proteins and rescued by drugs stimulating autophagy. Acta Neuropathol. 2019, 138, 67–84. [Google Scholar] [CrossRef] [PubMed]
- Fornai, F.; Longone, P.; Cafaro, L.; Kastsiuchenka, O.; Ferrucci, M.; Manca, M.L.; Lazzeri, G.; Spalloni, A.; Bellio, N.; Lenzi, P.; et al. Lithium delays progression of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 2008, 105, 2052–2057. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Zhang, S.; Tian, X.; Wu, C. Mask mitigates MAPT- and FUS-induced degeneration by enhancing autophagy through lysosomal acidification. Autophagy 2017, 13, 1924–1938. [Google Scholar] [CrossRef]
- Harman, C.A.; Monteiro, M.J. The specificity of ubiquitin binding to ubiquilin-1 is regulated by sequences besides its UBA domain. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 1568–1574. [Google Scholar] [CrossRef] [PubMed]
- Hjerpe, R.; Bett, J.S.; Keuss, M.J.; Solovyova, A.; McWilliams, T.G.; Johnson, C.; Sahu, I.; Varghese, J.; Wood, N.; Wightman, M.; et al. UBQLN2 Mediates Autophagy-Independent Protein Aggregate Clearance by the Proteasome. Cell 2016, 166, 935–949. [Google Scholar] [CrossRef]
- Şentürk, M.; Lin, G.; Zuo, Z.; Mao, D.; Watson, E.; Mikos, A.G.; Bellen, H.J. Ubiquilins regulate autophagic flux through mTOR signalling and lysosomal acidification. Nat. Cell Biol. 2019, 21, 384–396. [Google Scholar] [CrossRef]
- Kim, S.H.; Stiles, S.G.; Feichtmeier, J.M.; Ramesh, N.; Zhan, L.; Scalf, M.A.; Smith, L.M.; Pandey, U.B.; Tibbetts, R.S. Mutation-dependent aggregation and toxicity in a Drosophila model for UBQLN2-associated ALS. Hum. Mol. Genet. 2018, 27, 322–337. [Google Scholar] [CrossRef]
- Osaka, M.; Ito, D.; Suzuki, N. Disturbance of proteasomal and autophagic protein degradation pathways by amyotrophic lateral sclerosis-linked mutations in ubiquilin 2. Biochem. Biophys. Res. Commun. 2016, 472, 324–331. [Google Scholar] [CrossRef]
- Shen, W.C.; Li, H.Y.; Chen, G.C.; Chern, Y.; Tu, P.H. Mutations in the ubiquitin-binding domain of OPTN/optineurin interfere with autophagy-mediated degradation of misfolded proteins by a dominant-negative mechanism. Autophagy 2015, 11, 685–700. [Google Scholar] [CrossRef]
- Mao, J.; Xia, Q.; Liu, C.; Ying, Z.; Wang, H.; Wang, G. A critical role of Hrd1 in the regulation of optineurin degradation and aggresome formation. Hum. Mol. Genet. 2017, 26, 1877–1889. [Google Scholar] [CrossRef] [PubMed]
- Wardman, J.H.; Henriksen, E.E.; Marthaler, A.G.; Nielsen, J.E.; Nielsen, T.T. Enhancement of Autophagy and Solubilization of Ataxin-2 Alleviate Apoptosis in Spinocerebellar Ataxia Type 2 Patient Cells. Cerebellum 2020, 19, 165–181. [Google Scholar] [CrossRef] [PubMed]
- Nalbandian, A.; Donkervoort, S.; Dec, E.; Badadani, M.; Katheria, V.; Rana, P.; Nguyen, C.; Mukherjee, J.; Caiozzo, V.; Martin, B.; et al. The multiple faces of valosin-containing protein-associated diseases: Inclusion body myopathy with Paget’s disease of bone, frontotemporal dementia, and amyotrophic lateral sclerosis. J. Mol. Neurosci. 2011, 45, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Altmann, C.; Hardt, S.; Fischer, C.; Heidler, J.; Lim, H.Y.; Häussler, A.; Albuquerque, B.; Zimmer, B.; Möser, C.; Behrends, C.; et al. Progranulin overexpression in sensory neurons attenuates neuropathic pain in mice: Role of autophagy. Neurobiol. Dis. 2016, 96, 294–311. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Ma, Q.; Peng, P.; Yu, Y.; Xu, S.; Wang, G.; Ying, Z.; Wang, H. Autophagy and Ubiquitin-Proteasome System Coordinate to Regulate the Protein Quality Control of Neurodegenerative Disease-Associated DCTN1. Neurotox. Res. 2020, 37, 48–57. [Google Scholar] [CrossRef]
