Micro-Transcriptome Analysis Reveals Immune-Related MicroRNA Regulatory Networks of Paralichthys olivaceus Induced by Vibrio anguillarum Infection

Abstract
:1. Introduction
2. Results
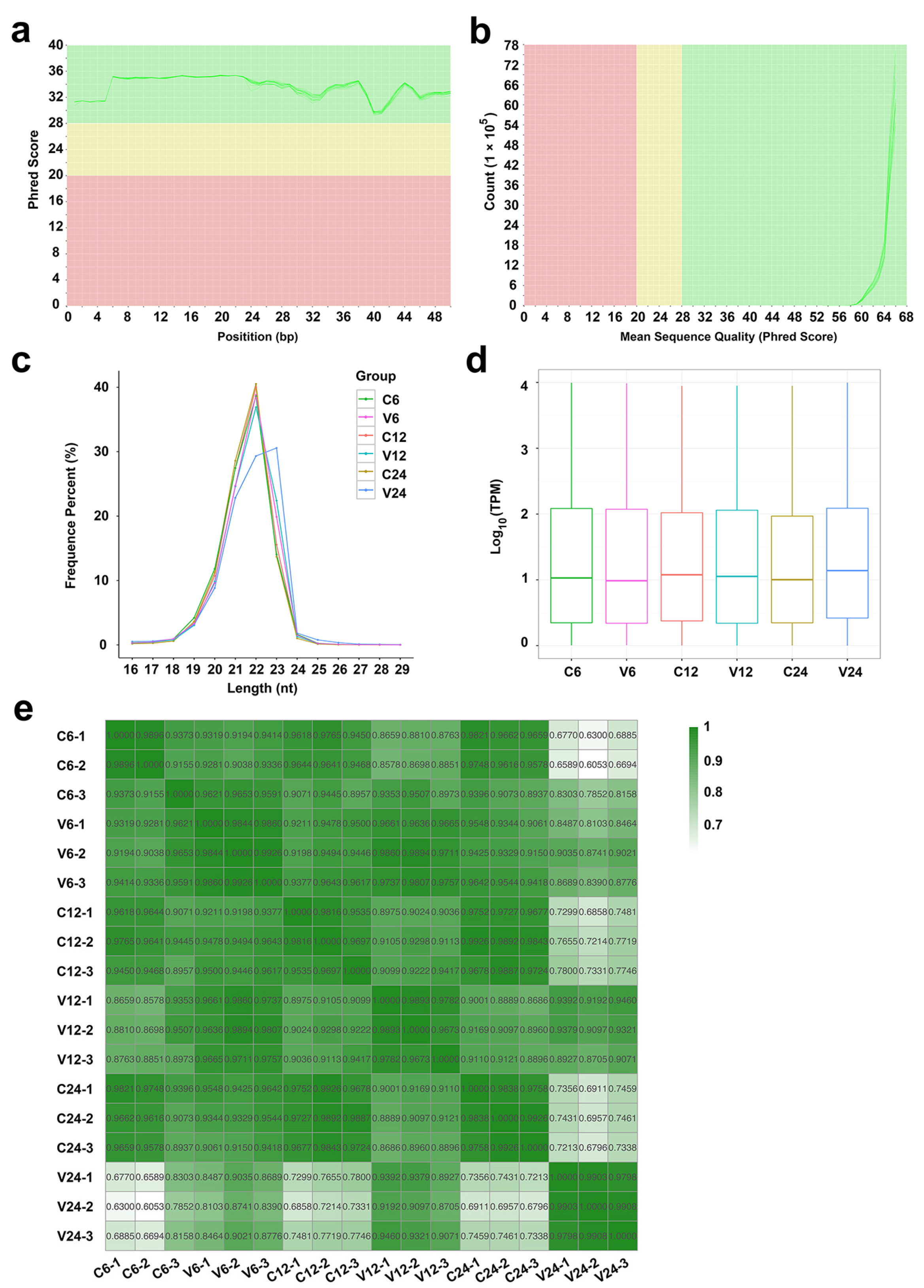
2.1. Data Processing and miRNA Identification
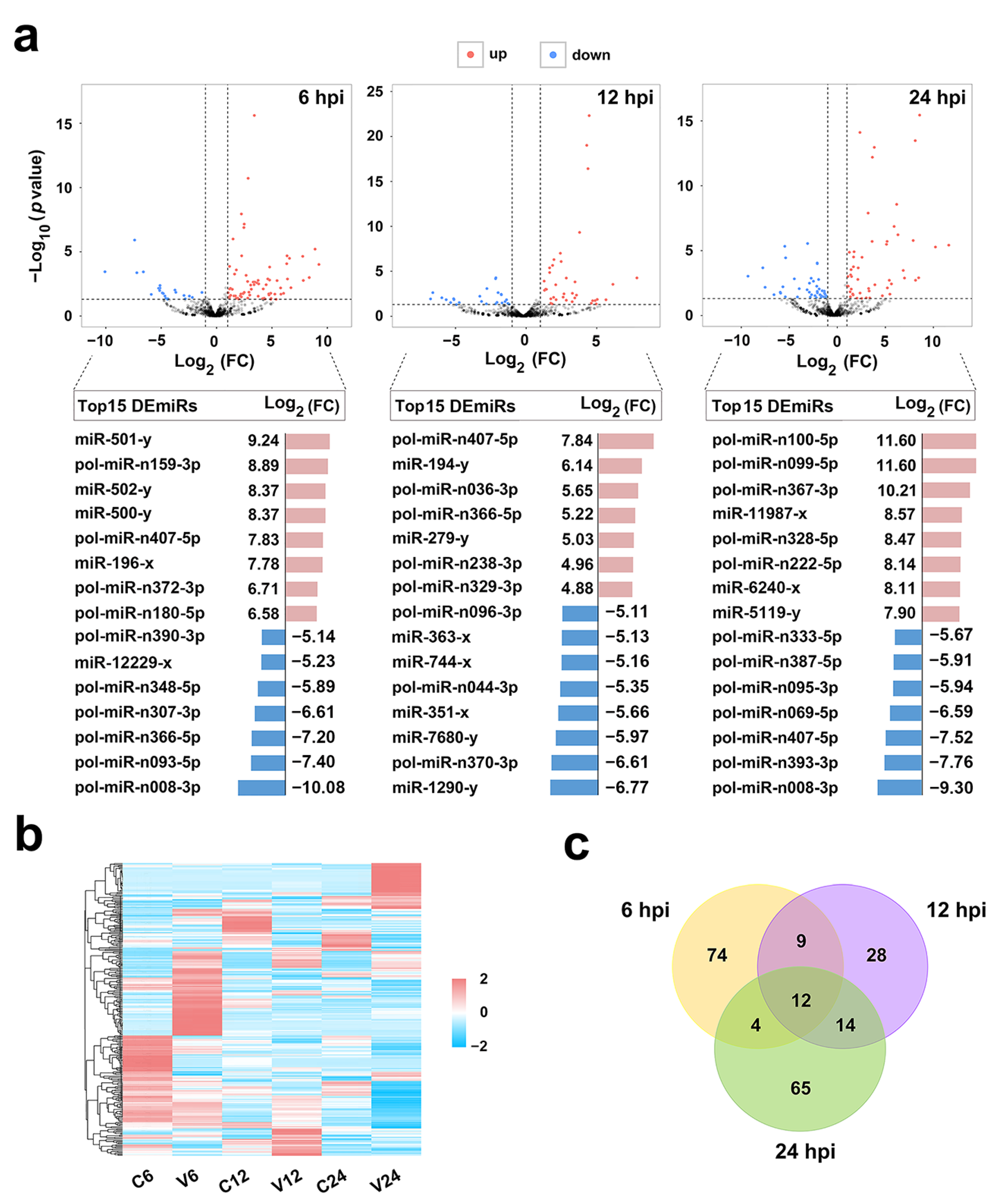
2.2. Identification and Validation of DEmiRs Induced by V. anguillarum
2.3. Identification of the Target Genes of the DEmiRs
2.4. Functional Enrichment of the DETGs
2.5. Immune-Related DEmiR-DETG Network Analysis
2.6. The Networks Formed by DEmiRs and V. anguillarum-Induced Hub Genes
3. Discussion
4. Materials and Methods
4.1. Sample Preparation
4.2. Small RNA Library Construction and Sequencing
4.3. Data Processing and miRNA Identification
4.4. Differential Expression Analysis
4.5. Experimental Validation of DEmiRs
4.6. Identification and Functional Enrichment Analysis of the Target Genes of DEmiRs
4.7. Interaction Network Construction
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cheng, A.M.; Byrom, M.; Shelton, J.; Ford, L.P. Antisense inhibition of human miRNAs and indications for an involvement of miRNA in cell growth and apoptosis. Nucleic Acids Res. 2005, 33, 1290–1297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mciver, S.C.; Roman, S.D.; Nixon, B.; Mclaughlin, E.A. miRNA and mammalian male germ cells. Hum. Reprod. Update 2012, 18, 44–59. [Google Scholar] [CrossRef] [Green Version]
- Bagga, S.; Bracht, J.R.; Hunter, S.; Massirer, K.B.; Holtz, J.; Eachus, R.; Pasquinelli, A.E. Regulation by let-7 and lin-4 miRNAs results in target mRNA degradation. Cell 2005, 122, 553–563. [Google Scholar] [CrossRef] [Green Version]
- Ge, Y.X.; Yan, X.D.; Jin, Y.G.; Yang, X.Y.; Yu, X.; Zhou, L.Q.; Han, S.C.; Yuan, Q.P.; Yang, M. fMiRNA-192 and miRNA-204 directly suppress lncRNA HOTTIP and interrupt GLS1-mediated glutaminolysis in hepatocellular carcinoma. PLoS Genet. 2015, 11, e1005726. [Google Scholar] [CrossRef]
