The Role of Estrogen Receptors in Cardiovascular Disease


 and
and
Abstract
:1. Introduction
2. Role of Estrogen Receptors in Vascular Pathology
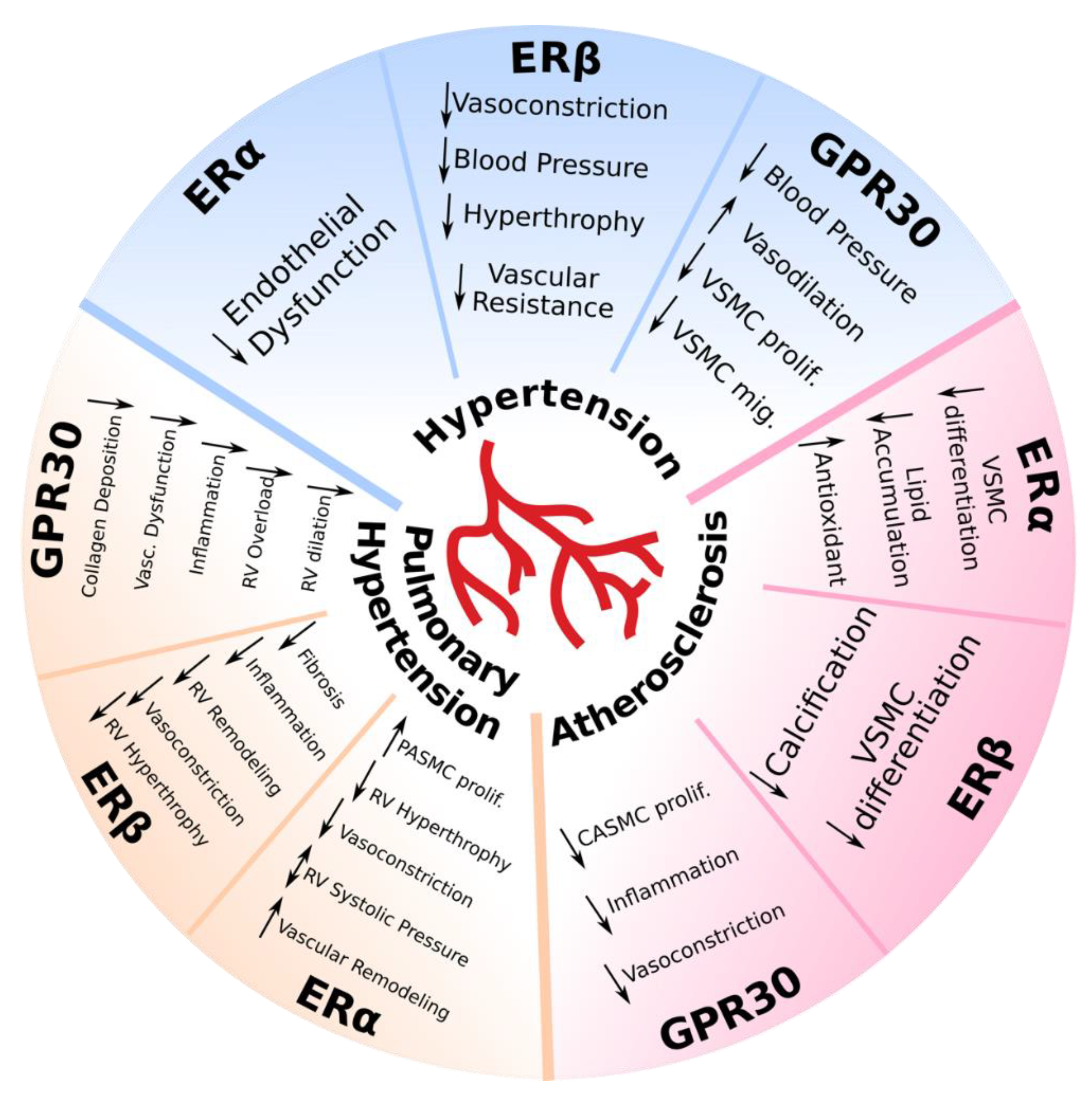
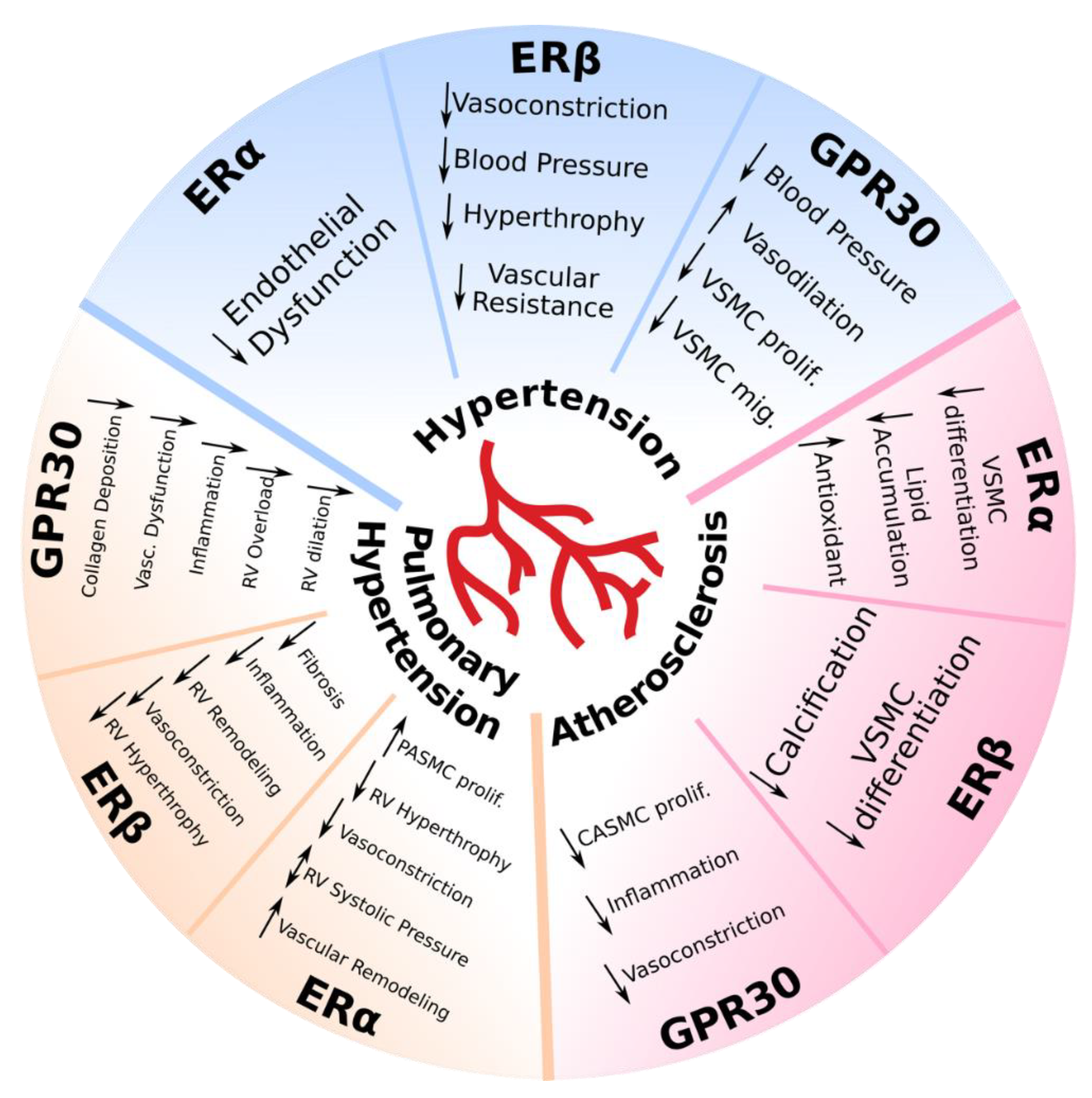
2.1. Hypertension
2.2. Pulmonary Hypertension
2.3. Atherosclerosis
3. Role of Estrogen Receptors in Heart Pathology
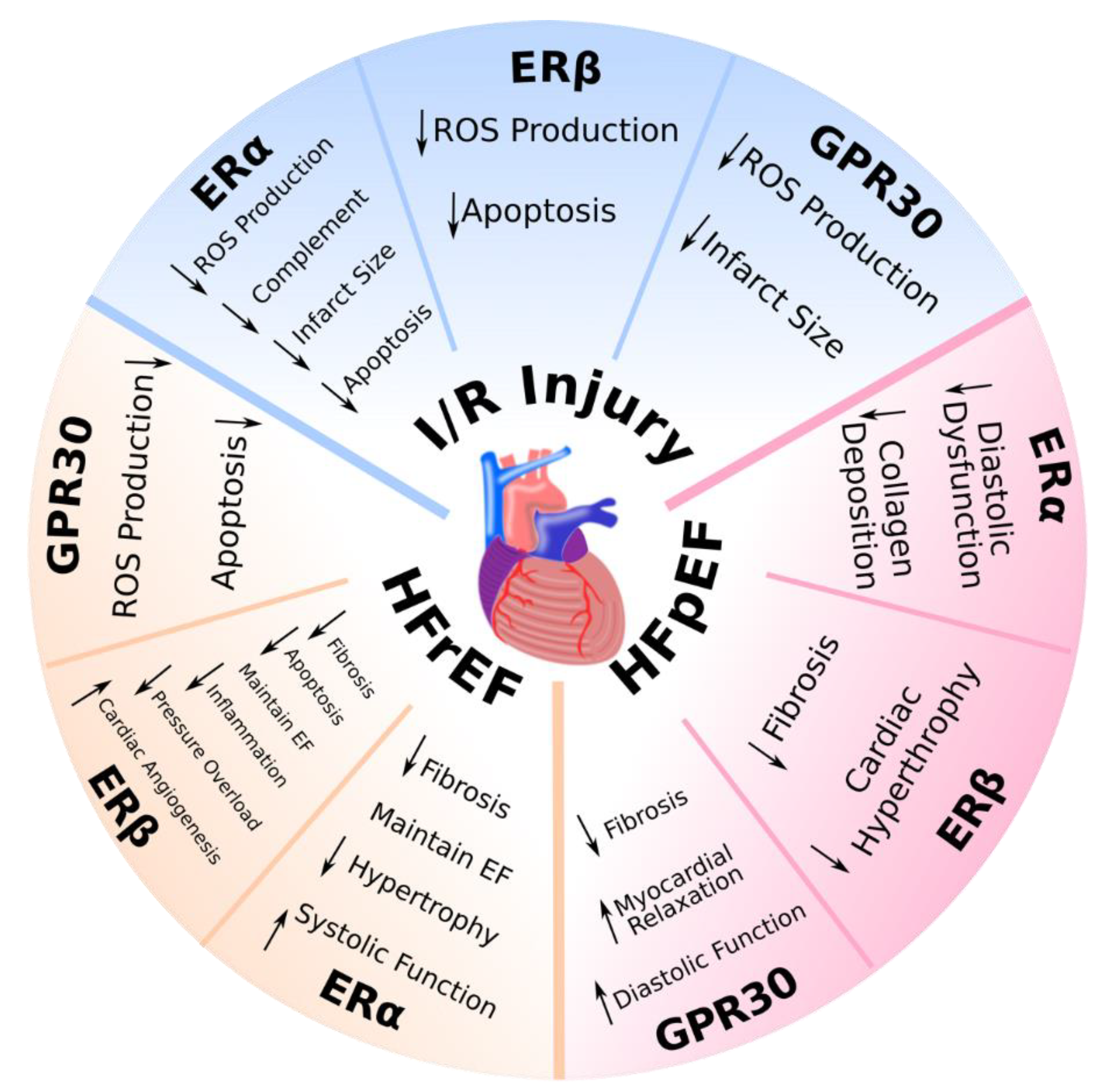
3.1. Ischemia/Reperfusion Injury
3.2. Heart Failure with Reduced Ejection Fraction
3.3. Heart Failure with Preserved Ejection Fraction
4. Future Perspectives and Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ADAM | a disintegrin and metalloproteinases |
| AF1 | activation function 1 |
| AF2 | activation function 2 |
| Akt | protein kinase B |
| Ang II | angiotensin II |
| ApoE | apolipoprotein E-deficient |
| BMPR2 | bone morphogenetic protein receptor type 2 |
| BP | blood pressure |
| c-SRC | proto-oncogene tyrosine-protein kinase |
| CVD | cardiovascular disease |
| EF | ejection fraction |
| EGFR | epidermal growth factor receptor |
| ERK1/2 | extracellular signal-regulated protein kinases 1 and 2 |
| eNOS | endothelial nitric oxide synthase |
| ER | estrogen receptor |
| ERα | estrogen receptor alpha |
| ERβ | estrogen receptor beta |
| ERE | estrogen response element |
| ERK | extracellular-signal-regulated kinase |
| E2 | 17beta-estradiol |
| GPER | G protein–coupled estrogen receptor 1 |
| GPR30 | G-protein-coupled estrogen receptor |
| GSK-3β | glycogen synthase kinase 3 |
| HF | heart failure |
| HFrEF | heart failure with reduced ejection fraction |
| HFpEF | heart failure with preserved ejection Fraction |
| HIF-1α | hypoxia-inducible factor 1α |
| HTN | hypertension |
| iNOS | inducible nitric oxide synthase |
| I/R injury | ischemia-reperfusion injury |
| JNK | c-Jun N-terminal kinase |
| KO | knockout |
| LDL | low-density lipoprotein |
| LV | left ventricular |
| LVEF | left ventricular ejection fraction |
| MAPK | mitogen-activated protein kinase |
| MCT | monocrotaline |
| NO | nitric oxide |
| NOS | nitric oxide synthase |
| NOX4 | NAD(P)H oxidase |
| OVX | ovariectomized |
| PAH | pulmonary arterial hypertension |
| PASMC | pulmonary artery smooth muscle cell |
| PH | pulmonary hypertension |
| PI3K | phosphoinositide-3-kinase |
| P53 | tumor protein p53 |
| RV | right ventricular |
| RVEF | right ventricular ejection fraction |
| RAS | renin-angiotensin system |
| ROS | reactive oxygen species |
| SERT | serotonin transporter |
| Su/Hx PH | SU5416/hypoxia-induced pulmonary hypertension |
| TAC | transverse aortic constriction |
| TGF | transforming growth factor |