- Yang, Y.; Halliday, G.M.; Kiernan, M.C.; Tan, R.H. TDP-43 levels in the brain tissue of ALS cases with and without C9ORF72 or ATXN2 gene expansions. Neurology 2019, 93, e1748–e1755. [Google Scholar] [CrossRef]
- Yang, Y.S.; Kato, M.; Wu, X.; Litsios, A.; Sutter, B.M.; Wang, Y.; Hsu, C.H.; Wood, N.E.; Lemoff, A.; Mirzaei, H.; et al. Yeast Ataxin-2 Forms an Intracellular Condensate Required for the Inhibition of TORC1 Signaling during Respiratory Growth. Cell 2019, 177, 697–710.e17. [Google Scholar] [CrossRef]
- Tresse, E.; Salomons, F.A.; Vesa, J.; Bott, L.C.; Kimonis, V.; Yao, T.P.; Dantuma, N.P.; Taylor, J.P. VCP/p97 is essential for maturation of ubiquitin-containing autophagosomes and this function is impaired by mutations that cause IBMPFD. Autophagy 2010, 6, 217–227. [Google Scholar] [CrossRef]
- Papadopoulos, C.; Kirchner, P.; Bug, M.; Grum, D.; Koerver, L.; Schulze, N.; Poehler, R.; Dressler, A.; Fengler, S.; Arhzaouy, K.; et al. VCP/p97 cooperates with YOD1, UBXD1 and PLAA to drive clearance of ruptured lysosomes by autophagy. EMBO J. 2017, 36, 135–150. [Google Scholar] [CrossRef]
- Liu, G.; Byrd, A.; Warner, A.N.; Pei, F.; Basha, E.; Buchanan, A.; Buchan, J.R. Cdc48/VCP and Endocytosis Regulate TDP-43 and FUS Toxicity and Turnover. Mol. Cell. Biol. 2020, 40, e00256-19. [Google Scholar] [CrossRef]
- Sleegers, K.; Brouwers, N.; Maurer-Stroh, S.; van Es, M.A.; Van Damme, P.; van Vught, P.W.; van der Zee, J.; Serneels, S.; De Pooter, T.; Van den Broeck, M.; et al. Progranulin genetic variability contributes to amyotrophic lateral sclerosis. Neurology 2008, 71, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Pegat, A.; Bouhour, F.; Mouzat, K.; Vial, C.; Pegat, B.; Leblanc, P.; Broussolle, E.; Millecamps, S.; Lumbroso, S.; Bernard, E. Electrophysiological Characterization of C9ORF72-Associated Amyotrophic Lateral Sclerosis: A Retrospective Study. Eur. Neurol. 2019, 82, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Swinnen, B.; Bento-Abreu, A.; Gendron, T.F.; Boeynaems, S.; Bogaert, E.; Nuyts, R.; Timmers, M.; Scheveneels, W.; Hersmus, N.; Wang, J.; et al. A zebrafish model for C9orf72 ALS reveals RNA toxicity as a pathogenic mechanism. Acta Neuropathol. 2018, 135, 427–443. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Zhou, B.; Lin, M.Y.; Wang, S.; Foust, K.D.; Sheng, Z.H. Endolysosomal Deficits Augment Mitochondria Pathology in Spinal Motor Neurons of Asymptomatic fALS Mice. Neuron 2015, 87, 355–370. [Google Scholar] [CrossRef]
- Natale, G.; Lenzi, P.; Lazzeri, G.; Falleni, A.; Biagioni, F.; Ryskalin, L.; Fornai, F. Compartment-dependent mitochondrial alterations in experimental ALS, the effects of mitophagy and mitochondriogenesis. Front. Cell. Neurosci. 2015, 9, 434. [Google Scholar] [CrossRef]
- Vaughan, S.K.; Kemp, Z.; Hatzipetros, T.; Vieira, F.; Valdez, G. Degeneration of proprioceptive sensory nerve endings in mice harboring amyotrophic lateral sclerosis-causing mutations. J. Comp. Neurol. 2015, 523, 2477–2494. [Google Scholar] [CrossRef]
- Limanaqi, F.; Gambardella, S.; Lazzeri, G.; Ferrucci, M.; Ruggieri, S.; Fornai, F. Revisiting the gamma loop in ALS. Arch. Ital. Biol. 2017, 155, 118–130. [Google Scholar] [CrossRef]
- Antonini, G.; Gragnani, F.; Romaniello, A.; Pennisi, E.M.; Morino, S.; Ceschin, V.; Santoro, L.; Cruccu, G. Sensory involvement in spinal-bulbar muscular atrophy (Kennedy’s disease). Muscle Nerve 2000, 23, 252–258. [Google Scholar] [CrossRef]
- Meng, L.; Liu, J.; Liu, X.; Wang, Z.; Yuan, Y.; Zhang, W. Pathological features of muscles and peripheral nerves of Kennedy’s disease: A report of 12 cases. Zhonghua Yi Xue Za Zhi 2015, 95, 1681–1685. [Google Scholar]
