Oxidative Stress and Thrombosis during Aging: The Roles of Oxidative Stress in RBCs in Venous Thrombosis


Abstract
:
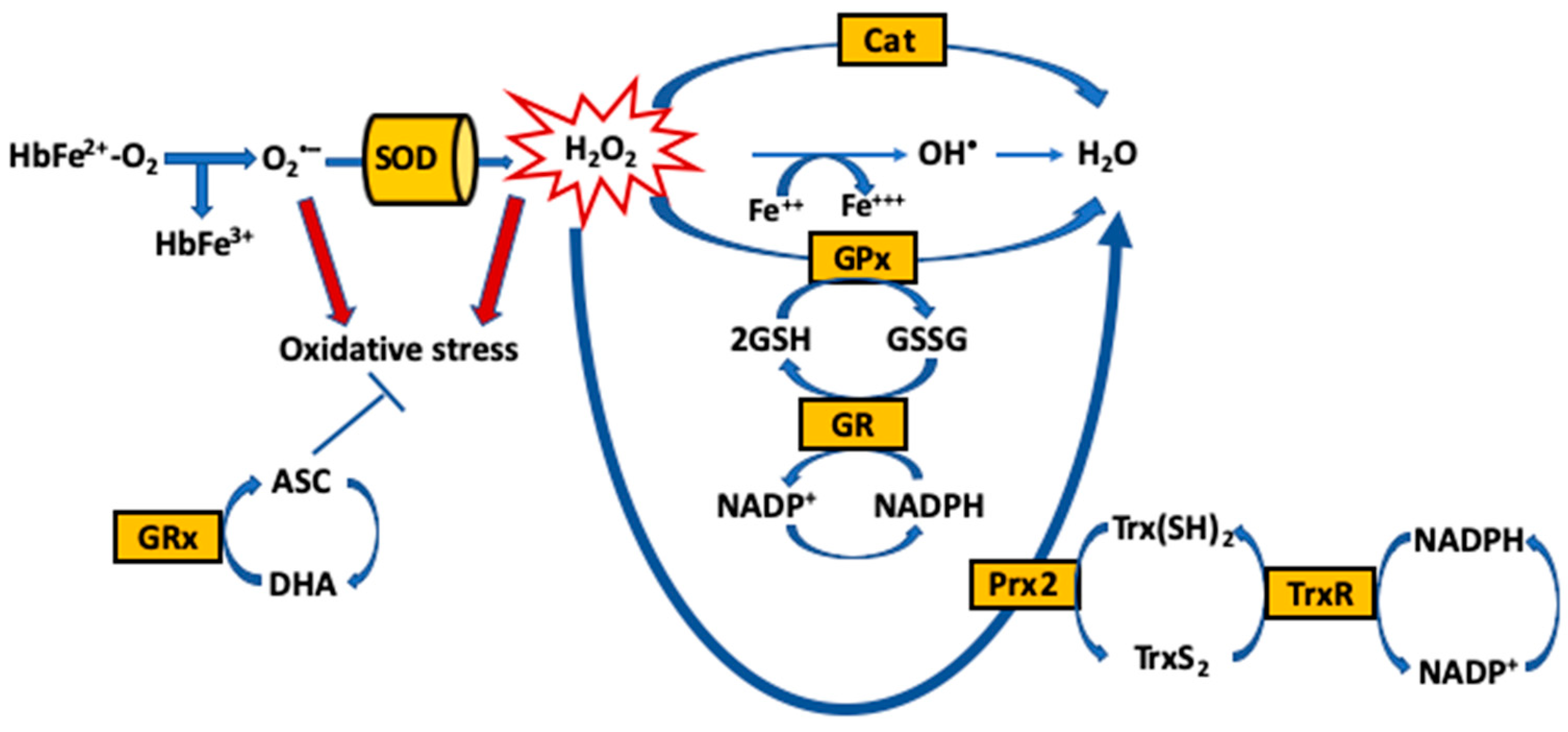
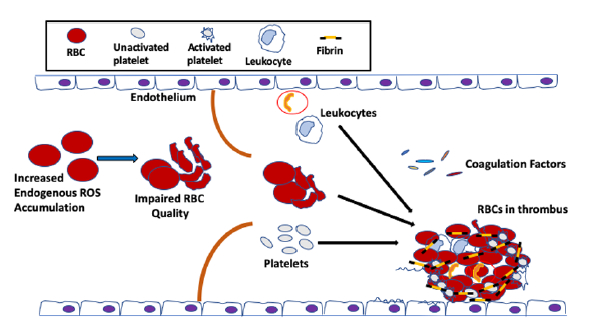{kind=link}
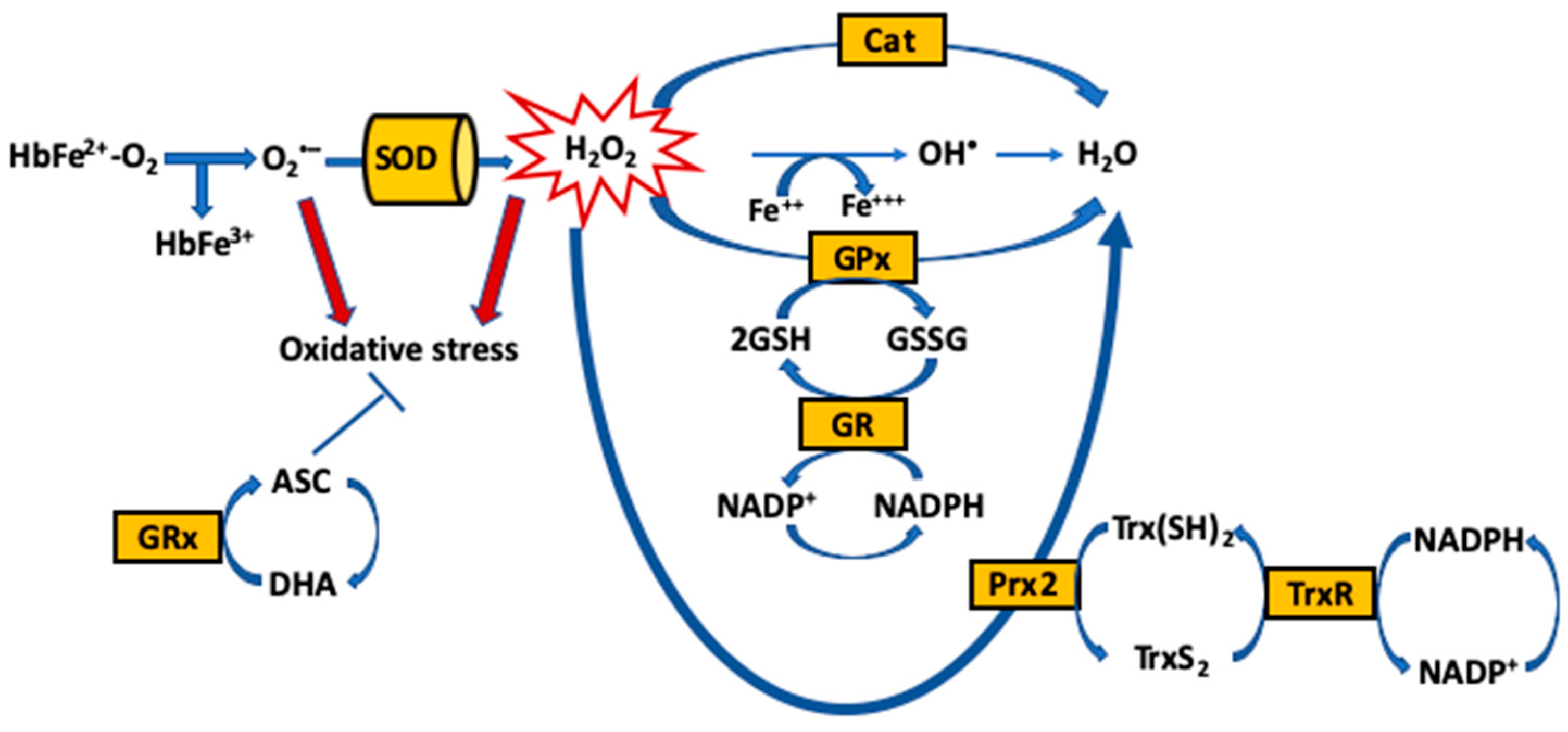{kind=link}
{kind=link}
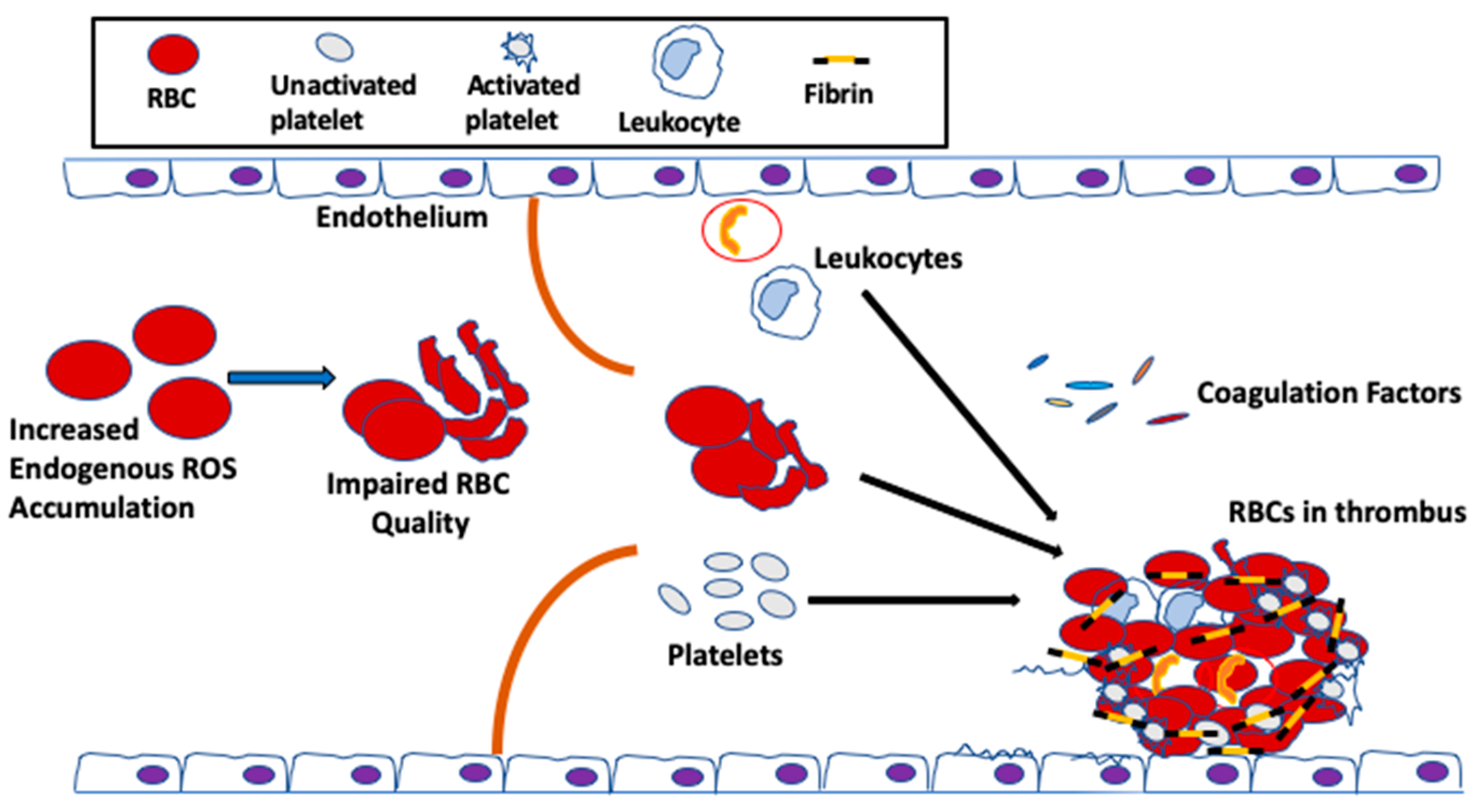{kind=link}
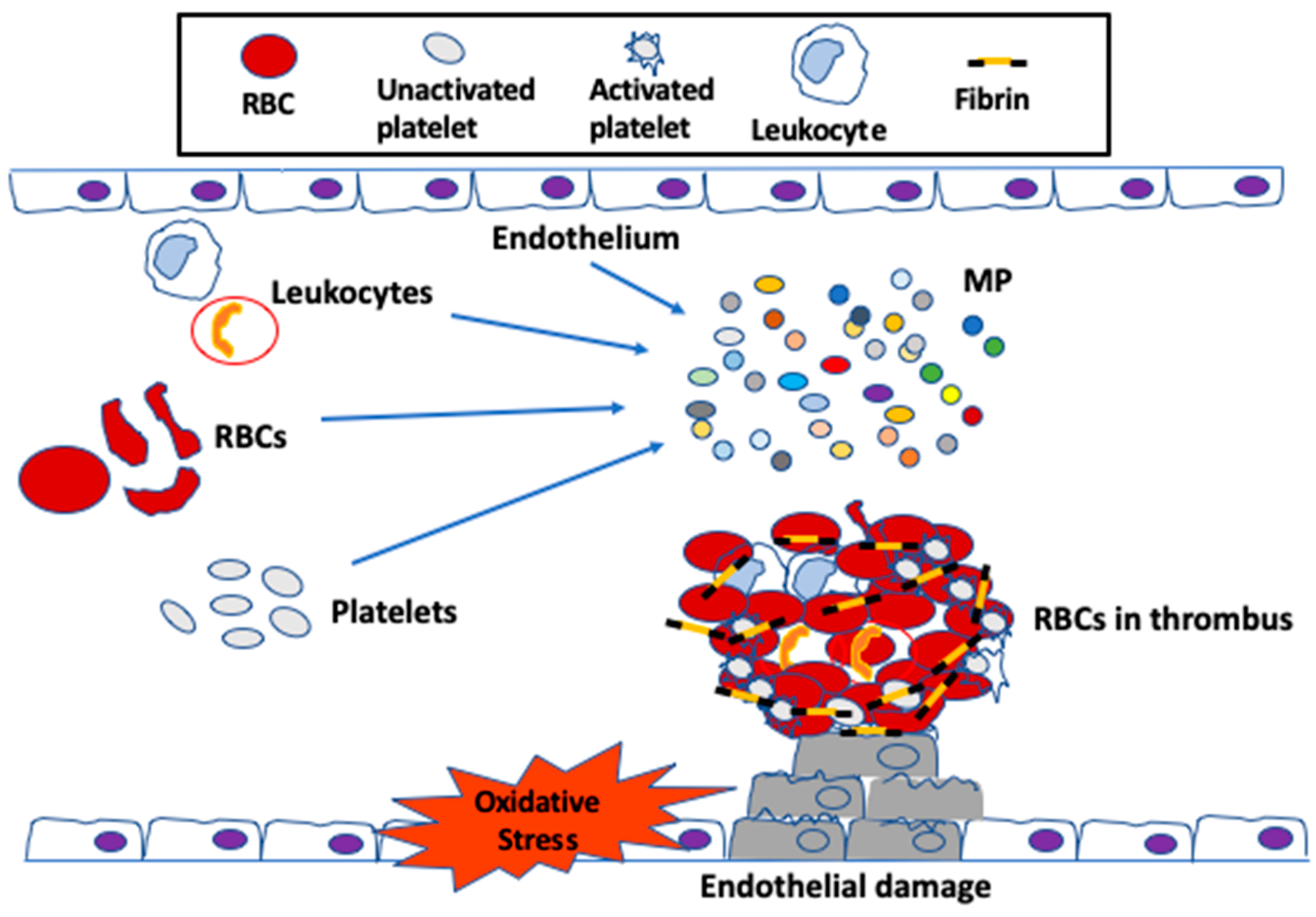
1. Introduction
2. Oxidative Stress Contribution to Venous Thrombosis during Aging
3. RBC Oxidative Stress and Its Effect on RBC Quality during Aging
4. Oxidative Stress in RBCs and Its Effect on Venous Thrombosis during Aging
5. RBCs as a Major Source of Oxidative Stress-Associated Thrombosis during Aging
6. Genetic Risk Factors and Venous Thrombosis
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Raskob, G.E.; Angchaisuksiri, P.; Blanco, A.N.; Buller, H.; Gallus, A.; Hunt, B.J.; Hylek, E.M.; Kakkar, A.; Konstantinides, S.V.; McCumber, M.; et al. Thrombosis: A major contributor to global disease burden. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2363–2371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacic, J.C.; Moreno, P.; Hachinski, V.; Nabel, E.G.; Fuster, V. Cellular senescence, vascular disease, and aging: Part 1 of a 2-part review. Circulation 2011, 123, 1650–1660. [Google Scholar] [CrossRef] [PubMed]
- Tsai, A.W.; Cushman, M.; Rosamond, W.D.; Heckbert, S.R.; Polak, J.F.; Folsom, A.R. Cardiovascular risk factors and venous thromboembolism incidence: The longitudinal investigation of thromboembolism etiology. Arch. Intern. Med. 2002, 162, 1182–1189. [Google Scholar] [CrossRef] [PubMed]
- Cushman, M.; Tsai, A.W.; White, R.H.; Heckbert, S.R.; Rosamond, W.D.; Enright, P.; Folsom, A.R. Deep vein thrombosis and pulmonary embolism in two cohorts: The longitudinal investigation of thromboembolism etiology. Am. J. Med. 2004, 117, 19–25. [Google Scholar] [CrossRef]
- Wakefield, T.W.; Strieter, R.M.; Wilke, C.A.; Kadell, A.M.; Wrobleski, S.K.; Burdick, M.D.; Schmidt, R.; Kunkel, S.L.; Greenfield, L.J. Venous thrombosis-associated inflammation and attenuation with neutralizing antibodies to cytokines and adhesion molecules. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 258–268. [Google Scholar] [CrossRef]
- Versteeg, H.H.; Heemskerk, J.W.; Levi, M.; Reitsma, P.H. New fundamentals in hemostasis. Physiol. Rev. 2013, 93, 327–358. [Google Scholar] [CrossRef] [Green Version]
- Hamada, T.; Kurachi, S.; Kurachi, K. Heterogeneous nuclear ribonucleoprotein A3 is the liver nuclear protein binding to age related increase element RNA of the factor IX gene. PLoS ONE 2010, 5, e12971. [Google Scholar] [CrossRef] [Green Version]
- Favaloro, E.J.; Franchini, M.; Lippi, G. Aging hemostasis: Changes to laboratory markers of hemostasis as we age—A narrative review. Semin. Thromb. Hemost. 2014, 40, 621–633. [Google Scholar] [CrossRef]
- Syed, F.A.; Ng, A.C. The pathophysiology of the aging skeleton. Curr. Osteoporos. Rep. 2010, 8, 235–240. [Google Scholar] [CrossRef]
- Sevitt, S. The structure and growth of valve-pocket thrombi in femoral veins. J. Clin. Pathol. 1974, 27, 517–528. [Google Scholar] [CrossRef] [Green Version]
- Darbousset, R.; Thomas, G.M.; Mezouar, S.; Frere, C.; Bonier, R.; Mackman, N.; Renne, T.; Dignat-George, F.; Dubois, C.; Panicot-Dubois, L. Tissue factor-positive neutrophils bind to injured endothelial wall and initiate thrombus formation. Blood 2012, 120, 2133–2143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rumley, A.; Emberson, J.R.; Wannamethee, S.G.; Lennon, L.; Whincup, P.H.; Lowe, G.D. Effects of older age on fibrin D-dimer, C-reactive protein, and other hemostatic and inflammatory variables in men aged 60-79 years. J. Thromb. Haemost. 2006, 4, 982–987. [Google Scholar] [CrossRef] [PubMed]
- Silva, G.C.; Abbas, M.; Khemais-Benkhiat, S.; Burban, M.; Ribeiro, T.P.; Toti, F.; Idris-Khodja, N.; Cortes, S.F.; Schini-Kerth, V.B. Replicative senescence promotes prothrombotic responses in endothelial cells: Role of NADPH oxidase- and cyclooxygenase-derived oxidative stress. Exp. Gerontol. 2017, 93, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Dayal, S.; Wilson, K.M.; Motto, D.G.; Miller, F.J., Jr.; Chauhan, A.K.; Lentz, S.R. Hydrogen peroxide promotes aging-related platelet hyperactivation and thrombosis. Circulation 2013, 127, 1308–1316. [Google Scholar] [CrossRef] [Green Version]
- Taddei, S.; Virdis, A.; Ghiadoni, L.; Salvetti, G.; Bernini, G.; Magagna, A.; Salvetti, A. Age-related reduction of NO availability and oxidative stress in humans. Hypertension 2001, 38, 274–279. [Google Scholar] [CrossRef]
- Space, S.L.; Lane, P.A.; Pickett, C.K.; Weil, J.V. Nitric oxide attenuates normal and sickle red blood cell adherence to pulmonary endothelium. Am. J. Hematol. 2000, 63, 200–204. [Google Scholar] [CrossRef]
- Loscalzo, J. Nitric oxide insufficiency, platelet activation, and arterial thrombosis. Circ. Res. 2001, 88, 756–762. [Google Scholar] [CrossRef] [Green Version]
- Chou, T.C.; Yen, M.H.; Li, C.Y.; Ding, Y.A. Alterations of nitric oxide synthase expression with aging and hypertension in rats. Hypertension 1998, 31, 643–648. [Google Scholar] [CrossRef] [Green Version]
- Tschudi, M.R.; Barton, M.; Bersinger, N.A.; Moreau, P.; Cosentino, F.; Noll, G.; Malinski, T.; Luscher, T.F. Effect of age on kinetics of nitric oxide release in rat aorta and pulmonary artery. J. Clin. Investig. 1996, 98, 899–905. [Google Scholar] [CrossRef]
- Miyazaki, H.; Matsuoka, H.; Cooke, J.P.; Usui, M.; Ueda, S.; Okuda, S.; Imaizumi, T. Endogenous nitric oxide synthase inhibitor: A novel marker of atherosclerosis. J. Cardiol. 1999, 33, 105–106. [Google Scholar] [CrossRef] [Green Version]
- Dimmeler, S.; Haendeler, J.; Nehls, M.; Zeiher, A.M. Suppression of apoptosis by nitric oxide via inhibition of interleukin-1beta-converting enzyme (ICE)-like and cysteine protease protein (CPP)-32-like proteases. J. Exp. Med. 1997, 185, 601–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mannick, J.B.; Miao, X.Q.; Stamler, J.S. Nitric oxide inhibits Fas-induced apoptosis. J. Biol. Chem. 1997, 272, 24125–24128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, J.; Haendeler, J.; Aicher, A.; Rossig, L.; Vasa, M.; Zeiher, A.M.; Dimmeler, S. Aging enhances the sensitivity of endothelial cells toward apoptotic stimuli: Important role of nitric oxide. Circ. Res. 2001, 89, 709–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bombeli, T.; Karsan, A.; Tait, J.F.; Harlan, J.M. Apoptotic vascular endothelial cells become procoagulant. Blood 1997, 89, 2429–2442. [Google Scholar] [CrossRef] [Green Version]
- Mackman, N.; Antoniak, S. Tissue factor and oxidative stress. Blood 2018, 131, 2094–2095. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Loscalzo, J. Regulation of tissue factor expression in human microvascular endothelial cells by nitric oxide. Circulation 2000, 101, 2144–2148. [Google Scholar] [CrossRef] [Green Version]