| VSMC | vascular smooth muscle cells |
| WT | wildtype |
References
- Mosca, L.; Barrett-Connor, E.; Wenger, N.K. Sex/gender differences in cardiovascular disease prevention: What a difference a decade makes. Circulation 2011, 124, 2145–2154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.-P.; Reckelhoff, J.F. Estrogen, hormonal replacement therapy and cardiovascular disease. Curr. Opin. Nephrol. Hypertens. 2011, 20, 133–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, E. Estrogen signaling and cardiovascular disease. Circ. Res. 2011, 109, 687–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, D.; Guallar, E.; Ouyang, P.; Subramanya, V.; Vaidya, D.; Ndumele, C.E.; Lima, J.A.; Allison, M.A.; Shah, S.J.; Bertoni, A.G.; et al. Endogenous Sex Hormones and Incident Cardiovascular Disease in Post-Menopausal Women. J. Am. Coll. Cardiol. 2018, 71, 2555–2566. [Google Scholar] [CrossRef]
- Marino, M.; Galluzzo, P.; Ascenzi, P. Estrogen Signaling Multiple Pathways to Impact Gene Transcription. Curr. Genom. 2006, 7, 497–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuentes, N.; Silveyra, P. Estrogen receptor signaling mechanisms. Adv. Protein Chem. Struct. Biol. 2019, 116, 135–170. [Google Scholar]
- Revankar, C.M. A Transmembrane Intracellular Estrogen Receptor Mediates Rapid Cell Signaling. Science 2005, 307, 1625–1630. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Sun, X.; Chou, J.; Lin, M.; Ferrario, C.M.; Zapata-Sudo, G.; Groban, L. Cardiomyocyte-specific deletion of the G protein-coupled estrogen receptor (GPER) leads to left ventricular dysfunction and adverse remodeling: A sex-specific gene profiling analysis. Biochim. Biophys. Acta Mol. Basis Dis. 2016, 1863, 1870–1882. [Google Scholar] [CrossRef]
- Deschamps, A.; Murphy, E. Activation of a novel estrogen receptor, GPER, is cardioprotective in male and female rats. Am. J. Physiol. Circ. Physiol. 2009, 297, H1806–H1813. [Google Scholar] [CrossRef] [Green Version]
- Menazza, S.; Murphy, E. The Expanding Complexity of Estrogen Receptor Signaling in the Cardiovascular System. Circ. Res. 2016, 118, 994–1007. [Google Scholar] [CrossRef]
- Iorga, A.; Cunningham, C.M.; Moazeni, S.; Ruffenach, G.; Umar, S.; Eghbali, M. The protective role of estrogen and estrogen receptors in cardiovascular disease and the controversial use of estrogen therapy. Boil. Sex. Differ. 2017, 8, 33. [Google Scholar] [CrossRef] [PubMed]
- 2017 Hypertension Clinical Guidelines. Available online: https://professional.heart.org/professional/ScienceNews/UCM_496965_2017-Hypertension-Clinical-Guidelines.jsp (accessed on 5 June 2020).