- Laird, A.S.; Van Hoecke, A.; De Muynck, L.; Timmers, M.; Van den Bosch, L.; Van Damme, P.; Robberecht, W. Progranulin is neurotrophic in vivo and protects against a mutant TDP-43 induced axonopathy. PLoS ONE 2010, 5, e13368. [Google Scholar] [CrossRef]
- Rusmini, P.; Cortese, K.; Crippa, V.; Cristofani, R.; Cicardi, M.E.; Ferrari, V.; Vezzoli, G.; Tedesco, B.; Meroni, M.; Messi, E.; et al. Trehalose induces autophagy via lysosomal-mediated TFEB activation in models of motoneuron degeneration. Autophagy 2019, 15, 631–651. [Google Scholar] [CrossRef] [PubMed]
- Castillo, K.; Nassif, M.; Valenzuela, V.; Rojas, F.; Matus, S.; Mercado, G.; Court, F.A.; van Zundert, B.; Hetz, C. Trehalose delays the progression of amyotrophic lateral sclerosis by enhancing autophagy in motoneurons. Autophagy 2013, 9, 1308–1320. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, S.; Song, L.; Tang, Y.; Shen, Y.; Jia, L.; Le, W. MTOR-independent, autophagic enhancer trehalose prolongs motor neuron survival and ameliorates the autophagic flux defect in a mouse model of amyotrophic lateral sclerosis. Autophagy 2014, 10, 588–602. [Google Scholar] [CrossRef] [PubMed]
- Holler, C.J.; Taylor, G.; McEachin, Z.T.; Deng, Q.; Watkins, W.J.; Hudson, K.; Easley, C.A.; Hu, W.T.; Hales, C.M.; Rossoll, W.; et al. Trehalose upregulates progranulin expression in human and mouse models of GRN haploinsufficiency: A novel therapeutic lead to treat frontotemporal dementia. Mol. Neurodegener. 2016, 11, 46. [Google Scholar] [CrossRef] [PubMed]
- Haidar, M.; Timmerman, V. Autophagy as an emerging common pathomechanism in inherited peripheral neuropathies. Front. Mol. Neurosci. 2017, 10, 143. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, K.R.; Godena, V.K.; Hewitt, V.L.; Whitworth, A.J. Axonal transport defects are a common phenotype in Drosophila models of ALS. Hum. Mol. Genet. 2016, 25, 2378–2392. [Google Scholar] [CrossRef]
- Ikenaka, K.; Katsuno, M.; Kawai, K.; Ishigaki, S.; Tanaka, F.; Sobue, G. Disruption of axonal transport in motor neuron diseases. Int. J. Mol. Sci. 2012, 13, 1225–1238. [Google Scholar] [CrossRef]
- Ström, A.L.; Gal, J.; Shi, P.; Kasarskis, E.J.; Hayward, L.J.; Zhu, H. Retrograde axonal transport and motor neuron disease. J. Neurochem. 2008, 106, 495–505. [Google Scholar] [CrossRef]
- Hafezparast, M.; Klocke, R.; Ruhrberg, C.; Marquardt, A.; Ahmad-Annuar, A.; Bowen, S.; Lalli, G.; Witherden, A.S.; Hummerich, H.; Nicholson, S.; et al. Mutations in dynein link motor neuron degeneration to defects in retrograde transport. Science 2003, 300, 808–812. [Google Scholar] [CrossRef]
- Puls, I.; Oh, S.J.; Sumner, C.J.; Wallace, K.E.; Floeter, M.K.; Mann, E.A.; Kennedy, W.R.; Wendelschafer-Crabb, G.; Vortmeyer, A.; Powers, R.; et al. Distal spinal and bulbar muscular atrophy caused by dynactin mutation. Ann. Neurol. 2005, 57, 687–694. [Google Scholar] [CrossRef]
- Yu, J.; Lai, C.; Shim, H.; Xie, C.; Sun, L.; Long, C.X.; Ding, J.; Li, Y.; Cai, H. Genetic ablation of dynactin p150(Glued) in postnatal neurons causes preferential degeneration of spinal motor neurons in aged mice. Mol. Neurodegener. 2018, 13, 10. [Google Scholar] [CrossRef] [PubMed]
- Montie, H.L.; Cho, M.S.; Holder, L.; Liu, Y.; Tsvetkov, A.S.; Finkbeiner, S.; Merry, D.E. Cytoplasmic retention of polyglutamine-expanded androgen receptor ameliorates disease via autophagy in a mouse model of spinal and bulbar muscular atrophy. Hum. Mol. Genet. 2009, 18, 1937–1950. [Google Scholar] [CrossRef] [PubMed]
