Abstract
p53 is a well-known tumor suppressor gene and one of the most extensively studied genes in cancer research. p53 functions largely as a transcription factor and can trigger a variety of antiproliferative programs via induction of its target genes. We identified PHLDA3 as a p53 target gene and found that its protein product is a suppressor of pancreatic neuroendocrine tumors (PanNETs) and a repressor of Akt function. PHLDA3 is frequently inactivated by loss of heterozygosity (LOH) and methylation in human PanNETs, and LOH at the PHLDA3 gene locus correlates with PanNET progression and poor prognosis. In addition, in PHLDA3-deficient mice, pancreatic islet cells proliferate abnormally and acquire resistance to apoptosis. In this article, we briefly review the roles of p53 and Akt in human neuroendocrine tumors (NETs) and describe the relationship between the p53-PHLDA3 and Akt pathways. We also discuss the role of PHLDA3 as a tumor suppressor in various NETs and speculate on the possibility that loss of PHLDA3 function may be a useful prognostic marker for NET patients indicating particular drug therapies. These results suggest that targeting the downstream PHLDA3-Akt pathway might provide new therapies to treat NETs.
1. Introduction
p53 is a well-known tumor suppressor gene that is mutated in more than half of human cancers of various types and contributes to their development [1,2,3,4]. p53 is a transcription factor that induces its target genes in accordance with the type and intensity of cell damage. Since the transactivation function of p53 is essential to its ability to suppress cancer, the identification and characterization of its target genes and associated pathways is critical to understanding p53-mediated tumorigenesis. We have previously identified several novel p53 target genes and downstream pathways that modulate tumorigenesis: PHLDA3, PHLDA1, IER5, AEN, FUCA1, RPRM, and NOXA [5,6,7,8,9,10,11,12,13,14]. In this review, we will focus on PHLDA3, a tumor suppressor gene that encodes a repressor of the Akt oncoprotein and is important for the development of neuroendocrine tumors (NETs). We will summarize the function of the p53-PHLDA3-Akt axis in NETs and discuss the potential importance of PHLDA3 in NET diagnosis and treatment.
2. p53-PHLDA3-Akt Axis
2.1. PHLDA3, a Target Gene of p53
p53 is a well-known tumor suppressor gene normally present in all human cells that is usually activated by various types of stress, such as DNA damage, hypoxia, or oncogene activation [15,16,17,18,19,20,21,22]. In response to these signals, p53 undergoes various post-translational modifications including phosphorylation and acetylation and both its expression levels and subcellular localization are altered. Consequently, p53 binds to the promoters of its various target genes and activates their transcription [16,23,24,25,26]. The effector functions of p53 have yet to be fully elucidated, and a comprehensive identification and functional analysis of p53 target genes remains a critical research goal.
In previous studies we comprehensively screened genes that are directly induced by p53 and focused in particular on genes of hitherto unknown function. Among these genes, we were particularly interested in PHLDA3, which is strongly induced by wild-type p53 [13,27,28]. In-silico analysis revealed that there is a p53 binding site near the PHLDA3 gene transcription site, and ChIP-chip analysis and ChIP-sequence analysis confirmed that p53 binds to this sequence [13]. These results thus identified PHLDA3 as a p53 target gene.
2.2. Oncogene Akt and p53-Akt Network
Akt is a particularly well-known oncogene that is abnormally activated in many tumors by a variety of extracellular stimuli, such as growth factors and hormones [29,30,31,32]. Akt activation occurs downstream of phosphoinositide 3-kinase (PI3K), a lipid kinase that catalyzes the phosphorylation of phosphatidylinositol 4,5-bisphosphate (PIP2) to produce phosphatidylinositol 3,4,5-trisphosphate (PIP3) and is linked to cellular transformation and human cancers [33,34]. The activation of Akt requires the binding of PIP3 and this binding is mediated by a Pleckstrin Homology (PH) domain found in the amino terminus of Akt [35]. When PIP3 appears in the membrane, Akt can re-localize to the membrane by binding to PIP3 via its PH domain, and this induces Akt phosphorylation and consequent activation. Activated Akt is a serine threonine kinase that regulates cell proliferation by phosphorylating various downstream substrates such as Mouse Double Minute 2 homolog (MDM2), Glycogen Synthase Kinase 3 (GSK3), Tuberous Sclerosis Complex 2 (TSC2), and Mechanistic Target of Rapamycin Complex 1 (mTORC1), and this regulation plays a central role in signaling required for cell survival [33,36].
Since p53 and Akt are major but opposing regulators in the signaling pathways determining cell survival and death, it is not surprising that there is a signaling network integrating the functions of these two proteins [37,38,39,40]. For example, activated Akt negatively regulates p53 expression by phosphorylating MDM2, which translocates to the nucleus and facilitates p53 degradation [41]. On the other hand, p53 induces the expression of PTEN, which terminates PI3K/PIP3 signaling by dephosphorylating PIP3 and thereby inhibiting Akt activation. Disruption of the p53-Akt network resulting from the functional loss of p53 or PTEN or the over-activation of Akt or MDM2 has been frequently observed in cancer cells [2,29,30,42,43,44,45]. This suggests that the balance of the p53-Akt network plays an essential role in tumorigenesis and could be one of the most important targets in cancer therapy.
2.3. PHLDA3 Functions as an Endogenous Dominant Negative Regulator of Akt
PHLDA3 (Pleckstrin Homology Like Domain Family A Member 3) is a gene that encodes a protein consisting of 127 amino acids, most of which consists of a PH domain (amino acids 7-108; Figure 1A). PHLDA3 binds to various phosphatidylinositol phosphates (PIPs) through this domain and thereby localizes to the cell membrane. Overexpression of PHLDA3 results in activation of caspase-3 and increased cell death, indicating that PHLDA3 can induce apoptosis. Akt also possess a PH domain, and we found that PHLDA3 suppresses Akt activation by competitively inhibiting the binding of Akt to PIPs. Thus, PHLDA3 functions as an endogenous dominant-negative repressor of Akt that blocks Akt-mediated survival signaling by suppressing Akt activation (Figure 1B). The identification and analysis of PHLDA3 has revealed a novel p53 downstream pathway that suppresses Akt’s oncogenic function [13].
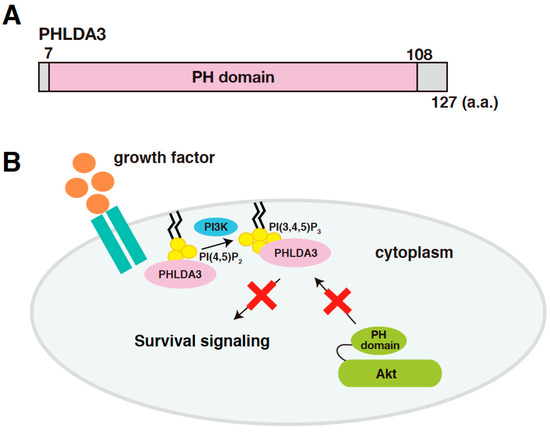
Figure 1.
PHLDA3 is a target gene of p53 and encodes a protein that functions as an endogenous inhibitor of Akt. (A) A schematic diagram of PHLDA3. Pleckstrin Homology (PH) domain is the only known protein domain in PHLDA3. (B) A model of Akt repression by PHLDA3. PHLDA3 functions as a dominant negative repressor of Akt. PHLDA3 localizes to the plasma membrane via binding to phosphatidylinositol phosphates (PIPs), thereby competing with Akt binding to PIPs and suppressing Akt-mediated survival signaling.
3. p53 Mutations in Neuroendocrine Tumors
3.1. Neuroendocrine Tumors
Neuroendocrine tumors (NETs) are a relatively rare and heterogeneous tumor type [46,47,48,49]. Neuroendocrine cells are distributed throughout the entire body, and NETs have been found in lung, pancreatic islet, pituitary, thyroid gland, parathyroid gland, rectum, and many other neuroendocrine tissues. NETs are generally low-grade tumors with a good prognosis and low risk of distant metastases. Histopathological classification is important in determining the nature of the NET, its prognosis, and recommended therapies. NETs synthesize and secrete peptide hormones and depending on the effects of these tumor-secreted hormones on the body, NETs are classified into functional NETs with abnormal hormonal symptoms and non-functional NETs without any symptoms. Functional NETs cause a variety of symptoms depending on the type of hormone being over-produced. Non-functional NETs usually are a larger size and often compress nearby organs.
There is a relatively higher incidence of NETs derived from lung and pancreatic islet compared to other sites [48]. Nearly one-fourth of lung neoplasms are lung NETs. Based on the cell shape, Lung NETs are divided into two types, carcinoids and neuroendocrine carcinomas. In addition, carcinoids are categorized into low-grade typical carcinoids (TC) and atypical carcinoids (AC), and neuroendocrine carcinomas are divided into high-grade small-cell lung cancer (SCLC) and large-cell neuroendocrine carcinomas (LCNEC).
Pancreatic neuroendocrine tumors (PanNETs) are the second most common type of pancreatic epithelial tumor, with an incidence of 2~3 per 100,000 [50]. PanNETs tend to grow slowly and can possibly spread to other parts of the body. The 2017 WHO classification grouped well-differentiated PanNETs into various grades depending on their Ki-67 proliferation index, i.e., Grade 1 (Ki-67 index <3%), Grade 2 (Ki-67 index 3~20%), and Grade 3 (Ki-67 index >20%). Poorly-differentiated pancreatic neuroendocrine carcinomas (PanNECs) usually have a Ki-67 proliferation index of more than 20%, tend to grow and spread quickly, and can spread to other parts of the body. It has been reported that about 85% of PanNETs are nonfunctional tumors, about 10% are insulinomas, which are the most common type of functional PanNET and produce insulin, and the remaining PanNETs produce other hormones such as glucagon, somatostatin, or gastrin [51,52].
The gastrointestinal (GI) tract is another site where most of the NETs develop in. Rectal NETs are the most common type of GI-NET, with a 5-year overall survival rate of 88% [53,54,55]. Rectal NETs are classified into grade 1 (Ki-67 index<3%), grade 2 (Ki-67 index 3~20%), and grade 3 (Ki-67 index >20%) according to the 2016 WHO classification.
Although the incidence of NETs has strikingly increased in the last decades, there are still limited data about the pathogenesis and treatment in NETs. The mechanisms elucidation of NET tumorigenesis and the development of a prognostic marker may be helpful in predicting tumor behavior and guiding therapy.
3.2. p53 Mutations Are Rare in Lung Neuroendocrine Tumors (NETs), Pancreatic Neuroendocrine Tumors (PanNETs), and Rectal NETs
As NETs are rare cancers and very difficult to study, the mechanisms of NET tumorigenesis and development are still not well understood. It has been reported that p53 mutations occur in many types of cancer and are associated with poorer clinical outcomes, greater resistance to treatment, and higher degrees of metastases [2,56,57,58]. Several groups have investigated the status of p53 in NET specimens and shown that p53 mutations are infrequent in low-grade lung NETs [59,60,61]. More recently, Vollbrecht et al. looked for mutations in 221 mutational hot spots within 48 tumor-relevant genes in lung NETs and found that KIT, PTEN, HNF1A, and SMO were altered in ACs, while the SMAD4 mutation was found in TC subtypes. However, they did not find any p53 mutations in these specimens [62]. In addition, Simbolo et al. screened 148 lung NETs, consisting of 53 TCs, 35 ACs, 33 SCLCs, and 27 LCNECs, using next-generation sequencing and whole-exome sequencing and found MEN1 alterations are almost exclusive to low-grade carcinoids, whereas alterations in both p53 and PI3K/Akt/mTOR pathway genes were found more commonly in carcinomas, which are a higher grade of NET [63]. In 2018, George et al. performed a comprehensive genomic and transcriptomic analysis of 75 LCNECs and identified two molecular subgroups: “type I LCNECs” and “type II LCNECs”, both of which are enriched for inactivated p53 [64]. In summary, p53 mutations are frequently found in high-grade carcinomas, but are rarely found in low-grade carcinoids.
Mutations of p53 are also rare in PanNETs [65,66]. Jiao et al. screened the most commonly mutated cancer genes, including p53, in well-differentiated somatic PanNETs by whole exome sequencing and found a high frequency of mutations in MEN1 (multiple Endocrine Neoplasia Type 1), ATRX (alpha thalassemia/mental retardation syndrome X-linked), and DAXX (death-domain associated protein), but a low frequency of mutations in p53 [67]. Recently, Scarpa et al. have performed whole-genome sequencing and found that clinically sporadic PanNETs contain frequent germline mutations, including mutations in MUTYH, CHEK2, and BRCA2, which are the DNA repair genes. They reported that sporadic PanNETs also usually bear mutations in MEN1, the chromatin remodeling pathway (SETD2, MLL3), the telomere maintenance pathway (DAXX, ATRX) and activators of the mTOR signaling pathway (PTEN, DEPDC5, TSC1, TSC2), but rarely have mutations in p53. In addition to these gene mutations, they also identified a subgroup of tumors that are associated with hypoxia and HIF signaling [50].
There is also limited data about the incidence of driver mutations in rectal NETs. Ha et al. sequenced 69 primary low-grade rectal NETs and found only 10% of these tumors bore p53 mutations [68].
Therefore, while mutations of p53 are frequently found in many cancer types, they are rarely observed in low-grade lung NETs, PanNETs, and rectal NETs.
4. PHLDA3 Functions as a Tumor Suppressor in Various NETs
4.1. Functional Loss of PHLDA3 Is Frequently Found in Lung NETs
Abnormal activation of the PI3K/Akt pathway is observed in many cases of lung cancer. Since PHLDA3 is a repressor of Akt, it is conceivable that functional loss of PHLDA3 could contribute to the tumorigenesis and development of lung cancers. We analyzed various types of lung cancers and found a high frequency of PHLDA3 gene defects in lung NETs compared to other types of lung cancer [13]. In these lung NETs, PHLDA3 gene expression was lower and Akt activation was higher compared to normal tissues, raising the possibility that loss of PHLDA3 function caused Akt activation in these cancers.
The PTEN tumor suppressor is an upstream factor and major suppressor of the Akt pathway. It has been shown that PHLDA3 and PTEN inhibit the PI3K/Akt pathway by different mechanisms. We also found that in LCNEC samples, loss of PTEN and loss of PHLDA3 are not mutually exclusive, but rather are additive with respect to Akt activation [13].
As PHLDA3 is a p53 target gene, we analyzed the association between PHLDA3 loss and p53 status in these lung NET specimens. We found that among the lung NETs having wild-type p53, 63% exhibited PHLDA3 loss, whereas among specimens having a nonfunctional p53 only 13% exhibited PHLDA3 loss (Figure 2A). Collectively, 91% of lung NETs have a functional loss of either p53 or PHLDA3 (Figure 2A), suggesting that defects in this p53-PHLDA3 pathway play a major role in lung NET tumorigenesis [13].
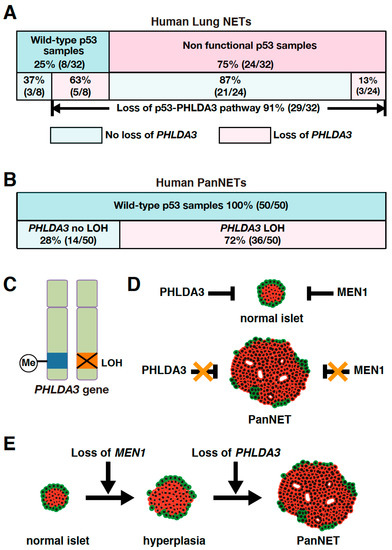
Figure 2.
PHLDA3 gene defects are observed in lung and pancreatic neuroendocrine tumors (NETs). (A) Among lung NETs having wild-type p53, 63% exhibited loss of PHLDA3, whereas among lung NETs having a nonfunctional p53, only 13% exhibited loss of PHLDA3. Nonfunctional p53 can be caused by deletion as well as mutation. Abnormalities in p53 can result in upregulated expression of the protein, which may be detected by immunohistochemistry. Genomic sequencing is also preformed to analyze p53 mutations. Chromosome copy number alterations in PHLDA3 are analyzed by comparative genomic hybridization (CGH). (B) High frequency of PHLDA3 loss of heterozygosity (LOH) is found in pancreatic neuroendocrine tumors (PanNETs), which commonly have wild-type p53. (C) Two-hit inactivation of PHLDA3 in PanNETs. One of the PHLDA3 loci undergoes LOH and the other undergoes methylation. (D) PanNET tumorigenesis requires the functional loss of both PHLDA3 and MEN1. PHLDA3 and MEN1 suppress cell proliferation in normal islet cells. Loss of both PHLDA3 and MEN1 function is necessary for PanNET tumorigenesis. (E) Functional loss of PHLDA3 contributes to PanNET progression. Loss of MEN1 function leads to islet hyperplasia and/or atypia over time, and additional loss of PHLDA3 function is required for tumor formation and progression.
4.2. PHLDA3 Is a Tumor Suppressor of PanNETs
4.2.1. PHLDA3 Is Inactivated by Both Loss of Heterozygosity (LOH) and Methylation in Human PanNETs
It has been reported that loss of heterozygosity (LOH) is frequently found at the 1q31 locus in PanNETs [69,70]. Since the PHLDA3 gene is located at 1q31 and LOH at the PHLDA3 gene locus is frequently found in lung NETs, we speculated that the PHLDA3 gene may also undergo LOH in PanNETs. We therefore analyzed the LOH of the PHLDA3 gene in PanNETs using a microsatellite marker near the PHLDA3 gene locus. We found that LOH at the PHLDA3 gene locus was detected in 72% of human PanNETs (Figure 2B). We further found that PHLDA3 gene expression level in PanNET specimens was significantly decreased in specimens that have LOH at the PHLDA3 gene locus compared to those that do not. Since LOH involves the loss of one allele, we analyzed the status of the remaining allele in PanNET specimens and found that the remaining PHLDA3 allele invariably underwent methylation in its transcriptional regulatory region. Thus, PanNETs are characterized by a two-hit inactivation of the PHLDA3 gene, i.e., LOH and methylation (Figure 2C). These results identify PHLDA3 as a novel tumor suppressor gene of PanNETs [14].
4.2.2. Functional Loss of Both PHLDA3 and MEN1 Is Essential for PanNET Tumorigenesis
The LOH frequency of the PHLDA3 gene is similar to that reported for the MEN1 gene, another important tumor suppressor gene in PanNETs. The MEN1 gene is known to be causative for multiple endocrine neoplasia type 1, which undergoes frequent LOH and mutation in PanNETs [50,67,71,72]. We therefore investigated the relationship between the PHLDA3 gene and MEN1 gene in PanNETs. We first performed LOH analysis of the MEN1 gene and found that its frequency of LOH was 67% in human PanNETs. Interestingly, LOH at the PHLDA3 and MEN1 loci were not mutually exclusive, as would be expected if PHLDA3 and MEN1 were on the same tumor-suppressing pathway. We also observed a significantly high frequency of LOH at both the PHLDA3 and MEN1 loci. These results suggest that the functional loss of both the PHLDA3 and MEN1 genes is necessary for the development of human PanNETs and inactivation of these pathways cooperatively contribute to PanNET development (Figure 2D) [14].
4.2.3. LOH at PHLDA3 Locus Is Related to PanNET Malignancy and Prognosis
The PHLDA3 gene locus has been reported to have a high frequency of LOH in PanNETs and correlate with tumor progression [14,69,70]. We next examined the relationship between LOH at the PHLDA3 gene locus, tumor malignancy, and patient prognosis. We observed that PanNETs with LOH at the PHLDA3 gene locus displayed a higher malignancy, and prognosis was worse versus those without LOH. On the other hand, LOH at the MEN1 gene locus was not associated with tumor malignancy or prognosis. These results suggest that the PHLDA3 gene pathway suppresses PanNET progression (Figure 2E) [14].
4.3. The Function of PHLDA3 in Other NETs
In addition to human lung NETs and PanNETs, the incidence of rectal NETs has been increasing in recent years [55,73,74,75]. In many cases, rectal NETs are very small lesions and very difficult to find unless accompanied by lymph node metastasis, and further research is needed to clarify the risk factors for rectal NET tumorigenesis and lymph node metastasis. We analyzed PHLDA3 status in 55 rectal NET specimens and reported that 60% of them showed LOH at the PHLDA3 gene locus. This indicates that PHLDA3 has an important function in tumor suppression in rectal NETs. In addition to PHLDA3, a high frequency of LOH was also observed at the MEN1 gene locus in these specimens. LOH at the PHLDA3 and MEN1 loci were not mutually exclusive and occurred together in both loci at a high frequency. Thus, similar to PanNETs, the tumor suppressing pathways involving PHLDA3 and MEN1 are distinct in rectal NETs, and their development involves the functional loss of both pathways [11].
We therefore regard PHLDA3 as a common tumor suppressor of various NETs, although analysis of NETs from other neuroendocrine tissues is still required to be conclusive.
5. Functional Analysis of PHLDA3 Using a PHLDA3-Deficient Mouse Model
5.1. Functional Loss of PHLDA3 Induces Akt Pathway Activation and Cell Proliferation in Islet β Cells
PanNETs are tumors that develop from pancreatic islet cells. Previous studies have shown the proliferation of pancreatic islet β cells involves the Akt pathway activated by glucose and/or growth factor stimulation [76]. Moreover, Bernal-Mizrachi et al. have noted that transgenic mice expressing a constitutively activated Akt in pancreatic islet β cells exhibit an increase in islet mass due to enhanced proliferation of β cells [77]. These results suggest that the Akt pathway plays a central role in promoting cell proliferation and inhibiting cell death in islet β cells. Since PHLDA3 inhibits Akt activation, we analyzed PHLDA3 function in islet cells using normal rat islet cells and RIN cells derived from a rat insulinoma. Inhibition of PHLDA3 gene expression enhanced Akt activation and promoted cell proliferation in these cells. In addition, overexpression of the PHLDA3 gene in MIN6 cells, a cell line derived from mouse pancreatic β cells, led to decreased Akt activity and phosphorylation of factors downstream of Akt. These results show that the PHLDA3 gene negatively regulates Akt pathway activation and cell proliferation in islet β cells [14].
5.2. Functional Loss of PHLDA3 Induces Hyperplasia of Pancreatic Islets and Increased Insulin Secreting
In order to analyze the function of PHLDA3 in vivo, islet cells from PHLDA3-deficient mice were analyzed for activation of Akt and its downstream substrates. We found that phosphorylation of Akt and its downstream substrates such as p70S6 kinase, ribosomal protein S6, GSK-3β, and MDM2 increased, indicating that the Akt pathway is abnormally activated by PHLDA3 deficiency. Since the activation of Akt pathway is known to cause proliferation of islet β cells and hyperplasia of pancreatic islets [77,78], we examined the pancreas of wild-type and PHLDA3-deficient mice and found that pancreatic islets taken from 10-month-old PHLDA3-deficient mice displayed islet hyperplasia (Figure 3A).
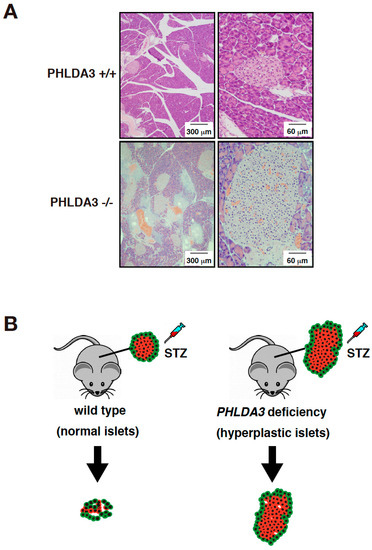
Figure 3.
PHLDA3-deficient mice exhibit islet hyperplasia and resistance of β cells to apoptosis. (A) HE staining of islets from wild type and PHLDA3-deficient 10 month-old mice. (B) Apoptosis of β cells is observed in wild type mice treated with streptozotocin (STZ) but is suppressed in PHLDA3-deficient mice.
Pancreatic islets contain several types of cells, such as β cells that produce insulin and α cells that produce glucagon. When pancreatic sections from wild-type and PHLDA3-deficient mice were double-stained for insulin and Ki-67, a cell proliferation marker, it was found that most Ki-67-positive cells were β cells in PHLDA3-deficient islets, indicating that β cell proliferation is enhanced in PHLDA3-deficient islets. Since β cells produce and secrete insulin, which regulates blood glucose levels, we measured the plasma insulin levels and the blood glucose levels in wild-type and PHLDA3-deficient mice. We found higher plasma insulin and lower blood glucose levels in PHLDA3-deficient mice compared with wild-type mice. These results show that loss of PHLDA3 expression results in enhanced proliferation of islet β cells and increased insulin secretion.
As it is also known that cells become enlarged when Akt is activated, we checked the size of the islet cells from PHLDA3-deficient mice and found that β cells are indeed larger than those from wild-type mice. These data show that functional loss of PHLDA3 leads to β cell hypertrophy.
Taken together, these results show that loss of PHLDA3 induces Akt hyperactivation leading to the enhanced proliferation and enlargement of β cells, increased insulin production, and pancreatic islets hyperplasia [14].
5.3. PHLDA3 Deficiency Causes Apoptosis Resistance in Pancreatic Islet β Cells
Streptozotocin is a drug that induces apoptosis specifically in pancreatic islet β cells and is used experimentally to cause type 1 diabetes in animal models [79]. We found that when streptozotocin was administered to wild-type and PHLDA3-deficient mice, the resulting increase in blood glucose levels was significantly suppressed in PHLDA3-deficient mice compared to wild-type mice. In addition, the decrease in β cells was dramatically suppressed (Figure 3B). These results show that PHLDA3 deficiency causes islet β cells to become resistant to apoptosis [14].
5.4. PHLDA3-Deficient Islets May Have Application in Diabetes Therapy
Interestingly, PHLDA3-deficient mice do not show increased tumor development, even though PHLDA3 deficiency activates the PI3K/Akt/mTOR pathway and promotes the proliferation of islet β cells, increased insulin production and decreased apoptosis. Another characteristic of PHLDA3 deficiency is increased resistance to stress such as hypoxia, which is a problem in islet transplantation, compromising islet isolation and inducing damage during the early stages of transplantation [10]. We have found that transplantation of PHLDA3-deficient islets into a diabetic mouse model leads to a better outcome than those transplanted with wild-type islets [10]. These results suggest that PHLDA3 deficiency contributes to islet cell survival, suggesting that such cells could be useful in the treatment of diabetes by islets transplantation. We are hopeful that the transplantation of PHLDA3-deficient islets may be used as a novel therapy for the treatment of diabetes in the future.
6. Conclusions
We have shown that the PHLDA3 gene is a p53 target gene and encodes a repressor of Akt. In addition to our works, several other groups have described the significance of PHLDA3 as a p53 target gene. For example, Allen et al. have revealed that PHLDA3 is one of the genes most strongly induced by p53 [27]. Brady et al. have found that PHLDA3 is one of the most important p53 target genes involved in tumor suppression [28]. In our studies, we have shown that PHLDA3 is a tumor suppressor of human PanNETs, and loss of PHLDA3 function is achieved by 2-hit inactivation in PanNETs, i.e., LOH and methylation. PHLDA3 has been found to be functionally deficient not only in PanNETs but also in lung NETs and rectal NETs, suggesting that it may be a common tumor suppressor of NETs [11,13,14]. Analysis of the function of PHLDA3 in other types of NETs including pituitary NETs, esophageal NETs, thyroid NETs, and parathyroid NETs will be important in future studies. Although we expect this area of research will be of interest to numerous researchers, most of the results to date come from our own studies. Further studies by many researchers, including our group, will be necessary to assess the functions of PHLDA3 in tumorigenesis.
Although PHLDA3 is a target gene regulated by p53, the mutation of p53 itself is rarely found in several NETs including low-grade lung NETs, PanNETs, and rectal NETs. We have noted that functional loss of PHLDA3 and p53 occur in a mutually exclusive pattern in lung NETs and PanNETs (Figs. 2A, 2B). This indicates that PHLDA3 is an important downstream tumor-suppressive mediator of p53, and its functional loss is critical to tumorigenesis in NETs having wild-type p53. Further analysis should clarify whether loss of p53 and PHLDA3 function also occur in a mutually exclusive pattern in other types of NETs, which would confirm the importance of the p53-PHLDA3 pathway in NET tumorigenesis. We would also note that there are many other types of cancer besides NETs that retain wild-type p53 [80,81]. Therefore, it would be of great interest to ask if PHLDA3 function is defective in these cancers. We could imagine that PHLDA3 plays a role as a tumor suppressor in a broad range of cancers having wild-type p53, and this p53-PHLDA3 tumor-suppressive pathway could be a novel target for cancer therapy.
We also have showed that functional loss of PHLDA3 in PanNETs correlates with higher malignant tumor progression and poorer patient prognosis. These results indicate that PHLDA3 is very important in the progression of PanNETs and suggest that analysis of PHLDA3 LOH in PanNETs could be a useful diagnostic marker providing information regarding the tumor’s metastatic potential and patient prognosis.
In recent years, streptozotocin has been used as a PanNET therapy [82,83,84,85,86]. However, islet β cells from PHLDA3-deficient mice were found to be resistant to streptozotocin [14]. Based on this result, PanNET patients having PHLDA3 LOH may be expected to be non-responsive to streptozotocin therapy. Another effective drug for PanNETs is everolimus, a PI3K/Akt/mTOR pathway inhibitor [87]. We may expect everolimus to be more effective in PanNET patients having PHLDA3 LOH, since loss of PHLDA3 results in the abnormal activation of the PI3K/Akt/mTOR pathway. On the other hand, the PI3K/Akt/mTOR pathway has been reported to be activated in many other cancers [29,30,31]. Since PHLDA3 regulates the PI3K/Akt/mTOR pathway by suppressing Akt activation, it is possible that new drugs that mimic PHLDA3 function may show efficacy not only in PanNETs but also in other cancers in which Akt is highly activated.
Analysis of a PHLDA3-deficient mouse model has revealed that Akt is abnormally activated due to functional loss of PHLDA3 in pancreatic islet β cells, leading to islet hyperplasia, but not to the development of PanNETs [14]. These results suggest that activation of Akt pathway alone is not sufficient to cause PanNETs. On the other hand, MEN1 knockout mice develop low-grade PanNETs at a late stage. This delay in PanNET formation implies that additional abnormal factors and/or pathways are required for PanNET tumorigenesis [88]. In human PanNETs, deficiencies of both the PHLDA3 and MEN1 pathway are required for tumorigenesis and progression, indicating that the combined loss of PHLDA3 and MEN1 function promotes PanNET tumorigenesis (Figure 4). The above-mentioned results suggest that mice lacking both the PHLDA3 and MEN1 genes might be a good model for development of PanNETs at an early age, and for analyzing the mechanisms of PanNET tumorigenesis and progression.
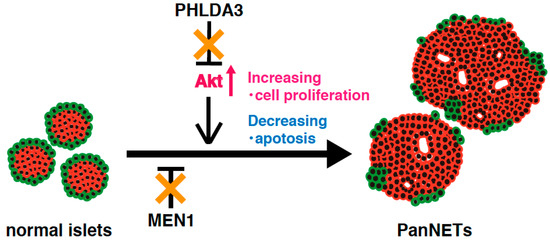
Figure 4.
A model of NET tumorigenesis resulting from functional loss of both PHLDA3 and MEN1. PHLDA3 is a repressor of Akt. Loss of PHLDA3 leads to Akt activation, increased cell proliferation and decreased apoptosis. PHLDA3 functions independent of MEN1, and PanNET tumorigenesis therefore requires the functional loss of both PHLDA3 and MEN1.
In summary, PHLDA3 is a p53 target gene that functions as an endogenic inhibitor of Akt. Functional loss of PHLDA3 is found in PanNETs, and LOH at PHLDA3 locus is frequently found in lung and rectal NETs. Together these results suggest that loss of PHLDA3 function disrupts the balance of p53-PHLDA3-Akt axis and promotes NET tumorigenesis and progression.
Author Contributions
Y.C. and R.O. drafted the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This study was partly supported by a Grant-in-Aid for Scientific Research (B) (#17H03587) (R.O.), Grant-in-Aid for Scientific Research (C) (#20K07605) (R.O.), Grant-in-Aid for Young Scientist (B) (#19K16732) (Y.C.) and Grant-in-Aid for JSPS Fellows (#18F18758) from the Ministry of Education, Culture, Sports, Science and Technology of Japan; an Extramural Collaborative Research Grant of the Cancer Research Institute, Kanazawa University, Japan (Y.C.); research grants from Pancreas Research Foundation of Japan (Y.C.); the Ichiro Kanehara Foundation for the promotion of Medical Sciences and Medical Care (Y.C.); research grants from Applied Research for Innovative Treatment of Cancer from the Ministry of Health, Labour and Welfare (R.O.), Project for Development of Innovative Research on Cancer Therapeutics (P-Direct) and Project for Cancer Research and Therapeutic Evolution (P-Create) from Japan Agency for Medical Research and Development (R.O.); research grants from Research Grant of the Princess Takamatsu Cancer Research Fund (R.O.); the Mitsubishi Foundation (to R.O.); the NOVARTIS Foundation (Japan) for the Promotion of Science (to R.O.); the Project Mirai Cancer Research Grants (R.O.); the Okinaka Memorial Institute for Medical Research (to R.O.); the Life Science Foundation of Japan (to R.O.); and Foundation for Promotion of Cancer Research in Japan (to R.O.).
Acknowledgments
We thank Marc Lamphier for critical reading of the manuscript.
Conflicts of Interest
The authors declare that they have no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Hainaut, P.; Hollstein, M. P53 and human cancer: The first ten thousand mutations. Adv. Cancer Res. 2000, 77, 81–137. [Google Scholar]
- Hollstein, M.; Sidransky, D.; Vogelstein, B.; Harris, C.C. P53 mutations in human cancers. Science 1991, 253, 49–53. [Google Scholar] [CrossRef]
- Levine, A.J.; Momand, J.; Finlay, C.A. The p53 tumour suppressor gene. Nature 1991, 351, 453–456. [Google Scholar] [CrossRef] [PubMed]
- Greenblatt, M.; Bennett, W.P.; Hollstein, M.; Harris, C. Mutations in the p53 tumor suppressor gene: Clues to cancer etiology and molecular pathogenesis. Cancer Res. 1994, 54, 4855–4878. [Google Scholar] [PubMed]
- Ohki, R.; Nemoto, J.; Murasawa, H.; Oda, E.; Inazawa, J.; Tanaka, N.; Taniguchi, T. Reprimo, a new candidate mediator of the p53-mediated cell cycle arrest at the G2 phase. J. Biol. Chem. 2000, 275, 22627–22630. [Google Scholar] [CrossRef] [PubMed]
- Ohki, R.; Kawase, T.; Ohta, T.; Ichikawa, H.; Taya, Y. Dissecting functional roles of p53 N-terminal transactivation domains by microarray expression analysis. Cancer Sci. 2007, 98, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Kawase, T.; Ichikawa, H.; Ohta, T.; Nozaki, N.; Tashiro, F.; Ohki, R.; Taya, Y. p53 target gene AEN is a nuclear exonuclease required for p53-dependent apoptosis. Oncogene 2008, 27, 3797–3810. [Google Scholar] [CrossRef]
- Asano, Y.; Kawase, T.; Okabe, A.; Tsutsumi, S.; Ichikawa, H.; Tatebe, S.; Kitabayashi, I.; Tashiro, F.; Namiki, H.; Kondo, T.; et al. IER5 generates a novel hypo-phosphorylated active form of HSF1 and contributes to tumorigenesis. Sci. Rep. 2016, 6, 19174. [Google Scholar] [CrossRef]
- Ezawa, I.; Sawai, Y.; Kawase, T.; Okabe, A.; Tsutsumi, S.; Ichikawa, H.; Kobayashi, Y.; Tashiro, F.; Namiki, H.; Kondo, T.; et al. Novel p53 target gene FUCA1 encodes a fucosidase and regulates growth and survival of cancer cells. Cancer Sci. 2016, 107, 734–745. [Google Scholar] [CrossRef]
- Sakata, N.; Yamaguchi, Y.; Chen, Y.; Shimoda, M.; Yoshimatsu, G.; Unno, M.; Sumi, S.; Ohki, R. Pleckstrin homology-like domain family a, member 3 (PHLDA3) deficiency improves islets engraftment through the suppression of hypoxic damage. PLoS ONE 2017, 12, e0187927. [Google Scholar] [CrossRef]
- Kojima, M.; Chen, Y.; Ikeda, K.; Tsukada, Y.; Takahashi, D.; Kawano, S.; Amemiya, K.; Ito, M.; Ohki, R.; Ochiai, A. Recommendation of long-term and systemic management according to the risk factors in rectal NETs patients. Sci. Rep. 2019, 9, 2404. [Google Scholar] [CrossRef] [PubMed]
- Yamano, S.; Kimura, M.; Chen, Y.; Imamoto, N.; Ohki, R. Nuclear import of IER5 is mediated by a classical bipartite nuclear localization signal and is required for HSF1 full activation. Exp. Cell Res. 2020, 386, 111686. [Google Scholar] [CrossRef] [PubMed]
- Kawase, T.; Ohki, R.; Shibata, T.; Tsutsumi, S.; Kamimura, N.; Inazawa, J.; Ohta, T.; Ichikawa, H.; Aburatani, H.; Tashiro, F.; et al. PH domain-only protein PHLDA3 is a p53-regulated repressor of Akt. Cell 2009, 136, 535–550. [Google Scholar] [CrossRef]
- Ohki, R.; Saito, K.; Chen, Y.; Kawase, T.; Hiraoka, N.; Saigawa, R.; Minegishi, M.; Aita, Y.; Yanai, G.; Shimizu, H.; et al. PHLDA3 is a novel tumor suppressor of pancreatic neuroendocrine tumors. Proc. Natl. Acad. Sci. USA 2014, 111, E2404–E2413. [Google Scholar] [CrossRef]
- Levine, A.J. p53, the cellular gatekeeper for growth and division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef]
- Banin, S.; Moyal, L.; Shieh, S.-Y.; Taya, Y.; Anderson, C.; Chessa, L.; Smorodinsky, N.; Prives, C.; Reiss, Y.; Shiloh, Y. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 1998, 281, 1674–1677. [Google Scholar] [CrossRef]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef]
- Carr, A.M. Piecing together the p53 puzzle. Science 2000, 287, 1765–1766. [Google Scholar] [CrossRef]
- Sherr, C.J.; Weber, J.D. The ARF/p53 pathway. Curr. Opin. Genet. Dev. 2000, 10, 94–99. [Google Scholar] [CrossRef]
- Lowe, S.W.; Lin, A.W. Apoptosis in cancer. Carcinogenesis 2000, 21, 485–495. [Google Scholar] [CrossRef]
- Meek, D.W. Mechanisms of switching on p53: A role for covalent modification? Oncogene 1999, 18, 7666–7675. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Levine, A.; Hu, W.; Feng, Z. The P53 pathway: What questions remain to be explored? Cell Death Differ. 2006, 13, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Bode, A.M.; Dong, Z. Post-translational modification of p53 in tumorigenesis. Nat. Rev. Cancer 2004, 4, 793–805. [Google Scholar] [CrossRef] [PubMed]
- Barlev, N.A.; Liu, L.; Chehab, N.H.; Mansfield, K.; Harris, K.G.; Halazonetis, T.D.; Berger, S.L. Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. Mol. Cell 2001, 8, 1243–1254. [Google Scholar] [CrossRef]
- Chehab, N.H.; Malikzay, A.; Stavridi, E.S.; Halazonetis, T.D. Phosphorylation of ser-20 mediates stabilization of human p53 in response to DNA damage. Proc. Natl. Acad. Sci. USA 1999, 96, 13777–13782. [Google Scholar] [CrossRef]
- Meek, D.W.; Anderson, C.W. Posttranslational modification of p53: Cooperative integrators of function. Cold Spring Harb. Perspect. Biol. 2009, 1, a000950. [Google Scholar] [CrossRef]
- Allen, M.A.; Andrysik, Z.; Dengler, V.L.; Mellert, H.S.; Guarnieri, A.; Freeman, J.A.; Sullivan, K.D.; Galbraith, M.D.; Luo, X.; Kraus, W.L. Global analysis of p53-regulated transcription identifies its direct targets and unexpected regulatory mechanisms. Elife 2014, 3, e02200. [Google Scholar] [CrossRef] [PubMed]
- Brady, C.A.; Jiang, D.; Mello, S.S.; Johnson, T.M.; Jarvis, L.A.; Kozak, M.M.; Broz, D.K.; Basak, S.; Park, E.J.; McLaughlin, M.E. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell 2011, 145, 571–583. [Google Scholar] [CrossRef]
- Luo, J.; Manning, B.D.; Cantley, L.C. Targeting the PI3K-Akt pathway in human cancer: Rationale and promise. Cancer Cell 2003, 4, 257–262. [Google Scholar] [CrossRef]
- Altomare, D.A.; Testa, J.R. Perturbations of the AKT signaling pathway in human cancer. Oncogene 2005, 24, 7455–7464. [Google Scholar] [CrossRef]
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-kinase–AKT pathway in human cancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar] [CrossRef]
- Bellacosa, A.; Kumar, C.C.; Di Cristofano, A.; Testa, J.R. Activation of AKT kinases in cancer: Implications for therapeutic targeting. Adv. Cancer Res. 2005, 94, 29–86. [Google Scholar]
- Manning, B.D.; Toker, A. AKT/PKB signaling: Navigating the network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef]
- Yuan, T.; Cantley, L. PI3K pathway alterations in cancer: Variations on a theme. Oncogene 2008, 27, 5497–5510. [Google Scholar] [CrossRef]
- James, S.R.; Peter DOWNES, C.; Gigg, R.; Grove, S.J.; Holmes, A.B.; Alessi, D.R. Specific binding of the Akt-1 protein kinase to phosphatidylinositol 3, 4, 5-trisphosphate without subsequent activation. Biochem. J. 1996, 315, 709–713. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef]
- Mayo, L.D.; Donner, D.B. The PTEN, Mdm2, p53 tumor suppressor–oncoprotein network. Trends Biochem. Sci. 2002, 27, 462–467. [Google Scholar] [CrossRef]
- Brooks, C.L.; Gu, W. p53 ubiquitination: Mdm2 and beyond. Mol. Cell 2006, 21, 307–315. [Google Scholar] [CrossRef]
- Gottlieb, T.M.; Leal, J.F.M.; Seger, R.; Taya, Y.; Oren, M. Cross-talk between Akt, p53 and Mdm2: Possible implications for the regulation of apoptosis. Oncogene 2002, 21, 1299–1303. [Google Scholar] [CrossRef]
- Momand, J.; Zambetti, G.P.; Olson, D.C.; George, D.; Levine, A.J. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 1992, 69, 1237–1245. [Google Scholar] [CrossRef]
- Mayo, L.D.; Donner, D.B. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc. Natl. Acad. Sci. USA 2001, 98, 11598–11603. [Google Scholar] [CrossRef]
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, R. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275, 1943–1947. [Google Scholar] [CrossRef]
- Maier, D.; Zhang, Z.; Taylor, E.; Hamou, M.-F.; Gratzl, O.; Van Meir, E.G.; Scott, R.J.; Merlo, A. Somatic deletion mapping on chromosome 10 and sequence analysis of PTEN/MMAC1 point to the 10q25-26 region as the primary target in low-grade and high-grade gliomas. Oncogene 1998, 16, 3331–3335. [Google Scholar] [CrossRef][Green Version]
- Juven-Gershon, T.; Oren, M. Mdm2: The ups and downs. Mol. Med. 1999, 5, 71–83. [Google Scholar] [CrossRef]
- Momand, J.; Jung, D.; Wilczynski, S.; Niland, J. The MDM2 gene amplification database. Nucleic Acids Res. 1998, 26, 3453–3459. [Google Scholar] [CrossRef]
- Oronsky, B.; Ma, P.C.; Morgensztern, D.; Carter, C.A. Nothing but NET: A review of neuroendocrine tumors and carcinomas. Neoplasia 2017, 19, 991–1002. [Google Scholar] [CrossRef]
- Hauso, O.; Gustafsson, B.I.; Kidd, M.; Waldum, H.L.; Drozdov, I.; Chan, A.K.; Modlin, I.M. Neuroendocrine tumor epidemiology: Contrasting Norway and North America. Cancer 2008, 113, 2655–2664. [Google Scholar] [CrossRef]
- Yao, J.C.; Hassan, M.; Phan, A.; Dagohoy, C.; Leary, C.; Mares, J.E.; Abdalla, E.K.; Fleming, J.B.; Vauthey, J.N.; Rashid, A.; et al. One hundred years after "carcinoid": Epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the united states. J. Clin. Oncol. 2008, 26, 3063–3072. [Google Scholar] [CrossRef]
- Hamilton, S.R.; Aaltonen, L.A. (Eds.) World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of the Digestive System; IARC Press: Lyon, France, 2000. [Google Scholar]
- Scarpa, A.; Chang, D.K.; Nones, K.; Corbo, V.; Patch, A.M.; Bailey, P.; Lawlor, R.T.; Johns, A.L.; Miller, D.K.; Mafficini, A.; et al. Whole-genome landscape of pancreatic neuroendocrine tumours. Nature 2017, 543, 65–71. [Google Scholar] [CrossRef]
- Franko, J.; Feng, W.; Yip, L.; Genovese, E.; Moser, A.J. Non-functional neuroendocrine carcinoma of the pancreas: Incidence, tumor biology, and outcomes in 2,158 patients. J. Gastrointest. Surg. 2010, 14, 541–548. [Google Scholar] [CrossRef]
- Bilimoria, K.Y.; Tomlinson, J.S.; Merkow, R.P.; Stewart, A.K.; Ko, C.Y.; Talamonti, M.S.; Bentrem, D.J. Clinicopathologic features and treatment trends of pancreatic neuroendocrine tumors: Analysis of 9821 patients. J. Gastrointest. Surg. 2007, 11, 1460–1469. [Google Scholar] [CrossRef]
- Modlin, I.M.; Oberg, K.; Chung, D.C.; Jensen, R.T.; de Herder, W.W.; Thakker, R.V.; Caplin, M.; Delle Fave, G.; Kaltsas, G.A.; Krenning, E.P. Gastroenteropancreatic neuroendocrine tumours. Lancet Oncol. 2008, 9, 61–72. [Google Scholar] [CrossRef]
- Soga, J. Early-stage carcinoids of the gastrointestinal tract: An analysis of 1914 reported cases. Cancer Interdiscip. Int. J. Am. Cancer Soc. 2005, 103, 1587–1595. [Google Scholar] [CrossRef]
- Modlin, I.M.; Lye, K.D.; Kidd, M. A 5-decade analysis of 13,715 carcinoid tumors. Cancer Interdiscip. Int. J. Am. Cancer Soc. 2003, 97, 934–959. [Google Scholar] [CrossRef] [PubMed]
- Goh, A.M.; Coffill, C.R.; Lane, D.P. The role of mutant p53 in human cancer. J. Pathol. 2011, 223, 116–126. [Google Scholar] [CrossRef]
- Morton, J.P.; Timpson, P.; Karim, S.A.; Ridgway, R.A.; Athineos, D.; Doyle, B.; Jamieson, N.B.; Oien, K.A.; Lowy, A.M.; Brunton, V.G. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 246–251. [Google Scholar] [CrossRef]
- Muller, P.A.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef]
- Przygodzki, R.M.; Finkelstein, S.D.; Langer, J.C.; Swalsky, P.A.; Fishback, N.; Bakker, A.; Guinee, D.G.; Koss, M.; Travis, W.D. Analysis of p53, k-ras-2, and c-raf-1 in pulmonary neuroendocrine tumors. Correlation with histological subtype and clinical outcome. Am. J. Pathol. 1996, 148, 1531–1541. [Google Scholar]
- Couce, M.E.; Bautista, D.; Costa, J.; Carter, D. Analysis of k-ras, n-ras, h-ras, and p53 in lung neuroendocrine neoplasms. Diagn. Mol. Pathol. 1999, 8, 71–79. [Google Scholar] [CrossRef]
- Lohmann, D.R.; Fesseler, B.; Pütz, B.; Reich, U.; Böhm, J.; Präuer, H.; Wünsch, P.H.; Höfler, H. Infrequent mutations of the p53 gene in pulmonary carcinoid tumors. Cancer Res. 1993, 53, 5797–5801. [Google Scholar]
- Vollbrecht, C.; Werner, R.; Walter, R.F.; Christoph, D.C.; Heukamp, L.C.; Peifer, M.; Hirsch, B.; Burbat, L.; Mairinger, T.; Schmid, K.W.; et al. Mutational analysis of pulmonary tumours with neuroendocrine features using targeted massive parallel sequencing: A comparison of a neglected tumour group. Br. J. Cancer 2015, 113, 1704–1711. [Google Scholar] [CrossRef] [PubMed]
- Simbolo, M.; Mafficini, A.; Sikora, K.O.; Fassan, M.; Barbi, S.; Corbo, V.; Mastracci, L.; Rusev, B.; Grillo, F.; Vicentini, C.; et al. Lung neuroendocrine tumours: Deep sequencing of the four World Health Organization histotypes reveals chromatin-remodelling genes as major players and a prognostic role for TERT, RB1, MEN1 and KMT2D. J. Pathol. 2017, 241, 488–500. [Google Scholar] [CrossRef] [PubMed]
- George, J.; Walter, V.; Peifer, M.; Alexandrov, L.B.; Seidel, D.; Leenders, F.; Maas, L.; Müller, C.; Dahmen, I.; Delhomme, T.M.; et al. Integrative genomic profiling of large-cell neuroendocrine carcinomas reveals distinct subtypes of high-grade neuroendocrine lung tumors. Nat. Commun. 2018, 9, 1048. [Google Scholar] [CrossRef]
- Bartz, C.; Ziske, C.; Wiedenmann, B.; Moelling, K. p53 tumour suppressor gene expression in pancreatic neuroendocrine tumour cells. Gut 1996, 38, 403–409. [Google Scholar] [CrossRef][Green Version]
- Lohmann, D.R.; Funk, A.; Niedermeyer, H.P.; Häupel, S.; Höfler, H. Identification of p53 gene mutations in gastrointestinal and pancreatic carcinoids by nonradioisotopic SSCA. Virchows Arch. B 1993, 64, 293–296. [Google Scholar] [CrossRef]
- Jiao, Y.; Shi, C.; Edil, B.H.; De Wilde, R.F.; Klimstra, D.S.; Maitra, A.; Schulick, R.D.; Tang, L.H.; Wolfgang, C.L.; Choti, M.A. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 2011, 331, 1199–1203. [Google Scholar] [CrossRef]
- Park, H.Y.; Kwon, M.J.; Kang, H.S.; Kim, Y.J.; Kim, N.Y.; Kim, M.J.; Min, K.-W.; Choi, K.C.; Nam, E.S.; Cho, S.J. Targeted next-generation sequencing of well-differentiated rectal, gastric, and appendiceal neuroendocrine tumors to identify potential targets. Hum. Pathol. 2019, 87, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-J.; Vortmeyer, A.; Zhuang, Z.; Huang, S.; Jensen, R.T. Loss of heterozygosity of chromosome 1q in gastrinomas: Occurrence and prognostic significance. Cancer Res. 2003, 63, 817–823. [Google Scholar]
- Yang, Y.M.; Liu, T.H.; Chen, Y.J.; Jiang, W.J.; Qian, J.M.; Lu, X.; Gao, J.; Wu, S.F.; Sang, X.T.; Chen, J. Chromosome 1q loss of heterozygosity frequently occurs in sporadic insulinomas and is associated with tumor malignancy. Int. J. Cancer 2005, 117, 234–240. [Google Scholar] [CrossRef]
- Pannett, A.; Thakker, R. Multiple endocrine neoplasia type 1. Endocr. -Relat. Cancer 1999, 6, 449–473. [Google Scholar] [CrossRef]
- Corbo, V.; Dalai, I.; Scardoni, M.; Barbi, S.; Beghelli, S.; Bersani, S.; Albarello, L.; Doglioni, C.; Schott, C.; Capelli, P. MEN1 in pancreatic endocrine tumors: Analysis of gene and protein status in 169 sporadic neoplasms reveals alterations in the vast majority of cases. Endocr. -Relat. Cancer 2010, 17, 771–783. [Google Scholar] [CrossRef]
- Konishi, T.; Watanabe, T.; Kishimoto, J.; Kotake, K.; Muto, T.; Nagawa, H. Prognosis and risk factors of metastasis in colorectal carcinoids: Results of a nationwide registry over 15 years. Gut 2007, 56, 863–868. [Google Scholar] [CrossRef]
- Kojima, M.; Ikeda, K.; Saito, N.; Sakuyama, N.; Koushi, K.; Kawano, S.; Watanabe, T.; Sugihara, K.; Ito, M.; Ochiai, A. Neuroendocrine tumors of the large intestine: Clinicopathological features and predictive factors of lymph node metastasis. Front. Oncol. 2016, 6, 173. [Google Scholar] [CrossRef] [PubMed]
- Bernick, P.; Klimstra, D.; Shia, J.; Minsky, B.; Saltz, L.; Shi, W.; Thaler, H.; Guillem, J.; Paty, P.; Cohen, A. Neuroendocrine carcinomas of the colon and rectum. Dis. Colon Rectum 2004, 47, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, R.L.; Gill, N.S.; Pugh, W.; Lee, J.-P.; Koeberlein, B.; Furth, E.E.; Polonsky, K.S.; Naji, A.; Birnbaum, M.J. Regulation of pancreatic β-cell growth and survival by the serine/threonine protein kinase Akt1/PKBα. Nat. Med. 2001, 7, 1133–1137. [Google Scholar] [CrossRef] [PubMed]
- Bernal-Mizrachi, E.; Wen, W.; Stahlhut, S.; Welling, C.M.; Permutt, M.A. Islet β cell expression of constitutively active Akt1/PKBα induces striking hypertrophy, hyperplasia, and hyperinsulinemia. J. Clin. Investig. 2001, 108, 1631–1638. [Google Scholar] [CrossRef] [PubMed]
- Balcazar Morales, N.; Aguilar de Plata, C. Role of AKT/mTORC1 pathway in pancreatic β-cell proliferation. Colomb. Médica 2012, 43, 235–243. [Google Scholar] [CrossRef]
- Lenzen, S. The mechanisms of alloxan-and streptozotocin-induced diabetes. Diabetologia 2008, 51, 216–226. [Google Scholar] [CrossRef]
- Crook, T.; Wrede, D.; Tidy, J.; Vousden, K.; Tidy, J.; Mason, W.; Evans, D. Clonal p53 mutation in primary cervical cancer: Association with human-papillomavirus-negative tumours. Lancet 1992, 339, 1070–1073. [Google Scholar] [CrossRef]
- Hall, M.C.; Navone, N.M.; Troncoso, P.; Pollack, A.; Zagars, G.K.; von Eschenbach, A.C.; Conti, C.J.; Chung, L.W. Frequency and characterization of p53 mutations in clinically localized prostate cancer. Urology 1995, 45, 470–475. [Google Scholar] [CrossRef]
- Antonodimitrakis, P.C.; Sundin, A.; Wassberg, C.; Granberg, D.; Skogseid, B.; Eriksson, B. Streptozocin and 5-fluorouracil for the treatment of pancreatic neuroendocrine tumors: Efficacy, prognostic factors and toxicity. Neuroendocrinology 2016, 103, 345–353. [Google Scholar] [CrossRef]
- Murray-Lyon, I.; Eddleston, A.; Williams, R.; Brown, M.; Hogbin, B.; Bennett, A.; Edwards, J.; Taylor, K. Treatment of multiple-hormone-producing malignant islet-cell tumour with streptozotocin. Lancet 1968, 292, 895–898. [Google Scholar] [CrossRef]
- Broder, L.E.; Carter, S.K. Pancreatic islet cell carcinoma. II. Results of therapy with streptozotocin in 52 patients. Ann. Intern. Med. 1973, 79, 108–118. [Google Scholar] [CrossRef]
- Delaunoit, T.; Ducreux, M.; Boige, V.; Dromain, C.; Sabourin, J.-C.; Duvillard, P.; Schlumberger, M.; De Baere, T.; Rougier, P.; Ruffie, P. The doxorubicin-streptozotocin combination for the treatment of advanced well-differentiated pancreatic endocrine carcinoma: A judicious option? Eur. J. Cancer 2004, 40, 515–520. [Google Scholar] [CrossRef]
- Dilz, L.-M.; Denecke, T.; Steffen, I.G.; Prasad, V.; von Weikersthal, L.F.; Pape, U.-F.; Wiedenmann, B.; Pavel, M. Streptozocin/5-fluorouracil chemotherapy is associated with durable response in patients with advanced pancreatic neuroendocrine tumours. Eur. J. Cancer 2015, 51, 1253–1262. [Google Scholar] [CrossRef]
- Yao, J.C.; Shah, M.H.; Ito, T.; Bohas, C.L.; Wolin, E.M.; Van Cutsem, E.; Hobday, T.J.; Okusaka, T.; Capdevila, J.; De Vries, E.G. Everolimus for advanced pancreatic neuroendocrine tumors. New Engl. J. Med. 2011, 364, 514–523. [Google Scholar] [CrossRef]
- Crabtree, J.S.; Scacheri, P.C.; Ward, J.M.; McNally, S.R.; Swain, G.P.; Montagna, C.; Hager, J.H.; Hanahan, D.; Edlund, H.; Magnuson, M.A. Of mice and MEN1: Insulinomas in a conditional mouse knockout. Mol. Cell. Biol. 2003, 23, 6075–6085. [Google Scholar]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).