- Writing Group Members; Roger, V.L.; Go, A.S.; Lloyd-Jones, N.M.; Benjamin, E.J.; Berry, J.D.; Borden, W.B.; Bravata, D.M.; Dai, S.; Ford, E.S.; et al. Heart Disease and Stroke Statistics—2012 Update. Circulation 2012, 125. [Google Scholar] [CrossRef]
- Lawes, C.M.; Hoorn, S.V.; Rodgers, A. Global burden of blood-pressure-related disease. Lancet 2008, 371, 1513–1518. [Google Scholar] [CrossRef]
- Ahmad, A.; Oparil, S. Hypertension in Women. Hypertension 2017, 70, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.S.; Carroll, M.D.; Fryar, C.D. Hypertension Prevalence and Control among Adults: United States, 2011–2014. NCHS Data Brief 2015, 220, 1–8. [Google Scholar]
- Maranon, R.; Reckelhoff, J.F. Sex and gender differences in control of blood pressure. Clin. Sci. 2013, 125, 311–318. [Google Scholar] [CrossRef] [Green Version]
- Meyer, M.; Prossnitz, E.R.; Barton, M. GPER/GPR30 and Regulation of Vascular Tone and Blood Pressure. Immunol. Endocr. Metab. Agents Med. Chem. 2011, 11, 255–261. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.K.; Levin, E.R. Estrogen signaling in the cardiovascular system. Nucl. Recept. Signal. 2006, 4, e013. [Google Scholar] [CrossRef]
- Weiner, C.P.; Lizasoain, I.; Baylis, S.A.; Knowles, R.G.; Charles, I.G.; Moncada, S. Induction of calcium-dependent nitric oxide synthases by sex hormones. Proc. Natl. Acad. Sci. USA 1994, 91, 5212–5216. [Google Scholar] [CrossRef] [Green Version]
- Xue, B.; Pamidimukkala, J.; Lubahn, D.B.; Hay, M. Estrogen receptor-α mediates estrogen protection from angiotensin II-induced hypertension in conscious female mice. Am. J. Physiol. Circ. Physiol. 2007, 292, H1770–H1776. [Google Scholar] [CrossRef] [Green Version]
- Guivarc’H, E.; Guihot, A.; Favre, J.; Vessières, E.; Wakim, J.; Arnal, J.-F.; Lenfant, F.; Loufrani, L.; Henrion, D. CO-46: Protective role of the estrogen receptor alpha during hypertension. Ann. Cardiol. D’angéiologie 2015, 64, S21–S22. [Google Scholar] [CrossRef]
- Widder, J.; Pelzer, T.; Von Poser-Klein, C.; Hu, K.; Jazbutyte, V.; Fritzemeier, K.-H.; Hegele-Hartung, C.; Neyses, L.; Bauersachs, J. Improvement of Endothelial Dysfunction by Selective Estrogen Receptor-α Stimulation in Ovariectomized SHR. Hypertension 2003, 42, 991–996. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Bian, Z.; Lu, P.; Karas, R.H.; Bao, L.; Cox, D.; Hodgin, J.; Shaul, P.W.; Thorén, P.; Smithies, O.; et al. Abnormal Vascular Function and Hypertension in Mice Deficient in Estrogen Receptor beta. Science 2002, 295, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Jazbutyte, V.; Arias-Loza, P.A.; Hu, K.; Widder, J.; Govindaraj, V.; Von Poser-Klein, C.; Bauersachs, J.; Fritzemeier, K.-H.; Hegele-Hartung, C.; Neyses, L.; et al. Ligand-dependent activation of ER lowers blood pressure and attenuates cardiac hypertrophy in ovariectomized spontaneously hypertensive rats. Cardiovasc. Res. 2007, 77, 774–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, E.; Bhattacharya, I.; Brailoiu, E.; Damjanović, M.; Brailoiu, G.C.; Gao, X.; Mueller-Guerre, L.; Marjon, N.A.; Gut, A.; Minotti, R.; et al. Regulatory role of G protein-coupled estrogen receptor for vascular function and obesity. Circ. Res. 2009, 104, 288–291. [Google Scholar] [CrossRef] [Green Version]
- Owens, G.K.; Kumar, M.S.; Wamhoff, B.R. Molecular Regulation of Vascular Smooth Muscle Cell Differentiation in Development and Disease. Physiol. Rev. 2004, 84, 767–801. [Google Scholar] [CrossRef]
- Pare, G.; Krust, A.; Karas, R.H.; Dupont, S.; Aronovitz, M.; Chambon, P.; Mendelsohn, M.E. Estrogen Receptor-α Mediates the Protective Effects of Estrogen against Vascular Injury. Circ. Res. 2002, 90, 1087–1092. [Google Scholar] [CrossRef] [Green Version]
- Smirnova, N.; Fontaine, C.; Buscato, M.; Lupieri, A.; Vinel, A.; Valera, M.-C.; Guillaume, M.; Malet, N.; Foidart, J.-M.; Raymond-Letron, I.; et al. The Activation Function-1 of Estrogen Receptor Alpha Prevents Arterial Neointima Development through a Direct Effect on Smooth Muscle Cells. Circ. Res. 2015, 117, 770–778. [Google Scholar] [CrossRef]
- Fredette, N.C.; Meyer, M.R.; Prossnitz, E.R. Role of GPER in estrogen-dependent nitric oxide formation and vasodilation. J. Steroid Biochem. Mol. Boil. 2018, 176, 65–72. [Google Scholar] [CrossRef] [Green Version]
- Aryan, L.; Medzikovic, L.; Umar, S.; Eghbali, M. Pregnancy-associated cardiac dysfunction and the regulatory role of microRNAs. Boil. Sex. Differ. 2020, 11, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Large, M.J.; Wetendorf, M.; Lanz, R.B.; Hartig, S.M.; Creighton, C.J.; Mancini, M.A.; Kovanci, E.; Lee, K.-F.; Threadgill, D.W.; Lydon, J.P.; et al. The Epidermal Growth Factor Receptor Critically Regulates Endometrial Function during Early Pregnancy. PLoS Genet. 2014, 10, e1004451. [Google Scholar] [CrossRef] [Green Version]
- Nadadur, R.D.; Umar, S.; Wong, G.; Eghbali, M.; Iorga, A.; Matori, H.; Partow-Navid, R.; Eghbali, M. Reverse right ventricular structural and extracellular matrix remodeling by estrogen in severe pulmonary hypertension. J. Appl. Physiol. 2012, 113, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Rabinovitch, M.; Gamble, W.J.; Miettinen, O.S.; Reid, L. Age and sex influence on pulmonary hypertension of chronic hypoxia and on recovery. Am. J. Physiol. Circ. Physiol. 1981, 240, H62–H72. [Google Scholar] [CrossRef] [PubMed]
- Umar, S.; Rabinovitch, M.; Eghbali, M. Estrogen Paradox in Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2012, 186, 125–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Memon, H.A.; Park, M.H. Pulmonary Arterial Hypertension in Women. Methodist DeBakey Cardiovasc. J. 2017, 13, 224–237. [Google Scholar] [PubMed]
- Resta, T.C.; Kanagy, N.L.; Walker, B.R. Estradiol-induced attenuation of pulmonary hypertension is not associated with altered eNOS expression. Am. J. Physiol. Cell. Mol. Physiol. 2001, 280, L88–L97. [Google Scholar] [CrossRef]
- Farhat, M.Y.; Chen, M.-F.; Bhatti, T.; Iqbal, A.; Cathapermal, S.; Ramwell, P.W. Protection by oestradiol against the development of cardiovascular changes associated with monocrotaline pulmonary hypertension in rats. Br. J. Pharmacol. 1993, 110, 719–723. [Google Scholar] [CrossRef] [Green Version]
- Umar, S.; Iorga, A.; Matori, H.; Nadadur, R.D.; Li, J.; Maltese, F.; Van Der Laarse, A.; Eghbali, M. Estrogen Rescues Preexisting Severe Pulmonary Hypertension in Rats. Am. J. Respir. Crit. Care Med. 2011, 184, 715–723. [Google Scholar] [CrossRef]
