N-Acetylcysteine Reduces Skeletal Muscles Oxidative Stress and Improves Grip Strength in Dysferlin-Deficient Bla/J Mice

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
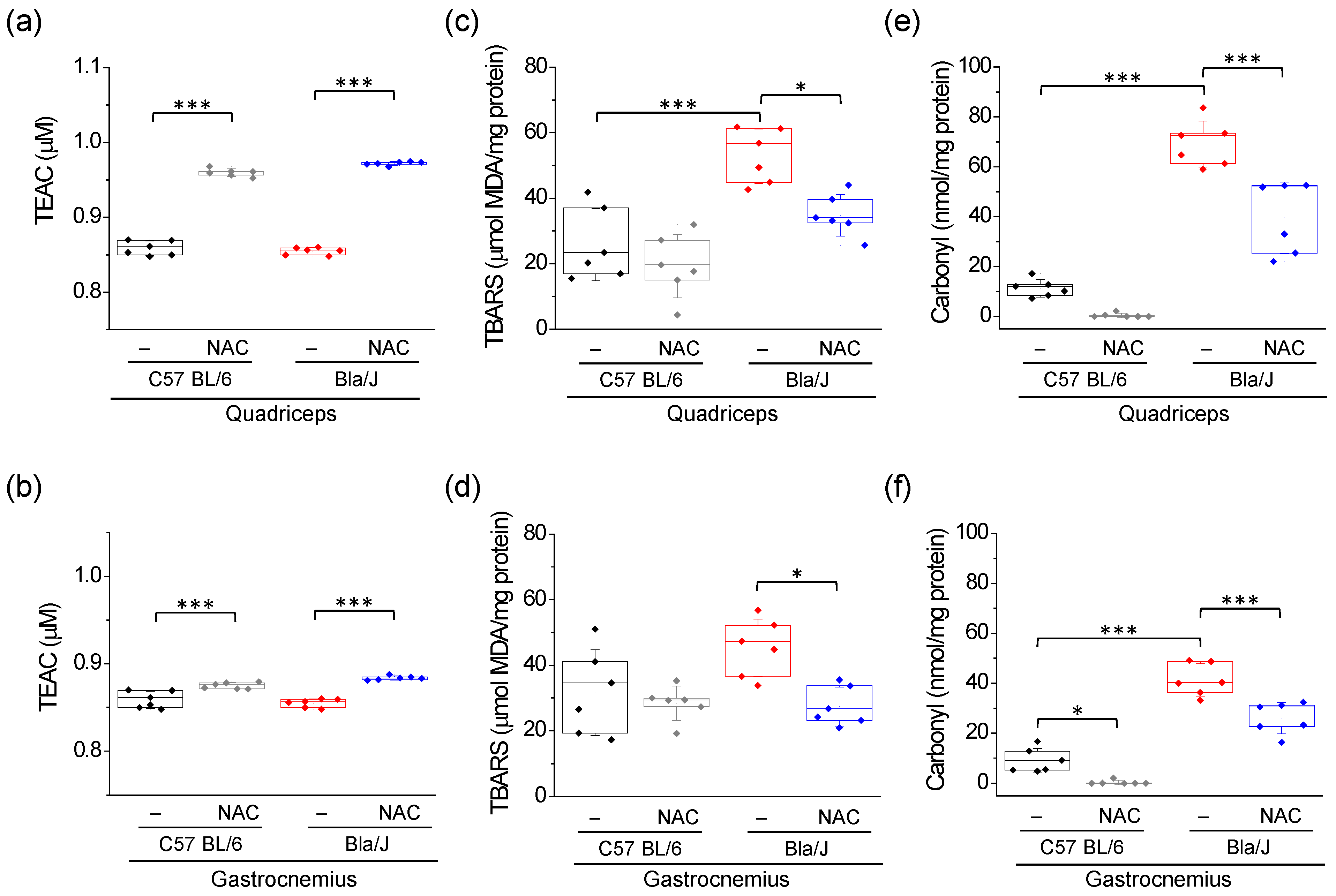
2.1. Increased Lipid Peroxidation and Carbonyl Protein in Quadriceps and Gastrocnemius of Dysferlin-Deficient Bla/J Mice Are Ameliorated by NAC Supplementation
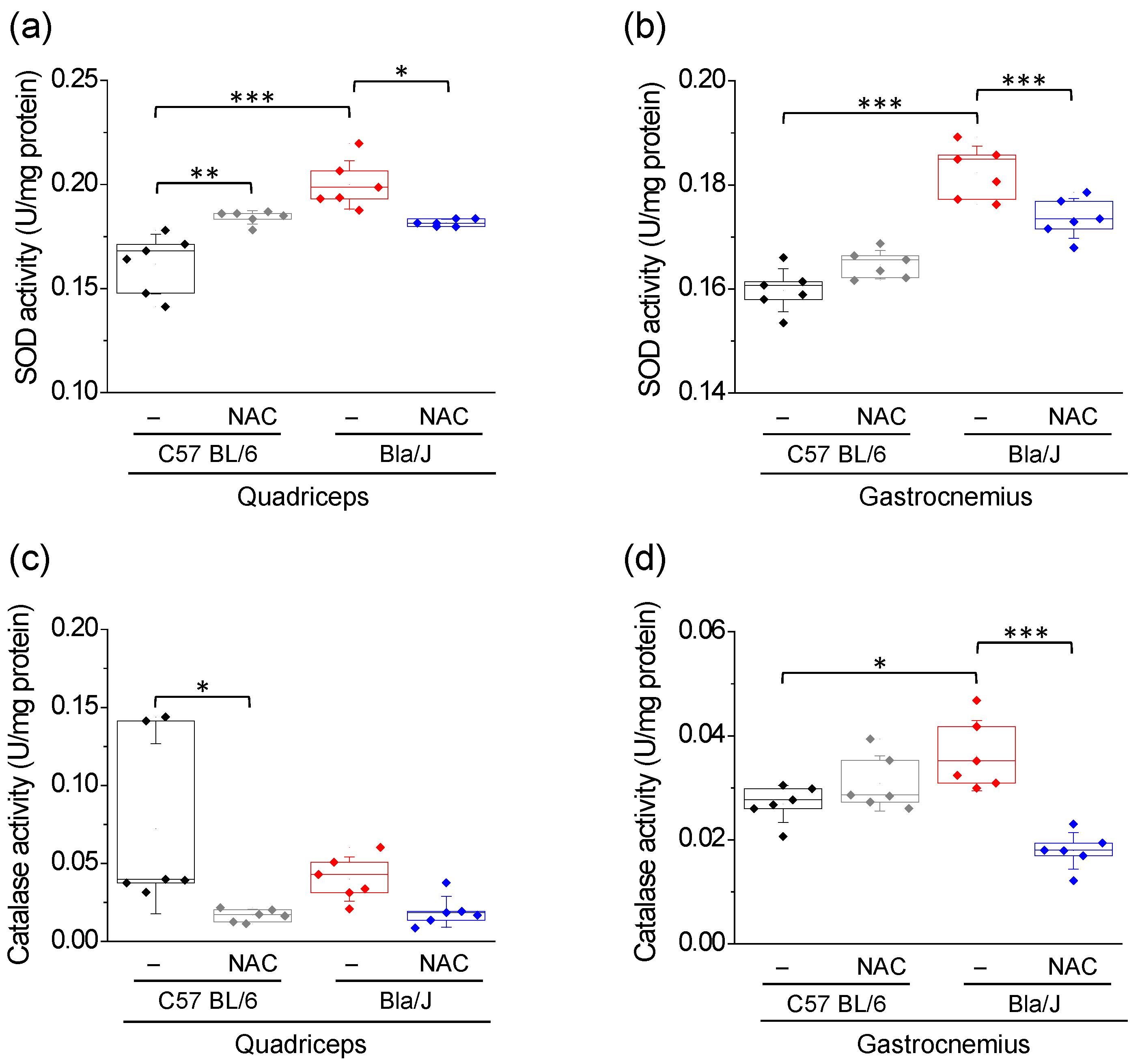
2.2. Effects of NAC Supplementation on SOD and Catalase Activity in Quadriceps and Gastrocnemius of Dysferlin-Deficient Bla/J Mice