- Abugo, O.O.; Rifkind, J.M. Oxidation of hemoglobin and the enhancement produced by nitroblue tetrazolium. J. Biol. Chem. 1994, 269, 24845–24853. [Google Scholar]
- Alayash, A.I.; Patel, R.P.; Cashon, R.E. Redox reactions of hemoglobin and myoglobin: Biological and toxicological implications. Antioxid. Redox Signal. 2001, 3, 313–327. [Google Scholar] [CrossRef]
- Brownlee, N.R.; Huttner, J.J.; Panganamala, R.V.; Cornwell, D.G. Role of vitamin E in glutathione-induced oxidant stress: Methemoglobin, lipid peroxidation, and hemolysis. J. Lipid Res. 1977, 18, 635–644. [Google Scholar]
- George, A.; Pushkaran, S.; Konstantinidis, D.G.; Koochaki, S.; Malik, P.; Mohandas, N.; Zheng, Y.; Joiner, C.H.; Kalfa, T.A. Erythrocyte NADPH oxidase activity modulated by Rac GTPases, PKC, and plasma cytokines contributes to oxidative stress in sickle cell disease. Blood 2013, 121, 2099–2107. [Google Scholar] [CrossRef] [Green Version]
- Sonveaux, P.; Lobysheva, I.I.; Feron, O.; McMahon, T.J. Transport and peripheral bioactivities of nitrogen oxides carried by red blood cell hemoglobin: Role in oxygen delivery. Physiology 2007, 22, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, J.G.; Nagababu, E.; Rifkind, J.M. Red blood cell oxidative stress impairs oxygen delivery and induces red blood cell aging. Front. Physiol. 2014, 5, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagababu, E.; Mohanty, J.G.; Friedman, J.S.; Rifkind, J.M. Role of peroxiredoxin-2 in protecting RBCs from hydrogen peroxide-induced oxidative stress. Free Radic. Res. 2013, 47, 164–171. [Google Scholar] [CrossRef] [Green Version]
- Gonzales, R.; Auclair, C.; Voisin, E.; Gautero, H.; Dhermy, D.; Boivin, P. Superoxide dismutase, catalase, and glutathione peroxidase in red blood cells from patients with malignant diseases. Cancer Res. 1984, 44, 4137–4139. [Google Scholar] [PubMed]
- Nagababu, E.; Chrest, F.J.; Rifkind, J.M. Hydrogen-peroxide-induced heme degradation in red blood cells: The protective roles of catalase and glutathione peroxidase. Biochim. Biophys. Acta 2003, 1620, 211–217. [Google Scholar] [CrossRef]
- Lee, T.H.; Kim, S.U.; Yu, S.L.; Kim, S.H.; Park, D.S.; Moon, H.B.; Dho, S.H.; Kwon, K.S.; Kwon, H.J.; Han, Y.H.; et al. Peroxiredoxin II is essential for sustaining life span of erythrocytes in mice. Blood 2003, 101, 5033–5038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsantes, A.E.; Bonovas, S.; Travlou, A.; Sitaras, N.M. Redox imbalance, macrocytosis, and RBC homeostasis. Antioxid. Redox Signal. 2006, 8, 1205–1216. [Google Scholar] [CrossRef]
- Labunskyy, V.M.; Gladyshev, V.N. Role of reactive oxygen species-mediated signaling in aging. Antioxid. Redox Signal. 2013, 19, 1362–1372. [Google Scholar] [CrossRef] [Green Version]
- Cebe, T.; Atukeren, P.; Yanar, K.; Kuruc, A.I.; Ozan, T.; Kunbaz, A.; Sitar, M.E.; Mirmaroufizibandeh, R.; Aydin, S.; Cakatay, U. Oxidation scrutiny in persuaded aging and chronological aging at systemic redox homeostasis level. Exp. Gerontol. 2014, 57, 132–140. [Google Scholar] [CrossRef]
- The RANTTAS Investigators. A randomized trial of tirilazad mesylate in patients with acute stroke (RANTTAS). Stroke 1996, 27, 1453–1458. [Google Scholar] [CrossRef]
- Rizvi, S.I.; Maurya, P.K. Markers of oxidative stress in erythrocytes during aging in humans. Ann. N. Y. Acad. Sci. 2007, 1100, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Maurya, P.K.; Kumar, P.; Siddiqui, N.; Tripathi, P.; Rizvi, S.I. Age-associated changes in erythrocyte glutathione peroxidase activity: Correlation with total antioxidant potential. Indian J. Biochem. Biophys. 2010, 47, 319–321. [Google Scholar] [PubMed]
- Kiefer, C.R.; Snyder, L.M. Oxidation and erythrocyte senescence. Curr. Opin. Hematol. 2000, 7, 113–116. [Google Scholar] [CrossRef] [PubMed]
- Rifkind, J.M.; Ajmani, R.S.; Heim, J. Impaired hemorheology in the aged associated with oxidative stress. Adv. Exp. Med. Biol. 1997, 428, 7–13. [Google Scholar] [CrossRef]
- Nagababu, E.; Rifkind, J.M. Formation of fluorescent heme degradation products during the oxidation of hemoglobin by hydrogen peroxide. Biochem. Biophys. Res. Commun. 1998, 247, 592–596. [Google Scholar] [CrossRef]
- Aoshiba, K.; Nakajima, Y.; Yasui, S.; Tamaoki, J.; Nagai, A. Red blood cells inhibit apoptosis of human neutrophils. Blood 1999, 93, 4006–4010. [Google Scholar] [CrossRef]
- Hsu, C.P.; Lin, C.H.; Kuo, C.Y. Endothelial-cell inflammation and damage by reactive oxygen species are prevented by propofol via ABCA1-mediated cholesterol efflux. Int. J. Med. Sci. 2018, 15, 978–985. [Google Scholar] [CrossRef] [Green Version]
- Rajagopalan, S.; Meng, X.P.; Ramasamy, S.; Harrison, D.G.; Galis, Z.S. Reactive oxygen species produced by macrophage-derived foam cells regulate the activity of vascular matrix metalloproteinases in vitro. Implications for atherosclerotic plaque stability. J. Clin. Investig. 1996, 98, 2572–2579. [Google Scholar] [CrossRef] [Green Version]
- Tian, L.; Cai, Q.; Wei, H. Alterations of antioxidant enzymes and oxidative damage to macromolecules in different organs of rats during aging. Free Radic. Biol. Med. 1998, 24, 1477–1484. [Google Scholar] [CrossRef]
- Fujino, T.; Kojima, M.; Beppu, M.; Kikugawa, K.; Yasuda, H.; Takahashi, K. Identification of the cleavage sites of oxidized protein that are susceptible to oxidized protein hydrolase (OPH) in the primary and tertiary structures of the protein. J. Biochem. 2000, 127, 1087–1093. [Google Scholar] [CrossRef]
- Fujino, T.; Watanabe, K.; Beppu, M.; Kikugawa, K.; Yasuda, H. Identification of oxidized protein hydrolase of human erythrocytes as acylpeptide hydrolase. Biochim. Biophys. Acta 2000, 1478, 102–112. [Google Scholar] [CrossRef]
- Niki, E. Biomarkers of lipid peroxidation in clinical material. Biochim. Biophys. Acta 2014, 1840, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Repka, T.; Hebbel, R.P. Hydroxyl radical formation by sickle erythrocyte membranes: Role of pathologic iron deposits and cytoplasmic reducing agents. Blood 1991, 78, 2753–2758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, D.; Rizvi, S.I. A critical period in lifespan of male rats coincides with increased oxidative stress. Arch. Gerontol. Geriatr. 2014, 58, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Allagui, M.S.; Feriani, A.; Saoudi, M.; Badraoui, R.; Bouoni, Z.; Nciri, R.; Murat, J.C.; Elfeki, A. Effects of melatonin on aluminium-induced neurobehavioral and neurochemical changes in aging rats. Food Chem. Toxicol. 2014, 70, 84–93. [Google Scholar] [CrossRef]
- Pizzimenti, S.; Ciamporcero, E.; Daga, M.; Pettazzoni, P.; Arcaro, A.; Cetrangolo, G.; Minelli, R.; Dianzani, C.; Lepore, A.; Gentile, F.; et al. Interaction of aldehydes derived from lipid peroxidation and membrane proteins. Front. Physiol. 2013, 4, 242. [Google Scholar] [CrossRef] [Green Version]
- Mandal, D.; Baudin-Creuza, V.; Bhattacharyya, A.; Pathak, S.; Delaunay, J.; Kundu, M.; Basu, J. Caspase 3-mediated proteolysis of the N-terminal cytoplasmic domain of the human erythroid anion exchanger 1 (band 3). J. Biol. Chem. 2003, 278, 52551–52558. [Google Scholar] [CrossRef] [Green Version]