- Wang, Y.D.; Li, Y.D.; Ding, X.Y.; Wu, X.P.; Li, C.; Guo, D.C.; Shi, Y.P.; Lv, X.Z. 17β-estradiol preserves right ventricular function in rats with pulmonary arterial hypertension An echocardiographic and histochemical study. Int. J. Cardiovasc. Imaging 2018, 35, 441–450. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, W.; Van De Veerdonk, M.C.; Trip, P.; Man, F.H.D.; Heymans, M.W.; Marcus, J.T.; Kawut, S.M.; Bogaard, H.J.; Boonstra, A.; Noordegraaf, A.V. The right ventricle explains sex differences in survival in idiopathic pulmonary arterial hypertension. Chest 2014, 145, 1230–1236. [Google Scholar] [CrossRef] [Green Version]
- Frump, A.; Goss, K.N.; Vayl, A.; Albrecht, M.; Fisher, A.J.; Tursunova, R.; Fierst, J.; Whitson, J.; Cucci, A.R.; Brown, M.B.; et al. Estradiol improves right ventricular function in rats with severe angioproliferative pulmonary hypertension Effects of endogenous and exogenous sex hormones. Am. J. Physiol. Cell. Mol. Physiol. 2015, 308, L873–L890. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.F.; Ewart, M.-A.; Mair, K.; Nilsen, M.; Dempsie, Y.; Loughlin, L.; MacLean, M.R. Oestrogen receptor alpha in pulmonary hypertension. Cardiovasc. Res. 2015, 106, 206–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Austin, E.D.; Hamid, R.; Hemnes, A.; Loyd, J.; Blackwell, T.; Yu, C.; Phillips, J.A.; Gaddipati, R.; Gladson, S.; Gu, E.; et al. BMPR2 expression is suppressed by signaling through the estrogen receptor. Boil. Sex. Differ. 2012, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrell, N.W. Pulmonary Hypertension Due to BMPR2 Mutation: A New Paradigm for Tissue Remodeling? Proc. Am. Thorac. Soc. 2006, 3, 680–686. [Google Scholar] [CrossRef]
- Lane, K.B.; Machado, R.D.; Pauciulo, M.W.; Thomson, J.R.; Phillips, J.A.; Loyd, J.; Nichols, W.C.; Trembath, R.C. Heterozygous germline mutations in BMPR2, encoding a TGF-β receptor, cause familial primary pulmonary hypertension. Nat. Genet. 2000, 26, 81–84. [Google Scholar] [CrossRef]
- Hamid, R.; Cogan, J.D.; Hedges, L.K.; Austin, E.; Phillips, J.A.; Newman, J.H.; Loyd, J. Penetrance of pulmonary arterial hypertension is modulated by the expression of normalBMPR2allele. Hum. Mutat. 2009, 30, 649–654. [Google Scholar] [CrossRef] [Green Version]
- White, K.; Dempsie, Y.; Nilsen, M.; Wright, A.F.; Loughlin, L.; MacLean, M.R. The serotonin transporter, gender, and 17 oestradiol in the development of pulmonary arterial hypertension. Cardiovasc. Res. 2010, 90, 373–382. [Google Scholar] [CrossRef]
- Frump, A.; Selej, M.; A Wood, J.; Albrecht, M.; Yakubov, B.; Petrache, I.; Lahm, T. Hypoxia Upregulates Estrogen Receptor β in Pulmonary Artery Endothelial Cells in a HIF-1α–Dependent Manner. Am. J. Respir. Cell Mol. Boil. 2018, 59, 114–126. [Google Scholar] [CrossRef]
- Pedram, A.; Razandi, M.; Narayanan, R.; Levin, E.R. Estrogen receptor beta signals to inhibition of cardiac fibrosis. Mol. Cell. Endocrinol. 2016, 434, 57–68. [Google Scholar] [CrossRef]
- Matori, H.; Umar, S.; Nadadur, R.D.; Sharma, S.; Partow-Navid, R.; Afkhami, M.; Amjadi, M.; Eghbali, M. Genistein, a soy phytoestrogen, reverses severe pulmonary hypertension and prevents right heart failure in rats. Hypertension 2012, 60, 425–430. [Google Scholar] [CrossRef]
- Umar, S.; Partow-Navid, R.; Ruffenach, G.; Iorga, A.; Moazeni, S.; Eghbali, M. Severe pulmonary hypertension in aging female apolipoprotein E-deficient mice is rescued by estrogen replacement therapy. Boil. Sex. Differ. 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lahm, T.; Albrecht, M.; Fisher, A.J.; Selej, M.; Patel, N.G.; Brown, J.A.; Justice, M.J.; Brown, M.B.; Van Demark, M.; Trulock, K.M.; et al. 17β-Estradiol Attenuates Hypoxic Pulmonary Hypertension via Estrogen Receptor–mediated Effects. Am. J. Respir. Crit. Care Med. 2012, 185, 965–980. [Google Scholar] [CrossRef] [PubMed]
- Lahm, T.; Crisostomo, P.R.; Markel, T.A.; Wang, M.; Wang, Y.; Tan, J.; Meldrum, D.R. Selective estrogen receptor-alpha and estrogen receptor-beta agonists rapidly decrease pulmonary artery vasoconstriction by a nitric oxide-dependent mechanism. Am. J. Physiol. Integr. Comp. Physiol. 2008, 295, R1486–R1493. [Google Scholar] [CrossRef] [PubMed]
- Alencar, A.K.; Montes, G.C.; Montagnoli, T.; Silva, A.M.S.; Martinez, S.T.; Fraga, A.G.; Wang, H.; Groban, L.; Sudo, R.T.; Zapata-Sudo, G. Activation of GPER ameliorates experimental pulmonary hypertension in male rats. Eur. J. Pharm. Sci. 2016, 97, 208–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barton, M. Not lost in translation: Emerging clinical importance of the G protein-coupled estrogen receptor GPER. Steroids 2016, 111, 37–45. [Google Scholar] [CrossRef] [Green Version]
- Alencar, A.K.N.; Montes, G.C.; Costa, D.G.; Mendes, L.V.P.; Silva, A.M.S.; Martinez, S.T.; Trachez, M.M.; Cunha, V.D.M.N.; Montagnoli, T.; Fraga, A.G.M.; et al. Cardioprotection Induced by Activation of GPER in Ovariectomized Rats with Pulmonary Hypertension. J. Gerontol. Ser. A Boil. Sci. Med. Sci. 2018, 73, 1158–1166. [Google Scholar] [CrossRef] [Green Version]
- Insull, W. The Pathology of Atherosclerosis: Plaque Development and Plaque Responses to Medical Treatment. Am. J. Med. 2009, 122, S3–S14. [Google Scholar] [CrossRef]
- Shen, D.; Tian, L.; Shen, T.; Sun, H.; Liu, P. Alpha-Lipoic Acid Protects Human Aortic Endothelial Cells against H2O2-Induced Injury and Inhibits Atherosclerosis in Ovariectomized Low Density Lipoprotein Receptor Knock-out Mice. Cell. Physiol. Biochem. 2018, 47, 2261–2277. [Google Scholar] [CrossRef]
- Fairweather, D. Sex Differences in Inflammation during Atherosclerosis. Clin. Med. Insights: Cardiol. 2014, 8, 49–59. [Google Scholar] [CrossRef]