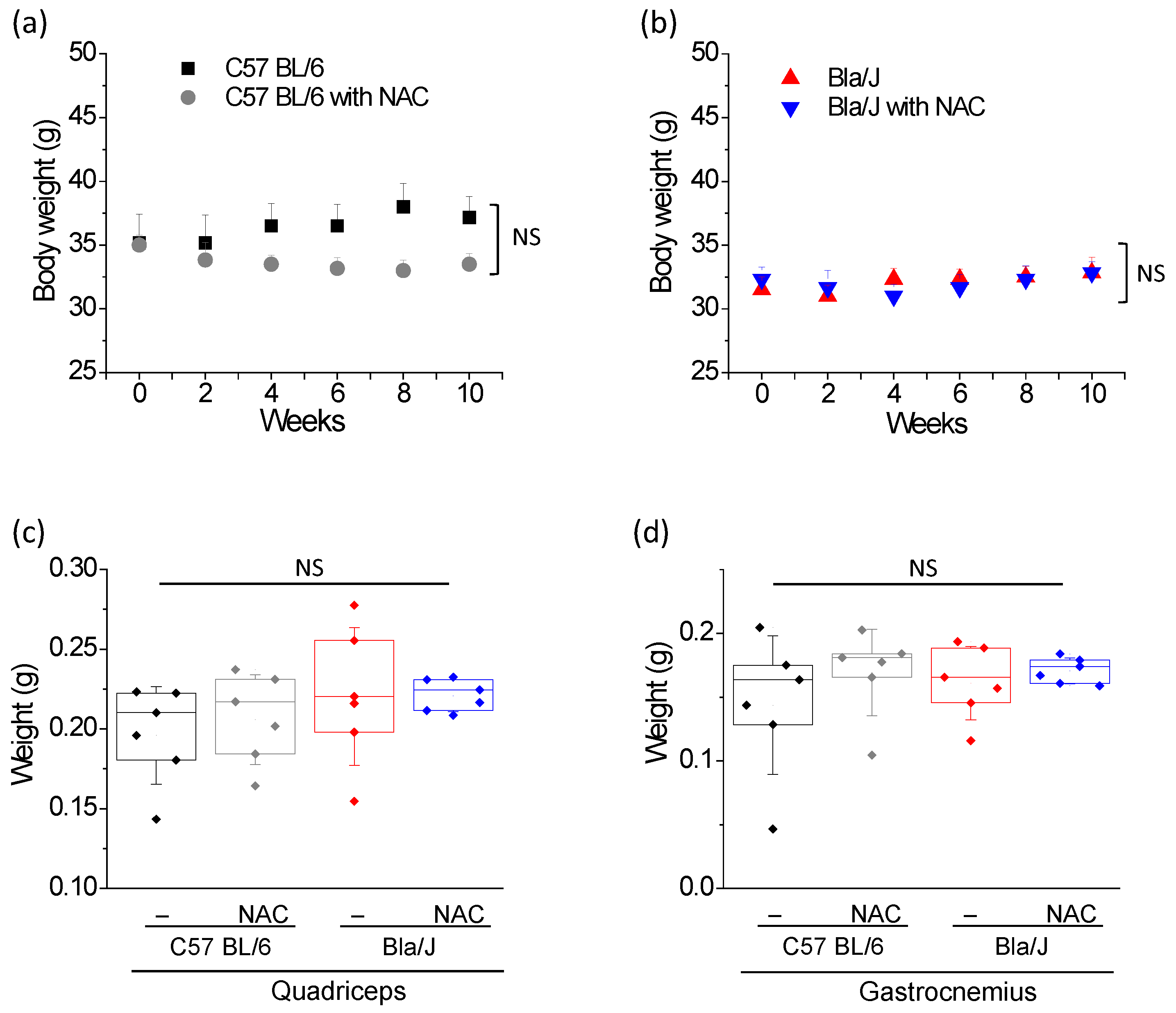
2.3. NAC Treatment Has no Effect on Muscle Mass of Dysferlin Deficient Bla/J Mice
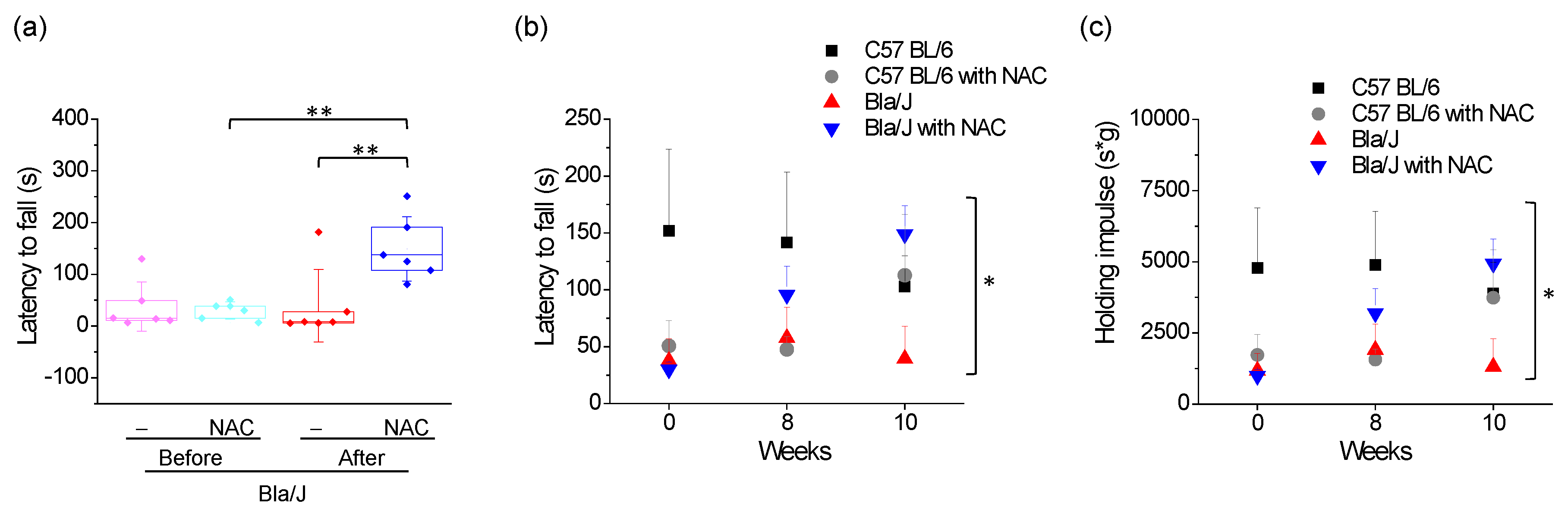
2.4. Effects of NAC Supplementation on Grip Strength in Dysferlin Deficient Bla/J Mice
3. Discussion
3.1. NAC Restores the Redox Balance in Dysferlin-Deficient Skeletal Muscle
3.2. Effect of NAC Supplementation on Body Mass and Muscle Weight
3.3. Effects of NAC Supplementation on Grip Strength in Dysferlin Deficient Bla/J Mice
3.4. Comparison of the Effect of NAC in Bla/J and mdx Mice
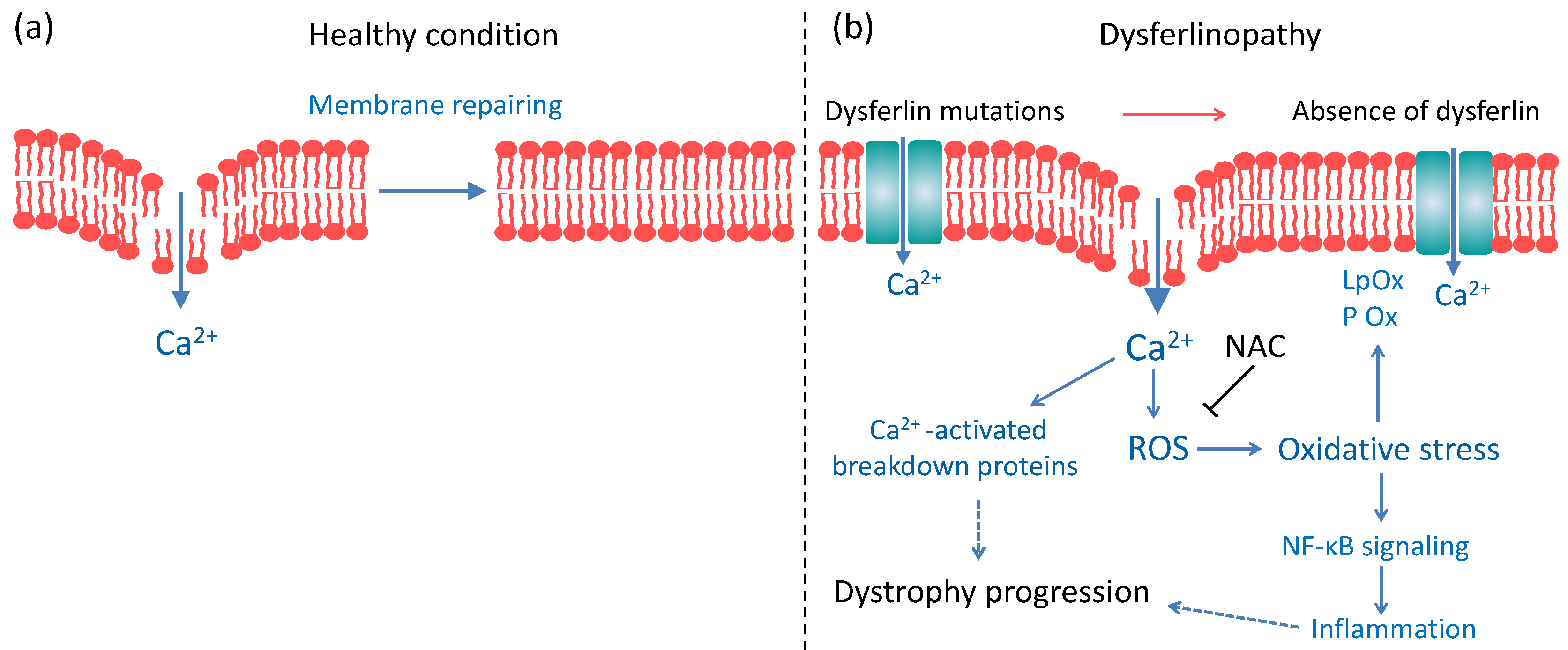
3.5. Would NAC Be Useful as a Therapy for Dysferlinopathies?
4. Materials and Methods
4.1. Animals and NAC Treatments
4.2. Tissue Collections
4.3. Total Antioxidant Capacity (TAC)
4.4. Lipid Peroxidation Measurement
4.5. Protein Carbonyl Content Assay
4.6. Total Protein Measurement
4.7. Superoxide Dismutase (SOD) Activity
4.8. Catalase Activity
4.9. Grip Strength
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BSA | Bovine serum albumin |
| MDA | Malondialdehyde |
| NAC | N-Acetylcysteine |
| Nox2 | NADPH Oxidase 2 |
| Nuclear factor κappa B | NF-κB |
| OS | Oxidative stress |
| ROS | Reactive oxygen species |
| SD | Standard deviation |
| SE | Standard error |
| SOD | Superoxide dismutase |
| TAC | Total antioxidant capacity |
| TBA | Thiobarbituric acid |
| TBARS | Thiobarbituric acid reactive substances |
| TEAC | Trolox equivalent antioxidant capacity |
| TCA | Trichloroacetic acid |
| Trolox | 6-Hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid |
| WT | Wild-type |
| X-ROS | Nox2-dependent ROS generation |
References
- Canton, M.; Menazza, S.; Di Lisa, F. Oxidative stress in muscular dystrophy: From generic evidence to specific sources and targets. J. Muscle Res. Cell Motil. 2014, 35, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.H.; Ow, J.R.; Yang, N.D.; Taneja, R. Oxidative Stress-Mediated Skeletal Muscle Degeneration: Molecules, Mechanisms, and Therapies. Oxid. Med. Cell. Longev. 2016, 2016, 6842568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renjini, R.; Gayathri, N.; Nalini, A.; Srinivas Bharath, M. Oxidative damage in muscular dystrophy correlates with the severity of the pathology: Role of glutathione metabolism. Neurochem. Res. 2012, 37, 885–898. [Google Scholar] [CrossRef] [PubMed]
- Terrill, J.R.; Radley-Crabb, H.G.; Iwasaki, T.; Lemckert, F.A.; Arthur, P.G.; Grounds, M.D. Oxidative stress and pathology in muscular dystrophies: Focus on protein thiol oxidation and dysferlinopathies. FEBS J. 2013, 280, 4149–4164. [Google Scholar] [CrossRef]
- Rajakumar, D.; Senguttuvan, S.; Alexander, M.; Oommen, A. Involvement of oxidative stress, nuclear factor kappa B and the ubiquitin proteasomal pathway in dysferlinopathy. Life Sci. 2014, 108, 54–61. [Google Scholar] [CrossRef]
- Cárdenas, A.M.; González-Jamett, A.M.; Cea, L.A.; Bevilacqua, J.A.; Caviedes, P. Dysferlin function in skeletal muscle: Possible pathological mechanisms and therapeutical targets in dysferlinopathies. Exp. Neurol. 2016, 283, 246–254. [Google Scholar] [CrossRef]