- Giorgetti, E.; Rusmini, P.; Crippa, V.; Cristofani, R.; Boncoraglio, A.; Cicardi, M.E.; Galbiati, M.; Poletti, A. Synergic prodegradative activity of Bicalutamide and trehalose on the mutant androgen receptor responsible for spinal and bulbar muscular atrophy. Hum. Mol. Genet. 2015, 24, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Waza, M.; Adachi, H.; Katsuno, M.; Minamiyama, M.; Sang, C.; Tanaka, F.; Inukai, A.; Doyu, M.; Sobue, G. 17-AAG, an Hsp90 inhibitor, ameliorates polyglutamine-mediated motor neuron degeneration. Nat. Med. 2005, 11, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Rusmini, P.; Simonini, F.; Crippa, V.; Bolzoni, E.; Onesto, E.; Cagnin, M.; Sau, D.; Ferri, N.; Poletti, A. 17-AAG increases autophagic removal of mutant androgen receptor in spinal and bulbar muscular atrophy. Neurobiol. Dis. 2011, 41, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Rusmini, P.; Crippa, V.; Giorgetti, E.; Boncoraglio, A.; Cristofani, R.; Carra, S.; Poletti, A. Clearance of the mutant androgen receptor in motoneuronal models of spinal and bulbar muscular atrophy. Neurobiol. Aging 2013, 34, 2585–2603. [Google Scholar] [CrossRef]
- Beitel, L.K.; Alvarado, C.; Mokhtar, S.; Paliouras, M.; Trifiro, M. Mechanisms mediating spinal and bulbar muscular atrophy: Investigations into polyglutamine-expanded androgen receptor function and dysfunction. Front. Neurol. 2013, 4, 53. [Google Scholar] [CrossRef]
- Stenoien, D.L.; Cummings, C.J.; Adams, H.P.; Mancini, M.G.; Patel, K.; DeMartino, G.N.; Marcelli, M.; Weigel, N.L.; Mancini, M.A. Polyglutamine-expanded androgen receptors form aggregates that sequester heat shock proteins, proteasome components and SRC-1, and are suppressed by the HDJ-2 chaperone. Hum. Mol. Genet. 1999, 8, 731–741. [Google Scholar] [CrossRef]
- Heine, E.M.; Berger, T.R.; Pluciennik, A.; Orr, C.R.; Zboray, L.; Merry, D.E. Proteasome-mediated proteolysis of the polyglutamine-expanded androgen receptor is a late event in spinal and bulbar muscular atrophy (SBMA) pathogenesis. J. Biol. Chem. 2015, 290, 12572–12584. [Google Scholar] [CrossRef]
- Wang, Y.; Su, G.; Huang, Z.; Fan, J.; Wang, Y. Cepharanthine hydrochloride degrades polyglutamine-expanded androgen receptor proteins through an autophagy pathway in neuron cells. Eur. J. Pharmacol. 2019, 861, 172534. [Google Scholar] [CrossRef]
- Pandey, U.B.; Nie, Z.; Batlevi, Y.; McCray, B.A.; Ritson, G.P.; Nedelsky, N.B.; Schwartz, S.L.; DiProspero, N.A.; Knight, M.A.; Schuldiner, O.; et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 2007, 447, 859–863. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhai, B.; Gygi, S.P.; Goldberg, A.L. mTOR inhibition activates overall protein degradation by the ubiquitin proteasome system as well as by autophagy. Proc. Natl. Acad. Sci. USA 2015, 112, 15790–15797. [Google Scholar] [CrossRef] [PubMed]
- Lazzeri, G.; Biagioni, F.; Fulceri, F.; Busceti, C.L.; Scavuzzo, M.C.; Ippolito, C.; Salvetti, A.; Lenzi, P.; Fornai, F. mTOR Modulates Methamphetamine-Induced Toxicity through Cell Clearing Systems. Oxid. Med. Cell. Longev. 2018, 2018, 6124745. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Katsuno, M.; Banno, H.; Takeuchi, Y.; Atsuta, N.; Ito, M.; Watanabe, H.; Yamashita, F.; Hori, N.; Nakamura, T.; et al. CAG repeat size correlates to electrophysiological motor and sensory phenotypes in SBMA. Brain 2008, 131, 229–239. [Google Scholar] [CrossRef]
- Li, M.; Sobue, G.; Doyu, M.; Mukai, E.; Hashizume, Y.; Mitsuma, T. Primary sensory neurons in X-linked recessive bulbospinal neuropathy: Histopathology and androgen receptor gene expression. Muscle Nerve 1995, 18, 301–308. [Google Scholar] [CrossRef]
- Lieberman, A.P. Spinal and bulbar muscular atrophy. Handb. Clin. Neurol. 2018, 148, 625–632. [Google Scholar] [CrossRef]