- Billon-Galés, A.; Fontaine, C.; Douin-Echinard, V.; Delpy, L.; Berges, H.; Calippe, B.; Lenfant, F.; Laurell, H.; Guéry, J.-C.; Gourdy, P.; et al. Endothelial Estrogen Receptor-α Plays a Crucial Role in the Atheroprotective Action of 17β-Estradiol in Low-Density Lipoprotein Receptor–Deficient Mice. Circulation 2009, 120, 2567–2576. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Shi, J.; Luu, T.N.; Neuman, J.C.; Trefts, E.; Yu, S.; Palmisano, B.T.; Wasserman, D.H.; Linton, M.F.; Stafford, J.M. Hepatocyte estrogen receptor alpha mediates estrogen action to promote reverse cholesterol transport during Western-type diet feeding. Mol. Metab. 2017, 8, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Campesi, I.; Marino, M.; Montella, A.; Pais, S.; Franconi, F. Sex Differences in Estrogen Receptor α and β Levels and Activation Status in LPS-Stimulated Human Macrophages. J. Cell. Physiol. 2016, 232, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Min, J.; Weitian, Z.; Peng, C.; Yan, P.; Bo, Z.; Wang, Y.; Bai, Y.; Wang, X. Correlation between insulin-induced estrogen receptor methylation and atherosclerosis. Cardiovasc. Diabetol. 2016, 15, 156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortmann, J.; Veit, M.; Zingg, S.; Di Santo, S.; Traupe, T.; Yang, Z.; Völzmann, J.; Dubey, R.K.; Christen, S.; Baumgartner, I. Estrogen receptor-α but not -β or GPER inhibits high glucose-induced human VSMC proliferation Potential role of ROS and ERK. J. Clin. Endocrinol. Metab. 2010, 96, 220–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyle, A.; Griendling, K.K. Modulation of Vascular Smooth Muscle Signaling by Reactive Oxygen Species. Physiology 2006, 21, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Billon-Galés, A.; Krust, A.; Fontaine, C.; Abot, A.; Flouriot, G.; Toutain, C.; Berges, H.; Gadeau, A.-P.; Lenfant, F.; Gourdy, P.; et al. Activation function 2 (AF2) of estrogen receptor-α is required for the atheroprotective action of estradiol but not to accelerate endothelial healing. Proc. Natl. Acad. Sci. USA 2011, 108, 13311–13316. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; Zhang, H.; Luo, Y.; Zhang, J.; Wang, M.; Liao, P.; Cao, L.; Guo, P.; Sun, G.; Sun, X. Gypenoside XVII Prevents Atherosclerosis by Attenuating Endothelial Apoptosis and Oxidative Stress: Insight into the ERα-Mediated PI3K/Akt Pathway. Int. J. Mol. Sci. 2017, 18, 77. [Google Scholar] [CrossRef] [Green Version]
- Villablanca, A.; Lubahn, D.; Shelby, L.; Lloyd, K.; Barthold, S. Susceptibility to Early Atherosclerosis in Male Mice Is Mediated by Estrogen Receptor α. Arter. Thromb. Vasc. Boil. 2004, 24, 1055–1061. [Google Scholar] [CrossRef] [Green Version]
- Christian, R.C.; Liu, P.Y.; Harrington, S.; Ruan, M.; Miller, V.M.; Fitzpatrick, L.A. Intimal Estrogen Receptor (ER)β, but Not ERα Expression, Is Correlated with Coronary Calcification and Atherosclerosis in Pre- and Postmenopausal Women. J. Clin. Endocrinol. Metab. 2006, 91, 2713–2720. [Google Scholar] [CrossRef] [Green Version]
- Mendelsohn, M.E.; Karas, R.H. The Protective Effects of Estrogen on the Cardiovascular System. N. Engl. J. Med. 1999, 340, 1801–1811. [Google Scholar] [CrossRef]
- Villard, C.; Eriksson, P.; Kronqvist, M.; Lengquist, M.; Jorns, C.; Hartman, J.; Roy, J.; Hultgren, R. Differential expression of sex hormone receptors in abdominal aortic aneurysms. Maturitas 2017, 96, 39–44. [Google Scholar] [CrossRef]
- Kim, J.; Kim, J.Y.; Song, K.S.; Lee, Y.H.; Seo, J.S.; Jelinek, J.; Goldschmidt-Clermont, P.J.; Issa, J.-P. Epigenetic changes in estrogen receptor β gene in atherosclerotic cardiovascular tissues and in-vitro vascular senescence. Biochim. Biophys. Acta Mol. Basis Dis. 2007, 1772, 72–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McRobb, L.S.; McGrath, K.; Tsatralis, T.; Liong, E.C.; Tan, J.; Hughes, G.; Handelsman, D.J.; Heather, A. Estrogen Receptor Control of Atherosclerotic Calcification and Smooth Muscle Cell Osteogenic Differentiation. Arter. Thromb. Vasc. Boil. 2017, 37, 1127–1137. [Google Scholar] [CrossRef] [Green Version]
- Meyer, M.R.; Fredette, N.C.; Howard, T.A.; Hu, C.; Ramesh, C.; Daniel, C.; Amann, K.; Arterburn, J.B.; Barton, M.; Prossnitz, E.R. G Protein-coupled Estrogen Receptor Protects from Atherosclerosis. Sci. Rep. 2014, 4, 7564. [Google Scholar] [CrossRef] [Green Version]
- Barton, M.; Prossnitz, E.R. Emerging roles of GPER in diabetes and atherosclerosis. Trends Endocrinol. Metab. 2015, 26, 185–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Yu, X.; Szynkarski, C.K.; Meng, C.; Zhou, B.; Barhoumi, R.; White, R.E.; Heaps, C.L.; Stallone, J.N.; Han, G. Activation of GPER Induces Differentiation and Inhibition of Coronary Artery Smooth Muscle Cell Proliferation. PLoS ONE 2013, 8, e64771. [Google Scholar] [CrossRef]
- Ghaffari, S.; Nabi, F.N.; Sugiyama, M.G.; Lee, W.L. Estrogen Inhibits LDL (Low-Density Lipoprotein) Transcytosis by Human Coronary Artery Endothelial Cells via GPER (G-Protein–Coupled Estrogen Receptor) and SR-BI (Scavenger Receptor Class B Type 1). Arter. Thromb. Vasc. Boil. 2018, 38, 2283–2294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Cell biology of ischemia/reperfusion injury. Int. Rev. Cell Mol. Boil. 2012, 298, 229–317. [Google Scholar] [CrossRef] [Green Version]
- Al-Salam, S.; Hashmi, S. Myocardial Ischemia Reperfusion Injury: Apoptotic, Inflammatory and Oxidative Stress Role of Galectin-3. Cell. Physiol. Biochem. 2018, 50, 1123–1139. [Google Scholar] [CrossRef]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Ischemia/Reperfusion. Compr. Physiol. 2016, 7, 113–170. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Crisostomo, P.; Wairiuko, G.M.; Meldrum, D.R. Estrogen receptor-α mediates acute myocardial protection in females. Am. J. Physiol. Circ. Physiol. 2006, 290, H2204–H2209. [Google Scholar] [CrossRef]
- Murphy, E.; Steenbergen, C. Gender-based differences in mechanisms of protection in myocardial ischemia–reperfusion injury. Cardiovasc. Res. 2007, 75, 478–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, S.; Zhang, L. Gender Differences in Cardioprotection against Ischemia/Reperfusion Injury in Adult Rat Hearts: Focus on Akt and Protein Kinase C Signaling. J. Pharmacol. Exp. Ther. 2005, 315, 1125–1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabir, M.E.; Singh, H.; Lu, R.; Olde, B.; Leeb-Lundberg, L.M.F.; Bopassa, J.C. G Protein-Coupled Estrogen Receptor 1 Mediates Acute Estrogen-Induced Cardioprotection via MEK/ERK/GSK-3β Pathway after Ischemia/Reperfusion. PLoS ONE 2015, 10, e0135988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Booth, E.A.; Obeid, N.; Lucchesi, B.R. Activation of estrogen receptor-α protects the in vivo rabbit heart from ischemia-reperfusion injury. Am. J. Physiol. Circ. Physiol. 2005, 289, H2039–H2047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, P.; Eurell, T.E.; Cooke, P.S.; Lubahn, D.B.; Gross, D.R. Myocardial ischemia-reperfusion injury in estrogen receptor-α knockout and wild-type mice. Am. J. Physiol. Circ. Physiol. 2000, 278, H1640–H1647. [Google Scholar] [CrossRef] [PubMed]
- Iorga, A.; Umar, S.; Ruffenach, G.; Aryan, L.; Li, J.; Sharma, S.; Motayagheni, N.; Nadadur, R.D.; Bopassa, J.C.; Eghbali, M. Estrogen rescues heart failure through estrogen receptor Beta activation. Boil. Sex. Differ. 2018, 9, 48. [Google Scholar] [CrossRef]