- Clementi, M.E.; Giardina, B.; Colucci, D.; Galtieri, A.; Misiti, F. Amyloid-beta peptide affects the oxygen dependence of erythrocyte metabolism: A role for caspase 3. Int. J. Biochem. Cell Biol. 2007, 39, 727–735. [Google Scholar] [CrossRef]
- Grey, J.L.; Kodippili, G.C.; Simon, K.; Low, P.S. Identification of contact sites between ankyrin and band 3 in the human erythrocyte membrane. Biochemistry 2012, 51, 6838–6846. [Google Scholar] [CrossRef] [Green Version]
- Samaja, M.; Rubinacci, A.; Motterlini, R.; De Ponti, A.; Portinaro, N. Red cell aging and active calcium transport. Exp. Gerontol. 1990, 25, 279–286. [Google Scholar] [CrossRef] [Green Version]
- Ney, P.A.; Christopher, M.M.; Hebbel, R.P. Synergistic effects of oxidation and deformation on erythrocyte monovalent cation leak. Blood 1990, 75, 1192–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barodka, V.; Mohanty, J.G.; Mustafa, A.K.; Santhanam, L.; Nyhan, A.; Bhunia, A.K.; Sikka, G.; Nyhan, D.; Berkowitz, D.E.; Rifkind, J.M. Nitroprusside inhibits calcium-induced impairment of red blood cell deformability. Transfusion 2014, 54, 434–444. [Google Scholar] [CrossRef] [PubMed]
- Shan, G.; Yang, F.; Zhou, L.; Tang, T.; Okoro, E.U.; Yang, H.; Guo, Z. Increase in Blood Glutathione and Erythrocyte Proteins Related to Glutathione Generation, Reduction and Utilization in African-American Old Women with Diabetes. J. Sci. Technol. Environ. 2015, 5, 3000251. [Google Scholar] [PubMed]
- Carroll, J.; Raththagala, M.; Subasinghe, W.; Baguzis, S.; D’Amico Oblak, T.; Root, P.; Spence, D. An altered oxidant defense system in red blood cells affects their ability to release nitric oxide-stimulating ATP. Mol. Biosyst. 2006, 2, 305–311. [Google Scholar] [CrossRef]
- Prall, Y.G.; Gambhir, K.K.; Ampy, F.R. Acetylcholinesterase: An enzymatic marker of human red blood cell aging. Life Sci. 1998, 63, 177–184. [Google Scholar] [CrossRef]
- Herz, F.; Kaplan, E. A review: Human erythrocyte acetylcholinesterase. Pediatric Res. 1973, 7, 204–214. [Google Scholar] [CrossRef] [Green Version]
- Aloni, B.L.A. Acetycholinesterase as a probe for erythrocyte-membrane intactness. Biochim. Biophys. Acta 1974, 339, 359–366. [Google Scholar] [CrossRef]
- Furchgott, R.F.; Vanhoutte, P.M. Endothelium-derived relaxing and contracting factors. FASEB J. 1989, 3, 2007–2018. [Google Scholar] [CrossRef]
- Zhou, Y.; Varadharaj, S.; Zhao, X.; Parinandi, N.; Flavahan, N.A.; Zweier, J.L. Acetylcholine causes endothelium-dependent contraction of mouse arteries. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H1027–H1032. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Paik, H.D.; Yoon, Y.C.; Park, E. Whey protein inhibits iron overload-induced oxidative stress in rats. J. Nutr. Sci. Vitam. 2013, 59, 198–205. [Google Scholar] [CrossRef] [Green Version]
- Kiffin, R.; Christian, C.; Knecht, E.; Cuervo, A.M. Activation of chaperone-mediated autophagy during oxidative stress. Mol. Biol. Cell 2004, 15, 4829–4840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortensen, M.; Ferguson, D.J.; Edelmann, M.; Kessler, B.; Morten, K.J.; Komatsu, M.; Simon, A.K. Loss of autophagy in erythroid cells leads to defective removal of mitochondria and severe anemia in vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 832–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erwig, L.P.; Henson, P.M. Immunological consequences of apoptotic cell phagocytosis. Am. J. Pathol. 2007, 171, 2–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napoli, I.; Neumann, H. Microglial clearance function in health and disease. Neuroscience 2009, 158, 1030–1038. [Google Scholar] [CrossRef]
- Li, W. Eat-me signals: Keys to molecular phagocyte biology and “appetite” control. J. Cell. Physiol. 2012, 227, 1291–1297. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, A.; Agrawal, S.; Cao, J.N.; Su, H.; Osann, K.; Gupta, S. Altered innate immune functioning of dendritic cells in elderly humans: A role of phosphoinositide 3-kinase-signaling pathway. J. Immunol. 2007, 178, 6912–6922. [Google Scholar] [CrossRef] [Green Version]
- Aprahamian, T.; Takemura, Y.; Goukassian, D.; Walsh, K. Ageing is associated with diminished apoptotic cell clearance in vivo. Clin. Exp. Immunol. 2008, 152, 448–455. [Google Scholar] [CrossRef]
- Duke, W.W. The relation of blood platelets to hemorrhagic disease. J. Am. Med. Assoc. 1983, 250, 1201–1209. [Google Scholar] [CrossRef]
- Goel, M.S.; Diamond, S.L. Adhesion of normal erythrocytes at depressed venous shear rates to activated neutrophils, activated platelets, and fibrin polymerized from plasma. Blood 2002, 100, 3797–3803. [Google Scholar] [CrossRef]
- Barr, J.D.; Chauhan, A.K.; Schaeffer, G.V.; Hansen, J.K.; Motto, D.G. Red blood cells mediate the onset of thrombosis in the ferric chloride murine model. Blood 2013, 121, 3733–3741. [Google Scholar] [CrossRef]
- Litvinov, R.I.; Weisel, J.W. Role of red blood cells in haemostasis and thrombosis. ISBT Sci. Ser. 2017, 12, 176–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Ami, R.; Barshtein, G.; Mardi, T.; Deutch, V.; Elkayam, O.; Yedgar, S.; Berliner, S. A synergistic effect of albumin and fibrinogen on immunoglobulin-induced red blood cell aggregation. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H2663–H2669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spring, F.A.; Parsons, S.F.; Ortlepp, S.; Olsson, M.L.; Sessions, R.; Brady, R.L.; Anstee, D.J. Intercellular adhesion molecule-4 binds alpha(4)beta(1) and alpha(V)-family integrins through novel integrin-binding mechanisms. Blood 2001, 98, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Hermand, P.; Gane, P.; Huet, M.; Jallu, V.; Kaplan, C.; Sonneborn, H.H.; Cartron, J.P.; Bailly, P. Red cell ICAM-4 is a novel ligand for platelet-activated alpha IIbbeta 3 integrin. J. Biol. Chem. 2003, 278, 4892–4898. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, F.A.; Connell, S.; Miltenberger-Miltenyi, G.; Pereira, S.V.; Tavares, A.; Ariens, R.A.; Santos, N.C. Atomic force microscopy-based molecular recognition of a fibrinogen receptor on human erythrocytes. ACS Nano 2010, 4, 4609–4620. [Google Scholar] [CrossRef]
- Austin, H.; Key, N.S.; Benson, J.M.; Lally, C.; Dowling, N.F.; Whitsett, C.; Hooper, W.C. Sickle cell trait and the risk of venous thromboembolism among blacks. Blood 2007, 110, 908–912. [Google Scholar] [CrossRef]
- Helley, D.; Eldor, A.; Girot, R.; Ducrocq, R.; Guillin, M.C.; Bezeaud, A. Increased procoagulant activity of red blood cells from patients with homozygous sickle cell disease and beta-thalassemia. Thromb. Haemost. 1996, 76, 322–327. [Google Scholar]
- Helley, D.; Girot, R.; Guillin, M.C.; Bezeaud, A. Sickle cell disease: Relation between procoagulant activity of red blood cells from different phenotypes and in vivo blood coagulation activation. Br. J. Haematol. 1997, 99, 268–272. [Google Scholar] [CrossRef] [Green Version]