- Gray, A.L.; Annan, L.; Dick, J.R.T.; La Spada, A.R.; Hanna, M.G.; Greensmith, L.; Malik, B. Deterioration of muscle force and contractile characteristics are early pathological events in spinal and bulbar muscular atrophy mice. Dis. Model Mech. 2020. [Google Scholar] [CrossRef]
- Wang, L.; Lin, H.K.; Hu, Y.C.; Xie, S.; Yang, L.; Chang, C. Suppression of androgen receptor-mediated transactivation and cell growth by the glycogen synthase kinase 3 beta in prostate cells. J. Biol. Chem. 2004, 279, 32444–32452. [Google Scholar] [CrossRef]
- Sarkar, S.; Krishna, G.; Imarisio, S.; Saiki, S.; O’Kane, C.J.; Rubinsztein, D.C. A rational mechanism for combination treatment of Huntington’s disease using lithium and rapamycin. Hum. Mol. Genet. 2008, 17, 170–178. [Google Scholar] [CrossRef]
- Pasquali, L.; Busceti, C.L.; Fulceri, F.; Paparelli, A.; Fornai, F. Intracellular pathways underlying the effects of lithium. Behav. Pharmacol. 2010, 21, 473–492. [Google Scholar] [CrossRef]
- Limanaqi, F.; Biagioni, F.; Ryskalin, L.; Busceti, C.L.; Fornai, F. Molecular Mechanisms Linking ALS/FTD and Psychiatric Disorders, the Potential Effects of Lithium. Front. Cell. Neurosci. 2019, 13, 450. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Cho, S.I.; Lim, H.R.; Lee, J.K.; Lee, Y.A.; Noh, J.S.; Joo, I.S.; Kim, K.W.; Gwag, B.J. Concurrent administration of Neu2000 and lithium produces marked improvement of motor neuron survival, motor function, and mortality in a mouse model ofamyotrophic lateral sclerosis. Mol. Pharmacol. 2007, 71, 965–975. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.L.; Leng, Y.; Ma, C.H.; Zhang, J.; Ren, M.; Chuang, D.M. Combined lithium and valproate treatment delays disease onset, reduces neurological deficits and prolongs survival in an amyotrophic lateral sclerosis mouse model. Neuroscience 2008, 155, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Calderó, J.; Brunet, N.; Tarabal, O.; Piedrafita, L.; Hereu, M.; Ayala, V.; Esquerda, J.E. Lithium prevents excitotoxic cell death of motoneurons in organotypic slice cultures of spinal cord. Neuroscience 2010, 165, 1353–1369. [Google Scholar] [CrossRef]
- Wang, S.Y.; Ren, M.; Jiang, H.Z.; Wang, J.; Jiang, H.Q.; Yin, X.; Qi, Y.; Wang, X.D.; Dong, G.T.; Wang, T.H.; et al. Notch pathway is activated in cell culture and mouse models of mutant SOD1-related familial amyotrophic lateral sclerosis, with suppression of its activation as an additional mechanism of neuroprotection for lithium and valproate. Neuroscience 2015, 301, 276–288. [Google Scholar] [CrossRef]
- Boll, M.C.; Bayliss, L.; Vargas-Cañas, S.; Burgos, J.; Montes, S.; Peñaloza-Solano, G.; Rios, C.; Alcaraz-Zubeldia, M. Clinical and biological changes under treatment with lithium carbonate and valproic acid in sporadic amyotrophic lateral sclerosis. J. Neurol. Sci. 2014, 340, 103–108. [Google Scholar] [CrossRef]
- Van Eijk, R.P.A.; Jones, A.R.; Sproviero, W.; Shatunov, A.; Shaw, P.J.; Leigh, P.N.; Young, C.A.; Shaw, C.E.; Mora, G.; Mandrioli, J.; et al. Meta-analysis of pharmacogenetic interactions in amyotrophic lateral sclerosis clinical trials. Neurology 2017, 89, 1915–1922. [Google Scholar] [CrossRef]
- Terracciano, C.; Nogalska, A.; Engel, W.K.; Askanas, V. In AbetaPP-overexpressing cultured human muscle fibers proteasome inhibition enhances phosphorylation of AbetaPP751 and GSK3beta activation: Effects mitigated by lithium and apparently relevant to sporadic inclusion-body myositis. J. Neurochem. 2010, 112, 389–396. [Google Scholar] [CrossRef]
- Nath, S.R.; Yu, Z.; Gipson, T.A.; Marsh, G.B.; Yoshidome, E.; Robins, D.M.; Todi, S.V.; Housman, D.E.; Lieberman, A.P. Androgen receptor polyglutamine expansion drives age-dependent quality control defects and muscle dysfunction. J. Clin. Investig. 2018, 128, 3630–3641. [Google Scholar] [CrossRef]