- Gabel, S.A.; Walker, V.R.; London, R.E.; Steenbergen, C.; Korach, K.S.; Murphy, E. Estrogen receptor beta mediates gender differences in ischemia/reperfusion injury. J. Mol. Cell. Cardiol. 2005, 38, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Schubert, C.; Raparelli, V.; Westphal, C.; Dworatzek, E.; Petrov, G.; Kararigas, G.; Regitz-Zagrosek, V. Reduction of apoptosis and preservation of mitochondrial integrity under ischemia/reperfusion injury is mediated by estrogen receptor β. Boil. Sex. Differ. 2016, 7, 53. [Google Scholar] [CrossRef] [Green Version]
- Nikolic, I.; Liu, D.; Bell, J.A.; Collins, J.; Steenbergen, C.; Murphy, E. Treatment with an estrogen receptor-beta-selective agonist is cardioprotective. J. Mol. Cell. Cardiol. 2007, 42, 769–780. [Google Scholar] [CrossRef]
- Liu, H.; Pedram, A.; Kim, J.K. Oestrogen prevents cardiomyocyte apoptosis by suppressing p38α-mediated activation of p53 and by down-regulating p53 inhibition on p38β. Cardiovasc. Res. 2010, 89, 119–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bopassa, J.C.; Eghbali, M.; Toro, L.; Stefani, E. A novel estrogen receptor GPER inhibits mitochondria permeability transition pore opening and protects the heart against ischemia-reperfusion injury. Am. J. Physiol. Circ. Physiol. 2009, 298, H16–H23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; Madungwe, N.; JunHo, C.C.; Bopassa, J.C. Activation of G protein-coupled oestrogen receptor 1 at the onset of reperfusion protects the myocardium against ischemia/reperfusion injury by reducing mitochondrial dysfunction and mitophagy. Br. J. Pharmacol. 2017, 174, 4329–4344. [Google Scholar] [CrossRef] [PubMed]
- Tschöpe, C.; Birner, C.; Böhm, M.; Bruder, O.; Frantz, S.; Luchner, A.; Maier, L.; Störk, S.; Kherad, B.; Laufs, U. Heart failure with preserved ejection fraction: Current management and future strategies. Clin. Res. Cardiol. 2017, 107, 1–19. [Google Scholar] [CrossRef]
- Savarese, G.; Lund, L.H. Global Public Health Burden of Heart Failure. Card. Fail. Rev. 2017, 3, 7–11. [Google Scholar] [CrossRef]
- Sickinghe, A.A.; Korporaal, S.J.A.; Ruijter, H.M.D.; Kessler, E.L. Estrogen Contributions to Microvascular Dysfunction Evolving to Heart Failure with Preserved Ejection Fraction. Front. Endocrinol. 2019, 10, 442. [Google Scholar] [CrossRef] [Green Version]
- Westphal, C.; Schubert, C.; Prelle, K.; Penkalla, A.; Fliegner, D.; Petrov, G.; Regitz-Zagrosek, V. Effects of Estrogen, an ERα Agonist and Raloxifene on Pressure Overload Induced Cardiac Hypertrophy. PLoS ONE 2012, 7, e50802. [Google Scholar] [CrossRef] [Green Version]
- Mahmoodzadeh, S.; Eder, S.; Nordmeyer, J.; Ehler, E.; Huber, O.; Martus, P.; Weiske, J.; Pregla, R.; Hetzer, R.; Regitz-Zagrosek, V. Estrogen receptor alpha up-regulation and redistribution in human heart failure. FASEB J. 2006, 20, 926–934. [Google Scholar] [CrossRef] [Green Version]
- Kararigas, G.; Fliegner, D.; Gustafsson, J.-Å.; Regitz-Zagrosek, V. Role of the estrogen/estrogen-receptor-beta axis in the genomic response to pressure overload-induced hypertrophy. Physiol. Genom. 2011, 43, 438–446. [Google Scholar] [CrossRef]
- Kararigas, G.; Fliegner, D.; Forler, S.; Klein, O.; Schubert, C.; Gustafsson, J.-Å.; Klose, J.; Regitz-Zagrosek, V. Comparative Proteomic Analysis Reveals Sex and Estrogen Receptor β Effects in the Pressure Overloaded Heart. J. Proteome Res. 2014, 13, 5829–5836. [Google Scholar] [CrossRef]
- Skavdahl, M.; Steenbergen, C.; Clark, J.; Myers, P.; Demianenko, T.; Mao, L.; Rockman, H.A.; Korach, K.S.; Murphy, E. Estrogen receptor-β mediates male-female differences in the development of pressure overload hypertrophy. Am. J. Physiol. Circ. Physiol. 2005, 288, H469–H476. [Google Scholar] [CrossRef] [PubMed]
- Fliegner, D.; Schubert, C.; Penkalla, A.; Witt, H.; Kararigas, G.; Dworatzek, E.; Staub, E.; Martus, P.; Noppinger, P.R.; Kintscher, U.; et al. Female sex and estrogen receptor-β attenuate cardiac remodeling and apoptosis in pressure overload. Am. J. Physiol. Integr. Comp. Physiol. 2010, 298, R1597–R1606. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Arenas, I.A.; Armstrong, S.J.; Davidge, S.T. Estrogen modulation of left ventricular remodeling in the aged heart. Cardiovasc. Res. 2003, 57, 388–394. [Google Scholar] [CrossRef] [Green Version]
- Delbeck, M.; Golz, S.; Janssen, W.; Schäfer, S.; Vonk, R.; Hucho, T.; Isensee, J.; Otto, C. Impaired left-ventricular cardiac function in male GPR30-deficient mice. Mol. Med. Rep. 2010, 4, 37–40. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.; Liu, Y.; Sun, D.; Zhou, C.; Liu, A.; Xu, C.; Hao, Y.; Li, D.; Yan, C.; Sun, H. Chronic Activation of the G Protein-Coupled Receptor 30 with Agonist G-1 Attenuates Heart Failure. PLoS ONE 2012, 7, e48185. [Google Scholar] [CrossRef]
- Valero-Munoz, M.; Backman, W.; Sam, F. Murine Models of Heart Failure with Preserved Ejection Fraction. JACC: Basic Transl. Sci. 2017, 2, 770–789. [Google Scholar] [CrossRef]
- Conceição, G.; Heinonen, I.; Lourenço, A.P.; Duncker, D.J.; Falcão-Pires, I. Animal models of heart failure with preserved ejection fraction. Neth. Hear. J. 2016, 24, 275–286. [Google Scholar] [CrossRef] [Green Version]
- Scantlebury, D.; Borlaug, B.A. Why are women more likely than men to develop heart failure with preserved ejection fraction? Curr. Opin. Cardiol. 2011, 26, 562–568. [Google Scholar] [CrossRef]
- Oktay, A.A.; Rich, J.D.; Shah, S.J. The emerging epidemic of heart failure with preserved ejection fraction. Curr. Hear. Fail. Rep. 2013, 10, 401–410. [Google Scholar] [CrossRef] [Green Version]
- Michalson, K.T.; Groban, L.; Howard, T.D.; Shively, C.A.; Sophonsritsuk, A.; Appt, S.E.; Cline, J.M.; Clarkson, T.B.; Carr, J.J.; Kitzman, D.W.; et al. Estradiol Treatment Initiated Early after Ovariectomy Regulates Myocardial Gene Expression and Inhibits Diastolic Dysfunction in Female Cynomolgus Monkeys: Potential Roles for Calcium Homeostasis and Extracellular Matrix Remodeling. J. Am. Hear. Assoc. 2018, 7, 009769. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.; Wang, H.; Jessup, J.A.; Lindsey, S.H.; Chappell, M.C.; Groban, L. Role of estrogen in diastolic dysfunction. Am. J. Physiol. Circ. Physiol. 2014, 306, H628–H640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, T.; Philip, J.L.; Tabima, D.; Frump, A.L.; Bellofiore, A.; Hacker, T.; Sun, X.; Lahm, T.; Chesler, N.C.; Biomedical Engineering, University of Wisconsin Madison; et al. Estrogen Receptor Alpha Protects against Diastolic Dysfunction and Fibrosis in Female Rats with Right Ventricular Pressure Overload. 2020. Available online: https://www.atsjournals.org/doi/pdf/10.1164/ajrccm-conference.2020.201.1_MeetingAbstracts.A6069 (accessed on 5 June 2020).