- Kulcheski, F.R.; Christoff, A.P.; Margis, R. Circular RNAs are miRNA sponges and can be used as a new class of biomarker. J. Biotechnol. 2016, 238, 42–51. [Google Scholar] [CrossRef]
- Li, Y.; Wang, F.F.; Xu, J.F.; Ye, F.; Shen, Y.M.; Zhou, J.S.; Lu, W.G.; Wan, X.Y.; Ma, D.; Xie, X. Progressive miRNA expression profiles in cervical carcinogenesis and identification of HPV-related target genes for miR-29. J. Pathol. 2011, 224, 484–495. [Google Scholar] [CrossRef]
- Sarma, N.J.; Tiriveedhi, V.; Crippin, J.S.; Chapman, W.C.; Mohanakumar, T. Hepatitis C virus-induced changes in microRNA 107 (miRNA-107) and miRNA-449a modulate CCL2 by targeting the interleukin-6 receptor complex in hepatitis. J. Virol. 2014, 88, 3733–3743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.J.; Lu, H.; Meng, Q.L.; Wang, J.F.; Wang, W.M.; Yang, L.; Lin, L. Profilings of microRNAs in the liver of common carp (Cyprinus carpio) infected with Flavobacterium columnare. Int. J. Mol. Sci. 2016, 17, 566. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Gan, Z.; Cai, S.H.; Wang, Z.L.; Yu, D.P.; Lin, Z.W.; Lu, Y.S.; Wu, Z.H.; Jian, J.C. Comprehensive identification and profiling of Nile tilapia (Oreochromis niloticus) microRNAs response to Streptococcus agalactiae infection through high-throughput sequencing. Fish. Shellfish Immun. 2016, 54, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Sha, Z.X.; Gong, G.Y.; Wang, S.L.; Lu, Y.; Wang, L.; Wang, Q.L.; Chen, S.L. Identification and characterization of Cynoglossus semilaevis microRNA response to Vibrio anguillarum infection through high-throughput sequencing. Dev. Comp. Immunol. 2014, 44, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.L.; Han, J.J.; Xu, T.J. Comparative analysis of the small RNA transcriptomes of miiuy croaker revealed microRNA-mediated regulation of TLR signaling pathway response to Vibrio anguillarum infection. Fish. Shellfish Immun. 