- Díaz, J.; Woudt, L.; Suazo, L.; Garrido, C.; Caviedes, P.; Cárdenas, A.M.; Castiglioni, C.; Bevilacqua, J.A. Broadening the imaging phenotype of dysferlinopathy at different disease stages. Muscle Nerve 2016, 54, 203–210. [Google Scholar] [CrossRef]
- Bashir, R.; Britton, S.; Strachan, T.; Keers, S.; Vafiadaki, E.; Lako, M.; Richard, I.; Marchand, S.; Bourg, N.; Argov, Z.; et al. A gene related to Caenorhabditis elegans spermatogenesis factor fer-1 is mutated in limb-girdle muscular dystrophy type 2B. Nat. Genet. 1998, 20, 37–42. [Google Scholar] [CrossRef]
- Liu, J.; Aoki, M.; Illa, I.; Wu, C.; Fardeau, M.; Angelini, C.; Serrano, C.; Urtizberea, J.A.; Hentati, F.; Hamida, M.B.; et al. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat. Genet. 1998, 20, 31–36. [Google Scholar] [CrossRef]
- Illarioshkin, S.N.; Ivanova-Smolenskaya, I.A.; Greenberg, C.R.; Nylen, E.; Sukhorukov, V.S.; Poleshchuk, V.V.; Markova, E.D.; Wrogemann, K. Identical dysferlin mutation in limb-girdle muscular dystrophy type 2B and distal myopathy. Neurology 2000, 55, 1931–1933. [Google Scholar] [CrossRef]
- Cenacchi, G.; Fanin, M.; De Giorgi, L.B.; Angelini, C. Ultrastructural changes in dysferlinopathy support defective membrane repair mechanism. J. Clin. Pathol. 2005, 58, 190–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vincent, A.E.; Rosa, H.S.; Alston, C.L.; Grady, J.P.; Rygiel, K.A.; Rocha, M.C.; Barresi, R.; Taylor, R.W.; Turnbull, D.M. Dysferlin mutations and mitochondrial dysfunction. Neuromuscul. Disord. 2016, 26, 782–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kombairaju, P.; Kerr, J.P.; Roche, J.A.; Pratt, S.J.P.; Lovering, R.M.; Sussan, T.E.; Kim, J.H.; Shi, G.; Biswal, S.; Ward, C.W. Genetic silencing of Nrf2 enhances X-ROS in dysferlin-deficient muscle. Front. Physiol. 2014, 5, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhanarajan, R.; Patil, A.B.; Alexander, M.; Chacko, G.; Oommen, A. Degradation of myofibrillar proteins and inadequate antioxidants in selective muscle wasting of limb girdle muscular dystrophy. Int. J. Case Rep. Images 2011, 2, 6–11. [Google Scholar] [CrossRef] [Green Version]
- Prosser, B.L.; Khairallah, R.J.; Ziman, A.P.; Ward, C.W.; Lederer, W.J. X-ROS signaling in the heart and skeletal muscle: Stretch-dependent local ROS regulates [Ca2+]i. J. Mol. Cell. Cardiol. 2013, 58, 172–181. [Google Scholar] [CrossRef] [Green Version]
- Potgieter, M.; Pretorius, E.; Van der Merwe, C.; Beukes, M.; Vieira, W.; Auer, R.; Auer, M.; Meyer, S. Histological assessment of SJL/J mice treated with the antioxidants coenzyme Q10 and resveratrol. Micron 2011, 42, 275–282. [Google Scholar] [CrossRef]
- Van der Spuy, W.J.; Pretorius, E. The qualitative effects of resveratrol and coenzyme Q10 administration on the gluteus complex muscle morphology of SJL/J mice with dysferlinopathy. Int. J. Morphol. 2011, 29, 876–884. [Google Scholar] [CrossRef] [Green Version]
- Lostal, W.; Bartoli, M.; Bourg, N.; Roudaut, C.; Bentaïb, A.; Miyake, K.; Guerchet, N.; Fougerousse, F.; McNeil, P.; Richard, I. Efficient recovery of dysferlin deficiency by dual adeno-associated vector-mediated gene transfer. Hum. Mol. Genet. 2010, 19, 1897–1907. [Google Scholar] [CrossRef] [Green Version]