- Palazzolo, I.; Stack, C.; Kong, L.; Musaro, A.; Adachi, H.; Katsuno, M.; Sobue, G.; Taylor, J.P.; Sumner, C.J.; Fischbeck, K.H.; et al. Overexpression of IGF-1 in muscle attenuates disease in a mouse model of spinal and bulbar muscular atrophy. Neuron 2009, 63, 316–328. [Google Scholar] [CrossRef]
- Rusmini, P.; Polanco, M.J.; Cristofani, R.; Cicardi, M.E.; Meroni, M.; Galbiati, M.; Piccolella, M.; Messi, E.; Giorgetti, E.; Lieberman, A.P.; et al. Aberrant Autophagic Response in The Muscle of A Knock-in Mouse Model of Spinal and Bulbar Muscular Atrophy. Sci. Rep. 2015, 5, 15174. [Google Scholar] [CrossRef] [PubMed]
- Chevalier-Larsen, E.S.; Wallace, K.E.; Pennise, C.R.; Holzbaur, E.L. Lysosomal proliferation and distal degeneration in motor neurons expressing the G59S mutation in the p150Glued subunit of dynactin. Hum. Mol. Genet. 2008, 17, 1946–1955. [Google Scholar] [CrossRef] [PubMed]
- Cox, L.E.; Ferraiuolo, L.; Goodall, E.F.; Heath, P.R.; Higginbottom, A.; Mortiboys, H.; Hollinger, H.C.; Hartley, J.A.; Brockington, A.; Burness, C.E.; et al. Mutations in CHMP2B in lower motor neuron predominant amyotrophic lateral sclerosis (ALS). PLoS ONE 2010, 5, e9872. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Ganetzky, B. Autophagy promotes synapse development in Drosophila. J. Cell Biol. 2009, 187, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Stavoe, A.K.; Hill, S.E.; Hall, D.H.; Colón-Ramos, D.A. KIF1A/UNC-104 Transports ATG-9 to Regulate Neurodevelopment and Autophagy at Synapses. Dev. Cell 2016, 38, 171–185. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, M.R.C.; Geisler, S.M.; Pittman, S.K.; Doan, R.A.; Weihl, C.C.; Milbrandt, J.; DiAntonio, A. TMEM184b Promotes Axon Degeneration and Neuromuscular Junction Maintenance. J. Neurosci. 2016, 36, 4681–4689. [Google Scholar] [CrossRef]
- Ju, J.S.; Fuentealba, R.A.; Miller, S.E.; Jackson, E.; Piwnica-Worms, D.; Baloh, R.H.; Weihl, C.C. Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J. Cell. Biol. 2009, 187, 875–888. [Google Scholar] [CrossRef]
- Malik, I.; Turk, J.; Mancuso, D.J.; Montier, L.; Wohltmann, M.; Wozniak, D.F.; Schmidt, R.E.; Gross, R.W.; Kotzbauer, P.T. Disrupted membrane homeostasis and accumulation of ubiquitinated proteins in a mouse model of infantile neuroaxonal dystrophy caused by PLA2G6 mutations. Am. J. Pathol. 2008, 172, 406–416. [Google Scholar] [CrossRef]
- Sumi-Akamaru, H.; Beck, G.; Kato, S.; Mochizuki, H. Neuroaxonal dystrophy in PLA2G6 knockout mice. Neuropathology 2015, 35, 289–302. [Google Scholar] [CrossRef]
- Chen, P.C.; Qin, L.N.; Li, X.M.; Walters, B.J.; Wilson, J.A.; Mei, L.; Wilson, S.M. The proteasome-associated deubiquitinating enzyme Usp14 is essential for the maintenance of synaptic ubiquitin levels and the development of neuromuscular junctions. J. Neurosci. 2009, 29, 10909–10919. [Google Scholar] [CrossRef]
- Limanaqi, F.; Biagioni, F.; Busceti, C.L.; Ryskalin, L.; Fornai, F. The effects of proteasome on baseline and methamphetamine-dependent dopamine transmission. Neurosci. Biobehav. Rev. 2019, 102, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Limanaqi, F.; Biagioni, F.; Busceti, C.L.; Ryskalin, L.; Soldani, P.; Frati, A.; Fornai, F. Cell Clearing Systems Bridging Neuro-Immunity and Synaptic Plasticity. Int. J. Mol. Sci. 2019, 20, 2197. [Google Scholar] [CrossRef] [PubMed]