2016, 52, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.C.; Zhang, J.; Sun, L. In-depth profiling and analysis of host and viral microRNAs in Japanese flounder (Paralichthys olivaceus) infected with megalocytivirus reveal involvement of microRNAs in host-virus interaction in teleost fish. BMC Genomics 2014, 15, 878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.R.; Hu, Y.H.; Jiang, S.; Sun, L. Global profiling and characterization of Japanese flounder (Paralichthys olivaceus) kidney microRNAs regulated by Edwardsiella tarda infection in a time-dependent fashion. Fish. Shellfish Immun. 2019, 93, 766–780. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Ning, X.H.; Guan, X.L.; Li, X.P.; Sun, L. Characterization of Streptococcus iniae-induced microRNA profiles in Paralichthys olivaceus and identification of pol-3p-10740_175 as a regulator of antibacterial immune response. Fish. Shellfish Immun. 2020, 98, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.H.; Zhang, B.C.; Zhou, H.Z.; Guan, X.L.; Sun, L. Edwardsiella tarda-induced miRNAs in a teleost host: Global profile and role in bacterial infection as revealed by integrative miRNA-mRNA analysis. Virulence 2017, 8, 1457–1464. [Google Scholar] [CrossRef] [Green Version]
- Valenzuela-Muñoz, V.; Novoa, B.; Figueras, A.; Gallardo-Escárate, C. Modulation of Atlantic salmon miRNome response to sea louse infestation. Dev. Comp. Immunol. 2017, 76, 380–391. [Google Scholar] [CrossRef] [Green Version]
- Andreassen, R.; Woldemariam, N.T.; Egeland, I.O.; Agafonov, O.; Sindre, H.; Hoyheim, B. Identification of differentially expressed Atlantic salmon miRNAs responding to salmonid alphavirus (SAV) infection. BMC Genomics 2017, 18, 349. [Google Scholar] [CrossRef]