- Schafer, A.I. Bleeding and thrombosis in the myeloproliferative disorders. Blood 1984, 64, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Peduzzi, M.; Codeluppi, L.; Poggi, M.; Baraldi, P. Abnormal blood viscosity and erythrocyte deformability in retinal vein occlusion. Am. J. Ophthalmol. 1983, 96, 399–400. [Google Scholar] [CrossRef]
- Martinez, M.; Vaya, A.; Labios, M.; Gabriel, F.; Guiral, V.; Aznar, J. The effect of long-term treatment with hypotensive drugs on blood viscosity and erythrocyte deformability in patients with essential arterial hypertension. Clin. Hemorheol. Microcirc. 1997, 17, 193–198. [Google Scholar] [PubMed]
- Hayakawa, M.; Kuzuya, F. Effects of ticlopidine on erythrocyte aggregation in thrombotic disorders. Angiology 1991, 42, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Garg, G.; Singh, S.; Rizvi, S.I. Synergistic effect of rapamycin and metformin against age-dependent oxidative stress in rat erythrocyte. Rejuvenation Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Singh, S.; Singh, A.K.; Rizvi, S.I. Pomegranate (Punica granatum) peel extract provides protection against mercuric chloride-induced oxidative stress in Wistar strain rats. Pharm. Biol. 2013, 51, 441–446. [Google Scholar] [CrossRef]
- Holland, J.A.; Meyer, J.W.; Chang, M.M.; O’Donnell, R.W.; Johnson, D.K.; Ziegler, L.M. Thrombin stimulated reactive oxygen species production in cultured human endothelial cells. Endothelium 1998, 6, 113–121. [Google Scholar] [CrossRef]
- Kovalski, N.N.; de Lamirande, E.; Gagnon, C. Reactive oxygen species generated by human neutrophils inhibit sperm motility: Protective effect of seminal plasma and scavengers. Fertil Steril 1992, 58, 809–816. [Google Scholar] [CrossRef]
- MacKinney, A.; Woska, E.; Spasojevic, I.; Batinic-Haberle, I.; Zennadi, R. Disrupting the vicious cycle created by NOX activation in sickle erythrocytes exposed to hypoxia/reoxygenation prevents adhesion and vasoocclusion. Redox Biol. 2019, 101097. [Google Scholar] [CrossRef]
- Thamilarasan, M.; Estupinan, R.; Batinic-Haberle, I.; Zennadi, R. Mn porphyrins as a novel treatment targeting sickle cell NOXs to reverse and prevent acute vaso-occlusion in vivo. Blood Adv. 2020, 4, 2372–2386. [Google Scholar] [CrossRef]
- Banerjee, T.; Kuypers, F.A. Reactive oxygen species and phosphatidylserine externalization in murine sickle red cells. Br. J. Haematol. 2004, 124, 391–402. [Google Scholar] [CrossRef]
- Setty, B.N.; Kulkarni, S.; Stuart, M.J. Role of erythrocyte phosphatidylserine in sickle red cell-endothelial adhesion. Blood 2002, 99, 1564–1571. [Google Scholar] [CrossRef] [Green Version]
- Gayen Betal, S.; Setty, B.N. Phosphatidylserine-positive erythrocytes bind to immobilized and soluble thrombospondin-1 via its heparin-binding domain. Transl. Res. J. Lab. Clin. Med. 2008, 152, 165–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, R.S.; Tanaka, Y.; Fidler, I.J.; Chiu, D.T.; Lubin, B.; Schroit, A.J. Increased adherence of sickled and phosphatidylserine-enriched human erythrocytes to cultured human peripheral blood monocytes. J. Clin. Investig. 1985, 75, 1965–1972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semeraro, F.; Ammollo, C.T.; Esmon, N.L.; Esmon, C.T. Histones induce phosphatidylserine exposure and a procoagulant phenotype in human red blood cells. J. Thromb. Haemost. 2014, 12, 1697–1702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wautier, M.P.; Heron, E.; Picot, J.; Colin, Y.; Hermine, O.; Wautier, J.L. Red blood cell phosphatidylserine exposure is responsible for increased erythrocyte adhesion to endothelium in central retinal vein occlusion. J. Thromb. Haemost. 2011, 9, 1049–1055. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Xie, R.; Yu, C.; Wang, Q.; Shi, F.; Yao, C.; Xie, R.; Zhou, J.; Gilbert, G.E.; Shi, J. Procoagulant activity of erythrocytes and platelets through phosphatidylserine exposure and microparticles release in patients with nephrotic syndrome. Thromb. Haemost. 2012, 107, 681–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whelihan, M.F.; Zachary, V.; Orfeo, T.; Mann, K.G. Prothrombin activation in blood coagulation: The erythrocyte contribution to thrombin generation. Blood 2012, 120, 3837–3845. [Google Scholar] [CrossRef] [Green Version]
- Bonomini, M.; Sirolli, V.; Merciaro, G.; Antidormi, T.; Di Liberato, L.; Brummer, U.; Papponetti, M.; Cappelli, P.; Di Gregorio, P.; Arduini, A. Red blood cells may contribute to hypercoagulability in uraemia via enhanced surface exposure of phosphatidylserine. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc. Eur. Ren. Assoc. 2005, 20, 361–366. [Google Scholar] [CrossRef] [Green Version]
- Weiss, E.; Rees, D.C.; Gibson, J.S. Role of calcium in phosphatidylserine externalisation in red blood cells from sickle cell patients. Anemia 2011, 2011, 379894. [Google Scholar] [CrossRef] [Green Version]
- Kamp, D.; Sieberg, T.; Haest, C.W. Inhibition and stimulation of phospholipid scrambling activity. Consequences for lipid asymmetry, echinocytosis, and microvesiculation of erythrocytes. Biochemistry 2001, 40, 9438–9446. [Google Scholar] [CrossRef]
- Gordeeva, A.V.; Zvyagilskaya, R.A.; Labas, Y.A. Cross-talk between reactive oxygen species and calcium in living cells. Biochemistry 2003, 68, 1077–1080. [Google Scholar] [CrossRef]
- Gorlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bookchin, R.M.; Lew, V.L.; Roth, E.F., Jr. Elevated red cell calcium: Innocent bystander or kiss of death? Prog. Clin. Biol. Res. 1985, 195, 369–380. [Google Scholar] [PubMed]
- Clark, M.R. Senescence of red blood cells: Progress and problems. Physiol. Rev. 1988, 68, 503–554. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.J.; Romero, E.A. The role of calcium metabolism in human red blood cell ageing: A proposal. Blood Cells Mol. Dis. 1999, 25, 9–19. [Google Scholar] [CrossRef]
- Makhro, A.; Hanggi, P.; Goede, J.S.; Wang, J.; Bruggemann, A.; Gassmann, M.; Schmugge, M.; Kaestner, L.; Speer, O.; Bogdanova, A. N-methyl-D-aspartate receptors in human erythroid precursor cells and in circulating red blood cells contribute to the intracellular calcium regulation. Am. J. Physiol. Cell Physiol. 2013, 305, C1123–C1138. [Google Scholar] [CrossRef] [Green Version]
- Friederichs, E.; Meiselman, H.J. Effects of calcium permeabilization on RBC rheologic behavior. Biorheology 1994, 31, 207–215. [Google Scholar] [CrossRef]
- Steffen, P.; Jung, A.; Nguyen, D.B.; Muller, T.; Bernhardt, I.; Kaestner, L.; Wagner, C. Stimulation of human red blood cells leads to Ca2+-mediated intercellular adhesion. Cell Calcium 2011, 50, 54–61. [Google Scholar] [CrossRef] [Green Version]
- Kaestner, L.; Steffen, P.; Nguyen, D.B.; Wang, J.; Wagner-Britz, L.; Jung, A.; Wagner, C.; Bernhardt, I. Lysophosphatidic acid induced red blood cell aggregation in vitro. Bioelectrochemistry 2012, 87, 89–95. [Google Scholar] [CrossRef]