- Nuedling, S.; Karas, R.H.; Mendelsohn, M.E.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S.; Meyer, R.; Vetter, H.; Grohé, C. Activation of estrogen receptor β is a prerequisite for estrogen-dependent upregulation of nitric oxide synthases in neonatal rat cardiac myocytes. FEBS Lett. 2001, 502, 103–108. [Google Scholar] [CrossRef] [Green Version]
- Pedram, A.; Razandi, M.; Lubahn, D.; Liu, J.; Vannan, M.; Levin, E.R. Estrogen inhibits cardiac hypertrophy: Role of estrogen receptor-beta to inhibit calcineurin. Endocrinology 2008, 149, 3361–3369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jessup, J.A.; Zhang, L.; Chen, A.F.; Presley, T.D.; Kim-Shapiro, D.B.; Chappell, M.C.; Wang, H.; Groban, L. Neuronal nitric oxide synthase inhibition improves diastolic function and reduces oxidative stress in ovariectomized mRen2.Lewis rats. Menopause 2011, 18, 698–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Silva, J.S.; Sun, X.; Ahmad, S.; Wang, H.; Sudo, R.T.; Varagic, J.; Ferrario, C.M.; Zapata-Sudo, G.; Groban, L. G-Protein–Coupled Estrogen Receptor Agonist G1 Improves Diastolic Function and Attenuates Cardiac Renin–Angiotensin System Activation in Estrogen-Deficient Hypertensive Rats. J. Cardiovasc. Pharmacol. 2019, 74, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Alencar, A.K.; Da Silva, J.S.; Lin, M.; Silva, A.M.; Sun, X.; Ferrario, C.M.; Cheng, C.; Sudo, R.T.; Zapata-Sudo, G.; Wang, H.; et al. Effect of Age, Estrogen Status, and Late-Life GPER Activation on Cardiac Structure and Function in the Fischer344×Brown Norway Female Rat. J. Gerontol. Ser. A Boil. Sci. Med. Sci. 2016, 72, 152–162. [Google Scholar] [CrossRef]
- Wang, H.; Jessup, J.A.; Lin, M.S.; Chagas, C.; Lindsey, S.H.; Groban, L. Activation of GPR30 attenuates diastolic dysfunction and left ventricle remodelling in oophorectomized mRen2.Lewis rats. Cardiovasc. Res. 2012, 94, 96–104. [Google Scholar] [CrossRef] [Green Version]
- Jessup, J.A.; Lindsey, S.H.; Wang, H.; Chappell, M.C.; Groban, L. Attenuation of Salt-Induced Cardiac Remodeling and Diastolic Dysfunction by the GPER Agonist G-1 in Female mRen2.Lewis Rats. PLoS ONE 2010, 5, e15433. [Google Scholar] [CrossRef]
- Tong, D.; Schiattarella, G.G.; Jiang, N.; May, H.I.; Lavandero, S.; Gillette, T.G.; Hill, J.A. Female Sex Is Protective in a Preclinical Model of Heart Failure with Preserved Ejection Fraction. Circulation 2019, 140, 1769–1771. [Google Scholar] [CrossRef]
- Umar, S.; Cunningham, C.M.; Itoh, Y.; Moazeni, S.; Vaillancourt, M.; Sarji, S.; Centala, A.; Arnold, A.P.; Eghbali, M. The Y Chromosome Plays a Protective Role in Experimental Hypoxic Pulmonary Hypertension. Am. J. Respir. Crit. Care Med. 2017, 197, 952–955. [Google Scholar] [CrossRef]
- Arnold, A.P.; Reue, K.; Eghbali, M.; Vilain, E.; Chen, X.; Ghahramani, N.; Itoh, Y.; Li, J.; Link, J.C.; Ngun, T.; et al. The importance of having two X chromosomes. Philos. Trans. R. Soc. B Boil. Sci. 2016, 371, 20150113. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
| ER | Intervention | Model | Mechanism and Outcome | Ref |
|---|---|---|---|---|
| ERα | ERα KO | Ang II, OVX female mice | ↑ sympathetic outflow → ↑ HTN | [21] |
| ERα KO | Ang II, Female mice lacking membrane ERα | ↓ Activation Factor 2-dependent transcription → ↑ HTN3 | [22] | |
| ERα agonist | Spontaneously hypertensive OVX rats with reduced aortic eNOS | Normalized aortic eNOS → ↓ endothelial dysfunction | [23] | |
| ERα transfection | E2, Primary human VSMC | ↓ iNOS activity → ↑ HTN | [24] | |
| ERβ | ERβ KO | Male mice with VSMC ERβ KO | ↑ abnormal vascular contraction, ↑ ion channel dysfunction, ↑ vasoconstriction, ↑BP → ↑ HTN | [24] |
| ERβ agonist | Spontaneously hypertensive OVX rats | ↓ BP, ↓ cardiac hypertrophy, ↓ vascular resistance → ↓ HTN | [25] | |
| GPR30 | GPR30 agonist | Human endothelial cells | ↑ eNOS → ↑ c-Src/EGFR/PI3K/ERK → ↑ vasodilation → ↓ HTN | [30] |
| GPR30 antagonist | Human endothelial cells | ↓ NO production → ↓ vasodilation → ↑ HTN | [30] |
| ER | Intervention | Model | Mechanism and Outcome | Ref |
|---|---|---|---|---|
| ERα | E2 repletion | OVX SuHx-induced female PH rats with decreased RV ERα expression | E2 repletion → ↑ ERα → ↓ RV systolic pressure → ↓ RV hypertrophy → ↓ PH | [42] |
| ERα antagonist | Serotonin transporter with PH and mice with hypoxia-induced PH | ↓ ERα → ↓ pulmonary vascular remodeling, ↓ RV systolic pressure, ↓ PASMC proliferation | [43] | |
| ERα agonist | Human PASMC in vitro | ↑ ERα → ↑ Akt, ↑ MAPK → ↑ proliferation | [43] | |
| ERα transfection | Male mice lungs and OVX female mice lungs | ↑ ERα in OVX females → ↓ BMPR2 gene expression → ↑ PH | [44] | |
| ERβ | ERβ agonist | Male rats with monocrotaline-induced PH | ↑ ERβ → ↓ fibrosis, ↓ inflammation, ↓ RV hypertrophy → ↓ PH | [39] |
| Deferoxamine, HIF-1α stabilizer | Male rat lungs with hypoxia-induced PH | ↑ hypoxia → ↑ ERβ in lung → ↓ HIF-1α, ↓ pulmonary vascular remodeling → ↓ PH | [49] | |
| ERβ KO | Male rat lungs with hypoxia-induced PH | ↑ HIF-2α → ↓ response to E2 → ↑ PH | [49] | |
| E2 therapy | Male and female rats with MCT-induced PH | ↑ E2 → ↑ ERβ → ↓ fibrosis, ↑ ADAM15/ADAM17/osteopontin the RV → ↓ RV remodeling | [33] | |
| E2 therapy | Female ApoE deficient mice with MCT-induced PH | ↑ E2 → restore ERβ → ↓ PAH | [52] | |
| ERα and ERβ | ERα agonist and EEβ agonist | Adult male rats | ↑ ERα and ERβ → ↓ pulmonary artery vasoconstriction (attenuated with NOS inhibitor) | [54] |
| ERα antagonist and ERβ antagonist | Male rats with hypoxia-induced PH | ↓ ERα and ERβ → ↓ pulmonary RV remodeling | [53] | |
| E2 treatment | Male rats with hypoxia-induced PH | ↑ E2 treatment → ↓ ERK1/2 activation in the lung and RV (attenuated with co-treatment of ERα and ERβ antagonist) | [53] | |
| GPR30 | GPR30 agonist | Male rats with MCT-induced PH | ↑ GPR30 → ↑ eNOS, ↓ collagen deposition in pulmonary and cardiac fibroblasts, ↑ Ca2+ handling regulation and ↓ inflammation in cardiomyocytes → ↑ pulmonary flow, → ↑ RV hypertrophy, ↑ LV dysfunction | [55] |
| GPR30 agonist | OVX female rats with MCT-induced PH | ↑ GPR30 → ↓ pulmonary artery dysfunction, ↓ RV overload, ↓ RV dilation, ↓ wall hypertrophy, ↓ collagen deposition, normalizes LV dysfunction | [57] |
| ER | Intervention | Model | Mechanism and Outcome | Ref |
|---|---|---|---|---|
| ERα | ERα KO | Male mice fed a high-fat, high-cholesterol diet | ↑ ERα → ↑ atheroma formation → ↑ susceptibility to early atherosclerosis & more extensive atherosclerotic lesions | [69] |
| Hepatocyte Erα deletion | Female mice fed a Western-type diet | ↓ ERα → ↑ serum cholesterol levels, increased HDL particle sizes → ↑ size of early atherosclerotic lesions | [62] | |