- Nagy, N.; Nonneman, R.J.; Llanga, T.; Dial, C.F.; Riddick, N.V.; Hampton, T.; Moy, S.S.; Lehtimäki, K.K.; Ahtoniemi, T.; Puoliväli, J.; et al. Hip region muscular dystrophy and emergence of motor deficits in dysferlin-deficient Bla/J mice. Physiol. Rep. 2017, 5, e13173. [Google Scholar] [CrossRef] [Green Version]
- Aruoma, O.I.; Halliwell, B.; Hoey, B.M.; Butler, J. The antioxidant action of N-acetylcysteine: Its reaction with hydrogen peroxide, hydroxyl radical, superoxide, and hypochlorous acid. Free Radic. Biol. Med. 1989, 6, 593–597. [Google Scholar] [CrossRef]
- Mokhtari, V.; Afsharian, P.; Shahhoseini, M.; Kalantar, S.M.; Moini, A. A Review on Various Uses of N-Acetyl Cysteine. Cell J. 2017, 19, 11–17. [Google Scholar]
- Whitehead, N.P.; Pham, C.; Gervasio, O.L.; Allen, D.G. N-Acetylcysteine ameliorates skeletal muscle pathophysiology in mdx mice. J Physiol. 2008, 586, 2003–2014. [Google Scholar] [CrossRef]
- Terrill, J.R.; Radley-Crabb, H.G.; Grounds, M.D.; Arthur, P.G. N-Acetylcysteine treatment of dystrophic mdx mice results in protein thiol modifications and inhibition of exercise induced myofibre necrosis. Neuromuscul. Disord. 2012, 22, 427–434. [Google Scholar] [CrossRef]
- Hornsey, M.A.; Laval, S.H.; Barresi, R.; Lochmüller, H.; Bushby, K. Muscular dystrophy in dysferlin-deficient mouse models. Neuromuscul. Disord. 2013, 23, 377–387. [Google Scholar] [CrossRef]
- Miller, N.J.; Rice-Evans, C.; Davies, M.J.; Gopinathan, V.; Milner, A. A novel method for measuring antioxidant capacity and its application to monitoring the antioxidant status in premature neonates. Clin. Sci. (Lond.) 1993, 84, 407–412. [Google Scholar] [CrossRef] [Green Version]
- Schnorr, C.E.; Morrone Mda, S.; Simões-Pires, A.; Bittencourt Lda, S.; Zeidán-Chuliá, F.; Moreira, J.C. Supplementation of adult rats with moderate amounts of β-carotene modulates the redox status in plasma without exerting pro-oxidant effects in the brain: A safer alternative to food fortification with vitamin A? Nutrients 2014, 6, 5572–5582. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.D.; Bock, P.M.; Becker, G.F.; Moreira, J.C.F.; Bello-Klein, A.; Oliveira, A.R. Comparison of the effects of two antioxidant diets on oxidative stress markers in triathletes. Biol. Sport. 2018, 35, 181–189. [Google Scholar]
- Gonos, E.S.; Kapetanou, M.; Sereikaite, J.; Bartosz, G.; Naparło, K.; Grzesik, M.; Sadowska-Bartosz, I. Origin and pathophysiology of protein carbonylation, nitration and chlorination in age-related brain diseases and aging. Aging (Albany NY) 2018, 10, 868–901. [Google Scholar] [CrossRef]
- Kozakowska, M.; Pietraszek-Gremplewicz, K.; Jozkowicz, A.; Dulak, J. The role of oxidative stress in skeletal muscle injury and regeneration: Focus on antioxidant enzymes. J. Muscle Res. Cell. Motil. 2015, 36, 377–393. [Google Scholar] [CrossRef] [Green Version]
- González-Jamett, A.M.; Bevilacqua, J.A.; Cárdenas, A.M. Hereditary Myopathies. In Muscle Cell and Tissue edited by Kunihiro Sakuma; IntechOpen: Rijeka, Croatia, 2018; pp. 252–268. [Google Scholar]
- Moore, U.; Jacobs, M.; James, M.K.; Mayhew, A.G.; Fernandez-Torron, R.; Feng, J.; Cnaan, A.; Eagle, M.; Bettinson, K.; Rufibach, L.E.; et al. Assessment of disease progression in dysferlinopathy: A 1-year cohort study. Neurology 2019. [Google Scholar] [CrossRef] [Green Version]