- Willekens, F.L.; Werre, J.M.; Groenen-Dopp, Y.A.; Roerdinkholder-Stoelwinder, B.; de Pauw, B.; Bosman, G.J. Erythrocyte vesiculation: A self-protective mechanism? Br. J. Haematol. 2008, 141, 549–556. [Google Scholar] [CrossRef]
- Willekens, F.L.; Roerdinkholder-Stoelwinder, B.; Groenen-Dopp, Y.A.; Bos, H.J.; Bosman, G.J.; van den Bos, A.G.; Verkleij, A.J.; Werre, J.M. Hemoglobin loss from erythrocytes in vivo results from spleen-facilitated vesiculation. Blood 2003, 101, 747–751. [Google Scholar] [CrossRef] [Green Version]
- Diamant, M.; Tushuizen, M.E.; Sturk, A.; Nieuwland, R. Cellular microparticles: New players in the field of vascular disease? Eur. J. Clin. Investig. 2004, 34, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Rubin, O.; Delobel, J.; Prudent, M.; Lion, N.; Kohl, K.; Tucker, E.I.; Tissot, J.D.; Angelillo-Scherrer, A. Red blood cell-derived microparticles isolated from blood units initiate and propagate thrombin generation. Transfusion 2013, 53, 1744–1754. [Google Scholar] [CrossRef] [PubMed]
- Schifferli, J.A. Microvesicles are messengers. Semin. Immunopathol. 2011, 33, 393–394. [Google Scholar] [CrossRef]
- Tissot, J.D.; Rubin, O.; Canellini, G. Analysis and clinical relevance of microparticles from red blood cells. Curr. Opin. Hematol. 2010, 17, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Bosman, G.J. Erythrocyte aging in sickle cell disease. Cell Mol. Biol. 2004, 50, 81–86. [Google Scholar] [PubMed]
- van Beers, E.J.; Schaap, M.C.; Berckmans, R.J.; Nieuwland, R.; Sturk, A.; van Doormaal, F.F.; Meijers, J.C.; Biemond, B.J.; group, C.S. Circulating erythrocyte-derived microparticles are associated with coagulation activation in sickle cell disease. Haematologica 2009, 94, 1513–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westerman, M.; Pizzey, A.; Hirschman, J.; Cerino, M.; Weil-Weiner, Y.; Ramotar, P.; Eze, A.; Lawrie, A.; Purdy, G.; Mackie, I.; et al. Microvesicles in haemoglobinopathies offer insights into mechanisms of hypercoagulability, haemolysis and the effects of therapy. Br. J. Haematol. 2008, 142, 126–135. [Google Scholar] [CrossRef]
- Setty, B.N.; Rao, A.K.; Stuart, M.J. Thrombophilia in sickle cell disease: The red cell connection. Blood 2001, 98, 3228–3233. [Google Scholar] [CrossRef] [Green Version]
- Horne, M.K., 3rd; Cullinane, A.M.; Merryman, P.K.; Hoddeson, E.K. The effect of red blood cells on thrombin generation. Br. J. Haematol. 2006, 133, 403–408. [Google Scholar] [CrossRef]
- Van Der Meijden, P.E.; Van Schilfgaarde, M.; Van Oerle, R.; Renne, T.; ten Cate, H.; Spronk, H.M. Platelet- and erythrocyte-derived microparticles trigger thrombin generation via factor XIIa. J. Thromb. Haemost. 2012, 10, 1355–1362. [Google Scholar] [CrossRef]
- Aleman, M.M.; Byrnes, J.R.; Wang, J.G.; Tran, R.; Lam, W.A.; Di Paola, J.; Mackman, N.; Degen, J.L.; Flick, M.J.; Wolberg, A.S. Factor XIII activity mediates red blood cell retention in venous thrombi. J. Clin. Investig. 2014, 124, 3590–3600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atici, A.G.; Kayhan, S.; Aydin, D.; Yilmaz, Y.A. Plasma viscosity levels in pulmonary thromboembolism. Clin. Hemorheol. Microcirc. 2013, 55, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Pawloski, J.R.; Hess, D.T.; Stamler, J.S. Export by red blood cells of nitric oxide bioactivity. Nature 2001, 409, 622–626. [Google Scholar] [CrossRef]
- Baskurt, O.K.; Yalcin, O.; Ozdem, S.; Armstrong, J.K.; Meiselman, H.J. Modulation of endothelial nitric oxide synthase expression by red blood cell aggregation. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H222–H229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chigaev, A.; Smagley, Y.; Sklar, L.A. Nitric oxide/cGMP pathway signaling actively down-regulates alpha4beta1-integrin affinity: An unexpected mechanism for inducing cell de-adhesion. BMC Immunol. 2011, 12, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gromotowicz-Poplawska, A.; Kloza, M.; Aleksiejczuk, M.; Marcinczyk, N.; Szemraj, J.; Kozlowska, H.; Chabielska, E. Nitric oxide as a modulator in platelet- and endothelium-dependent antithrombotic effect of eplerenone in diabetic rats. J. Physiol. Pharmacol. 2019, 70. [Google Scholar] [CrossRef]
- Xu, D.; Kaliviotis, E.; Munjiza, A.; Avital, E.; Ji, C.; Williams, J. Large scale simulation of red blood cell aggregation in shear flows. J. Biomech. 2013, 46, 1810–1817. [Google Scholar] [CrossRef]
- Ben-Ami, R.; Sheinman, G.; Yedgar, S.; Eldor, A.; Roth, A.; Berliner, A.S.; Barshtein, G. Thrombolytic therapy reduces red blood cell aggregation in plasma without affecting intrinsic aggregability. Thromb. Res. 2002, 105, 487–492. [Google Scholar] [CrossRef]
- Tokarev, A.A.; Butylin, A.A.; Ataullakhanov, F.I. Platelet adhesion from shear blood flow is controlled by near-wall rebounding collisions with erythrocytes. Biophys. J. 2011, 100, 799–808. [Google Scholar] [CrossRef] [Green Version]
- Joist, J.H.; Bauman, J.E.; Sutera, S.P. Platelet adhesion and aggregation in pulsatile shear flow: Effects of red blood cells. Thromb. Res. 1998, 92, S47–S52. [Google Scholar] [CrossRef]
- Klatt, C.; Kruger, I.; Zey, S.; Krott, K.J.; Spelleken, M.; Gowert, N.S.; Oberhuber, A.; Pfaff, L.; Luckstadt, W.; Jurk, K.; et al. Platelet-RBC interaction mediated by FasL/FasR induces procoagulant activity important for thrombosis. J. Clin. Investig. 2018, 128, 3906–3925. [Google Scholar] [CrossRef] [PubMed]
- Valles, J.; Santos, M.T.; Aznar, J.; Martinez, M.; Moscardo, A.; Pinon, M.; Broekman, M.J.; Marcus, A.J. Platelet-erythrocyte interactions enhance alpha(IIb)beta(3) integrin receptor activation and P-selectin expression during platelet recruitment: Down-regulation by aspirin ex vivo. Blood 2002, 99, 3978–3984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reimers, R.C.; Sutera, S.P.; Joist, J.H. Potentiation by red blood cells of shear-induced platelet aggregation: Relative importance of chemical and physical mechanisms. Blood 1984, 64, 1200–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahavi, J.; Jones, N.A.; Leyton, J.; Dubiel, M.; Kakkar, V.V. Enhanced in vivo platelet “release reaction” in old healthy individuals. Thromb. Res. 1980, 17, 329–336. [Google Scholar] [CrossRef]
- Bastyr, E.J., 3rd; Kadrofske, M.M.; Vinik, A.I. Platelet activity and phosphoinositide turnover increase with advancing age. Am. J. Med. 1990, 88, 601–606. [Google Scholar] [CrossRef] [Green Version]
- Tran, P.L.; Pietropaolo, M.G.; Valerio, L.; Brengle, W.; Wong, R.K.; Kazui, T.; Khalpey, Z.I.; Redaelli, A.; Sheriff, J.; Bluestein, D.; et al. Hemolysate-mediated platelet aggregation: An additional risk mechanism contributing to thrombosis of continuous flow ventricular assist devices. Perfusion 2016, 31, 401–408. [Google Scholar] [CrossRef]
- Reiter, C.D.; Wang, X.; Tanus-Santos, J.E.; Hogg, N.; Cannon, R.O., 3rd; Schechter, A.N.; Gladwin, M.T. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nat. Med. 2002, 8, 1383–1389. [Google Scholar] [CrossRef]