| NA | Human blood monocytes-derived macrophages | ↑ lipopolysaccharide-mediated inflammatory responses → ERα activation in males | [63] | |
| E2 treatment | OVX mice | ↑ ERα → ↑ Activation Factor 2 → ↑ atheroprotection | [67] | |
| ERα agonist | ApoE KO male mice | ↑ ERα → ↓ serum lipid levels, ↑ PI3K/Akt pathway → ↑ atherosclerotic lesions | [68] | |
| ERβ ERα and ERβ | NA | Coronary arteries of pre- and post-menopausal women | ↑ ERβ → ↑ coronary calcification → ↑ atherosclerosis | [70] |
| NA | Coronary atherosclerotic tissues | Epigenetic modification in the ERβ gene → ↑ methylation levels → ↓ vascular aging, → ↑ atherosclerosis | [73] | |
| E2 treatment | ApoE KO male and female mice | ↓ ERα and ERβ → ↑ differentiation of VSMC to osteoblast-like cells → ↑ calcification of advanced atherosclerotic lesions | [74] | |
| GPR30 | GPR30 KO | Ovary-intact mice with GPER deletion | ↓ GPR30 → ↑ LDL cholesterol levels, ↑ inflammation, ↓ vascular NO bioactivity → ↑ progression of atherosclerosis | [75] |
| GPR30 deletion | Male and female GPR30 KO mice | ↓ GPR30 → ↑ endothelium-dependent vasoconstriction, ↑ visceral obesity, ↑ LDL levels, ↑ inflammation | [76] | |
| NA | Human coronary artery endothelial cells | ↑ GPR30 → ↑ EGFR → ↓ endothelial scavenger receptor class B type I → ↓ LDL transcytosis | [78] |
| ER | Intervention | Model | Mechanism and Outcome | Ref |
|---|---|---|---|---|
| ERα | ERα agonist | Female rabbit model after regional I/R | ↑ ERα → ↓ infarct size → ↓ complement system | [86] |
| ERα overexpression | Isolated female mouse hearts after I/R | ↑ ERα → ↑ ERK1/2 activation → ↓ pro-apoptotic JNK → ↑ cardioprotection | [82] | |
| ERα KO | Male mice after I/R | ↓ ERα → impaired mitochondrial respiratory function, ↓ nitrite production, ↑ Ca2+ accumulation | [87] | |
| ERβ | ERβ agonist | Female mice prior to I/R | ↑ ERβ → ↓ apoptosis, preserved mitochondrial integrity, ↑ B-cell lymphoma 2, ↑ acetyl coenzyme A acetyltransferase 2, ↓ caspase 9 → ↑ cardiac recovery | [90] |
| ERβ agonist pretreatmen | Isolated OVX female mouse hearts | ↑ ERβ → ↑ cardioprotective genes (anti-apoptotic protein, heat shock protein, cyclooxygenase 2, growth arrest and DNA damage 45 beta) | [91] | |
| ERα and ERβ | E2 treatment | Cultured neonatal rat cardiomyocytes stimulated with hypoxia/reoxygen | ↑ ERα and ERβ → ↓ ROS, ↓ p53 dependent apoptosis → ↑ cardioprotection | [92] |
| GPR30 | Acute activation with GPR30 agonist | Male and female rats during I/R injury | ↑ GPR30 → ↑ PI3K/Akt pathway → ↑ cardioprotection | [9] |
| GPR30 agonist | Isolated hearts from male mice undergoing I/R via Langendorff | ↑ GPR30 → ↑ ERK pathway → ↓ Ca2+-induced mitochondrial permeability pore opening → ↓ cell death | [93] | |
| GPR30 agonist | In vivo rat hearts subjected to I/R | ↑ GPR30 → ↑ MEK/ERK → ↓ GSK-3β pathway → ↓ mitochondrial dysfunction and mitophagy | [94] |
| ER | Intervention | Model | Mechanism and Outcome | Ref |
|---|---|---|---|---|
| ERα | ERα agonist | OVX female mice with myocardial hypertrophy induced by TAC | ↑ ERα → smallest reduction in EF, ↓ fibrosis → improved systolic function | [98] |
| NA | Male and female humans with end-stage HF | Human HF → loss of colocalization between Erα and β-catenin, altered expression and localization of ERα | [99] | |
| ERβ | ERβ KO | Female mice induced by TAC | ↓ ERβ → ↑ inflammatory pathways | [100] |
| ERβ KO | Male and female WT vs. ERβ KO mice | ↑ ERβ = more prominent cardioprotective role in females → ↓ pressure overload | [101] | |
| ERβ KO | ERβ KO mice that underwent TAC | ↓ ERβ → ↑ hypertrophy | [102] | |
| ERβ KO | Female mice with TAC-induced pressure overload | ↓ ERβ → ↑ cardiac fibrosis, ↑ apoptosis → ↑ HF | [103] | |
| ERβ agonist | Male mice with TAC-induced HF | ↑ ERβ → ↓ cardiac fibrosis, ↑ EF, restoration of cardiac angiogenesis, normalization of hemodynamic parameters | [88] | |
| GPR30 | GPR30 agonist | Female OVX rats with isoproterenol-induced HF | ↑ GPR30 → mediates the expression of β1- and β2-adrenergic receptors → ↑ cardiac function, ↑ myocyte contractility, ↓ fibrosis | [106] |
| GPR30 KO | Male GPR30-deficient mice | ↓ GPR30 → impaired cardiac function in LV, LV enlargement, ↓ contractility/relaxation of LV → ↑ LV end-diastolic pressure | [105] |
| ER | Intervention | Model | Mechanism and Outcome | Ref |
|---|---|---|---|---|
| ERα | ERα mutant | Male and female rats via pulmonary artery banding-induced RV hypertrophy | ↓ ERα in females → ↑ collagen type I → ↑ diastolic dysfunction | [113] |
| ERβ | ERβ agonist | Ang II-induced cardiac hypertrophy in female mice | ↑ ERβ → ↓ transition from α to β myosin heavy chain production, ↓ ERK activation, ↓ calcineurin activity, ↓ interstitial fibrosis | [115] |
| ERβ antagonist | Neonatal rat cardiac myocytes transfected with ERβ antagonist | ↑ ERβ → ↑ expression of eNOS and iNOS | [114] | |
| GPR30 | GPR30 agonist | OVX spontaneously hypertensive rats | ↑ GPR30 → ↓ cardiac angiotensin-converting enzyme, ↓ angiotensin type I receptor expression, ↓ AngII immunoreactivity → improved vasorelaxant responsiveness | [117] |
| GPR30 agonist | Aged OVX female rats | ↑ GPR30 → ↑ cardiomyocyte sarcoplasmic reticulum Ca2+ ATPase expression, ↓ cardiac fibrosis → ameliorates impaired myocardial relaxation | [118] | |
| Chronic GPR30 agonist | OVX female rats | ↑ GPR30 → prevents increases in cardiac NOX4 expression → conserved diastolic function | [119] | |
| GPR30 agonist | Salt-induced diastolic dysfunction in female rats | ↑ GPR30 → ↑ myocardial relaxation, ↑ ratio of early-to-late LV filling | [120] | |
| GPR30 KO | Cardiomyocyte specific GPR30 KO mice | ↓ GPR30 → ↑ LV dysfunction, adverse cardiac remodeing | [8] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aryan, L.; Younessi, D.; Zargari, M.; Banerjee, S.; Agopian, J.; Rahman, S.; Borna, R.; Ruffenach, G.; Umar, S.; Eghbali, M. The Role of Estrogen Receptors in Cardiovascular Disease. Int. J. Mol. Sci. 2020, 21, 4314. https://doi.org/10.3390/ijms21124314
Aryan L, Younessi D, Zargari M, Banerjee S, Agopian J, Rahman S, Borna R, Ruffenach G, Umar S, Eghbali M. The Role of Estrogen Receptors in Cardiovascular Disease. International Journal of Molecular Sciences. 2020; 21(12):4314. https://doi.org/10.3390/ijms21124314
Chicago/Turabian StyleAryan, Laila, David Younessi, Michael Zargari, Somanshu Banerjee, Jacqueline Agopian, Shadie Rahman, Reza Borna, Gregoire Ruffenach, Soban Umar, and Mansoureh Eghbali. 2020. "The Role of Estrogen Receptors in Cardiovascular Disease" International Journal of Molecular Sciences 21, no. 12: 4314. https://doi.org/10.3390/ijms21124314