- Kondziela, W. Eine neue method zur messung der muskularen relaxation bei weissen mausen. Arch. Int. Pharmacodyn. 1964, 152, 277–284. [Google Scholar]
- De Araújo, R.F.F.; Martins, D.B.G.; Borba, M.A.C.S.M. Oxidative Stress and Disease. In The Transcription Factor Nrf2; Morales-Gonzalez, J.S., Morales-Gonzalez, A., Madrigal-Santillan, E.O., Eds.; IntechOpen: Rijeka, Croatia, 2016; pp. 185–189. [Google Scholar]
- Klover, P.; Chen, W.; Zhu, B.M.; Hennighausen, L. Skeletal muscle growth and fiber composition in mice are regulated through the transcription factors STAT5a/b: Linking growth hormone to the androgen receptor. FASEB J. 2009, 23, 3140–3148. [Google Scholar] [CrossRef] [Green Version]
- Mänttäri, S.; Järvilehto, M. Comparative analysis of mouse skeletal muscle fibre type composition and contractile responses to calcium channel blocker. BMC Physiol. 2005, 5, 4. [Google Scholar] [CrossRef] [Green Version]
- Powers, S.K.; Ji, L.L.; Kavazis, A.N.; Jackson, M.J. Reactive oxygen species: Impact on skeletal muscle. Compr. Physiol. 2011, 1, 941–969. [Google Scholar] [PubMed] [Green Version]
- Ho, M.; Post, C.M.; Donahue, L.R.; Lidov, H.G.W.; Bronson, R.T.; Goolsby, H.; Watkins, S.C.; Cox, G.A.; Brown, R.H., Jr. Disruption of muscle membrane and phenotype divergence in two novel mouse models of dysferlin deficiency. Hum. Mol. Genet. 2004, 13, 1999–2010. [Google Scholar] [CrossRef] [Green Version]
- Shin, J.; Tajrishi, M.M.; Ogura, Y.; Kumar, A. Wasting mechanisms in muscular dystrophy. Int. J. Biochem. Cell. Biol. 2013, 45, 2266–2279. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.R.; Ryu, H.H.; Chung, H.J.; Lee, J.H.; Kim, S.W.; Kwun, W.H.; Baek, S.H.; Kim, J.H. Association of anti-obesity activity of N-acetylcysteine with metallothionein-II down-regulation. Exp. Mol. Med. 2006, 38, 162–172. [Google Scholar] [CrossRef]
- Chang, Y.C.; Yu, Y.H.; Shew, J.Y.; Lee, W.J.; Hwang, J.J.; Chen, Y.H.; Chen, Y.R.; Wei, P.C.; Chuang, L.M.; Lee, W.H. Deficiency of NPGPx, an oxidative stress sensor, leads to obesity in mice and human. EMBO Mol. Med. 2013, 5, 1165–1179. [Google Scholar] [CrossRef]
- Ma, Y.; Gao, M.; Liu, D. N-acetylcysteine Protects Mice from High Fat Diet-induced Metabolic Disorders. Pharm. Res. 2016, 33, 2033–2042. [Google Scholar] [CrossRef] [Green Version]
- Pinniger, G.J.; Terrill, J.R.; Assan, E.B.; Grounds, M.D.; Arthur, P.G. Pre-clinical evaluation of N-acetylcysteine reveals side effects in the mdx mouse model of Duchenne muscular dystrophy. J. Physiol. 2017, 595, 7093–7107. [Google Scholar] [CrossRef]
- Smith, L.R.; Barton, E.R. Regulation of fibrosis in muscular dystrophy. Matrix Biol. 2018, 68–69, 602–615. [Google Scholar] [CrossRef] [PubMed]
- Mason, S.A.; Morrison, D.; McConell, G.K.; Wadley, G.D. Muscle redox signalling pathways in exercise. Role of antioxidants. Free Radic. Biol. Med. 2016, 98, 29–45. [Google Scholar] [CrossRef] [PubMed]
- McLeay, Y.; Stannard, S.; Houltham, S.; Starck, C. Dietary thiols in exercise: Oxidative stress defence, exercise performance, and adaptation. J. Int. Soc. Sports Nutr. 2017, 14, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Senzi Moraes Pinto, R.; Ferretti, R.; Moraes, L.H.; Neto, H.S.; Marques, M.J.; Minatel, E. N-acetylcysteine treatment reduces TNF-α levels and myonecrosis in diaphragm muscle of mdx mice. Clin. Nutr. 2013, 32, 472–475. [Google Scholar] [CrossRef]
- Burns, D.P.; Drummond, S.E.; Bolger, D.; Coiscaud, A.; Murphy, K.H.; Edge, D.; O’Halloran, K.D. N-acetylcysteine Decreases Fibrosis and Increases Force-Generating Capacity of mdx Diaphragm. Antioxidants (Basel) 2019, 8, 581. [Google Scholar] [CrossRef] [Green Version]
- De Medeiros, W.A.; da Silva, L.A.; Dall’Igna, D.M.; Michels, M.; Manfredini, A.; Santos Cardoso, L.D.; Constantino, L.; Scaini, G.; Vuolo, F.; Streck, E.L.; et al. N-acetylcysteine effects on a murine model of chronic critical limb ischemia. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 454–463. [Google Scholar] [CrossRef]
- Roseguini, B.T.; Silva, L.M.; Polotow, T.G.; Barros, M.P.; Souccar, C.; Han, S.W. Effects of N-acetylcysteine on skeletal muscle structure and function in a mouse model of peripheral arterial insufficiency. J. Vasc Surg. 2015, 61, 777–786. [Google Scholar] [CrossRef] [Green Version]
- Faulkner, J.A.; Ng, R.; Davis, C.S.; Li, S.; Chamberlain, J.S. Diaphragm muscle strip preparation for evaluation of gene therapies in mdx mice. Clin. Exp. Pharmacol. Physiol. 2008, 35, 725–729. [Google Scholar] [CrossRef]
- Barton, E.R.; Wang, B.J.; Brisson, B.K.; Sweeney, H.L. Diaphragm displays early and progressive functional deficits in dysferlin deficient mice. Muscle Nerve 2010, 42, 22–29. [Google Scholar] [CrossRef] [Green Version]
- Gumerson, J.D.; Michele, D.E. The dystrophin-glycoprotein complex in the prevention of muscle damage. J. Biomed. Biotechnol. 2011, 2011, 210797. [Google Scholar] [CrossRef]
- Allen, D.G.; Whitehead, N.P.; Froehner, S.C. Absence of dystrophin disrupts skeletal muscle signaling: Roles of Ca2+, reactive oxygen species, and nitric oxide in the development of muscular dystrophy. Physiol. Rev. 2016, 96, 253–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janke, A.; Upadhaya, R.; Snow, W.M.; Anderson, J.E. A new look at cytoskeletal NOS-1 and β-dystroglycan changes in developing muscle and brain in control and mdx dystrophic mice. Dev. Dyn. 2013, 242, 1369–1381. [Google Scholar] [CrossRef] [PubMed]
- Jelinkova, S.; Fojtik, P.; Kohutova, A.; Vilotic, A.; Markováe, L.; Pesl, M.; Jurakova, T.; Kruta, M.; Vrbsky, J.; Gaillyova, R.; et al. Dystrophin Deficiency Leads to Genomic Instability in Human Pluripotent Stem Cells via NO Synthase-Induced Oxidative Stress. Cells 2019, 8, 53. [Google Scholar] [CrossRef] [Green Version]
- Cea, L.A.; Bevilacqua, J.A.; Arriagada, C.; Cárdenas, A.M.; Bigot, A.; Mouly, V.; Sáez, J.C.; Caviedes, P. The absence of dysferlin induces the expression of functional connexin-based hemichannels in human myotubes. BMC Cell Biol. 2016, 17 (Suppl. 1), 15. [Google Scholar] [CrossRef] [Green Version]
- Fernández, G.; Arias-Bravo, G.; Bevilacqua, J.A.; Castillo-Ruiz, M.; Caviedes, P.; Sáez, J.C.; Cea, L.A. Myofibers deficient in connexins 43 and 45 expression protect mice from skeletal muscle and systemic dysfunction promoted by a dysferlin mutation. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165800. [Google Scholar] [CrossRef] [PubMed]