- Solum, N.O. Procoagulant expression in platelets and defects leading to clinical disorders. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2841–2846. [Google Scholar] [CrossRef] [Green Version]
- Chaar, V.; Picot, J.; Renaud, O.; Bartolucci, P.; Nzouakou, R.; Bachir, D.; Galacteros, F.; Colin, Y.; Le Van Kim, C.; El Nemer, W. Aggregation of mononuclear and red blood cells through an α4β1-Lu/basal cell adhesion molecule interaction in sickle cell disease. Haematologica 2010, 95, 1841–1848. [Google Scholar] [CrossRef] [Green Version]
- Zennadi, R.; Chien, A.; Xu, K.; Batchvarova, M.; Telen, M.J. Sickle red cells induce adhesion of lymphocytes and monocytes to endothelium. Blood 2008, 112, 3474–3483. [Google Scholar] [CrossRef] [Green Version]
- von Bruhl, M.L.; Stark, K.; Steinhart, A.; Chandraratne, S.; Konrad, I.; Lorenz, M.; Khandoga, A.; Tirniceriu, A.; Coletti, R.; Kollnberger, M.; et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J. Exp. Med. 2012, 209, 819–835. [Google Scholar] [CrossRef] [PubMed]
- Kenawy, H.I.; Boral, I.; Bevington, A. Complement-Coagulation Cross-Talk: A Potential Mediator of the Physiological Activation of Complement by Low pH. Front. Immunol. 2015, 6, 215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frimat, M.; Tabarin, F.; Dimitrov, J.D.; Poitou, C.; Halbwachs-Mecarelli, L.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement activation by heme as a secondary hit for atypical hemolytic uremic syndrome. Blood 2013, 122, 282–292. [Google Scholar] [CrossRef] [PubMed]
- Wiedmer, T.; Esmon, C.T.; Sims, P.J. Complement proteins C5b-9 stimulate procoagulant activity through platelet prothrombinase. Blood 1986, 68, 875–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiedmer, T.; Esmon, C.T.; Sims, P.J. On the mechanism by which complement proteins C5b-9 increase platelet prothrombinase activity. J. Biol. Chem. 1986, 261, 14587–14592. [Google Scholar]
- Ikeda, K.; Nagasawa, K.; Horiuchi, T.; Tsuru, T.; Nishizaka, H.; Niho, Y. C5a induces tissue factor activity on endothelial cells. Thromb. Haemost. 1997, 77, 394–398. [Google Scholar] [CrossRef]
- Ritis, K.; Doumas, M.; Mastellos, D.; Micheli, A.; Giaglis, S.; Magotti, P.; Rafail, S.; Kartalis, G.; Sideras, P.; Lambris, J.D. A novel C5a receptor-tissue factor cross-talk in neutrophils links innate immunity to coagulation pathways. J. Immunol. 2006, 177, 4794–4802. [Google Scholar] [CrossRef]
- Chudwin, D.S.; Papierniak, C.; Lint, T.F.; Korenblit, A.D. Activation of the alternative complement pathway by red blood cells from patients with sickle cell disease. Clin. Immunol. Immunopathol. 1994, 71, 199–202. [Google Scholar] [CrossRef]
- Chapin, J.; Terry, H.S.; Kleinert, D.; Laurence, J. The role of complement activation in thrombosis and hemolytic anemias. Transfus. Apher. Sci. 2016, 54, 191–198. [Google Scholar] [CrossRef]
- Camus, S.M.; Gausseres, B.; Bonnin, P.; Loufrani, L.; Grimaud, L.; Charue, D.; De Moraes, J.A.; Renard, J.M.; Tedgui, A.; Boulanger, C.M.; et al. Erythrocyte microparticles can induce kidney vaso-occlusions in a murine model of sickle cell disease. Blood 2012, 120, 5050–5058. [Google Scholar] [CrossRef] [Green Version]
- Merle, N.S.; Grunenwald, A.; Rajaratnam, H.; Gnemmi, V.; Frimat, M.; Figueres, M.L.; Knockaert, S.; Bouzekri, S.; Charue, D.; Noe, R.; et al. Intravascular hemolysis activates complement via cell-free heme and heme-loaded microvesicles. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Mari, D.; Coppola, R.; Provenzano, R. Hemostasis factors and aging. Exp. Gerontol. 2008, 43, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Balleisen, L.; Bailey, J.; Epping, P.H.; Schulte, H.; van de Loo, J. Epidemiological study on factor VII, factor VIII and fibrinogen in an industrial population: I. Baseline data on the relation to age, gender, body-weight, smoking, alcohol, pill-using, and menopause. Thromb. Haemost. 1985, 54, 475–479. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, S.; Vitorino de Almeida, V.; Calado, A.; Rosario, H.S.; Saldanha, C. Integrin-associated protein (CD47) is a putative mediator for soluble fibrinogen interaction with human red blood cells membrane. Biochim. Biophys. Acta 2012, 1818, 481–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, F.A.; de Oliveira, S.; Freitas, T.; Goncalves, S.; Santos, N.C. Variations on fibrinogen-erythrocyte interactions during cell aging. PLoS ONE 2011, 6, e18167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwata, H.; Kaibara, M. Activation of factor IX by erythrocyte membranes causes intrinsic coagulation. Blood Coagul. Fibrinolysis Int. J. Haemost. Thromb. 2002, 13, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Tolosano, E.; Fagoonee, S.; Morello, N.; Vinchi, F.; Fiorito, V. Heme scavenging and the other facets of hemopexin. Antioxid. Redox Signal. 2010, 12, 305–320. [Google Scholar] [CrossRef]
- Cooper, C.E.; Schaer, D.J.; Buehler, P.W.; Wilson, M.T.; Reeder, B.J.; Silkstone, G.; Svistunenko, D.A.; Bulow, L.; Alayash, A.I. Haptoglobin binding stabilizes hemoglobin ferryl iron and the globin radical on tyrosine beta145. Antioxid Redox Signal. 2013, 18, 2264–2273. [Google Scholar] [CrossRef]
- Smith, A.; McCulloh, R.J. Hemopexin and haptoglobin: Allies against heme toxicity from hemoglobin not contenders. Front. Physiol. 2015, 6, 187. [Google Scholar] [CrossRef]
- Smith, A.; Morgan, W.T. Haem transport to the liver by haemopexin. Receptor-mediated uptake with recycling of the protein. Biochem J. 1979, 182, 47–54. [Google Scholar] [CrossRef] [Green Version]
- Thomsen, J.H.; Etzerodt, A.; Svendsen, P.; Moestrup, S.K. The haptoglobin-CD163-heme oxygenase-1 pathway for hemoglobin scavenging. Oxid Med. Cell Longev. 2013, 2013, 523652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagy, E.; Eaton, J.W.; Jeney, V.; Soares, M.P.; Varga, Z.; Galajda, Z.; Szentmiklósi, J.; Méhes, G.; Csonka, T.; Smith, A.; et al. Red cells, hemoglobin, heme, iron, and atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1347–1353. [Google Scholar] [CrossRef] [PubMed]
- Balla, J.; Jacob, H.S.; Balla, G.; Nath, K.; Eaton, J.W.; Vercellotti, G.M. Endothelial-cell heme uptake from heme proteins: Induction of sensitization and desensitization to oxidant damage. Proc. Natl. Acad. Sci. USA 1993, 90, 9285–9289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woollard, K.J.; Sturgeon, S.; Chin-Dusting, J.P.; Salem, H.H.; Jackson, S.P. Erythrocyte hemolysis and hemoglobin oxidation promote ferric chloride-induced vascular injury. J. Biol. Chem. 2009, 284, 13110–13118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belcher, J.D.; Chen, C.; Nguyen, J.; Milbauer, L.; Abdulla, F.; Alayash, A.I.; Smith, A.; Nath, K.A.; Hebbel, R.P.; Vercellotti, G.M. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood 2014, 123, 377–390. [Google Scholar] [CrossRef] [Green Version]
- Silva, G.; Jeney, V.; Chora, A.; Larsen, R.; Balla, J.; Soares, M.P. Oxidized hemoglobin is an endogenous proinflammatory agonist that targets vascular endothelial cells. J. Biol. Chem. 2009, 284, 29582–29595. [Google Scholar] [CrossRef] [Green Version]
- Ogasawara, N.; Oguro, T.; Sakabe, T.; Matsushima, M.; Takikawa, O.; Isobe, K.; Nagase, F. Hemoglobin induces the expression of indoleamine 2,3-dioxygenase in dendritic cells through the activation of PI3K, PKC, and NF-kappaB and the generation of reactive oxygen species. J. Cell Biochem. 2009, 108, 716–725. [Google Scholar] [CrossRef]
- Lin, S.; Yin, Q.; Zhong, Q.; Lv, F.L.; Zhou, Y.; Li, J.Q.; Wang, J.Z.; Su, B.Y.; Yang, Q.W. Heme activates TLR4-mediated inflammatory injury via MyD88/TRIF signaling pathway in intracerebral hemorrhage. J. Neuroinflamm. 2012, 9, 46. [Google Scholar] [CrossRef] [Green Version]
- Piazza, M.; Damore, G.; Costa, B.; Gioannini, T.L.; Weiss, J.P.; Peri, F. Hemin and a metabolic derivative coprohemin modulate the TLR4 pathway differently through different molecular targets. Innate Immun. 2011, 17, 293–301. [Google Scholar] [CrossRef] [Green Version]
- Lisk, C.; Kominsky, D.; Ehrentraut, S.; Bonaventura, J.; Nuss, R.; Hassell, K.; Nozik-Grayck, E.; Irwin, D.C. Hemoglobin-induced endothelial cell permeability is controlled, in part, via a myeloid differentiation primary response gene-88-dependent signaling mechanism. Am. J. Respir. Cell Mol. Biol. 2013, 49, 619–626. [Google Scholar] [CrossRef] [Green Version]
- Kupari, M.; Rapola, J. Reversible pulmonary hypertension associated with vitamin C deficiency. Chest 2012, 142, 225–227. [Google Scholar] [CrossRef] [PubMed]
- Tracz, M.J.; Juncos, J.P.; Grande, J.P.; Croatt, A.J.; Ackerman, A.W.; Katusic, Z.S.; Nath, K.A. Induction of heme oxygenase-1 is a beneficial response in a murine model of venous thrombosis. Am. J. Pathol. 2008, 173, 1882–1890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mustafa, S.; Weltermann, A.; Fritsche, R.; Marsik, C.; Wagner, O.; Kyrle, P.A.; Eichinger, S. Genetic variation in heme oxygenase 1 (HMOX1) and the risk of recurrent venous thromboembolism. J. Vasc. Surg. Off. Publ. Soc. Vasc. Surg. Int. Soc. Cardiovasc. Surg. N. Am. Chapter 2008, 47, 566–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavrovsky, Y.; Song, C.S.; Chatterjee, B.; Roy, A.K. Age-dependent increase of heme oxygenase-1 gene expression in the liver mediated by NFkappaB. Mech. Ageing Dev. 2000, 114, 49–60. [Google Scholar] [CrossRef]
- Schipper, H.M. Heme oxygenase-1: Role in brain aging and neurodegeneration. Exp. Gerontol. 2000, 35, 821–830. [Google Scholar] [CrossRef]
- Rattan, V.; Sultana, C.; Shen, Y.; Kalra, V.K. Oxidant stress-induced transendothelial migration of monocytes is linked to phosphorylation of PECAM-1. Am. J. Physiol. 1997, 273, E453–E461. [Google Scholar] [CrossRef]
- Saragih, H.; Zilian, E.; Jaimes, Y.; Paine, A.; Figueiredo, C.; Eiz-Vesper, B.; Blasczyk, R.; Larmann, J.; Theilmeier, G.; Burg-Roderfeld, M.; et al. PECAM-1-dependent heme oxygenase-1 regulation via an Nrf2-mediated pathway in endothelial cells. Thromb. Haemost. 2014, 111, 1077–1088. [Google Scholar] [CrossRef]
- Gladwin, M.T.; Schechter, A.N.; Ognibene, F.P.; Coles, W.A.; Reiter, C.D.; Schenke, W.H.; Csako, G.; Waclawiw, M.A.; Panza, J.A.; Cannon, R.O., 3rd. Divergent nitric oxide bioavailability in men and women with sickle cell disease. Circulation 2003, 107, 271–278. [Google Scholar] [CrossRef] [Green Version]
- Wood, K.C.; Hebbel, R.P.; Lefer, D.J.; Granger, D.N. Critical role of endothelial cell-derived nitric oxide synthase in sickle cell disease-induced microvascular dysfunction. Free Radic. Biol. Med. 2006, 40, 1443–1453. [Google Scholar] [CrossRef]
- Chen, G.; Zhang, D.; Fuchs, T.A.; Manwani, D.; Wagner, D.D.; Frenette, P.S. Heme-induced neutrophil extracellular traps contribute to the pathogenesis of sickle cell disease. Blood 2014, 123, 3818–3827. [Google Scholar] [CrossRef]
- Day, S.M.; Reeve, J.L.; Pedersen, B.; Farris, D.M.; Myers, D.D.; Im, M.; Wakefield, T.W.; Mackman, N.; Fay, W.P. Macrovascular thrombosis is driven by tissue factor derived primarily from the blood vessel wall. Blood 2005, 105, 192–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iacoviello, L.; Kolpakov, V.; Salvatore, L.; Amore, C.; Pintucci, G.; de Gaetano, G.; Donati, M.B. Human endothelial cell damage by neutrophil-derived cathepsin G. Role of cytoskeleton rearrangement and matrix-bound plasminogen activator inhibitor-1. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 2037–2046. [Google Scholar] [CrossRef] [PubMed]
- Saha, P.; Humphries, J.; Modarai, B.; Mattock, K.; Waltham, M.; Evans, C.E.; Ahmad, A.; Patel, A.S.; Premaratne, S.; Lyons, O.T.; et al. Leukocytes and the natural history of deep vein thrombosis: Current concepts and future directions. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 506–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D., Jr.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef] [Green Version]
- Singhal, R.; Annarapu, G.K.; Pandey, A.; Chawla, S.; Ojha, A.; Gupta, A.; Cruz, M.A.; Seth, T.; Guchhait, P. Hemoglobin interaction with GP1balpha induces platelet activation and apoptosis: A novel mechanism associated with intravascular hemolysis. Haematologica 2015, 100, 1526–1533. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Delaney, M.K.; O’Brien, K.A.; Du, X. Signaling during platelet adhesion and activation. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2341–2349. [Google Scholar] [CrossRef] [Green Version]
- Ezumi, Y.; Shindoh, K.; Tsuji, M.; Takayama, H. Physical and functional association of the Src family kinases Fyn and Lyn with the collagen receptor glycoprotein VI-Fc receptor gamma chain complex on human platelets. J. Exp. Med. 1998, 188, 267–276. [Google Scholar] [CrossRef]
- Du, X. Signaling and regulation of the platelet glycoprotein Ib-IX-V complex. Curr. Opin. Hematol. 2007, 14, 262–269. [Google Scholar] [CrossRef]
- Bourne, J.H.; Colicchia, M.; Di, Y.; Martin, E.; Slater, A.; Roumenina, L.T.; Dimitrov, J.D.; Watson, S.P.; Rayes, J. Heme induces human and mouse platelet activation through C-type-lectin-like receptor-2. Haematologica 2020. [Google Scholar] [CrossRef]
- Bahl, N.; Winarsih, I.; Tucker-Kellogg, L.; Ding, J.L. Extracellular haemoglobin upregulates and binds to tissue factor on macrophages: Implications for coagulation and oxidative stress. Thromb. Haemost. 2014, 111, 67–78. [Google Scholar] [CrossRef] [Green Version]