- Limanaqi, F.; Biagioni, F.; Gambardella, S.; Ryskalin, L.; Fornai, F. Interdependency Between Autophagy and Synaptic Vesicle Trafficking: Implications for Dopamine Release. Front. Mol. Neurosci. 2018, 11, 299. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, D.; Torres, C.A.; Setlik, W.; Cebrián, C.; Mosharov, E.V.; Tang, G.; Cheng, H.C.; Kholodilov, N.; Yarygina, O.; Burke, R.E.; et al. Regulation of presynaptic neurotransmission by macroautophagy. Neuron 2012, 74, 277–284. [Google Scholar] [CrossRef]
- Bachiller, S.; Rybkina, T.; Porras-García, E.; Pérez-Villegas, E.; Tabares, L.; Armengol, J.A.; Carrión, A.M.; Ruiz, R. The HERC1 E3 Ubiquitin Ligase is essential for normal development and for neurotransmission at the mouse neuromuscular junction. Cell. Mol. Life Sci. 2015, 72, 2961–2971. [Google Scholar] [CrossRef]
- Aravamudan, B.; Broadie, K. Synaptic Drosophila UNC-13 is regulated by antagonistic G-protein pathways via a proteasome-dependent degradation mechanism. J. Neurobiol. 2003, 54, 417–438. [Google Scholar] [CrossRef]
- Khan, M.M.; Strack, S.; Wild, F.; Hanashima, A.; Gasch, A.; Brohm, K.; Reischl, M.; Carnio, S.; Labeit, D.; Sandri, M.; et al. Role of autophagy, SQSTM1, SH3GLB1, and TRIM63 in the turnover of nicotinic acetylcholine receptors. Autophagy 2014, 10, 123–136. [Google Scholar] [CrossRef]
- Wild, F.; Khan, M.M.; Straka, T.; Rudolf, R. Progress of endocytic CHRN to autophagic degradation is regulated by RAB5-GTPase and T145 phosphorylation of SH3GLB1 at mouse neuromuscular junctions in vivo. Autophagy 2016, 12, 2300–2310. [Google Scholar] [CrossRef]
- Rezvani, K.; Teng, Y.; De Biasi, M. The ubiquitin-proteasome system regulates the stability of neuronal nicotinic acetylcholine receptors. J. Mol. Neurosci. 2010, 40, 177–184. [Google Scholar] [CrossRef]
- Rezvani, K.; Teng, Y.; Pan, Y.; Dani, J.A.; Lindstrom, J.; García Gras, E.A.; McIntosh, J.M.; De Biasi, M. UBXD4, a UBX-containing protein, regulates the cell surface number and stability of alpha3-containing nicotinic acetylcholine receptors. J. Neurosci. 2009, 29, 6883–6896. [Google Scholar] [CrossRef]
- Sabatelli, M.; Eusebi, F.; Al-Chalabi, A.; Conte, A.; Madia, F.; Luigetti, M.; Mancuso, I.; Limatola, C.; Trettel, F.; Sobrero, F.; et al. Rare missense variants of neuronal nicotinic acetylcholine receptor altering receptor function are associated with sporadic amyotrophic lateral sclerosis. Hum. Mol. Genet. 2009, 18, 3997–4006. [Google Scholar] [CrossRef] [PubMed]
- Monks, D.A.; Johansen, J.A.; Mo, K.; Rao, P.; Eagleson, B.; Yu, Z.; Lieberman, A.P.; Breedlove, S.M.; Jordan, C.L. Overexpression of wild-type androgen receptor in muscle recapitulates polyglutamine disease. Proc. Natl. Acad. Sci. USA 2007, 104, 18259–18264. [Google Scholar] [CrossRef] [PubMed]
- Poort, J.E.; Rheuben, M.B.; Breedlove, S.M.; Jordan, C.L. Neuromuscular junctions are pathological but not denervated in two mouse models of spinal bulbar muscular atrophy. Hum. Mol. Genet. 2016, 25, 3768–3783. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Je, H.S.; Young, P.; Gross, J.; Lu, B.; Feng, G. Regulation of synaptic growth and maturation by a synapse-associated E3 ubiquitin ligase at the neuromuscular junction. J. Cell. Biol. 2007, 177, 1077–1089. [Google Scholar] [CrossRef]
- Khan, M.M.; Lustrino, D.; Silveira, W.A.; Wild, F.; Straka, T.; Issop, Y.; O’Connor, E.; Cox, D.; Reischl, M.; Marquardt, T.; et al. Sympathetic innervation controls homeostasis of neuromuscular junctions in health and disease. Proc. Natl. Acad. Sci. USA 2016, 113, 746–750. [Google Scholar] [CrossRef]
- Salazar, G.; Cullen, A.; Huang, J.; Zhao, Y.; Serino, A.; Hilenski, L.; Patrushev, N.; Forouzandeh, F.; Hwang, H.S. SQSTM1/p62 and PPARGC1A/PGC-1alpha at the interface of autophagy and vascular senescence. Autophagy 2019. [Google Scholar] [CrossRef]