- Seikai, T. Flounder culture and its challenges in Asia. Rev. Fish. Sci. 2002, 10, 421–432. [Google Scholar] [CrossRef]
- Egidius, E. Vibriosis: Pathogenicity and pathology. A review. Aquaculture 1987, 67, 15–28. [Google Scholar] [CrossRef]
- Zhang, Y.X.; Chen, S.L.; Liu, Y.G.; Sha, Z.X.; Liu, Z.J. Major histocompatibility complex class IIB allele polymorphism and its association with resistance/susceptibility to Vibrio anguillarumin in Japanese flounder (Paralichthys olivaceus). Mar. Biotechnol. 2006, 8, 600–610. [Google Scholar] [CrossRef]
- Shao, C.W.; Niu, Y.C.; Rastas, P.; Liu, Y.; Xie, Z.Y.; Li, H.D.; Wang, L.; Jiang, Y.; Tai, S.S.; Tian, Y.S.; et al. Genome-wide SNP identification for the construction of a high-resolution genetic map of Japanese flounder (Paralichthys olivaceus): Applications to QTL mapping of Vibrio anguillarum disease resistance and comparative genomic analysis. DNA Res. 2015, 22, 161–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, J.; Xu, H.S.; Wang, Y.; Tang, X.Q.; Sheng, X.Z.; Zhan, W.B. Protective efficacy of six immunogenic recombinant proteins of Vibrio anguillarum and evaluation them as vaccine candidate for flounder (Paralichthys olivaceus). Microb Pathog. 2017, 107, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Ning, X.H.; Sun, L. Gene network analysis reveals a core set of genes involved in the immune response of Japanese flounder (Paralichthys olivaceus) against Vibrio anguillarum infection. Fish. Shellfish Immun. 2020, 98, 800–809. [Google Scholar] [CrossRef] [PubMed]
- Zapata, A.; Diez, B.; Cejalvo, T.; Frías, C.G.; Cortés, A. Ontogeny of the immune system of fish. Fish. Shellfish Immun. 2006, 20, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Gene Ontology. Available online: http://geneontology.org (accessed on 7 June 2020).
- Kyoto Encyclopedia of Genes and Genomes. Available online: http://www.genome.jp/kegg/ (accessed on 7 June 2020).
