Estrogen Receptors and Estrogen-Induced Uterine Vasodilation in Pregnancy

 , , ,
, , , 

Abstract
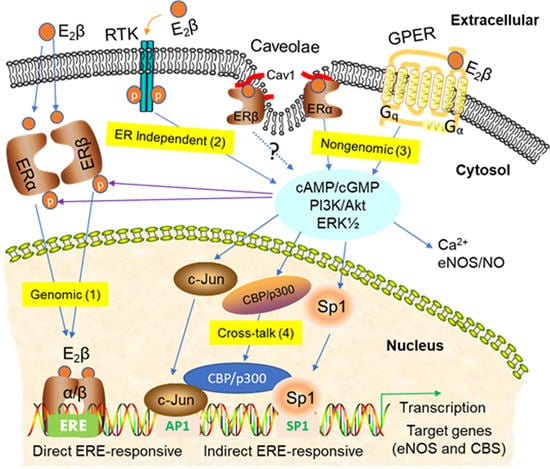
:
1. Introduction
2. Classical ERs—ERα and ERβ in Uterine Vasodilation in Pregnancy
Membrane ERs and Estrogen-Induced Uterine Vasodilation in Pregnancy
3. Signaling Pathways of Estrogen Action
3.1. Nuclear ER Actions in UA
3.2. Non-Nuclear ER Actions in UA
3.3. GPER-Mediated Nongenomic Effects in Uterine Vascular System
3.4. ER-Independent Estrogen Signaling and Estrogen-Independent ER Activation in UA
3.5. Posttranslational and Epigenetic Modification of ER Actions
4. Gasotransmitters in Estrogen-Induced Uterine Vasodilation in Pregnancy
4.1. Nitric Oxide
4.2. Carbon Monoxide
4.3. Hydrogen Sulfide
4.4. H2S Interactions with NO and CO in the Vasculature
5. Estrogens and Gasotransmitters in Preeclampsia
5.1. Pathophysiological Evidence
5.2. Therapeutic Considerations
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Garris, D.R.; Foreman, D. Follicular growth and atresia during the last half of the luteal phase of the guinea pig estrous cycle: Relation to serum progesterone and estradiol levels and utero-ovarian blood flow. Endocrinology 1984, 115, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Greiss, F.C., Jr.; Anderson, S.G. Uterine vascular changes during the ovarian cycle. Am. J. Obstet. Gynecol. 1969, 103, 629–640. [Google Scholar] [CrossRef]
- Magness, R.R.; Rosenfeld, C.R.; Carr, B.R. Protein kinase C in uterine and systemic arteries during ovarian cycle and pregnancy. Am. J. Physiol. 1991, 260, E464–E470. [Google Scholar] [CrossRef] [PubMed]
- Austin, C.E. Chronic and acute effects of oestrogens on vascular contractility. J. Hypertens. 2000, 18, 1365–1378. [Google Scholar] [CrossRef]
- Kostrzewska, A.; Laudanski, T.; Batra, S. Effect of ovarian steroids and diethylstilbestrol on the contractile responses of the human myometrium and intramyometrial arteries. Eur. J. Pharmacol. 1993, 233, 127–134. [Google Scholar] [CrossRef]
- