Abstract
Intracellular calcium (Ca2+) concentration ([Ca2+]i) is a key determinant of cell fate and is implicated in carcinogenesis. Membrane ion channels are structures through which ions enter or exit the cell, depending on the driving forces. The opening of transient receptor potential vanilloid 1 (TRPV1) ligand-gated ion channels facilitates transmembrane Ca2+ and Na+ entry, which modifies the delicate balance between apoptotic and proliferative signaling pathways. Proliferation is upregulated through two mechanisms: (1) ATP binding to the G-protein-coupled receptor P2Y2, commencing a kinase signaling cascade that activates the serine-threonine kinase Akt, and (2) the transactivation of the epidermal growth factor receptor (EGFR), leading to a series of protein signals that activate the extracellular signal-regulated kinases (ERK) 1/2. The TRPV1-apoptosis pathway involves Ca2+ influx and efflux between the cytosol, mitochondria, and endoplasmic reticulum (ER), the release of apoptosis-inducing factor (AIF) and cytochrome c from the mitochondria, caspase activation, and DNA fragmentation and condensation. While proliferative mechanisms are typically upregulated in cancerous tissues, shifting the balance to favor apoptosis could support anti-cancer therapies. TRPV1, through [Ca2+]i signaling, influences cancer cell fate; therefore, the modulation of the TRPV1-enforced proliferation–apoptosis balance is a promising avenue in developing anti-cancer therapies and overcoming cancer drug resistance. As such, this review characterizes and evaluates the role of TRPV1 in cell death and survival, in the interest of identifying mechanistic targets for drug discovery.
1. [Ca2+]i and the Critical Balance between Apoptosis and Proliferation
Molecular mechanisms that mediate cell death and proliferation exist in balance in functional physiological systems. Proliferation is involved in structural development and renewal, while programmed cell death is necessary to eliminate defective cells and prevent uncontrolled growth. Carcinogenesis results from imbalances in the described pathways, which favor proliferation and reduce apoptosis [1,2]. Therefore, anti-cancer therapies shift the balance in the opposite direction by reducing proliferation and upregulating apoptosis.
Apoptosis is defined as programmed cell death, characterized by fragmentation of inter-nucleosomal DNA [3]. Two major mechanisms of apoptosis are an extrinsic, death-receptor mediated mechanism, and an intrinsic, mitochondria-mediated mechanism [4]. The extrinsic mechanism involves the linking of membrane death receptors to adapter proteins, which bind and position pro-caspase 8 for conversion into caspase 8; the intrinsic mechanism is triggered by the release of cytochrome c from mitochondria, which promotes caspase 9 activation [4,5]. The Bcl-2 family of proteins, which includes the proapoptotic proteins Bax and Bak and the antiapoptotic protein Bcl-2, is implicated in the intrinsic mechanism of apoptosis [6]. Both the intrinsic and extrinsic apoptotic mechanisms lead to the activation of caspase 3, which mediates apoptosis through nuclear activity.
Calcium (Ca2+) is a second messenger that influences the proliferation–apoptosis balance. Intracellular Ca2+ ([Ca2+]i) is modulated by receptor-operated, store-operated (SOC), and voltage-sensitive ion channels, ion exchangers, pumps, Ca2+ binding proteins, mitochondrial Ca2+ ([Ca2+]m), and endoplasmic reticulum (ER) and sarcoplasmic reticulum (SR) Ca2+ ([Ca2+]ER and [Ca2+]SR) [7,8]. Intracellular Ca2+ release channels comprise one subset of ion channels; these include the ryanodine receptor (RyR) and inositol 1,4,5-triphosphosphate (IP3) receptor (IP3R) channels, both of which are localized to the ER and SR. RyR channels, which are activated by elevated [Ca2+]i or protein signaling, and IP3R channels, which are activated by IP3 binding, release Ca2+ from the ER and SR. Through [Ca2+]i signaling, these two channel types modulate muscle contraction and nerve impulse transmission [9,10]. Abberant Ca2+ transport from the ER or SR to the cytosol may elevate [Ca2+]m and consequently induce mitochondrial dysfunction [11,12].
Beyond locomotion and neurotransmission, shifts in [Ca2+]i homeostasis may also mediate cell death or proliferation. For instance, while [Ca2+]i signaling via IP3R contributes to proliferation and oncogenesis, RyR [Ca2+]i signaling supports apoptosis in lung cancer cells [10,13]. Furthermore, Ca2+ influx through T-type voltage-gated Ca2+ channels (VGCC) is implicated in the proliferation of cancerous and noncancerous cells, while the blockage of such channels promotes apoptosis in glioblastoma cells [14,15]. In contrast, Ca2+ influx through L-type VGCC causes death in bovine chromaffin cells [16]. Notably, [Ca2+]i-mediated cell death may be apoptotic or necrotic in nature, depending on the time of exposure and [Ca2+] i involved [17].
Significantly, [Ca2+]I signaling regulates proliferation, invasion, and metastasis in cancerous tissues [18]. A variety of oncologic therapies, including cisplatin, arsenic trioxide, trimethyltin chloride, and some candidate epigenetic drugs, induce their proapoptotic and anti-proliferative effects (in part or in whole) through the modulation of [Ca2+]i [19,20]. Therefore, specific [Ca2+]i-affecting proteins, including transmembrane ion channels, which mediate Ca2+ flow between the extracellular space and the cytosol, are potential targets for chemotherapeutic agents.
Transient receptor potential (TRP) channels comprise a large family of membrane Ca2+ channels, which respond to a wide variety of environmental stimuli [21,22,23]. Transient receptor potential vanilloid 1, or vanilloid receptor 1 (TRPV1/VR1), known as the capsaicin receptor, is a member of the TRPV subfamily of TRP channels. TRPV1 is a ligand-gated ion channel which is activated by capsaicin and capsaicin analogues (e.g., resiniferatoxin, RTX), heat, and endogenous cannabinoids such as anandamide (AEA); its antagonists include capsazepine and ruthenium red [24,25]. The stimulation of TRPV1 causes Ca2+ and Na+ influx through transmembrane ion channels. While these channels generally exhibit selectivity for Ca2+ over Na+, the precise nature of this selectivity depends on a variety of factors, including the nature and concentration of the agonist [26]. TRPV1 is involved in thermoregulation, circadian rhythms, energy intake and metabolism, and acute, chronic, and inflammatory nociception; as such, the ion channel receptor is a target in the development of analgesic therapies [27,28,29,30,31,32]. Furthermore, given its role in modulating [Ca2+]i, TRPV1 influences the balance between proliferation and apoptosis [33]. This review aims to characterize the molecular mechanisms through which TRPV1 exerts the mentioned effect in the interest of identifying potential targets for anti-cancer drug development.
2. Expression of TRPV1 in Cancerous and Healthy Tissues
TRPV1 mRNA and protein are expressed in optic, pulmonary, nervous, cardiac, skeletal, circulatory, and skin cells, as well as in numerous cancer cell lines (Table 1). Compared to healthy cells, TRPV1 mRNA and/or protein expression levels are downregulated in many cancerous tissues, including colorectal, nervous system, endometrial, renal and skin cancers. However, TRPV1 mRNA levels are upregulated in the U373 glioblastoma line, high-grade astrocytes, “brain tumors,” and the RT4 renal cell carcinoma line; likewise, upregulated TRPV1 protein expression is observed in the U373 and RT4 cell lines (Table 2).

Table 1.
mRNA and protein expression of the transient receptor potential vanilloid 1 (TRPV1) ligand-gated ion channel in a variety of cell lines. TRPV1 receptor expression is well characterized in the nervous and optic systems and less so in the muscular and skeletal systems. Cancerous cells lines are highlighted in gray.
Table 2.
mRNA and protein expression levels of the TRPV1 ligand-gated ion channel in cancerous cell lines, as compared to healthy tissues. The “TRPV1 mRNA vs. Normal” and “TRPV1 Protein vs. Normal” columns evaluate the mRNA and protein expression levels, respectively, observed in the cancerous cell lines relative to the corresponding control/healthy cells. “NC” indicates “No Change” in expression between the normal and cancerous tissues. Cancerous cells lines are highlighted in gray.
3. Balance Between Apoptosis and Proliferation Mediated by TRPV1
The activation of the TRPV1 ion channel is a critical signal involved in numerous intracellular processes, some of which trigger either apoptosis or proliferation (Figure 1). While the apoptotic effects of TRPV1 are well characterized, literature on TRPV1-related proliferation remains sparse. The binding of exogenous agonists to the TRPV1 receptor and subsequent Ca2+ influx from the cytosol into the cell are characteristics shared between the apoptotic and proliferative pathways. However, both the positive allosteric modulation of cell membrane TRPV1 receptors and the activation of endoplasmic reticulum-localized TRPV1 channels are associated exclusively with the pro-apoptotic pathway [43,55].
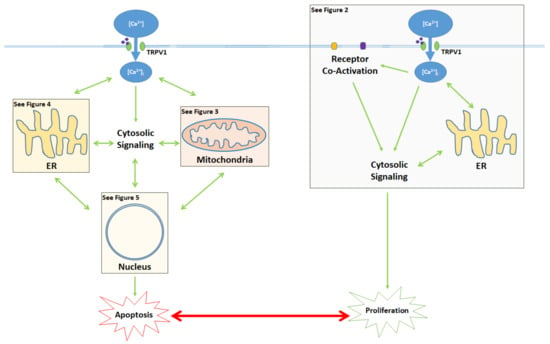
Figure 1.
Activation of the TRPV1 ligand-gated ion channel causes Ca2+ influx into the cytosol and influences the balance between proliferation and apoptosis. Apoptotic signaling occurs through the cytosol, mitochondria, endoplasmic reticulum (ER), and the nucleus. In contrast, the proliferative effects of TRPV1 are mediated by the activation of other cell membrane receptors, ER signaling, and cytosolic protein signaling cascades. The proliferative, proapoptotic mitochondrial, proapoptotic ER, and proapoptotic nuclear signaling mechanisms are highlighted in the colored boxes, and specified in Figure 2 Figure 3 Figure 4 Figure 5, respectively.
4. TRPV1-Mediated Proliferation
TRPV1 promotes proliferation upon activation by capsaicin, glycolic acid, anandamide (AEA), and its analogue SKM 4-45-1 (Figure 2) [40,41]. Glycolic acid stimulates Ca2+ influx into the cell and corresponding elevation of [Ca2+]i through TRPV1 channel opening. Furthermore, glycolic acid-TRPV1 interactions stimulate the release of intracellular ATP molecules into the cytosol, where they bind to the membrane G protein-coupled P2Y2 receptors [40]. Elevated [Ca2+]i and stimulated P2Y2 receptors activate phospholipase C (PLC), resulting in the upregulation of intracellular IP3 levels and subsequent store-operated Ca2+ entry [56,57,58]. Elevated [Ca2+]i and protein signals from the P2Y2 receptor activate the phosphoinisitide-3-kinase, PI3K [59,60]. PI3K subsequently activates the phosphoinositide-dependent kinases PDK 1 and 2. P2Y2 protein signals also activate protein kinase C (PKC), which activates the proto-oncogene protein kinase Src [61]. PDK 1 and 2 and Src then phosphorylate the Akt (serine/threonine protein kinase), promoting proliferation [59,60,61]. Concurrently, TRPV1 phosphorylates and thereby transactivates the epidermal growth factor receptor (EGFR) [62,63]. EGFR activates the Ras protein, a small GTPase, which in turn activates the serine-threonine protein kinase Raf; this kinase phosphorylates the mitogen-activated protein kinase (MAPK) kinases MEK 1/2. Finally, MEK 1/2 activate the extracellular signal-regulated kinases ERK 1/2 and their associated MAPKs, which further enhance proliferation [64].
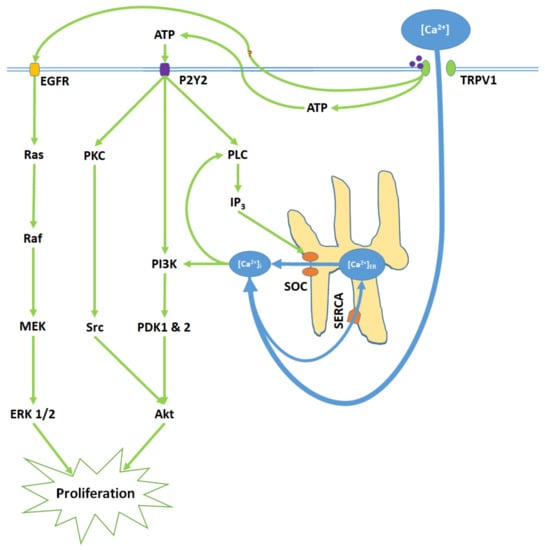
Figure 2.
TRPV1 induces proliferation through Ca2+ entry, ATP release and membrane P2Y2 receptor activation, and the transactivation of epidermal growth factor receptor (EGFR). Elevated [Ca2+]i and ATP-P2Y2 binding upregulate intracellular IP3 via phospholipase C (PLC); IP3 opens store-operated channels (SOC) and thereby causes Ca2+ release from the ER. Activated P2Y2 receptors also begin the PI3K/Akt pathway, a kinase signaling cascade that ultimately activates Akt. TRPV1 additionally transactivates EGFR; this prompts Ras/Raf/MAPK-ERK kinase (MEK)/extracellular signal-regulated kinase (ERK) signaling, which upregulates ERK 1/2 mitogen-activated protein kinases (MAPK). Akt and ERK 1/2 MAPK promote proliferation through nuclear activity.
Interestingly, in endothelial colony-forming cells (ECFC), capsaicin downregulates proliferation induced by AEA, potentially suggesting that the two TRPV1 agonists compete for binding sites and differentially influence the apoptosis–proliferation balance [41]. Additionally, despite its status as a TRPV1 antagonist, capsazepine upregulates proliferation in canine breast cancer cells [42] Another TRPV1 antagonist, AMG9810, enhances proliferation in human keratinocytes and murine skin cancer models [63]. As such, the precise functions of TRPV1 agonists and antagonists in proliferation may differ significantly between cell lines and therefore require further characterization (Table S1): ref. [35,41,42,62,63,65,66].
5. The Apoptotic Pathway and Upstream Cytosolic Effects
Apoptotic effects mediated by TRPV1 begin with the binding of the receptor to exogenous agonists or positive allosteric modulators, as well as the activation of the receptor through non-ligand means (such as magnetic fields). As TRPV1 is a ligand-gated cation channel, agonist binding results in Ca2+ and Na+ influx into the cell; notably, TRPV1 displays greater Ca2+ than Na+ affinity [67]. The mentioned Ca2+ influx elevates [Ca2+]i (Table S2): ref. [34,35,37,38,39,43,46,48,49,50,54,55,65,68,69,70,71,72,73,74,75,76,77,78,79]. The co-application of TRPV1 antagonists, including capsazepine, ruthenium red (RR) and idioresiniferatoxin (I-RTX), attenuates agonist-induced Ca2+ influx; furthermore, capsazepine alone reduces [Ca2+]i (Table S9): ref. [34,35,38,39,40,43,46,49,65,69,70,72,73,75,76,77,79].
6. Mitochondrial Pathway
The activation of TRPV1 by capsaicin leads to the phosphorylation and activation of the ATM serine-threonine kinase, which induces the downstream Fas pathway (Table S3): ref. [45,52]. ATM activation upregulates the Fas/CD95 death receptor, which co-clusters with TRPV1 to form a death signal complex. This function of this complex in TRPV1-mediated apoptosis is the cleavage of procaspase 8 into active caspase 8; caspase 8 transforms the BH3 interacting domain death agonist (BID) into its truncated form, which then contributes to the mitochondrial dysfunction [52]. The co-application of the TRPV1 antagonist capsazepine with capsaicin reduces the extent of TRPV1-Fas/CD95 co-clustering (Table S10): ref. [52].
Elevated [Ca2+]i, resulting from TRPV1 channel opening, causes an initial increase in [Ca2+]m and subsequent downstream effects, which can be attenuated by TRPV1 antagonists (Figure 3; Table S10): ref. [38,39,45,49,55,65,69,72,77]. Ca2+ entry into the mitochondrial matrix is mediated by the mitochondrial Ca2+ uniporter (MCU) [74,80]. To maintain mitochondrial homeostasis, some of the Ca2+ returns to the cytosol via the mitochondrial membrane Na+/Ca2+ exchanger (NCLX), which also transports Na+ from the cytosol into the matrix [74]. Additionally, reductions in intracellular glutathione ([GSH]i) levels (mediated by ER and nuclear action) decrease mitochondrial glutathione ([GSH]m) levels and mitochondrial ROS (ROSm) production, while Bax is upregulated via nuclear action; both GSH depletion and Bax binding to mitochondrial membrane voltage-dependent anion channels (VDAC) promote the opening of the mitochondrial permeability transition pore (PTP), through which further Ca2+ exit to the cytosol occurs [81,82]. Finally, Na+, which entered the matrix via the NCLX, is exported by the sodium–hydrogen exchanger (NHE), which also imports protons [83,84].
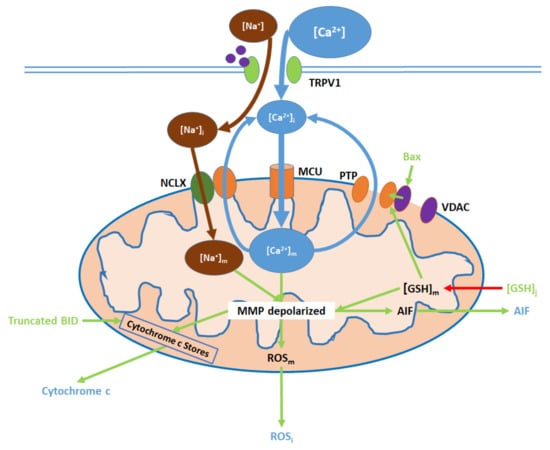
Figure 3.
TRPV1 induces mitochondrial dysfunction through Ca2+ and Na+ entry, membrane depolarization, ROS production, and the release of cytochrome c and apoptosis-inducing factor (AIF). Initial Ca2+ and Na+ influx causes the hyperpolarization of the mitochondrial membrane, while consequent Ca2+ export through the permeability transition pore (PTP) and active Ca2+ removal via the NCLX depolarize the membrane. As inputs, downregulated [GSH]i and upregulated Bax arise from nuclear activity. Upon their release (driven by membrane depolarization), AIF translocates directly to the nucleus, cytochrome c participates in intracellular caspase 9 activation, and intracellular ROS (ROSi) supports the activation of p38 MAPKs.
Initial elevations in [Ca2+]m and [Na+]m cause hyperpolarization of the membrane potential, followed by the depolarization upon PTP opening [85]. [GSH]m reduction contributes to mitochondrial membrane depolarization, which in turn upregulates ROSm generation. Intracellular ROS (ROSi) levels increase due to ROSm export to the cytosol [82,86]. Membrane depolarization also prompts the release of apoptosis-inducing factor (AIF) into the cytosol [87]. Furthermore, in conjunction with truncated BID activity, membrane depolarization promotes the export of cytochrome c from mitochondrial stores into the cytosol [52,87].
In the cytosol, ROSi upregulates p38 and associated MAPKs, which upregulate caspase 9 and contribute to ER stress [53]. ROSi levels are also elevated in the presence of Nerve Growth Factor [53]. Cytosolic cytochrome c upregulates caspase 9 activity while AIF translocates to the nucleus, binds to DNA, and induces DNA fragmentation and condensation [80,88]. See Table S4. ref. [38,39,45,47,49,52,55,65,72,74,77,87,89] for details.
7. Endoplasmic Reticulum (ER) Pathway
Within the pro-apoptotic pathway, the ER is an intracellular signaling center that modulates nuclear transcription factors, [Ca2+]i and kinase activity (Figure 4; Table S5): ref. [43,45,49,73,78,87,89]; Table S11: ref. [49,78]. Elevated [Ca2+]i promotes the activity of the SERCA(2) pump, which facilitates Ca2+ entry into the ER/SR [90]. To maintain homeostasis given elevated [Ca2+]ER, Ca2+ is exported to the cytosol via the RyR2 (ryanodine receptor (2) channels, which are activated by increases in [Ca2+]i [91]. As discussed for the mitochondria, increases in ROSi promote the upregulation of (p38) MAPK. MAPK, along with decreased Bcl-2 levels resulting from nuclear activity, causes the blockage of the SERCA(2) pump over time and therefore decreases cytosol-ER Ca2+ transfer [92].
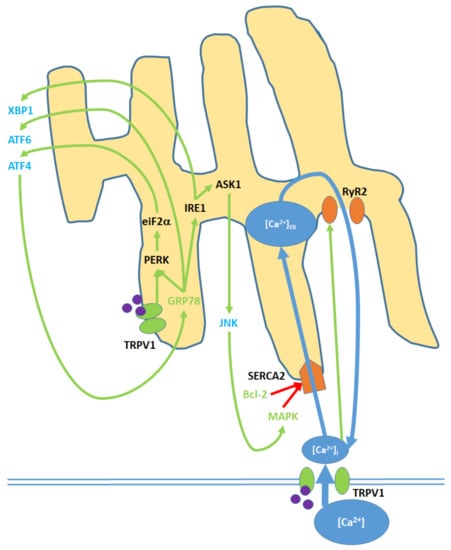
Figure 4.
Cell membrane and ER TRPV1 activation promote ER stress through the modulation of [Ca2+]ER, activation of various kinases, the upregulation of nuclear transcription factors, and the release of JNK into the cytosol. TRPV1 proteins localized to the ER membrane contribute only to protein signaling within the ER, while TRPV1 channels in the cell membrane promote both [Ca2+]i and protein signaling. Initial Ca2+ entry into the ER occurs through the SERCA2 pump, which is eventually blocked, causing net Ca2+ export via the RyR2 channels. GRP78 upregulation and Bcl-2 downregulation, as inputs, arise from nuclear activity. MAPK is both an input and output of ER stress, as it is upregulated via both mitochondrial activity and c Jun N-terminal kinases (JNK). ATF4, ATF6, and XBP1 are transcription factors that constitute the downstream nuclear targets of ER stress; ATF4, in particular, feeds back to the ER by upregulating GRP78.
Upon activation by endogenous agonists, TRPV1 protein units localized to the ER activate the eukaryotic translation initiation factor, eiF2-α, which promotes expression of activating transcription factor 4 (ATF4) [43,78]. ATF4, through nuclear activity, induces the expression of GRP78, or binding immunoglobin protein, which translocates to the ER and activates the endoplasmic reticulum protein kinase PERK; in turn, PERK further upregulates eiF2- α [93]. Activating transcription factor 6 (ATF6) and inositol requiring enzyme 1 (IRE1) are also upregulated by GRP78 [87]. IRE1, in turn, upregulates the apoptosis signal-regulating kinase, ASK1, which upregulates the c Jun N-terminal kinases (JNK) and prompts their release into the cytosol [94]. Cytosolic JNK contributes to MAPK upregulation [45]. IRE1 also upregulates X-box binding protein 1 (XBP1), a transcription factor [89].
8. Nuclear and Downstream Cytosolic Effects
Upstream proapoptotic nuclear activity results from transcription factor activation via cytosolic and ER protein signaling (Figure 5). [Ca2+]i elevation promotes the activation of calcineurin, a protein phosphatase [48,54]. Calcineurin upregulates the NFAT2 transcription factor while cytosolic ATM activation upregulates the myc proto-oncogene (MYC) and E2F1 transcription factors [45,52]. Together, NFAT2, MYC, and E2F1 upregulate the tumor suppressor protein p53 [48,52]. ATM also phosphorylates p53 in a nuclear activity-independent manner [95,96]. Activated p53 upregulates the cyclin dependent kinase (Cdk) inhibitors p16 and p21, and the proapoptotic Bcl-2 family protein Bax [48,54,71,87].
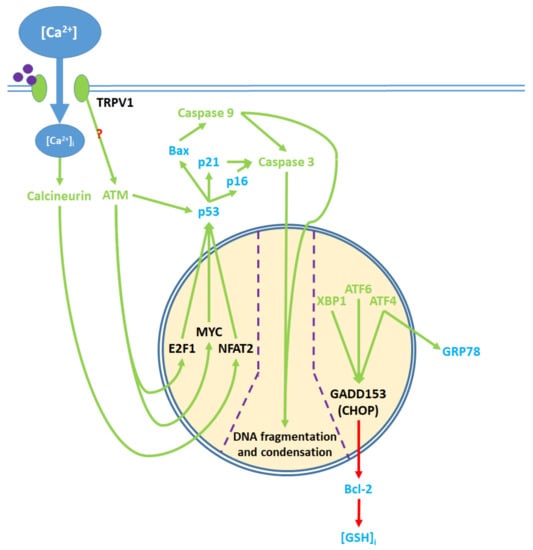
Figure 5.
Pro-apoptotic processes induced by TRPV1 localized in the nucleus. The nuclear component of ER stress occurs as ATF4, ATF6, and XBP1 are activated by ER stress and upregulate GADD153, which in turn downregulates Bcl-2 protein production. The upregulation of GRP78 by ATF4 feeds back to and enhances ER stress. The cytosolic activation of the ATM serine-threonine kinase by TRPV1 protein signaling and calcineurin by elevated [Ca2+]i promote the nuclear transcription factors E2F1, MYC, and NFAT2, which upregulate p53. The precise mechanism through which TRPV1 activates ATM remains unclear. p53 upregulates the apoptotic mediators Bax, p16, and p21, which activate caspase 9 and 3. Activated caspases translocate from the cytosol to the nucleus, where they mediate DNA fragmentation and condensation.
Through ER action, the transcription factors XBP1, ATF4 and ATF6 are upregulated. ATF4 complexes with another transcription factor, ATF1, to upregulate GRP78 protein expression [93]. XBP1, ATF4, and ATF6 together upregulate the GADD153/CHOP transcription factor, which in turn downregulates Bcl-2 protein expression [78,89]. Decreased Bcl-2 levels lead to [GSH]i depletion [82]. In notable contrast to this model, the stimulation of TRPV1 receptors with capsaicin upregulates Bcl-2 in RT4 bladder cancer cells [52].
Bax protein, which is upregulated by nuclear activity, and [GSH]i, which decreases, regulate PTP opening in the mitochondria [81,82]. The downregulation of Bcl-2 and upregulation of GRP78 contribute to ER stress [92,93]. Together, Bax, p16, and p21 promote caspase activity [87,97,98]. Caspase 9 activates caspase 3. Caspase 3 further contributes to the downregulation of Bcl-2 by converting Bcl-2 into the Bax-like analogue Bcl-2ΔN34 [99]. With some exceptions as discussed above, TRPV1 agonists upregulate these proapoptotic nuclear signaling mechanisms, while antagonists of the receptor downregulate said mechanisms (Table S6): ref. [45,48,52,54,71,78,87,89,100]; Table S12: ref. [78].
Caspase activity is well characterized in relation to TRPV1-mediated apoptosis (Table S7): ref. [45,47,48,49,52,54,55,65,69,71,72,73,75,77,78,100,101]; Table S13: ref. [49,55,65,72,75,77]. Activated caspase 9 and 3 interact with DNA in the nucleus and cause DNA fragmentation and condensation [39,45,49,100]. Subsequently, apoptosis ensues (Table S8): ref. [34,35,36,38,39,43,45,47,48,49,50,54,55,65,71,72,75,76,77,78,79,100,101,102]; Table S14: ref. [34,38,39,43,45,49,54,55,65,71,75,77,78,79].
9. TRPV1 as a Potential Target for Anti-Cancer Therapies
The collective objective of oncologic therapies is to restore and maintain a homeostatic balance between proliferation and apoptosis. In this interest, many anti-cancer compounds induce and enforce apoptosis through the targeting of p53 [103]. The accumulation of p53 amplifies the tumor suppressor protein’s downstream apoptotic and anticarcinogenic effects in vitro (e.g., HCT116 cell line) and in vivo (e.g., mice) [104]. Furthermore, independently of p53, some anti-cancer drugs bind directly to and damage DNA, inhibiting transcription and therefore downregulating proliferation [105]. While the modulation of individual components of the intrinsic apoptotic and proliferative mechanisms can constitute an effective means of tumor suppression, enormous potential exists for therapies that regulate both mechanisms to shift the balance.
In this regard, [Ca2+]i, a universal second messenger regulating cell death and survival, is a promising target. [Ca2+]i signaling is highly versatile, as it influences intracellular pathways for proliferation, differentiation, and apoptosis in neuroblastoma cells [106]. Within cancerous tissues, the molecular machinery involved in [Ca2+]i signaling is modified to promote proliferation and minimize apoptosis. Modifications of this nature may downregulate Ca2+ entry into the cytosol (e.g., blockade of Ca2+ release from ER stores) or eliminate downstream targets of [Ca2+]i signaling (e.g., enforced loss of p53) [107].
Numerous existing anti-cancer therapies, including metal compounds, anti-metabolites, and various natural and synthetic organic molecules, disrupt [Ca2+]i homeostasis (via elevation of [Ca2+]i due to transmembrane Ca2+ influx and release of Ca2+ from intracellular stores) and thereby promote apoptosis [8,108,109,110]. As such, [Ca2+]i affects mitochondrial dysfunction and ER stress along the proapoptotic pathway. [Ca2+]i is also an important regulator of the PI3K/Akt pathway, which promotes the proliferation of cancer cells [60].
TRPV1, a ligand-gated ion channel, modulates [Ca2+]i, [Ca2+]m, and [Ca2+]ER. While TRPV1 is well characterized with regards to cell death, the link between TRPV1 and proliferation has yet to be thoroughly investigated. The vast majority of studies pertaining to cell death elucidated a connection between TRPV1 activation and apoptosis in both cancerous and benign cell lines. However, some found TRPV1 activation in breast cancer cells (MCF-7) to result in necrotic cell death [102].
Notably, TRPV1 is expressed at the mRNA and protein levels in a wide variety of cancerous and non-cancerous cell lines (Table 1). In numerous nervous, colorectal, endometrial, renal, and dermal cancer cell lines, TRPV1 expression is reduced in comparison with healthy cells (Table 2). This pattern may indicate a primarily pro-apoptotic role for TRPV1 in the tumors. Under these conditions, the upregulation and subsequent stimulation of TRPV1 can potentiate the innate apoptotic pathway. It is therefore relevant that capsaicin upregulates the mRNA and protein expression of TRPV1 in certain healthy and cancerous (e.g., nasopharyngeal and skin cancer) cell lines [35,54,87]. In contrast, TRPV1 expression levels in high-grade astrocytes, “brain tumors,” U373 cells, and RT4 renal cell carcinoma cells are elevated in comparison to healthy controls (Table 2). These findings may hint that TRPV1 contributes primarily to proliferation rather than apoptosis in the cancerous cell lines in which its expression is upregulated. To combat carcinogenesis in these cases, it would be necessary to shift the TRPV1-mediated balance towards apoptosis, while native TRPV1 expression levels may be sufficient to induce the desired apoptotic effects once the aforementioned shift is achieved.
Similarly, in the vast majority of cell lines, apoptosis is increased by TRPV1 agonists and downregulated by TRPV1 antagonists. However, in canine breast cancer cells, agonists and antagonists both support proliferation; in human breast cancer cells, both types of molecules support apoptosis [42,76]. These results suggest that the nature of the proliferation–apoptosis balance may differ between cell lines.
The pathways through which TRPV1 channel activation affects apoptosis or proliferation are distinct and competitive (Figure 6). Signaling mechanisms that mediate the pro-apoptotic effects of TRPV1 act through the mitochondria, ER, nucleus, and cytosol. Ca2+ and Na+ influx into the mitochondria results in the depolarization of the mitochondrial membrane and subsequent release of cytochrome c, AIF, and ROS into the cytosol. Meanwhile, ER stress results in JNK release into the cytosol and the upregulation of the nuclear transcription factors ATF4, ATF6, and XBP1, which decrease intracellular Bcl-2 and [GSH]i. JNK and p38 (increased due ROSi) upregulate MAPK. Furthermore, TRPV1 [Ca2+]i and protein signaling activates calcineurin and ATM, which act through transcription factors to upregulate the tumor suppressor gene p53. p53 subsequently upregulates the proapoptotic proteins Bax, p16, and p21. Finally, MAPK, cytochrome c, p16, p21, and Bax activate caspases. Caspase 9 activates caspase 3; the two caspases and AIF cause DNA fragmentation and condensation in the nucleus, and ultimately apoptosis [39,45,49,87,97,98,99,100].
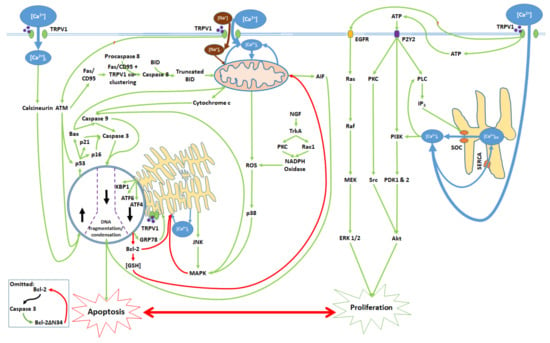
Figure 6.
Activation of the TRPV1 ligand-gated ion channel and subsequent [Ca2+]i and protein signaling influence the balance between proliferation and apoptosis. Mitochondrial dysfunction, ER stress, transcription factor activation, and nuclear activity promote caspase activation and the consequent DNA degradation, which are characteristic of apoptotic cell death. TRPV1 protein signaling upon activation induces the release of ATP, which binds to the P2Y2 receptor and activates a kinase signaling cascade, leading to ER Ca2+ release and Akt activation. TRPV1 also activates EGFR and its associated protein signaling pathway, which upregulates ERK 1/2. Akt and ERK 1/2 contribute to cell proliferation. All cytosolic signaling pathways associated with the proliferative and apoptotic effects of TRPV1 activation are shown.
The proliferative effects of TRPV1 activation are linked to ATP release into the extracellular space and transactivation of EGFR. ATP binding to cell membrane P2Y2 receptors starts an intracellular kinase signaling cascade that is enhanced by IP3-mediated Ca2+ release from ER stores and ultimately activates the Akt kinase; concurrently, EGFR activation prompts a separate series of protein signals to upregulate ERK 1/2. Both Akt and ERK 1/2 enhance proliferation. Akt is a serine/threonine protein kinase with oncogenic effects. The localization of active Akt to the cell membrane results in oncogenic transformation of chicken embryonic fibroblasts and hemangiosarcoma development in young chickens [111]. The linking of TRPV1 and P2Y2 signals to cell proliferation and oncogenicity suggests TRPV1 as a potential target for anti-cancer therapies, in the interest of downregulating proliferative protein signaling. As was demonstrated by Huang et al., the activation of TRPV1 promotes the proliferation and migration of esophageal squamous cell carcinoma cells. While capsaicin activates TRPV1, a TRPV1 inhibitor (AMG9810) antagonizes its effects [66].
TRPV1 mediates apoptosis primarily through [Ca2+]i and protein signaling; in contrast, the channel stimulates proliferation via ATP release, P2Y2 receptor activation, and EGFR transactivation, with a limited role for [Ca2+]i. As such, the activation of TRPV1 in the absence of P2Y2 and EGFR activation may result in the dominance of the apoptotic pathway. Exogenous and endogenous TRPV1 agonists, applied in conjunction with P2Y2 and EGFR antagonists, may therefore constitute a potential form of anti-cancer therapy.
Given the role of P2Y2 in activating PI3K and beginning the PI3K/Akt pathway, inactivation of the receptor may have implications in oncologic drug discovery. PI3K/Akt signaling is firstly implicated in cancer cell proliferation and tumorigenesis [112,113,114]. More importantly, the PI3K/Akt pathway is an important mediator of cancer drug resistance, such as that of breast cancer cells to trastuzumab [115]. As such, blockade of P2Y2 receptors has the potential to reduce PI3K activation and thereby impede the development of cancer drug resistance.
The inhibition of EGFR activity may likewise support effects to curb cancer drug resistance. EGFR is a transmembrane growth factor receptor which contributes to cell proliferation via the Ras/Raf/MEK/ERK signaling pathway [64]. This pathway promotes the resistance of hematopoietic cells to doxorubicin, and acts in conjunction with PI3K/Akt signaling in support of tumorigenesis [116]. Notably, functional crosstalk occurs between the PI3K/Akt and Ras/Raf/MEK/ERK pathways in colon cancer cells, as the downregulation of one pathway corresponds with the upregulation of the other; however, treatment with both patritumab (a PI3K inhibitor) and trametinib (an MEK inhibitor) reduces the viability of said cells [117]. Electroacupuncture (EA) therapy may be a viable avenue for the dual inhibition of PI3K/Akt and Ras/Raf/MEK/ERK signaling; recent evidence suggests that EA in the cerebellum downregulates PI3K, Akt, PKC, and ERK [31]. In this light, the activation of TRPV1 in the presence of PI3K/Akt and Ras/Raf/MEK/ERK pathway inhibitors—potentially including P2Y2 and EGFR antagonists—may promote cancer cell apoptosis through [Ca2+]i signaling without the induction of proliferative mechanisms.
Interestingly, PKC, a protein kinase, is involved in both the proliferative and apoptotic pathways. The activation of PKC as part of the Nerve Growth Factor pathway supports the elevation of ROSi levels via nicotinamide adenine dinucleotide phosphate (NADPH) oxidase upregulation [53,118]. However, PKC also activates the Src proto-oncogene, which promotes cell proliferation. Therefore, PKC and NGF have dual roles as determinants of cell fate. In the development of anti-cancer drugs, the presence or absence of NGF must be taken into account, as despite the inactivation/antagonism of the P2Y2 receptor, NGF may nevertheless stimulate cell proliferation [119].
It is important to note that TRPV1 modulates the apoptosis–proliferation balance through mechanisms beyond [Ca2+]i signaling and may therefore exert pleiotropic effects in cancerous tissues. In particular, the implications of TRPV1 for inflammation are multi-faceted (Figure S1). The activation of the membrane ion channel by formaldehyde and particulate matter (in an asthmatic murine model), and acidic solution (in human esophageal cells) enhances inflammation. Along this pro-inflammatory axis, TRPV1 induces the release of substance P and calcitonin gene-related peptide (CGRP), both of which are pro-inflammatory neuropeptides [120]. Substance P and CGRP activate the neurokinin 1 (NK1R) and CGRP (CGRPR) receptors, respectively, and promote the release of pro-inflammatory cytokines such as tumor necrosis factor alpha (TNF-α) and interleukins 1-beta (IL-1β), 6 (IL-6), and 8 (IL-8) [121,122]. These cytokines induce vasodilation and ultimately inflammation; significantly, inflammation is associated with cell proliferation and tumorigenesis [123,124].
In contrast, TRPV1 activation by capsaicin attenuates pro-inflammatory cytokine release and inflammation [120,125]. Within this anti-inflammatory mechanism, TRPV1 stimulation prompts the release of somatostatin (SST), an anti-inflammatory neuropeptide which binds to SST receptor 4 (sst4) and downregulates the aforementioned pro-inflammatory cytokines [126]. SST also attenuates substance P and CGRP release; this further downregulates said cytokines [127]. Capsaicin, through TRPV1, thereby decreases inflammation and may reduce inflammation-related cell proliferation.
TRPV1 is expressed in a wide variety of immune cells, such as macrophages, neutrophils, T cells, and dendritic cells [128]. As such, in attempting to upregulate apoptosis through TRPV1, care must be taken to prevent the inadvertent promotion of proliferation through inflammation. Further research is necessary to fully elucidate the dependence of TRPV1’s inflammatory effects on agonist type, cell/cancer type, and tumor microenvironment. Based on current knowledge, capsaicin and nonivamide are anti-inflammatory agents that exert their effects through TRPV1 channel activation [129,130]. These TRPV1 agonists may therefore have dual effects in upregulating apoptosis through [Ca2+]i signaling while suppressing proliferation through inflammation reduction.
In the development of anti-cancer drugs to affect TRPV1, it is in all cases necessary to first characterize the apoptosis–proliferation balance that exists in the target cell line, taking into account factors such as inflammation. Depending on the balance, oncologic therapies may upregulate and stimulate TRPV1 with carefully selected agonists, downregulate or otherwise inhibit TRPV1, or target specific aspects of the TRPV1-mediated mechanisms to enhance the protein’s apoptotic effects. Recent molecular and clinical developments suggest that these objectives are achievable.
Cancer management based on the targeting of TRPV1 currently encompasses a wide range of compounds with synthetic as well as natural origins. Arvanil, a synthetic TRPV1 agonist, decreases tumor weight and improves survival time in mice with implanted gliomas [43]. Moreover, bisphenol A (BPA) is a synthetic compound whose presence in the environment and in human tissues results from its widespread use and biological accumulation. BPA has hormone-like properties and can mimic estrogen and interact with its receptors; it can thereby modulate cell proliferation, apoptosis, and migration and contribute to carcinogenesis [131]. However, the proliferative and oxidative effects of BPA, enhanced through TRPV1 activation, are reduced by sodium selenite (Na-Se), which attenuates BPA-induced increases in cell number, mitochondrial oxidative stress, and modulation of TRPV1 channel activity in MCF-7 cells [132]. Interestingly, despite the function of both capsaicin and 6-gingerol as TRPV1 agonists, the two compounds exhibit opposing effects on cancerous cells. In a urethane-induced lung carcinogenic model, capsaicin enhances proliferation and epithelial-mesenchymal transitions via a decrease in TRPV1 expression and an increase in EGFR, followed by decreases in nuclear factor-κB (NF-κB) and cyclin D1. On the other hand, 6-gingerol reverses the carcinogenic effects of capsaicin by increasing TRPV1 expression and decreasing EGFR, NF-κB and cyclin D1 [133].
TRPV1 channels also modulate the efficacy of existing anti-cancer therapies. Alpha-lipoic acid and selenium enhance the cytotoxic efficacy of cisplatin, an existing platinum-derived anti-cancer drug, via TRPV1 stimulation [134,135]. Moreover, TRPV1 is differentially expressed in the bladder cancer cell lines 5637 and T24. TRPV1 mRNA and protein expression is low in T24 and high in 5637 cells; the activation of TRPV1 by capsaicin inhibits the growth of 5637 cells. Moreover, the activation of TRPV1 promotes the anti-proliferative efficacy of pirarubicin, an anthracycline agent used for intravesical chemotherapy of superficial bladder cancer. Therefore, TRPV1 stimulation may represent a strategy to increase the efficacy of traditional chemotherapeutic agents against bladder cancer [136]. In addition, the anticancer activity of selenium via TRPV1 was evaluated in the MCF-7 breast cancer cell line. Increases in mitochondrial membrane depolarization, apoptosis, and caspase 3 and caspase 9 levels occur after treatment with selenium alone and in combination with cisplatin. Therefore, the interaction of selenium and cisplatin with the same intracellular cascade through modulation of TRPV1 can bring about remarkable advantages in oncology [135]. Similarly, melatonin supports the anti-cancer effects of the chemotherapeutic agent doxorubicin via TRPV1 activation and subsequent apoptosis in MCF-7 cells [137]. It is notable that doxorubicin treatment may cause cardiomyopathy; the inhibition of the proapoptotic Bax protein is a potential avenue through which to prevent this side effect [138]. On the other hand, the antioxidant plant Hypericum perforatum (HP) does not support the antitumor effects of 5-Fluorouracil through TRPV1 modulation. The apoptotic effects of 5-Fluorouracil, mediated by TRPV1 activation in MCF-7 cells, are downregulated by treatment with HP [139].
Moreover, fibulin-5 is a multifunctional extracellular matrix protein with lower expression in colorectal cancer tissues than in peritumoral areas. Given its role in promoting apoptosis through TRPV1 downregulation and consequent ROS/MAPK and Akt signaling, fibulin-5 is a potential novel target in the treatment of colorectal cancer [140]. Furthermore, the endocannabinoid/endovanilloid system, composed of two G-protein coupled cannabinoid receptors (CB1 and CB2) and TRPV1, and their respective ligands and enzymes, is targeted in a novel therapeutic approach for T-cell acute lymphoblastic leukemia (T-ALL). Treatment of T-ALL lymphoblasts with the selective CB2 agonist JWH-133 and TRPV1 agonist RTX produces pro-apoptotic and anti-proliferative effects [141].
Recent Trends and Innovations in Oncologic Approaches Targeting TRPV1
Nanomedicine is associated with progress in anti-cancer research [142]. Distressing toxic effects are common consequences of chemotherapeutics. To mitigate this and other obstacles (e.g., poor biocompatibility, premature drug leakage, off-targeting), a [Ca2+]i signaling cascade for cancer therapy via photothermal TRPV1 activation has been developed. This cascade creates artificial Ca2+ overload stress, causing cell death via mitochondrial dysfunction (involving caspase 3 and cytochrome c upregulation, and Bcl-2 and ATP downregulation) in vitro. Given TRPV1 overexpression and near-infrared (NIR) irradiation at tumor sites, a CuS@CaCO3-PEG nanoplatform can initiate the [Ca2+]i cascade in both cancerous and non-cancerous tissues. Additionally, photothermal CuS nanoparticles involved in the nanoplatform allow for three-dimensional photoacoustic imaging in vivo [143]. Furthermore, an apoptosis-inducing TRPV1 nanoagonist comprised of semiconducting polymer nanoparticles (SPNs) as photothermally responsive nanocarriers and capsaicin as the agonist, has been developed. Under NIR irradiation, the nanoagonist releases capsaicin to activate cell membrane TRPV1 channels. Ionic influx into the mitochondria is followed by apoptosis in TRPV1-positive cancer cells. This photothermal mechanism allows for the release of high concentrations of TRPV1 agonist(s) at specific tumor sites with low systemic dosages [144]. Finally, nanoscale drug delivery systems based on single-walled carbon nanotubes can utilize TRPV1 channels for transmembrane drug transport [145].
Looking forward, natural products and drug conjugates can be evaluated for the treatment of prostate cancers with TRPV1 overexpression. The stoichiometric combination of a TRPV1 agonist with a small, positively charged cytotoxic agent constitutes a promising avenue for prostate cancer treatment [146]. Another potential oncologic approach utilizes a gold nanorod-assisted NIR light-activated tool to open TRPV1 channels and thereby induce apoptosis [147].
Beyond its potential as a therapeutic target, TRPV1 also has prognostic significance. It is, for instance, a biomarker in invasive breast carcinoma [148]. Moreover, TRPV1, alone and in combination with the Phosphatase and Tension Homolog (PTEN), is an important prognostic factor in epithelial ovarian and cervical cancers [149,150]. Finally, a non-invasive cancer detection method could utilize magnetoencephalography to measure cellular ion transport. In TRPV1-expressing HEK-293 cells, capsaicin induces a sudden change in the magnetic field signal, consistent with Ca2+ influx [151].
Targeting of TRPV1 represents an important avenue for cancer management, and current progress in related oncologic strategies is promising. However, the exact mechanisms by which the proliferation–apoptosis balance shifts in the described cases remain unclear; further investigations are therefore necessary to produce clinically applicable results.
10. Conclusions and Outlook
TRPV1, a ligand-activated membrane ion channel, functions in both apoptotic cell death and proliferation. Mitochondrial dysfunction and membrane depolarization, ER stress, caspase activation, and DNA damage are all implicated in TRPV1-mediated apoptosis. In contrast, TRPV1 supports proliferation through the activation of P2Y2 and EGFR, and the resulting intracellular protein signaling cascades. In healthy cells, a delicate and dynamic balance exists between the proliferative and apoptotic mechanisms. This balance is shifted in cancerous cells and tissues, in which proliferation dominates; however, the precise factors that modulate the balance remain largely uncharacterized.
As the objective of anti-cancer therapies is to hinder tumor growth through the enforcement of apoptosis and the minimization of proliferation, the TRPV1 ion channel constitutes a promising target. TRPV1 is constitutively expressed in a wide variety of cancerous cell lines, and the receptor’s differential expression in cancerous and healthy tissues provides insights into its role in the proliferation–apoptosis balance. Oncologic agents that target TRPV1 will exert their proapoptotic effects primarily through the modulation of [Ca2+]i, which has a dual role in that it regulates both proliferative and apoptotic mechanisms. Notably, the influence of TRPV1 on the PI3K/Akt signaling pathway may hold implications for the circumvention or minimization of cancer drug resistance. The activation of TRPV1 by agonists, in conjunction with a blockade of P13K/Akt signaling, may have therapeutic potential through the elevation of [Ca2+]i and consequent upregulation of the apoptotic pathways. The parallel involvement of TRPV1 in proliferation and apoptosis enhances the receptor’s relevance as an oncologic target. Several innovative anti-cancer strategies targeting TRPV1 are currently in development.
Supplementary Materials
The following are available online at https://www.mdpi.com/1422-0067/21/11/4177/s1, Table S1: Proliferative effects of the exogenous TRPV1 agonists capsaicin and glycolic acid, the endogenous agonist AEA and its analogue, SKM-4-45-1, and the antagonists capsazepine and AMG9810; Table S2: [Ca2+]i elevation induced by various endogenous and exogenous TRPV1 agonists and activators; Table S3: Effects of the TRPV1 agonist capsaicin on the Fas/CD95 cytosolic pathway; Table S4: Mitochondrial pathway effects induced by TRPV1 agonists; Table S5: ER effects induced by endogenous and exogenous TRPV1 agonists; Table S6: Nuclear effects of endogenous and exogenous TRPV1 agonists; Table S7: Effects of exogenous and endogenous TRPV1 agonists on caspase activity; Table S8: Downstream pro-apoptotic effects of endogenous and exogenous TRPV1 agonists and antagonists; Table S9: Effects of TRPV1 antagonists on transmembrane Ca2+ transport; Table S10: Effects of TRPV1 antagonists on mitochondrial dysfunction; Table S11: Effects of TRPV1 antagonists on ER stress; Table S12: Effects of TRPV1 antagonists on nuclear activity; Table S13: Effects of TRPV1 antagonists on caspase activity; Table S14: Effects of TRPV1 antagonists on agonist-induced apoptosis; Figure S1: Activation of TRPV1 modulates pro- and anti-inflammatory signaling pathways.
Author Contributions
Conceptualization, K.Z. and D.B.; Literature Review and Resources, K.Z.; Writing—Original Draft Preparation, K.Z.; Writing—Review and Editing, K.Z., A.L., P.K., and D.B.; Figure Preparation and Editing, K.Z. and D.B.; Visualization, K.Z. and D.B.; Supervision, D.B. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by a National Priorities Research Program grant (NPRP 11S-1214-170101; awarded to Dietrich Büsselberg, June 2019—current) from the Qatar National Research Fund (QNRF, a member of Qatar Foundation). The statements made herein are solely the responsibility of the authors.
Acknowledgments
We thank John Fowler (Weill Cornell Medicine-Qatar, Doha, Qatar) for language corrections and linguistic and grammatical suggestions. The publication costs of this article were covered by the Division of Student Affairs at Weill Cornell Medicine—Qatar.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| 13-HODE | 13-HydroxyOctaDecadEinoic acid |
| 2-AG | 2-ArachidonylGlycerol |
| AEA | Anandamide |
| Akt | Akt serine/threonine protein kinase |
| AIF | Apoptosis Inducing Factor |
| ASMC | Airway Smooth Muscle Cells |
| ATF1 | Activating Transcription Factor 1 |
| ATF3 | Activating Transcription Factor 3 |
| ATF4 | Activating Transcription Factor 4 |
| ATF6 | Activating Transcription Factor 6 |
| ATM | ATM serine-threonine kinase |
| ATP | Adenosine TriPhosphate |
| Bax | Bcl-2 associated X protein |
| Bcl-2 | B-cell lymphoma 2 |
| BID | BH3 Interacting-domain Death agonist |
| BPA | BisPhenol A |
| Ca2+ | Calcium |
| [Ca2+]i | Intracellular Calcium Concentration |
| [Ca2+]m | Mitochondrial Calcium Concentration |
| [Ca2+]ER | Endoplasmic Reticulum Calcium Concentration |
| CaM | CalModulin |
| CB1 | CannaBinoid receptor type 1 |
| CB2 | CannaBinoid receptor type 2 |
| CBD | CannaBiDiol |
| Cdk | Cyclin dependent kinase |
| CGRP | Calcitonin Gene-Related Peptide |
| CGRPR | Calcitonin Gene-Related Peptide Receptor |
| CRC | ColoRectal Cancer |
| DMBA | 7,12-DiMethylBenz[a]Anthracene |
| DNA | DeoxyriboNucleic Acid |
| DRG | Dorsal Root Ganglion |
| E2F1 | E2F transcription factor 1 |
| EA | ElectroAcupuncture |
| ECFC | Endothelial Colony Forming Cells |
| EET | EpoxyEicosaTrienoic acid |
| EGFR | Epidermal Growth Factor Receptor |
| eIF2 | eukaryotic Initiation Factor 2 |
| ER | Endoplasmic Reticulum |
| ERK | Extracellular signal-Regulated Kinase |
| FADD | Fas-Associated protein with Death Domain |
| Fas/CD95 | Fas cell surface death receptor/Cluster of Differentiation 95 |
| GADD153; DDIT3; CHOP | DNA-Damage Inducible Transcript 3; C/EBP HOmologous Protein |
| GSH | Glutathione |
| [GSH]i | Intracellular Glutathione Concentration |
| GRP78; BiP | Binding immunoglobulin Protein |
| HCEC | Human Corneal Epithelial Cells |
| HP | Hypericum perforatum |
| ICR | Institute for Cancer Research |
| IL-1β | InterLeukin 1β |
| IL-6 | InterLeukin 6 |
| IL-8 | InterLeukin 8 |
| IP3 | Inositol triPhosphate |
| IRE1 | Inositol-Requiring Enzyme 1 |
| IRTX | IodoResiniferaToXin |
| JNK | c-Jun N-terminal Kinase |
| MAPK | Mitogen-Activated Protein Kinase |
| MCU | Mitochondrial Calcium Uniporter |
| Mdm2 | Mouse double minute 2 homolog |
| MEK | MAPK-ERK Kinase |
| MET | (R)-METhanandamide |
| mRNA | messenger RiboNucleic Acid |
| MYC | Myc proto-oncogene |
| Na+ | Sodium |
| [Na+]i | Intracellular Sodium Concentration |
| [Na+]m | Mitochondrial Sodium Concentration |
| NADA | N-Arachidonoyl DopAmine |
| NADPH | Nicotinamide Adenine Dinucleotide PHosphate |
| NFAT2 | Nuclear Factor of Activated T-cells 2 |
| NF-κB | Nuclear Factor-Kappa light chain enhancer of activated B cells |
| NGF | Nerve Growth Factor |
| NHA | Normal Human Astrocytes |
| NHBE | Normal Human Bronchial Epithelial cells |
| NHEM | Normal Human Epidermal Melanocytes |
| NHUC | Normal Human Urothelial Cells |
| NIR | Near InfraRed |
| NK1R | NeuroKinin 1 Receptor |
| NPC | Neural Progenitor Cells |
| (m)NPC-CM | (murine) Neural Progenitor Cell-Culture Media |
| p16; CDKN2A | Cyclin-Dependent Kinase Inhibitor 2A |
| p21; CDKN1(A) | Cyclin-Dependent Kinase Inhibitor 1(A) |
| P2Y2 | P2Y purinoceptor 2 |
| p38 (MAPK) | p38 (Mitogen Activated Protein Kinase) |
| p53 | tumor protein p53 |
| PAM | Positive Allosteric Modulator |
| PCOS | PolyCystic Ovary Syndrome |
| PDK1 | Phosphoinositide Dependent Kinase 1 |
| PEG | PolyEthylene Glycol |
| PKC | Protein Kinase C |
| PLC | PhosphoLipase C |
| PI3K | Phosphoinositide-3-Kinase |
| PS | PhosphatidylSerine |
| PTEN | Phosphatase and TENsion homolog |
| PTP | Permeability Transition Pore |
| PTZ | PentyleneTetraZole |
| Rac1 | Ras-related C3 botulinum toxin substrate 1 |
| Raf | Rapidly accelerated fibrosarcoma |
| Ras | Rat sarcoma |
| RCC | Renal Cell Carcinoma |
| RGC | Retinal Ganglion Cells |
| ROS | Reactive Oxygen Species |
| ROSi | Intracellular Reactive Oxygen Species |
| ROSm | Mitochondrial Reactive Oxygen Species |
| RR | Ruthenium Red |
| RTX | ResiniferaToXin |
| RyR2 | Ryanodine Receptor 2 |
| SAEC | Small Airway Epithelial Cells |
| SCID-NOD | Severe Combined ImmunoDeficiency-NonObese Diabetic |
| SERCA | Sarco/Endoplasmic Reticulum Calcium ATPase |
| SGZ | SubGranular Zone |
| SMF | Static Magnetic Field |
| SNI | Sciatic Nerve Injury |
| SNP | Sodium NitroPrusside |
| SOC | Store-Operated Channel |
| SR | Sarcoplasmic Reticulum |
| Src | proto-oncogene tyrosine-protein kinase Src |
| SST | SomatoStaTin |
| sst4 | somatostatin receptor 4 |
| SVZ | SubVentricular Zone |
| T-ALL | T-cell Acute Lymphoblastic Leukemia |
| TG | Trigeminal Ganglia |
| TNF-α | Tumor Necrosis Factor α |
| TrkA | Tropomyosin receptor kinase A |
| TRPV1 | Transient Receptor Potential Vanilloid 1 |
| VDAC | Voltage Dependent Anion Channel |
| VGCC | Voltage Gated Calcium Channel |
| XBP1 | X-box Binding Protein 1 |
References
- Cvejic, D.; Selemetjev, S.; Savin, S.; Paunovic, I.; Tatic, S. Changes in the balance between proliferation and apoptosis during the progression of malignancy in thyroid tumours. Eur. J. Histochem. 2009, 53, e8. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.; Du, M.; Bishop, A.E.; Talbot, I.C. Imbalance between proliferation and apoptosis in the development of colorectal carcinoma. Virchows Arch. 1998, 433, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Afanas’ev, V.N.; Korol, B.A.; Mantsygin Yu, A.; Nelipovich, P.A.; Pechatnikov, V.A.; Umansky, S.R. Flow cytometry and biochemical analysis of DNA degradation characteristic of two types of cell death. FEBS Lett. 1986, 194, 347–350. [Google Scholar] [CrossRef]
- Reed, J.C. Mechanisms of apoptosis. Am. J. Pathol. 2000, 157, 1415–1430. [Google Scholar] [CrossRef]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef]
- Kuwana, T.; Newmeyer, D.D. Bcl-2-family proteins and the role of mitochondria in apoptosis. Curr. Opin. Cell Biol. 2003, 15, 691–699. [Google Scholar] [CrossRef]
- Orrenius, S.; Zhivotovsky, B.; Nicotera, P. Regulation of cell death: The calcium-apoptosis link. Nat. Rev. Mol. Cell Biol. 2003, 4, 552–565. [Google Scholar] [CrossRef]
- Varghese, E.; Samuel, S.M.; Sadiq, Z.; Kubatka, P.; Liskova, A.; Benacka, J.; Pazinka, P.; Kruzliak, P.; Busselberg, D. Anti-Cancer Agents in Proliferation and Cell Death: The Calcium Connection. Int. J. Mol. Sci. 2019, 20, 17. [Google Scholar] [CrossRef]
- Santulli, G.; Lewis, D.; des Georges, A.; Marks, A.R.; Frank, J. Ryanodine Receptor Structure and Function in Health and Disease. Subcell. Biochem. 2018, 87, 329–352. [Google Scholar] [CrossRef]
- Gambardella, J.; Lombardi, A.; Morelli, M.B.; Ferrara, J.; Santulli, G. Inositol 1,4,5-Trisphosphate Receptors in Human Disease: A Comprehensive Update. J. Clin. Med. 2020, 9, 96. [Google Scholar] [CrossRef]
- Santulli, G.; Nakashima, R.; Yuan, Q.; Marks, A.R. Intracellular calcium release channels: An update. J. Physiol. 2017, 595, 3041–3051. [Google Scholar] [CrossRef]
- Kania, E.; Roest, G.; Vervliet, T.; Parys, J.B.; Bultynck, G. IP3 Receptor-Mediated Calcium Signaling and Its Role in Autophagy in Cancer. Front. Oncol. 2017, 7, 140. [Google Scholar] [CrossRef]
- Shin, D.-H.; Leem, D.-G.; Shin, J.-S.; Kim, J.-I.; Kim, K.-T.; Choi, S.Y.; Lee, M.-H.; Choi, J.-H.; Lee, K.-T. Compound K induced apoptosis via endoplasmic reticulum Ca2+ release through ryanodine receptor in human lung cancer cells. J. Ginseng Res. 2018, 42, 165–174. [Google Scholar] [CrossRef]
- Panner, A.; Wurster, R.D. T-type calcium channels and tumor proliferation. Cell Calcium 2006, 40, 253–259. [Google Scholar] [CrossRef]
- Valerie, N.C.; Dziegielewska, B.; Hosing, A.S.; Augustin, E.; Gray, L.S.; Brautigan, D.L.; Larner, J.M.; Dziegielewski, J. Inhibition of T-type calcium channels disrupts Akt signaling and promotes apoptosis in glioblastoma cells. Biochem. Pharm. 2013, 85, 888–897. [Google Scholar] [CrossRef]
- Cano-Abad, M.A.F.; Villarroya, M.; Garcı‘a, A.G.; Gabilan, N.H.; Lo´pez, M.G. Calcium entry through L-type calcium channels causes mitochondrial disruption and chromaffin cell death. J. Biol. Chem. 2001, 276, 39695–39704. [Google Scholar] [CrossRef]
- Nicotera, P.; Orrenius, S. The role of calcium in apoptosis. Cell Calcium. 1998, 23, 173–180. [Google Scholar] [CrossRef]
- Stewart, T.A.; Yapa, K.T.; Monteith, G.R. Altered calcium signaling in cancer cells. Biochim. Biophys. Acta 2015, 1848, 2502–2511. [Google Scholar] [CrossRef]
- Florea, A.M.; Busselberg, D. Anti-cancer drugs interfere with intracellular calcium signaling. Neurotoxicology 2009, 30, 803–810. [Google Scholar] [CrossRef]
- Raynal, N.J.; Lee, J.T.; Wang, Y.; Beaudry, A.; Madireddi, P.; Garriga, J.; Malouf, G.G.; Dumont, S.; Dettman, E.J.; Gharibyan, V.; et al. Targeting Calcium Signaling Induces Epigenetic Reactivation of Tumor Suppressor Genes in Cancer. Cancer Res. 2016, 76, 1494–1505. [Google Scholar] [CrossRef]
- Clapham, D.E. TRP channels as cellular sensors. Nature 2003, 426, 517–524. [Google Scholar] [CrossRef]
- Huang, J.; Liu, J.; Qiu, L. Transient receptor potential vanilloid 1 promotes EGFR ubiquitination and modulates EGFR/MAPK signalling in pancreatic cancer cells. Cell Biochem. Funct. 2020. [Google Scholar] [CrossRef]
- So, C.L.; Milevskiy, M.J.G.; Monteith, G.R. Transient receptor potential cation channel subfamily V and breast cancer. Lab. Investig. 2020, 100, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Zygmunt, P.M.; Petersson, J.; Andersson, D.A.; Chuang, H.; Sorgard, M.; Di Marzo, V.; Julius, D.; Hogestatt, E.D. Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature 1999, 400, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Caterina, M.J.; Schumacher, M.A.; Tominaga, M.; Rosen, T.A.; Levine, J.D.; Julius, D. The capsaicin receptor: A heat-activated ion channel in the pain pathway. Nature 1997, 389, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Bautista, D.; Julius, D. Fire in the hole: Pore dilation of the capsaicin receptor TRPV1. Nat. Neurosci. 2008, 11, 528–529. [Google Scholar] [CrossRef]
- Satheesh, N.J.; Uehara, Y.; Fedotova, J.; Pohanka, M.; Büsselberg, D.; Kruzliak, P. TRPV currents and their role in the nociception and neuroplasticity. Neuropeptides 2016, 57, 1–8. [Google Scholar] [CrossRef]
- Szallasi, A.; Cruz, F.; Geppetti, P. TRPV1: A therapeutic target for novel analgesic drugs? Trends Mol. Med. 2006, 12, 545–554. [Google Scholar] [CrossRef]
- Christie, S.; Wittert, G.A.; Li, H.; Page, A.J. Involvement of TRPV1 Channels in Energy Homeostasis. Front. Endocrinol. (Lausanne) 2018, 9, 420. [Google Scholar] [CrossRef]
- Jeong, K.Y. Changes in TRPV1-Mediated Physiological Function in Rats Systemically Treated With Capsaicin on the Neonate. Int. J. Mol. Sci. 2020, 21, 3143. [Google Scholar] [CrossRef]
- Inprasit, C.; Lin, Y.-W. TRPV1 Responses in the Cerebellum Lobules V, VIa and VII Using Electroacupuncture Treatment for Inflammatory Hyperalgesia in Murine Model. Int. J. Mol. Sci. 2020, 21, 3312. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Carver, C.M.; Mullen, P.; Milne, S.; Lukacs, V.; Shapiro, M.S.; Gamper, N. Local Ca(2+) signals couple activation of TRPV1 and ANO1 sensory ion channels. Sci. Signal. 2020, 13. [Google Scholar] [CrossRef]
- Buch, T.R.H.; Buch, E.A.M.; Boekhoff, I.; Steinritz, D.; Aigner, A. Role of Chemosensory TRP Channels in Lung Cancer. Pharmaceuticals 2018, 11, 90. [Google Scholar] [CrossRef]
- Sappington, R.M.; Sidorova, T.; Long, D.J.; Calkins, D.J. TRPV1: Contribution to retinal ganglion cell apoptosis and increased intracellular Ca2+ with exposure to hydrostatic pressure. Invest. Ophthalmol. Vis. Sci. 2009, 50, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhang, X.; Kuang, H.; Wu, J.; Guo, Y.; Ma, L. Effect of TRPV1 channel on the proliferation and apoptosis in asthmatic rat airway smooth muscle cells. Exp. Lung Res. 2013, 39, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Shirakawa, H.; Yamaoka, T.; Sanpei, K.; Sasaoka, H.; Nakagawa, T.; Kaneko, S. TRPV1 stimulation triggers apoptotic cell death of rat cortical neurons. Biochem. Biophys. Res. Commun. 2008, 377, 1211–1215. [Google Scholar] [CrossRef] [PubMed]
- Stock, K.; Garthe, A.; de Almeida Sassi, F.; Glass, R.; Wolf, S.A.; Kettenmann, H. The capsaicin receptor TRPV1 as a novel modulator of neural precursor cell proliferation. Stem. Cells 2014, 32, 3183–3195. [Google Scholar] [CrossRef]
- Sun, Z.; Han, J.; Zhao, W.; Zhang, Y.; Wang, S.; Ye, L.; Liu, T.; Zheng, L. TRPV1 activation exacerbates hypoxia/reoxygenation-induced apoptosis in H9C2 cells via calcium overload and mitochondrial dysfunction. Int. J. Mol. Sci. 2014, 15, 18362–18380. [Google Scholar] [CrossRef]
- Hu, F.; Sun, W.W.; Zhao, X.T.; Cui, Z.J.; Yang, W.X. TRPV1 mediates cell death in rat synovial fibroblasts through calcium entry-dependent ROS production and mitochondrial depolarization. Biochem. Biophys. Res. Commun. 2008, 369, 989–993. [Google Scholar] [CrossRef]
- Denda, S.; Denda, M.; Inoue, K.; Hibino, T. Glycolic acid induces keratinocyte proliferation in a skin equivalent model via TRPV1 activation. J. Derm. Sci. 2010, 57, 108–113. [Google Scholar] [CrossRef]
- Hofmann, N.A.; Barth, S.; Waldeck-Weiermair, M.; Klec, C.; Strunk, D.; Malli, R.; Graier, W.F. TRPV1 mediates cellular uptake of anandamide and thus promotes endothelial cell proliferation and network-formation. Biol. Open 2014, 3, 1164–1172. [Google Scholar] [CrossRef] [PubMed]
- Vercelli, C.; Barbero, R.; Cuniberti, B.; Odore, R.; Re, G. Expression and functionality of TRPV1 receptor in human MCF-7 and canine CF.41 cells. Vet. Comp. Oncol. 2015, 13, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Stock, K.; Kumar, J.; Synowitz, M.; Petrosino, S.; Imperatore, R.; Smith, E.S.J.; Wend, P.; Purfürst, B.; Nuber, U.A.; Gurok, U.; et al. Neural precursor cells induce cell death of high-grade astrocytomas through stimulation of TRPV1. Nat. Med. 2012, 18, 1232. [Google Scholar] [CrossRef]
- Jambrina, E.; Alonso, R.; Alcalde, M.; del Carmen Rodrı́guez, M.; Serrano, A.; Martı́nez, A.C.; Garcı́a-Sancho, J.; Izquierdo, M. Calcium influx through receptor-operated channel induces mitochondria-triggered paraptotic cell death. J. Biol. Chem. 2003, 278, 14134–14145. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Wang, G.; Tao, H.; Yang, Z.; Wang, Y.; Meng, Z.; Zhou, J. Capsaicin mediates caspases activation and induces apoptosis through P38 and JNK MAPK pathways in human renal carcinoma. BMC Cancer 2016, 16, 790. [Google Scholar] [CrossRef]
- Sanchez, M.G.; Sanchez, A.M.; Collado, B.; Malagarie-Cazenave, S.; Olea, N.; Carmena, M.J.; Prieto, J.C.; Diaz-Laviada, I.I. Expression of the transient receptor potential vanilloid 1 (TRPV1) in LNCaP and PC-3 prostate cancer cells and in human prostate tissue. Eur. J. Pharm. 2005, 515, 20–27. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Basu, S. Fas-associated factor 1 is a negative regulator in capsaicin induced cancer cell apoptosis. Cancer Lett. 2010, 287, 142–149. [Google Scholar] [CrossRef]
- Hou, N.; He, X.; Yang, Y.; Fu, J.; Zhang, W.; Guo, Z.; Hu, Y.; Liang, L.; Xie, W.; Xiong, H.; et al. TRPV1 Induced Apoptosis of Colorectal Cancer Cells by Activating Calcineurin-NFAT2-p53 Signaling Pathway. Biomed. Res. Int. 2019, 2019, 6712536. [Google Scholar] [CrossRef]
- Amantini, C.; Mosca, M.; Nabissi, M.; Lucciarini, R.; Caprodossi, S.; Arcella, A.; Santoni, G. Capsaicin_induced apoptosis of glioma cells is mediated by TRPV1 vanilloid receptor and requires p38 MAPK activation. J. Neurochem. 2007, 102, 977–990. [Google Scholar] [CrossRef]
- Fonseca, B.M.; Correia-da-Silva, G.; Teixeira, N.A. Cannabinoid-induced cell death in endometrial cancer cells: Involvement of TRPV1 receptors in apoptosis. J. Physiol. Biochem. 2018, 74, 261–272. [Google Scholar] [CrossRef]
- Wu, Y.Y.; Liu, X.Y.; Zhuo, D.X.; Huang, H.B.; Zhang, F.B.; Liao, S.F. Decreased expression of TRPV1 in renal cell carcinoma: Association with tumor Fuhrman grades and histopathological subtypes. Cancer Manag. Res. 2018, 10, 1647–1655. [Google Scholar] [CrossRef] [PubMed]
- Amantini, C.; Ballarini, P.; Caprodossi, S.; Nabissi, M.; Morelli, M.B.; Lucciarini, R.; Cardarelli, M.A.; Mammana, G.; Santoni, G. Triggering of transient receptor potential vanilloid type 1 (TRPV1) by capsaicin induces Fas/CD95-mediated apoptosis of urothelial cancer cells in an ATM-dependent manner. Carcinogenesis 2009, 30, 1320–1329. [Google Scholar] [CrossRef] [PubMed]
- Puntambekar, P.; Mukherjea, D.; Jajoo, S.; Ramkumar, V. Essential role of Rac1/NADPH oxidase in nerve growth factor induction of TRPV1 expression. J. Neurochem. 2005, 95, 1689–1703. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Guo, W.; Ma, J.; Xu, P.; Zhang, W.; Guo, S.; Liu, L.; Ma, J.; Shi, Q.; Jian, Z.; et al. Downregulated TRPV1 Expression Contributes to Melanoma Growth via the Calcineurin-ATF3-p53 Pathway. J. Invest. Derm. 2018, 138, 2205–2215. [Google Scholar] [CrossRef] [PubMed]
- Nazıroğlu, M.; Çiğ, B.; Blum, W.; Vizler, C.; Buhala, A.; Marton, A.; Katona, R.; Jósvay, K.; Schwaller, B.; Oláh, Z.; et al. Targeting breast cancer cells by MRS1477, a positive allosteric modulator of TRPV1 channels. PLoS ONE 2017, 12, e0179950. [Google Scholar]
- Xie, R.; Xu, J.; Wen, G.; Jin, H.; Liu, X.; Yang, Y.; Ji, B.; Jiang, Y.; Song, P.; Dong, H.; et al. The P2Y2 nucleotide receptor mediates the proliferation and migration of human hepatocellular carcinoma cells induced by ATP. J. Biol. Chem. 2014, 289, 19137–19149. [Google Scholar] [CrossRef]
- Sabala, P.; Czajkowski, R.; Przybylek, K.; Kalita, K.; Kaczmarek, L.; Baranska, J. Two subtypes of G protein-coupled nucleotide receptors, P2Y1 and P2Y2 are involved in calcium signalling in glioma C6 cells. Br. J. Pharm. 2001, 132, 393–402. [Google Scholar] [CrossRef][Green Version]
- Liu, L.; Yudin, Y.; Rohacs, T. Diacylglycerol kinases regulate TRPV1 channel activity. J. Biol. Chem. 2020. [Google Scholar] [CrossRef]
- Heo, J.S.; Han, H.J. ATP stimulates mouse embryonic stem cell proliferation via protein kinase C, phosphatidylinositol 3-kinase/Akt, and mitogen-activated protein kinase signaling pathwats. Stem. Cells 2006, 24, 2637–2648. [Google Scholar] [CrossRef]
- Danciu, T.E.; Adam, R.M.; Naruse, K.; Freeman, M.R.; Hauschka, P.V. Calcium regulates the PI3K-Akt pathway in stretched osteoblasts. FEBS Lett. 2003, 536, 193–197. [Google Scholar] [CrossRef]
- Katz, S.; Ayala, V.; Santillán, G.; Boland, R. Activation of the PI3K/Akt signaling pathway through P2Y2 receptors by extracellular ATP is involved in osteoblastic cell proliferation. Arch. Biochem. Biophys. 2011, 513, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, Z.; Capó-Aponte, J.E.; Zhang, F.; Pan, Z.; Reinach, P.S. Epidermal growth factor recept or transactivation by the cannabinoid receptor (CB1) and transient receptor potential vanilloid 1 (TRPV1) induces differential responses in corneal epithelial cells. Exp. Eye Res. 2010, 91, 462–471. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Bode, A.M.; Zhu, F.; Liu, K.; Zhang, J.; Kim, M.O.; Langfald, A.K. TRPV1-antagonist AMG9810 promotes mouse skin tumorigenesis through EGFR/Akt signaling. Carcinogenesis 2011, 32, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Roberts, P.J.; Der, C.J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef]
- Uslusoy, F.; Nazıroğlu, M.; Çiğ, B. Inhibition of the TRPM2 and TRPV1 channels through Hypericum perforatum in sciatic nerve injury-induced rats demonstrates their key role in apoptosis and mitochondrial oxidative stress of sciatic nerve and dorsal root ganglion. Front. Physiol. 2017, 8, 335. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Wang, F.; Yang, Y.; Ma, W.; Lin, Z.; Cheng, N.; Long, Y.; Deng, S.; Li, Z. Recurrent activations of transient receptor potential vanilloid-1 and vanilloid-4 promote cellular proliferation and migration in esophageal squamous cell carcinoma cells. FEBS Open. Biol. 2019, 9, 206–225. [Google Scholar] [CrossRef]
- Chung, M.K.; Güler, A.D.; Caterina, M.J. TRPV1 shows dynamic ionic selectivity during agonist stimulation. Nat. Neurosci 2008, 11, 555. [Google Scholar] [CrossRef]
- Pereira, G.J.V.; Tavares, M.T.; Azevedo, R.A.; Martins, B.B.; Cunha, M.R.; Bhardwaj, R.; Cury, Y.; Zambelli, V.O.; Barbosa, E.G.; Hediger, M.A.; et al. Capsaicin-like analogue induced selective apoptosis in A2058 melanoma cells: Design, synthesis and molecular modeling. Bioorg Med. Chem. 2019, 27, 2893–2904. [Google Scholar] [CrossRef]
- Defo Deeh, P.B.; Watcho, P.; Wankeu_Nya, M.; Ngadjui, E.; Usman, U.Z. The methanolic extract of Guibourtia tessmannii (caesalpiniaceae) and selenium modulate cytosolic calcium accumulation, apoptosis and oxidative stress in R2C tumour Leydig cells: Involvement of TRPV 1 channels. Andrologia 2019, 51, e13216. [Google Scholar] [CrossRef]
- Köse, S.A.; Nazırolu, M. N-acetyl cysteine reduces oxidative toxicity, apoptosis, and calcium entry through TRPV1 channels in the neutrophils of patients with polycystic ovary syndrome. Free Radic Res. 2015, 49, 338–346. [Google Scholar] [CrossRef]
- Chen, W.T.; Lin, G.B.; Lin, S.H.; Lu, C.H.; Hsieh, C.H.; Ma, B.L.; Chao, C.Y. Static magnetic field enhances the anticancer efficacy of capsaicin on HepG2 cells via capsaicin receptor TRPV1. PLoS ONE 2018, 13, e0191078. [Google Scholar] [CrossRef]
- Pan, L.; Song, K.; Hu, F.; Sun, W.; Lee, I. Nitric oxide induces apoptosis associated with TRPV1 channel-mediated Ca2+ entry via S-nitrosylation in osteoblasts. Eur. J. Pharm. 2013, 715, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Lakshmi, S.; Joshi, P.G. Co-activation of P2Y2 receptor and TRPV channel by ATP: Implications for ATP induced pain. Cell Mol. Neurobiol. 2005, 25, 819–832. [Google Scholar] [CrossRef] [PubMed]
- Nita, I.I.; Caspi, Y.; Gudes, S.; Fishman, D.; Lev, S.; Hersfinkel, M.; Binshtok, A.M. Privileged crosstalk between TRPV1 channels and mitochondrial calcium shuttling machinery controls nociception. Bba-Mol. Cell Res. 2016, 1863, 2868–2880. [Google Scholar] [CrossRef]
- Nazırolu, M.; Övey, I.S. Involvement of apoptosis and calcium accumulation through TRPV1 channels in neurobiology of epilepsy. Neuroscience 2015, 293, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Vercelli, C.; Barbero, R.; Cuniberti, B.; Racca, S.; Abbadessa, G.; Piccione, F.; Re, G. Transient receptor potential vanilloid 1 expression and functionality in mcf-7 cells: A preliminary investigation. J. Breast Cancer 2014, 17, 332–338. [Google Scholar] [CrossRef]
- Ghazizadeh, V.; Nazırolu, M. Electromagnetic radiation (Wi-Fi) and epilepsy induce calcium entry and apoptosis through activation of TRPV1 channel in hippocampus and dorsal root ganglion of rats. Metab. Brain Dis. 2014, 29, 787–799. [Google Scholar] [CrossRef]
- Thomas, K.C.; Sabnis, A.S.; Johansen, M.E.; Lanza, D.L.; Moos, P.J.; Yost, G.S.; Reilly, C.A. Transient receptor potential vanilloid 1 agonists cause endoplasmic reticulum stress and cell death in human lung cells. J. Pharm. Exp. 2007, 321, 830–838. [Google Scholar] [CrossRef]
- Agopyan, N.; Head, J.; Yu, S.; Simon, S.A. TRPV1 receptors mediate particulate matter-induced apoptosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2004, 286, L563–L572. [Google Scholar] [CrossRef]
- He, L.; Poblenz, A.T.; Medrano, C.J.; Fox, D.A. Lead and calcium produce rod photoreceptor cell apoptosis by opening the mitochondrial permeability transition pore. J. Biol. Chem. 2000, 275, 12175–12184. [Google Scholar] [CrossRef]
- Narita, M.; Shimizu, S.; Ito, T.; Chittenden, T.; Lutz, R.J.; Matsuda, H.; Tsujimoto, Y. Bax interacts with the permeability transition pore to induce permeability transition and cytochrome c release in isolated mitochondria. Proc. Natl. Acad. Sci. USA 1998, 95, 14681–14686. [Google Scholar] [CrossRef]
- Armstrong, J.S.; Jones, D.P. Glutathione depletion enforces the mitochondrial permeability transition and causes cell death in Bcl-2 overexpressing HL60 cells. Faseb J. 2002, 16, 1263–1265. [Google Scholar] [CrossRef] [PubMed]
- Douglas, M.G.; Cockrell, R.S. Mitochondrial cation-hydrogen ion exchange. Sodium selective transport by mitochondria and submitochondrial particles. J. Biol. Chem. 1974, 249, 5464–5471. [Google Scholar] [PubMed]
- Numata, M.; Petrecca, K.; Lake, N.; Orlowski, J. Identification of a mitochondrial Na+/H+ exchanger. J. Biol. Chem. 1998, 273, 6951–6959. [Google Scholar] [CrossRef]
- Smaili, S.S.; Russell, J.T. Permeability transition pore regulates both mitochondrial membrane potential and agonist-evoked Ca2+ signals in oligodendrocyte progenitors. Cell Calcium. 1999, 26, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, J.S.; Steinauer, K.K.; Hornung, B.; Irish, J.M.; Lecane, P.; Birrell, G.W.; Peehl, D.M.; Knox, S.J. Role of glutathione depletion and reactive oxygen species generation in apoptotic signaling in a human B lymphoma cell line. Cell Death Differ. 2002, 9, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Ip, S.W.; Lan, S.H.; Lu, H.F.; Huang, A.C.; Yang, J.S.; Lin, J.P.; Wood, W.G. Capsaicin mediates apoptosis in human nasopharyngeal carcinoma NPC-TW 039 cells through mitochondrial depolarization and endoplasmic reticulum stress. Hum. Exp. Toxicol. 2012, 31, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Cande, C.; Stephanou, N.C.; Jiang, S.; Gurbuxani, S.; Larochette, N.; Daugas, E.; Garrido, C.; Kroemer, G.; Wu, H. DNA binding is required for the apoptogenic action of apoptosis inducing factor. Nat. Struct. Biol. 2002, 9, 680–684. [Google Scholar] [CrossRef]
- Krizanova, O.; Steliarova, I.; Csaderova, L.; Pastorek, M.; Hudecova, S. Capsaicin induces apoptosis in PC12 cells through ER stress. Oncol. Rep. 2014, 31, 581–588. [Google Scholar] [CrossRef]
- Dremina, E.S.; Sharov, V.S.; Kumar, K.; Zaidi, A.; Michaelis, E.K.; Schoneich, C. Anti-apoptotic protein Bcl-2 interacts with and destabilizes the sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SERCA). Biochem. J. 2004, 383, 361–370. [Google Scholar] [CrossRef]
- Laver, D.R.; Lamb, G.D. Inactivation of Ca2+ release channels (ryanodine receptors RyR1 and RyR2) with rapid steps in [Ca2+] and voltage. Biophys. J. 1998, 74, 2352–2364. [Google Scholar] [CrossRef]
- Andrews, C.; Ho, P.D.; Dillmann, W.H.; Glembotski, C.C.; McDonough, P.M. The MKK6-p38 MAPK pathway prolongs the cardiac contractile calcium transient, downregulates SERCA2, and activates NF-AT. Cardiovasc. Res. 2003, 59, 46–56. [Google Scholar] [CrossRef]
- Luo, S.; Baumeister, P.; Yang, S.; Abcouwer, S.F.; Lee, A.S. Induction of GRP78/BiP by translational block Activation of the Grp78 promoter by ATF4 through an upstream ATF/CRE site independent of the endoplasmic reticulum stress elements. J. Biol. Chem. 2003, 278, 37375–37385. [Google Scholar] [CrossRef]
- Lim, M.P.; Devi, L.A.; Rozenfeld, R. Cannabidiol causes activated hepatic stellate cell death through a mechanism of endoplasmic reticulum stress-induced apoptosis. Cell Death Dis. 2011, 2, e170. [Google Scholar] [CrossRef] [PubMed]
- Barlow, C.; Brown, K.D.; Deng, C.X.; Tagle, D.A.; Wynshaw-Boris, A. Atm selectively regulates distinct p53-dependent cell-cycle checkpoint and apoptotic pathways. Nat. Genet. 1997, 17, 453–456. [Google Scholar] [CrossRef] [PubMed]
- Canman, C.E.; Lim, D.S.; Cimprich, K.A.; Taya, Y.; Tamai, K.; Sakaguchi, K.; Appella, E.; Kastan, M.B.; Siliciano, J.D. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 1998, 281, 1677–1679. [Google Scholar] [CrossRef]
- Katsuda, K.; Kataoka, M.; Uno, F.; Murakami, T.; Kondo, T.; Roth, J.A.; Tanaka, N.; Fujiwara, T. Activation of caspase-3 and cleavage of Rb are associated with p16-mediated apoptosis in human non-small cell lung cancer cells. Oncogene 2002, 21, 2108–2113. [Google Scholar] [CrossRef]
- Hernandez, A.M.; Colvin, E.S.; Chen, Y.C.; Geiss, S.L.; Eller, L.E.; Fueger, P.T. Upregulation of p21 activates the intrinsic apoptotic pathway in beta-cells. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E1281–E1290. [Google Scholar] [CrossRef][Green Version]
- Cheng, E.H.; Kirsch, D.G.; Clem, R.J.; Ravi, R.; Kastan, M.B.; Bedi, A.; Ueno, K.; Hardwick, J.M. Conversion of Bcl-2 to a Bax-like death effector by caspases. Science 1997, 278, 1966–1968. [Google Scholar] [CrossRef]
- Gil, Y.G.; Kang, M.K. Capsaicin induces apoptosis and terminal differentiation in human glioma A172 cells. Life Sci. 2008, 82, 997–1003. [Google Scholar] [CrossRef]
- Song, J.; Lee, J.H.; Lee, S.H.; Park, K.A.; Lee, W.T.; Lee, J.E. TRPV1 Activation in Primary Cortical Neurons Induces Calcium-Dependent Programmed Cell Death. Exp. Neurobiol. 2013, 22, 51–57. [Google Scholar] [CrossRef]
- Wu, T.T.; Peters, A.A.; Tan, P.T.; Roberts-Thomson, S.J.; Monteith, G.R. Consequences of activating the calcium-permeable ion channel TRPV1 in breast cancer cells with regulated TRPV1 expression. Cell Calcium. 2014, 56, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. P53: An ubiquitous target of anticancer drugs. Int. J. Cancer 2002, 98, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Issaeva, N.; Bozko, P.; Enge, M.; Protopopova, M.; Verhoef, L.G.; Masucci, M.; Pramanik, A.; Selivanova, G. Small molecule RITA binds to p53, blocks p53-HDM-2 interaction and activates p53 function in tumors. Nat. Med. 2004, 10, 1321–1328. [Google Scholar] [CrossRef] [PubMed]
- Park, G.Y.; Wilson, J.J.; Song, Y.; Lippard, S.J. Phenanthriplatin, a monofunctional DNA-binding platinum anticancer drug candidate with unusual potency and cellular activity profile. Proc. Natl. Acad. Sci. USA 2012, 109, 11987–11992. [Google Scholar] [CrossRef]
- Satheesh, N.J.; Busselberg, D. The role of intracellular calcium for the development and treatment of neuroblastoma. Cancers 2015, 7, 823–848. [Google Scholar] [CrossRef]
- Roderick, H.L.; Cook, S.J. Ca2+ signalling checkpoints in cancer: Remodelling Ca2+ for cancer cell proliferation and survival. Nat. Rev. Cancer 2008, 8, 361–375. [Google Scholar] [CrossRef]
- Al-Taweel, N.; Varghese, E.; Florea, A.M.; Büsselberg, D. Cisplatin (CDDP) triggers cell death of MCF-7 cells following disruption of intracellular calcium ([Ca2+] i) homeostasis. J. Toxicol. Sci. 2014, 39, 765–774. [Google Scholar] [CrossRef]
- Varghese, E.; Busselberg, D. Auranofin, an anti-rheumatic gold compound, modulates apoptosis by elevating the intracellular calcium concentration ([ca2+]I) in mcf-7 breast cancer cells. Cancers 2014, 6, 2243–2258. [Google Scholar] [CrossRef]
- Günes, D.A.; Florea, A.-M.; Splettstoesser, F.; Büsselberg, D. Co-application of arsenic trioxide (As2O3) and cisplatin (CDDP) on human SY-5Y neuroblastoma cells has differential effects on the intracellular calcium concentration ([Ca2+]i) and cytotoxicity. NeuroToxicology 2009, 30, 194–202. [Google Scholar] [CrossRef]
- Aoki, M.; Batista, O.; Bellacosa, A.; Tsichlis, P.; Vogt, P.K. The akt kinase: Molecular determinants of oncogenicity. Proc. Natl. Acad. Sci. USA 1998, 95, 14950–14955. [Google Scholar] [CrossRef]
- Wu, Y.-R.; Qi, H.-J.; Deng, D.-F.; Luo, Y.-Y.; Yang, S.-L. MicroRNA-21 promotes cell proliferation, migration, and resistance to apoptosis through PTEN/PI3K/AKT signaling pathway in esophageal cancer. Tumor. Biol. 2016, 37, 12061–12070. [Google Scholar] [CrossRef]
- Jin, Y.; Feng, S.J.; Qiu, S.; Shao, N.; Zheng, J.H. LncRNA MALAT1 promotes proliferation and metastasis in epithelial ovarian cancer via the PI3K-AKT pathway. Eur. Rev. Med. Pharm. Sci. 2017, 21, 3176–3184. [Google Scholar]
- Gao, N.; Zhang, Z.; Jiang, B.H.; Shi, X. Role of PI3K/AKT/mTOR signaling in the cell cycle progression of human prostate cancer. Biochem. Biophys. Res. Commun. 2003, 310, 1124–1132. [Google Scholar] [CrossRef]
- Berns, K.; Horlings, H.M.; Hennessy, B.T.; Madiredjo, M.; Hijmans, E.M.; Beelen, K.; Linn, S.C.; Gonzalez-Angulo, A.M.; Stemke-Hale, K.; Hauptmann, M.; et al. A functional genetic approach identifies the PI3K pathway as a major determinant of trastuzumab resistance in breast cancer. Cancer Cell.
- Abrams, S.L.; Steelman, L.S.; Shelton, J.G.; Wong, E.W.T.; Chappell, W.H.; Bäsecke, J.; Stivala, F.; Donia, M.; Nicoletti, F.; Libra, M.; et al. The Raf/MEK/ERK pathway can govern drug resistance, apoptosis and sensitivity to targeted therapy. Cell Cycle 2010, 9, 1781–1791. [Google Scholar] [CrossRef]
- Bon, G.; Loria, R.; Amoreo, C.A.; Verdina, A.; Sperduti, I.; Mastrofrancesco, A.; Soddu, S.; Diodoro, M.G.; Mottolese, M.; Todaro, M.; et al. Dual targeting of HER3 and MEK may overcome HER3-dependent drug-resistance of colon cancers. Oncotarget 2017, 8, 108463–108479. [Google Scholar] [CrossRef]
- Dekker, L.V.; Leitges, M.; Altschuler, G.; Mistry, N.; McDermott, A.; Roes, J.; Segal, A.W. Protein kinase C-β contributes to NADPH oxidase activation in neutrophils. Biochem. J. 2000, 347, 285–289. [Google Scholar] [CrossRef]
- Descamps, S.; Toillon, R.-A.; Adriaenssens, E.; Pawlowski, V.R.; Cool, S.M.; Nurcombe, V.; Bourhis, X.L.; Boilly, B.n.; Peyrati, J.-P.; Hondermarck, H. Nerve Growth Factor stimulates proliferation and survival of human breast cancer cells through two distinct signaling pathways. J. Biol. Chem. 2001, 276, 17864–17870. [Google Scholar] [CrossRef]
- Bujak, J.K.; Kosmala, D.; Szopa, I.M.; Majchrzak, K.; Bednarczyk, P. Inflammation, Cancer and Immunity-Implication of TRPV1 Channel. Front. Oncol. 2019, 9, 1087. [Google Scholar] [CrossRef]
- Afroz, S.; Arakaki, R.; Iwasa, T.; Oshima, M.; Hosoki, M.; Inoue, M.; Baba, O.; Okayama, Y.; Matsuka, Y. CGRP Induces Differential Regulation of Cytokines from Satellite Glial Cells in Trigeminal Ganglia and Orofacial Nociception. Int. J. Mol. Sci. 2019, 20, 711. [Google Scholar] [CrossRef] [PubMed]
- Mashaghi, A.; Marmalidou, A.; Tehrani, M.; Grace, P.M.; Pothoulakis, C.; Dana, R. Neuropeptide substance P and the immune response. Cell Mol. Life Sci. 2016, 73, 4249–4264. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Georgescu, S.R.; Sarbu, M.I.; Matei, C.; Ilie, M.A.; Caruntu, C.; Constantin, C.; Neagu, M.; Tampa, M. Capsaicin: Friend or Foe in Skin Cancer and Other Related Malignancies? Nutrients 2017, 9, 1365. [Google Scholar] [CrossRef]
- Erin, N. Role of sensory neurons, neuroimmune pathways, and transient receptor potential vanilloid 1 (TRPV1) channels in a murine model of breast cancer metastasis. Cancer Immunol. Immunother. 2020, 69, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Peluso, G.; Petillo, O.; Melone, M.A.; Mazzarella, G.; Ranieri, M.; Tajana, G.F. Modulation of cytokine production in activated human monocytes by somatostatin. Neuropeptides 1996, 30, 443–451. [Google Scholar] [CrossRef]
- Pinter, E.; Helyes, Z.; Szolcsanyi, J. Inhibitory effect of somatostatin on inflammation and nociception. Pharm. Ther. 2006, 112, 440–456. [Google Scholar] [CrossRef]
- Li, Y.R.; Gupta, P. Immune aspects of the bi-directional neuroimmune facilitator TRPV1. Mol. Biol. Rep. 2019, 46, 1499–1510. [Google Scholar] [CrossRef]
- Fernandes, E.S.; Cerqueira, A.R.A.; Soares, A.G.; Costa, S.K.P. Capsaicin and Its Role in Chronic Diseases. In Drug Discovery from Mother Nature; Gupta, S.C., Prasad, S., Aggarwal, B.B., Eds.; Springer: Cham, Switzerland, 2016; Volume 929. [Google Scholar]
- Walker, J.; Ley, J.P.; Schwerzler, J.; Lieder, B.; Beltran, L.; Ziemba, P.M.; Hatt, H.; Hans, J.; Widder, S.; Krammer, G.E.; et al. Nonivamide, a capsaicin analogue, exhibits anti-inflammatory properties in peripheral blood mononuclear cells and U-937 macrophages. Mol. Nutr. Food Res. 2017, 61. [Google Scholar] [CrossRef]
- Gao, H.; Yang, B.J.; Li, N.; Feng, L.M.; Shi, X.Y.; Zhao, W.H.; Liu, S.J. Bisphenol A and hormone-associated cancers: Current progress and perspectives. Medicine (Baltimore) 2015, 94, e211. [Google Scholar] [CrossRef]
- Guzel, K.G.U.; Naziroglu, M.; Ceyhan, D. Bisphenol A-Induced Cell Proliferation and Mitochondrial Oxidative Stress Are Diminished via Modulation of TRPV1 Channel in Estrogen Positive Breast Cancer Cell by Selenium Treatment. Biol. Trace Elem. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Geng, S.; Zheng, Y.; Meng, M.; Guo, Z.; Cao, N.; Ma, X.; Du, Z.; Li, J.; Duan, Y.; Du, G. Gingerol Reverses the Cancer-Promoting Effect of Capsaicin by Increased TRPV1 Level in a Urethane-Induced Lung Carcinogenic Model. J. Agric. Food Chem. 2016, 64, 6203–6211. [Google Scholar] [CrossRef]
- Nur, G.; Naziroglu, M.; Deveci, H.A. Synergic prooxidant, apoptotic and TRPV1 channel activator effects of alpha-lipoic acid and cisplatin in MCF-7 breast cancer cells. J. Recept Signal. Transduct. Res. 2017, 37, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Çetin, E.S.; Nazıroğlu, M.; Çiğ, B.; Övey, İ.S.; Koşar, P.A. Selenium potentiates the anticancer effect of cisplatin against oxidative stress and calcium ion signaling-induced intracellular toxicity in MCF-7 breast cancer cells: Involvement of the TRPV1 channel. J. Recept. Sig. Transd. 2017, 37, 84–93. [Google Scholar] [CrossRef]
- Zheng, L.; Chen, J.; Ma, Z.; Liu, W.; Yang, F.; Yang, Z.; Wang, K.; Wang, X.; He, D.; Li, L.; et al. Capsaicin enhances anti-proliferation efficacy of pirarubicin via activating TRPV1 and inhibiting PCNA nuclear translocation in 5637 cells. Mol. Med. Rep. 2016, 13, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Kosar, P.A.; Naziroglu, M.; Ovey, I.S.; Cig, B. Synergic Effects of Doxorubicin and Melatonin on Apoptosis and Mitochondrial Oxidative Stress in MCF-7 Breast Cancer Cells: Involvement of TRPV1 Channels. J. Membr. Biol. 2016, 249, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Amgalan, D.; Garner, T.P.; Kitsis, R.N. A small-molecule allosteric inhibitor of BAX protects against doxorubicin-induced cardiomyopathy. Nat. Cancer 2020, 1, 315–328. [Google Scholar] [CrossRef]
- Deveci, H.A.; Naziroglu, M.; Nur, G. 5-Fluorouracil-induced mitochondrial oxidative cytotoxicity and apoptosis are increased in MCF-7 human breast cancer cells by TRPV1 channel activation but not Hypericum perforatum treatment. Mol. Cell Biochem. 2018, 439, 189–198. [Google Scholar] [CrossRef]
- Chen, Y.; Li, J.; Jin, L.; Lei, K.; Liu, H.; Yang, Y. Fibulin-5 contributes to colorectal cancer cell apoptosis via the ROS/MAPK and Akt signal pathways by downregulating transient receptor potential cation channel subfamily V member 1. J. Cell Biochem. 2019, 120, 17838–17846. [Google Scholar] [CrossRef]
- Punzo, F.; Manzo, I.; Tortora, C.; Pota, E.; Angelo, V.; Bellini, G.; Di Paola, A.; Verace, F.; Casale, F.; Rossi, F. Effects of CB2 and TRPV1 receptors’ stimulation in pediatric acute T-lymphoblastic leukemia. Oncotarget 2018, 9, 21244–21258. [Google Scholar] [CrossRef][Green Version]
- Mao, Y.; Liu, X. Bioresponsive Nanomedicine: The Next Step of Deadliest Cancers’ Theranostics. Front. Chem. 2020, 8, 257. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Zhang, J.; Zhang, W.; Foda, M.F.; Zhang, Y.; Ge, L.; Han, H. Intracellular Ca(2+) Cascade Guided by NIR-II Photothermal Switch for Specific Tumor Therapy. iScience 2020, 23, 101049. [Google Scholar] [CrossRef] [PubMed]
- Zhen, X.; Xie, C.; Jiang, Y.; Ai, X.; Xing, B.; Pu, K. Semiconducting Photothermal Nanoagonist for Remote-Controlled Specific Cancer Therapy. Nano Lett. 2018, 18, 1498–1505. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Guerrero, A.; Espinosa-Duran, J.M.; Velasco-Medina, J. TRPV1 channel as a target for cancer therapy using CNT-based drug delivery systems. Eur. Biophys. J. 2016, 45, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Baker, C.; Rodrigues, T.; de Almeida, B.P.; Barbosa-Morais, N.L.; Bernardes, G.J.L. Natural product-drug conjugates for modulation of TRPV1-expressing tumors. Bioorg. Med. Chem. 2019, 27, 2531–2536. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Pan, J.B.; Zhao, W.; Chen, H.Y.; Xu, J.J. Gold nanorod-assisted near-infrared light-mediated regulation of membrane ion channels activates apoptotic pathways. Chem. Commun. 2020. [Google Scholar] [CrossRef]
- Lozano, C.; Cordova, C.; Marchant, I.; Zuniga, R.; Ochova, P.; Ramirez-Barrantes, R.; Gonzalez-Arriagada, W.A.; Rodriguez, B.; Olivero, P. Intracellular aggregated TRPV1 is associated with lower survival in breast cancer patients. Breast Cancer 2018, 10, 161–168. [Google Scholar] [CrossRef]
- Han, G.H.; Chay, D.B.; Nam, S.; Cho, H.; Chung, J.Y.; Kim, J.H. Prognostic Significance of Transient Receptor Potential Vanilloid Type 1 (TRPV1) and Phosphatase and Tension Homolog (PTEN) in Epithelial Ovarian Cancer. Cancer Genom. Proteom. 2020, 17, 309–319. [Google Scholar] [CrossRef]
- Han, G.H.; Chay, D.B.; Nam, S.; Cho, H.; Chung, J.Y.; Kim, J.H. The Combination of Transient Receptor Potential Vanilloid Type 1 (TRPV1) and Phosphatase and Tension Homolog (PTEN) is an Effective Prognostic Biomarker in Cervical Cancer. Int. J. Gynecol. Pathol. 2020. [Google Scholar] [CrossRef]
- Sharma, S.K.; Vijay, S.; Gore, S.; Dore, T.M.; Jagannathan, R. Measuring Cellular Ion Transport by Magnetoencephalography. ACS Omega 2020, 5, 4024–4031. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).