- Mousavi, S.A.; Malerod, L.; Berg, T.; Kjeken, R. Clathrin-dependent endocytosis. Biochem. J. 2004, 377, 1–16. [Google Scholar] [CrossRef]
- Settembre, C.; Ballabio, A. Lysosomal adaptation: How the lysosome responds to external cues. CSH Perspect. Biol. 2014, 6, 176. [Google Scholar] [CrossRef] [Green Version]
- Toscano, M.A.; Ilarregui, J.M.; Bianco, G.A.; Campagna, L.; Croci, D.O.; Salatino, M.; Rabinovich, G.A. Dissecting the pathophysiologic role of endogenous lectins: Glycan-binding proteins with cytokine-like activity? Cytokine Growth Factor Rev. 2007, 18, 57–71. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J.; Murray, P.J. Cytokine signaling modules in inflammatory responses. Immunity 2008, 28, 477–487. [Google Scholar] [CrossRef] [Green Version]
- Weichhart, T.; Costantino, G.; Poglitsch, M.; Rosner, M.; Zeyda, M.; Stuhlmeier, K.M.; Kolbe, T.; Stulnig, T.M.; Horl, W.H.; Hengstschlager, M.; et al. The TSC-mTOR signaling pathway regulates the innate inflammatory response. Immunity 2008, 29, 565–577. [Google Scholar] [CrossRef] [Green Version]
- Tattoli, I.; Philpott, D.J.; Girardin, S.E. The bacterial and cellular determinants controlling the recruitment of mTOR to the Salmonella-containing vacuole. Biol. Open. 2012, 1, 1215–1225. [Google Scholar] [CrossRef] [Green Version]
- Gao, C.B.; Cai, X.; Fu, Q.; Yang, N.; Song, L.; Su, B.F.; Tan, F.H.; Liu, B.N.; Li, C. Dynamics of miRNA transcriptome in turbot (Scophthalmus maximus L.) intestine following Vibrio anguillarum infection. Mar. Biotechnol. 2019, 21, 550–564. [Google Scholar] [CrossRef] [PubMed]
- Herranz, H.; Cohen, S.M. MicroRNAs and gene regulatory networks: Managing the impact of noise in biological systems. Genes Dev. 2010, 24, 1339–1344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, E.X.; Obeng-Adjei, N.; Ying, Q.H.; Meertens, L.; Dragic, T.; Davey, R.A.; Ross, S.R. Mouse mammary tumor virus uses mouse but not human transferrin receptor 1 to reach a low pH compartment and infect cells. Virology 2008, 381, 230–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandenstein, L.; Schweizer, M.; Sedlacik, J.; Fiehler, J.; Storch, S. Lysosomal dysfunction and impaired autophagy in a novel mouse model deficient for the lysosomal membrane protein Cln7. Hum. Mol. Genet. 2015, 25, 777–791. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Qu, C.K. Protein tyrosine phosphatases in the JAK/STAT pathway. Front. Biosci. 2008, 13, 4925–4932. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.F.; Xia, J.Y.; Li, T.T.; Zhou, H.; Ouyang, W.; Hong, Z.P.; Ke, Y.H.; Qian, J.; Xu, F. Shp2 deficiency impairs the inflammatory response against Haemophilus influenzae by regulating macrophage polarization. J. Infect. Dis. 2016, 214, 625–633. [Google Scholar] [CrossRef] [Green Version]
- Dowd, G.C.; Bhalla, M.; Kean, B.; Thomas, R.R.; Ireton, K. Role of host type IA phosphoinositide 3-kinase pathway components in invasin-mediated internalization of Yersinia enterocolitica. Infect. Immun. 2016, 84, 1826–1841. [Google Scholar] [CrossRef] [Green Version]
- Cruz, R.; Pereira-Castro, I.; Almeida, M.T.; Moreira, A.; Cabanes, D.; Sousa, S. Epithelial keratins modulate cMet expression and signaling and promote InlB-mediated Listeria monocytogenes infection of hela cells. Front. Cell Infect. Microbiol. 2018, 8, 146. [Google Scholar] [CrossRef]
- Gong, G.Y.; Sha, Z.X.; Chen, S.L.; Li, C.; Yan, H.; Chen, Y.D.; Wang, T.Z. Expression profiling analysis of the microRNA response of Cynoglossus semilaevis to Vibrio anguillarum and other stimuli. Mar. Biotechnol. 2015, 17, 338–352. [Google Scholar] [CrossRef]
- Ordas, A.; Kanwal, Z.; Lindenberg, V.; Rougeot, J.; Mink, M.; Spaink, H.P.; Meijer, A.H. MicroRNA-146 function in the innate immune transcriptome response of zebrafish embryos to Salmonella typhimurium infection. BMC Genomics 2013, 14, 696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, C.B.; Fu, Q.; Yang, N.; Song, L.; Tan, F.H.; Zhu, J.J.; Li, C. Identification and expression profiling analysis of microRNAs in Nile tilapia (Oreochromis niloticus) in response to Streptococcus agalactiae infection. Fish. Shellfish Immun. 2019, 87, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.H.; Pan, C.Y.; Lin, M.C.; Hsieh, J.C.; Hui, C.F.; Chen, J.Y. In vivo screening of zebrafish microRNA responses to bacterial infection and their possible roles in regulating immune response genes after lipopolysaccharide stimulation. Fish Physiol. Biochem. 2012, 38, 1299–1310. [Google Scholar] [CrossRef] [PubMed]
- Guan, X.L.; Zhang, B.C.; Sun, L. pol-miR-194a of Japanese flounder (Paralichthys olivaceus) suppresses type I interferon response and facilitates Edwardsiella tarda infection. Fish. Shellfish Immun. 2019, 87, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Mayor, S.; Parton, R.G.; Donaldson, J.G. Clathrin-independent pathways of endocytosis. CSH Perspect. Biol. 2014, 6, a016758. [Google Scholar] [CrossRef] [Green Version]
- Tebar, F.; Sorkina, T.; Sorkin, A.; Ericsson, M.; Kirchhausen, T. Eps15 is a component of clathrin-coated pits and vesicles and is located at the rim of coated pits. J. Biol. Chem. 1996, 271, 28727–28730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, A.E.; Benmerah, A.; Guttman, J.A. Eps15 and Epsin1 are crucial for enteropathogenic Escherichia coli pedestal formation despite the absence of adaptor protein 2. J. Infect. Dis. 2011, 204, 695–703. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, I.M.; Cheng, G.F.; Wieland, S.; Volinia, S.; Croce, C.M.; Chisari, F.V.; David, M. Interferon modulation of cellular microRNAs as an antiviral mechanism. Nature 2007, 449, 919–922. [Google Scholar] [CrossRef]
- He, X.; Sun, Y.; Lei, N.H.; Fan, X.B.; Zhang, C.; Wang, Y.G.; Zheng, K.Y.; Zhang, D.M.; Pan, W.Q. MicroRNA-351 promotes schistosomiasis-induced hepatic fibrosis by targeting the vitamin D receptor. Proc. Natl. Acad. Sci. USA 2018, 115, 180–185. [Google Scholar] [CrossRef] [Green Version]
- Silva, W.D.; Santos, R.A.S.; Moraes, K.C.M. Mir-351-5p contributes to the establishment of a pro-inflammatory environment in the H9c2 cell line by repressing PTEN expression. Mol. Cell Biochem. 2015, 411, 1–9. [Google Scholar] [CrossRef]
- Garcia-Hernandez, V.; Sarmiento, N.; Sanchez-Bernal, C.; Covenas, R.; Hernandez-Hernandez, A.; Calvo, J.J.; Sanchez-Yague, J. Changes in the expression of LIMP-2 during cerulein-induced pancreatitis in rats: Effect of inhibition of leukocyte infiltration, cAMP and MAPKs early on in its development. Int. J. Biochem. Cell B 2016, 72, 109–117. [Google Scholar] [CrossRef]
- Tan, F.H.; Cao, M.; Ge, X.F.; Li, C.; Tian, M.Y.; Zhang, L.; Fu, Q.; Song, L.; Yang, N. Identification and initial functional characterization of lysosomal integral membrane protein type 2 (LIMP-2) in turbot (Scophthalmus maximus L.). Dev. Comp. Immunol. 2019, 99, 103412. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Tournier, C. The requirement of uncoordinated 51-like kinase 1 (ULK1) and ULK2 in the regulation of autophagy. Autophagy 2011, 7, 689–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saleiro, D.; Mehrotra, S.; Kroczynska, B.; Beauchamp, E.M.; Lisowski, P.; Majchrzak-Kita, B.; Bhagat, T.D.; Stein, B.L.; McMahon, B.; Altman, J.K.; et al. Central role of ULK1 in type I interferon signaling. Cell Rep. 2015, 11, 605–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.P.; Tan, S.R.; Yu, M.; Jundt, M.C.; Zhang, S.; Wu, M. Annexin A2 regulates autophagy in Pseudomonas aeruginosa infection through the Akt1-mTOR-ULK1/2 signaling pathway. J. Immunol. 2015, 195, 3901–3911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johns, J.L.; Borjesson, D.L. Downregulation of CXCL12 signaling and altered hematopoietic stem and progenitor cell trafficking in a murine model of acute Anaplasma phagocytophilum infection. Innate Immun. 2012, 18, 418–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, W.J.; Hu, Y.H.; Xiao, Z.Z.; Sun, L. Cloning and analysis of a ferritin subunit from turbot (Scophthalmus maximus). Fish. Shellfish Immun. 2010, 28, 829–836. [Google Scholar] [CrossRef]
- Chen, S.F.; Zhou, Y.Q.; Chen, Y.R.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- FastQC. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 7 June 2020).
- Rfam. Available online: http://rfam.xfam.org (accessed on 7 June 2020).
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST plus: Architecture and applications. BMC Bioinforma. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B. Aligning short sequencing reads with Bowtie. Curr. Protoc. Bioinform. 2010, 11, 7.1–7.14. [Google Scholar] [CrossRef]
- Chen, X.; Li, Q.B.; Wang, J.; Guo, X.; Jiang, X.R.; Ren, Z.J.; Weng, C.Y.; Sun, G.X.; Wang, X.Q.; Liu, Y.P.; et al. Identification and characterization of novel amphioxus microRNAs by Solexa sequencing. Genome Biol. 2009, 10, R78. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlidis, P. Using ANOVA for gene selection from microarray studies of the nervous system. Methods 2003, 31, 282–289. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads | Clean Tags | Clean Tag Ratio (%) | Known miRNAs | Novel miRNAs |
|---|---|---|---|---|---|
| C6-1 | 16,527,156 | 15,539,755 | 94.03 | 15,085,700 | 46,727 |
| C6-2 | 18,554,480 | 17,416,884 | 93.87 | 16,756,252 | 52,469 |
| C6-3 | 17,117,852 | 15,872,573 | 92.73 | 14,743,097 | 73,357 |
| V6-1 | 13,573,207 | 12,587,907 | 92.74 | 12,020,280 | 42,670 |
| V6-2 | 13,670,487 | 12,833,698 | 93.88 | 12,402,383 | 38,667 |
| V6-3 | 14,740,487 | 13,869,084 | 94.09 | 13,340,657 | 43,309 |
| C12-1 | 17,537,783 | 16,514,129 | 94.16 | 15,871,998 | 35,761 |
| C12-2 | 18,318,853 | 17,325,699 | 94.58 | 16,771,261 | 39,827 |
| C12-3 | 16,878,025 | 15,949,928 | 94.50 | 15,557,458 | 41,047 |
| V12-1 | 14,261,792 | 13,445,075 | 94.27 | 13,026,452 | 38,849 |
| V12-2 | 13,024,065 | 12,137,610 | 93.19 | 11,548,284 | 38,653 |
| V12-3 | 13,932,272 | 13,099,501 | 94.02 | 12,708,381 | 30,701 |
| C24-1 | 14,483,183 | 13,775,520 | 95.11 | 13,434,606 | 31,314 |
| C24-2 | 16,345,997 | 15,415,782 | 94.31 | 14,832,918 | 36,044 |
| C24-3 | 15,877,162 | 15,046,540 | 94.77 | 14,680,203 | 35,409 |
| V24-1 | 13,696,884 | 12,723,780 | 92.90 | 12,196,940 | 29,841 |
| V24-2 | 14,965,045 | 13,559,891 | 90.61 | 12,035,144 | 23,242 |
| V24-3 | 13,356,165 | 11,772,273 | 88.14 | 6,840,857 | 19,745 |
| MicroRNA | Primer | Sequence (5′ to 3′) |
|---|---|---|
| MiR-146-y | Stem-1 | GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACCAAAAG |
| M-146y-f | GCGCGTCTATGGGCTTAGTT | |
| M-146y-r | AGTGCAGGGTCCGAGGTATT | |
| MiR-221-x | Stem-2 | GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACACAGAA |
| M-221x-f | GCGACCTGGCATACAATGTAGAT | |
| M-221x-r | AGTGCAGGGTCCGAGGTATT | |
| MiR-29-x | Stem-3 | GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACTCTATG |
| M-29x-f | GCGCTGATTTCATCTGGTGA | |
| M-29x-r | AGTGCAGGGTCCGAGGTATT | |
| MiR-10400-x | Stem-4 | GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACCGTCCA |
| M-10400x-f | GCGGCGGCGGCGACTC | |
| M-10400x-r | AGTGCAGGGTCCGAGGTATT | |
| MiR-146-x | Stem-5 | GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACCCATCT |
| M-146x-f | CGCGTGAGAACTGAATTCCAT | |
| M-146x-r | AGTGCAGGGTCCGAGGTATT | |
| Pol-miR-n016-3p | Stem-6 | GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACCAAAAG |
| M-016-f | GCGCGATCTATGGGCTTAGTT | |
| M-016-r | AGTGCAGGGTCCGAGGTATT | |
| Pol-miR-n199-3p | Stem-7 | GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACGCCAGC |
| M-199-f | CGCGCAACACTGGTTTGTAA | |
| M-199-r | AGTGCAGGGTCCGAGGTATT | |
| Pol-miR-n127-3p | Stem-8 | GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACAGTGCA |
| M-127-f | CGCGCCTATGCTTGATTACT | |
| M-127-r | AGTGCAGGGTCCGAGGTATT | |
| Pol-miR-n273-5p | Stem-9 | GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACTGTTCA |
| M-273-f | CGCAGGACTTGACCCACATG | |
| M-273-r | AGTGCAGGGTCCGAGGTATT | |
| Pol-miR-n049-3p | Stem-10 | GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACAGCTAA |
| M-049-f | GCGCACCTACCATGTTAGCA | |
| M-049-r | AGTGCAGGGTCCGAGGTATT | |
| 5s | Stem-11 | CGGTCTCCCATCCAAGTA |
| 5s-f | CCATACCACCCTGAACAC | |
| 5s-r | CGGTCTCCCATCCAAGTA | |
| EDH2 | edh2-f | CCGCAAACTCAACCCTTTCG |
| edh2-r | GGAAGTCATAACCTCGGCTC | |
| EPS15 | eps15-f | CCAGCTTAGATGCAGATCCGT |
| eps15-r | ACTGGTCAGCCCCATTTGAC | |
| CLN7 | cln7-f | CATGTTCGCCTGGACAAGGA |
| cln7-r | GGACGATCCCCAACCCTTTG | |
| ARSB | arsb-f | GATTGGCTGCCTACCCTTGT |
| arsb-r | GAGGCAAACCCGTAGCTGAT | |
| CXCL12 | cxcl12-f | TCGTTCTACCCTCAACACGG |
| cxcl12-r | GCTCTTCAGTTTGGCAATGACT | |
| KDR | kdr-f | CAGCTCCACTATGACGACCC |
| kdr-r | CTGTGTGACCCTCCATGACG | |
| CREBBP | crebbp-f | GCTGTCACGCAGCTGTCTC |
| crebbp-r | TCCTCGGTCTCCATCTTGGT | |
| ULK1 | ulk1-f | GTGAGGACACCATTCGGGTT |
| ulk1-r | GAAGCCGAAGTCCGCAATCT | |
| PRKCA | prkca-f | CTTCAAGCCCAAAGTGTGTGG |
| prkca-r | TCTCATTCGGTGCTGCTGAG | |
| PIK3R3 | pik3r3-f | ACTGGTGGAATACAACGCCA |
| pik3r3-r | TCATCTGGATTTCCTGTGAGG | |
| TUBA | tuba-f | TGACATCACAAACGCCTGCTTC |
| tuba-r | GCACCACATCTCCACGGTACAG |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ning, X.; Sun, L. Micro-Transcriptome Analysis Reveals Immune-Related MicroRNA Regulatory Networks of Paralichthys olivaceus Induced by Vibrio anguillarum Infection. Int. J. Mol. Sci. 2020, 21, 4252. https://doi.org/10.3390/ijms21124252
Ning X, Sun L. Micro-Transcriptome Analysis Reveals Immune-Related MicroRNA Regulatory Networks of Paralichthys olivaceus Induced by Vibrio anguillarum Infection. International Journal of Molecular Sciences. 2020; 21(12):4252. https://doi.org/10.3390/ijms21124252
Chicago/Turabian StyleNing, Xianhui, and Li Sun. 2020. "Micro-Transcriptome Analysis Reveals Immune-Related MicroRNA Regulatory Networks of Paralichthys olivaceus Induced by Vibrio anguillarum Infection" International Journal of Molecular Sciences 21, no. 12: 4252. https://doi.org/10.3390/ijms21124252