- Lingappan, K. NF-κB in Oxidative Stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef]
- Fanin, M.; Nascimbeni, A.C.; Angelini, C. Muscle atrophy, ubiquitin-proteasome, and autophagic pathways in dysferlinopathy. Muscle Nerve 2014, 50, 340–347. [Google Scholar] [CrossRef]
- Huang, J.; Zhu, X. The molecular mechanisms of calpains action on skeletal muscle atrophy. Physiol. Res. 2016, 65, 547–560. [Google Scholar] [CrossRef]
- Pei, Y.; Liu, H.; Yang, Y.; Yang, Y.; Jiao, Y.; Tay, F.R.; Chen, J. Biological Activities and Potential Oral Applications of N-Acetylcysteine: Progress and Prospects. Oxid. Med. Cell Longev. 2018, 2018, 2835787. [Google Scholar] [CrossRef]
- Ooi, S.L.; Green, R.; Pak, S.C. N-Acetylcysteine for the Treatment of Psychiatric Disorders: A Review of Current Evidence. BioMed Res. Int. 2018, 2018, 2469486. [Google Scholar] [CrossRef] [Green Version]
- Janeczek, M.; Moy, L.; Riopelle, A.; Vetter, O.; Reserva, J.; Tung, R.; Swan, J. The Potential Uses of N-acetylcysteine in Dermatology: A Review. J. Clin. Aesthet. Dermatol. 2019, 12, 20–26. [Google Scholar] [PubMed]
- Feng, F.; Zhang, J.; Wang, Z.; Wu, Q.; Zhou, X. Efficacy and safety of N-acetylcysteine therapy for idiopathic pulmonary fibrosis: An updated systematic review and meta-analysis. Exp. Ther. Med. 2019, 18, 802–816. [Google Scholar] [PubMed]
- Koppen, A.; van Riel, A.; de Vries, I.; Meulenbelt, J. Recommendations for the paracetamol treatment nomogram and side effects of N-acetylcysteine. Neth. J. Med. 2014, 72, 251–257. [Google Scholar] [PubMed]
- Todd, J.J.; Lawal, T.A.; Witherspoon, J.W.; Chrismer, I.C.; Razaqyar, M.S.; Punjabi, M.; Elliott, J.S.; Tounkara, F.; Kuo, A.; Shelton, M.O.; et al. Randomized controlled trial of N-acetylcysteine therapy for RYR1-related myopathies. Neurology 2020, 94, e1434–e1444. [Google Scholar] [CrossRef] [Green Version]
- Crouch, B.I.; Caravati, E.M.; Dandoy, C. Effect of dilution with beverages on the smell and taste of oral acetylcysteine. Am. J. Health Syst. Pharm. 2007, 64, 1965–1968. [Google Scholar] [CrossRef]
- Waring, W.S. Novel acetylcysteine regimens for treatment of paracetamol overdose. Ther. Adv. Drug Saf. 2012, 3, 305–315. [Google Scholar] [CrossRef] [Green Version]
- Báez-Matus, X.; Figueroa-Cares, C.; Gónzalez-Jamett, A.M.; Almarza-Salazar, H.; Arriagada, C.; Maldifassi, M.C.; Guerra, M.J.; Mouly, V.; Bigot, A.; Caviedes, P.; et al. Defects in G-actin incorporation into filaments in myoblasts derived from dysferlinopathy patients are restored by dysferlin C2 domains. Int. J. Mol. Sci. 2019, 21, 37. [Google Scholar] [CrossRef] [Green Version]
- González-Jamett, A.M.; Baez-Matus, X.; Olivares, M.J.; Hinostroza, F.; Guerra-Fernández, M.J.; Vasquez-Navarrete, J.; Bui, M.T.; Guicheney, P.; Romero, N.B.; Bevilacqua, J.A.; et al. Dynamin-2 mutations linked to Centronuclear Myopathy impair actin-dependent trafficking in muscle cells. Sci. Rep. 2017, 7, 4580. [Google Scholar] [CrossRef] [Green Version]
- Potter, R.A.; Griffin, D.A.; Sondergaard, P.C.; Johnson, R.W.; Pozsgai, E.R.; Heller, K.N.; Peterson, E.L.; Lehtimäki, K.K.; Windish, H.P.; Mittal, P.J.; et al. Systemic Delivery of Dysferlin Overlap Vectors Provides Long-Term Gene Expression and Functional Improvement for Dysferlinopathy. Hum. Gene Ther. 2018, 29, 749–762. [Google Scholar] [CrossRef] [Green Version]
- Tabart, J.; Kevers, C.; Pincemail, J.; Defraigne, J.O.; Dommes, J. Comparative antioxidant capacities of phenolic compounds measured by various tests. Food Chem. 2009, 113, 1226–1233. [Google Scholar] [CrossRef]
- Romay, C.; Pascual, C.; Lissi, E.A. The reaction between ABTS radical cation and antioxidants and its use to evaluate the antioxidant status of serum samples. Braz. J. Med. Biol. Res. 1996, 29, 175–183. [Google Scholar] [PubMed]
- Esterbauer, H.; Cheeseman, K.H.; Dianzani, M.U.; Poli, G.; Slater, T.F. Separation and characterization of the aldehydic products of lipid peroxidation stimulated by ADP-Fe2+ in rat liver microsomes. Biochem. J. 1982, 208, 129–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, R.L.; Garland, D.; Oliver, C.N.; Amici, A.; Climent, I.; Lenz, A.G.; Ahn, B.W.; Shaltiel, S.; Stadtman, E.R. Determination of carbonyl content in oxidatively modified proteins. Methods Enzymol. 1990, 186, 464–478. [Google Scholar] [PubMed]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [PubMed]
- Beauchamp, C.; Fridovich, I. Superoxide dismutase: Improved assays and an assay applicable to acrylamide gels. Anal. Biochem. 1971, 44, 276–287. [Google Scholar] [CrossRef]
- Aebi, H. Catalase in vitro. Methods Enzymol. 1984, 105, 121–126. [Google Scholar]
- Deacon, R.M. Measuring the strength of mice. J. Vis. Exp. 2013, 2, 76. [Google Scholar] [CrossRef]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Campos, P.; Báez-Matus, X.; Jara-Gutiérrez, C.; Paz-Araos, M.; Astorga, C.; Cea, L.A.; Rodríguez, V.; Bevilacqua, J.A.; Caviedes, P.; Cárdenas, A.M. N-Acetylcysteine Reduces Skeletal Muscles Oxidative Stress and Improves Grip Strength in Dysferlin-Deficient Bla/J Mice. Int. J. Mol. Sci. 2020, 21, 4293. https://doi.org/10.3390/ijms21124293
García-Campos P, Báez-Matus X, Jara-Gutiérrez C, Paz-Araos M, Astorga C, Cea LA, Rodríguez V, Bevilacqua JA, Caviedes P, Cárdenas AM. N-Acetylcysteine Reduces Skeletal Muscles Oxidative Stress and Improves Grip Strength in Dysferlin-Deficient Bla/J Mice. International Journal of Molecular Sciences. 2020; 21(12):4293. https://doi.org/10.3390/ijms21124293
Chicago/Turabian StyleGarcía-Campos, Paz, Ximena Báez-Matus, Carlos Jara-Gutiérrez, Marilyn Paz-Araos, César Astorga, Luis A. Cea, Viviana Rodríguez, Jorge A. Bevilacqua, Pablo Caviedes, and Ana M. Cárdenas. 2020. "N-Acetylcysteine Reduces Skeletal Muscles Oxidative Stress and Improves Grip Strength in Dysferlin-Deficient Bla/J Mice" International Journal of Molecular Sciences 21, no. 12: 4293. https://doi.org/10.3390/ijms21124293
APA StyleGarcía-Campos, P., Báez-Matus, X., Jara-Gutiérrez, C., Paz-Araos, M., Astorga, C., Cea, L. A., Rodríguez, V., Bevilacqua, J. A., Caviedes, P., & Cárdenas, A. M. (2020). N-Acetylcysteine Reduces Skeletal Muscles Oxidative Stress and Improves Grip Strength in Dysferlin-Deficient Bla/J Mice. International Journal of Molecular Sciences, 21(12), 4293. https://doi.org/10.3390/ijms21124293