- Trausch-Azar, J.; Leone, T.C.; Kelly, D.P.; Schwartz, A.L. Ubiquitin proteasome-dependent degradation of the transcriptional coactivator PGC-1{alpha} via the N-terminal pathway. J. Biol. Chem. 2010, 285, 40192–40200. [Google Scholar] [CrossRef]
- Farah, B.L.; Sinha, R.A.; Wu, Y.; Singh, B.K.; Zhou, J.; Bay, B.H.; Yen, P.M. β-Adrenergic agonist and antagonist regulation of autophagy in HepG2 cells, primary mouse hepatocytes, and mouse liver. PLoS ONE 2014, 9, e98155. [Google Scholar] [CrossRef]
- Zhi, X.; Li, B.; Li, Z.; Zhang, J.; Yu, J.; Zhang, L.; Xu, Z. Adrenergic modulation of AMPK-dependent autophagy by chronic stress enhances cell proliferation and survival in gastric cancer. Int. J. Oncol. 2019, 54, 1625–1638. [Google Scholar] [CrossRef]
- Joassard, O.R.; Amirouche, A.; Gallot, Y.S.; Desgeorges, M.M.; Castells, J.; Durieux, A.C.; Berthon, P.; Freyssenet, D.G. Regulation of Akt-mTOR, ubiquitin-proteasome and autophagy-lysosome pathways in response to formoterol administration in rat skeletal muscle. Int. J. Biochem. Cell. Biol. 2013, 45, 2444–2455. [Google Scholar] [CrossRef]
- Cristofani, R.; Crippa, V.; Vezzoli, G.; Rusmini, P.; Galbiati, M.; Cicardi, M.E.; Meroni, M.; Ferrari, V.; Tedesco, B.; Piccolella, M.; et al. The small heat shock protein B8 (HSPB8) efficiently removes aggregating species of dipeptides produced in C9ORF72-related neurodegenerative diseases. Cell Stress Chaperones 2018, 23, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sakae, N.; Bieniek, K.F.; Zhang, Y.J.; Ross, K.; Gendron, T.F.; Murray, M.E.; Rademakers, R.; Petrucelli, L.; Dickson, D.W. Poly-GR dipeptide repeat polymers correlate with neurodegeneration and Clinicopathological subtypes in C9ORF72-related brain disease. Acta Neuropathol. Commun. 2018, 6, 63. [Google Scholar] [CrossRef] [PubMed]
- Scaramuzzino, C.; Monaghan, J.; Milioto, C.; Lanson, N.A., Jr.; Maltare, A.; Aggarwal, T.; Casci, I.; Fackelmayer, F.O.; Pennuto, M.; Pandey, U.B. Protein arginine methyltransferase 1 and 8 interact with FUS to modify its sub-cellular distribution and toxicity in vitro and in vivo. PLoS ONE 2013, 8, e61576. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Yang, P.; Tian, E.; Zhang, H. Arginine methylation modulates autophagic degradation of PGL granules in C. elegans. Mol. Cell 2013, 52, 421–433. [Google Scholar] [CrossRef] [PubMed][Green Version]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Limanaqi, F.; Busceti, C.L.; Biagioni, F.; Cantini, F.; Lenzi, P.; Fornai, F. Cell-Clearing Systems Bridging Repeat Expansion Proteotoxicity and Neuromuscular Junction Alterations in ALS and SBMA. Int. J. Mol. Sci. 2020, 21, 4021. https://doi.org/10.3390/ijms21114021
Limanaqi F, Busceti CL, Biagioni F, Cantini F, Lenzi P, Fornai F. Cell-Clearing Systems Bridging Repeat Expansion Proteotoxicity and Neuromuscular Junction Alterations in ALS and SBMA. International Journal of Molecular Sciences. 2020; 21(11):4021. https://doi.org/10.3390/ijms21114021
Chicago/Turabian StyleLimanaqi, Fiona, Carla Letizia Busceti, Francesca Biagioni, Federica Cantini, Paola Lenzi, and Francesco Fornai. 2020. "Cell-Clearing Systems Bridging Repeat Expansion Proteotoxicity and Neuromuscular Junction Alterations in ALS and SBMA" International Journal of Molecular Sciences 21, no. 11: 4021. https://doi.org/10.3390/ijms21114021
APA StyleLimanaqi, F., Busceti, C. L., Biagioni, F., Cantini, F., Lenzi, P., & Fornai, F. (2020). Cell-Clearing Systems Bridging Repeat Expansion Proteotoxicity and Neuromuscular Junction Alterations in ALS and SBMA. International Journal of Molecular Sciences, 21(11), 4021. https://doi.org/10.3390/ijms21114021