- Ohkura, N.; Hiraishi, S.; Itabe, H.; Hamuro, T.; Kamikubo, Y.; Takano, T.; Matsuda, J.; Horie, S. Oxidized phospholipids in oxidized low-density lipoprotein reduce the activity of tissue factor pathway inhibitor through association with its carboxy-terminal region. Antioxid. Redox Signal. 2004, 6, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Nalian, A.; Iakhiaev, A.V. Possible mechanisms contributing to oxidative inactivation of activated protein C: Molecular dynamics study. Thromb. Haemost. 2008, 100, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Glaser, C.B.; Morser, J.; Clarke, J.H.; Blasko, E.; McLean, K.; Kuhn, I.; Chang, R.J.; Lin, J.H.; Vilander, L.; Andrews, W.H.; et al. Oxidation of a specific methionine in thrombomodulin by activated neutrophil products blocks cofactor activity. A potential rapid mechanism for modulation of coagulation. J. Clin. Investig. 1992, 90, 2565–2573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Upchurch, G.R., Jr.; Ramdev, N.; Walsh, M.T.; Loscalzo, J. Prothrombotic Consequences of the Oxidation of Fibrinogen and their Inhibition by Aspirin. J. Thromb. Thrombolysis 1998, 5, 9–14. [Google Scholar] [CrossRef]
- De Cristofaro, R.; Landolfi, R. Oxidation of human alpha-thrombin by the myeloperoxidase-H2O2-chloride system: Structural and functional effects. Thromb. Haemost. 2000, 83, 253–261. [Google Scholar]
- Van Patten, S.M.; Hanson, E.; Bernasconi, R.; Zhang, K.; Manavalan, P.; Cole, E.S.; McPherson, J.M.; Edmunds, T. Oxidation of methionine residues in antithrombin. Effects on biological activity and heparin binding. J. Biol. Chem. 1999, 274, 10268–10276. [Google Scholar] [CrossRef] [Green Version]
- Ay, C.; Jungbauer, L.V.; Sailer, T.; Tengler, T.; Koder, S.; Kaider, A.; Panzer, S.; Quehenberger, P.; Pabinger, I.; Mannhalter, C. High concentrations of soluble P-selectin are associated with risk of venous thromboembolism and the P-selectin Thr715 variant. Clin. Chem. 2007, 53, 1235–1243. [Google Scholar] [CrossRef] [Green Version]
- Klyubin, I.V.; Kirpichnikova, K.M.; Gamaley, I.A. Hydrogen peroxide-induced chemotaxis of mouse peritoneal neutrophils. Eur. J. Cell Biol. 1996, 70, 347–351. [Google Scholar]
- Rosendaal, F.R. Venous thrombosis: A multicausal disease. Lancet 1999, 353, 1167–1173. [Google Scholar] [CrossRef] [Green Version]
- Rosendaal, F.R.; Koster, T.; Vandenbroucke, J.P.; Reitsma, P.H. High risk of thrombosis in patients homozygous for factor V Leiden (activated protein C resistance). Blood 1995, 85, 1504–1508. [Google Scholar] [CrossRef] [Green Version]
- Poort, S.R.; Rosendaal, F.R.; Reitsma, P.H.; Bertina, R.M. A common genetic variation in the 3’-untranslated region of the prothrombin gene is associated with elevated plasma prothrombin levels and an increase in venous thrombosis. Blood 1996, 88, 3698–3703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folsom, A.R.; Cushman, M.; Tsai, M.Y.; Aleksic, N.; Heckbert, S.R.; Boland, L.L.; Tsai, A.W.; Yanez, N.D.; Rosamond, W.D. A prospective study of venous thromboembolism in relation to factor V Leiden and related factors. Blood 2002, 99, 2720–2725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jick, H.; Slone, D.; Westerholm, B.; Inman, W.H.; Vessey, M.P.; Shapiro, S.; Lewis, G.P.; Worcester, J. Venous thromboembolic disease and ABO blood type. A cooperative study. Lancet 1969, 1, 539–542. [Google Scholar] [CrossRef]
- Ohira, T.; Cushman, M.; Tsai, M.Y.; Zhang, Y.; Heckbert, S.R.; Zakai, N.A.; Rosamond, W.D.; Folsom, A.R. ABO blood group, other risk factors and incidence of venous thromboembolism: The Longitudinal Investigation of Thromboembolism Etiology (LITE). J. Thromb. Haemost. 2007, 5, 1455–1461. [Google Scholar] [CrossRef]
- Dentali, F.; Sironi, A.P.; Ageno, W.; Turato, S.; Bonfanti, C.; Frattini, F.; Crestani, S.; Franchini, M. Non-O blood type is the commonest genetic risk factor for VTE: Results from a meta-analysis of the literature. Semin. Thromb. Hemost. 2012, 38, 535–548. [Google Scholar] [CrossRef]
- Karasu, A.; Engbers, M.J.; Cushman, M.; Rosendaal, F.R.; van Hylckama Vlieg, A. Genetic risk factors for venous thrombosis in the elderly in a case-control study. J. Thromb. Haemost. 2016, 14, 1759–1764. [Google Scholar] [CrossRef]
- de Visser, M.C.; van Minkelen, R.; van Marion, V.; den Heijer, M.; Eikenboom, J.; Vos, H.L.; Slagboom, P.E.; Houwing-Duistermaat, J.J.; Rosendaal, F.R.; Bertina, R.M. Genome-wide linkage scan in affected sibling pairs identifies novel susceptibility region for venous thromboembolism: Genetics In Familial Thrombosis study. J. Thromb. Haemost. 2013, 11, 1474–1484. [Google Scholar] [CrossRef]
- Bean, C.J.; Boulet, S.L.; Ellingsen, D.; Trau, H.; Ghaji, N.; Hooper, W.C.; Austin, H. Increased risk of venous thromboembolism is associated with genetic variation in heme oxygenase-1 in Blacks. Thromb. Res. 2012, 130, 942–947. [Google Scholar] [CrossRef] [Green Version]
- Munshi, R.; Panchal, F.; Kulkarni, V.; Chaurasia, A. Methylenetetrahydrofolate reductase polymorphism in healthy volunteers and its correlation with homocysteine levels in patients with thrombosis. Indian J. Pharmacol. 2019, 51, 248–254. [Google Scholar] [CrossRef]
- Hosseini, S.; Kalantar, E.; Hosseini, M.S.; Tabibian, S.; Shamsizadeh, M.; Dorgalaleh, A. Genetic risk factors in patients with deep venous thrombosis, a retrospective case control study on Iranian population. Thromb. J. 2015, 13, 35. [Google Scholar] [CrossRef] [Green Version]
- Tsiolakidou, G.; Koutroubakis, I.E. Thrombosis and inflammatory bowel disease-the role of genetic risk factors. World J. Gastroenterol. 2008, 14, 4440–4444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unruh, D.; Schwarze, S.R.; Khoury, L.; Thomas, C.; Wu, M.; Chen, L.; Chen, R.; Liu, Y.; Schwartz, M.A.; Amidei, C.; et al. Mutant IDH1 and thrombosis in gliomas. Acta Neuropathol. 2016, 132, 917–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes, R.A.; Neves, K.B.; Tostes, R.C.; Montezano, A.C.; Touyz, R.M. Downregulation of Nuclear Factor Erythroid 2-Related Factor and Associated Antioxidant Genes Contributes to Redox-Sensitive Vascular Dysfunction in Hypertension. Hypertension 2015, 66, 1240–1250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csiszar, A.; Pacher, P.; Kaley, G.; Ungvari, Z. Role of oxidative and nitrosative stress, longevity genes and poly(ADP-ribose) polymerase in cardiovascular dysfunction associated with aging. Curr. Vasc. Pharmacol. 2005, 3, 285–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Q.; Zennadi, R. Oxidative Stress and Thrombosis during Aging: The Roles of Oxidative Stress in RBCs in Venous Thrombosis. Int. J. Mol. Sci. 2020, 21, 4259. https://doi.org/10.3390/ijms21124259
Wang Q, Zennadi R. Oxidative Stress and Thrombosis during Aging: The Roles of Oxidative Stress in RBCs in Venous Thrombosis. International Journal of Molecular Sciences. 2020; 21(12):4259. https://doi.org/10.3390/ijms21124259
Chicago/Turabian StyleWang, Qinhong, and Rahima Zennadi. 2020. "Oxidative Stress and Thrombosis during Aging: The Roles of Oxidative Stress in RBCs in Venous Thrombosis" International Journal of Molecular Sciences 21, no. 12: 4259. https://doi.org/10.3390/ijms21124259
APA StyleWang, Q., & Zennadi, R. (2020). Oxidative Stress and Thrombosis during Aging: The Roles of Oxidative Stress in RBCs in Venous Thrombosis. International Journal of Molecular Sciences, 21(12), 4259. https://doi.org/10.3390/ijms21124259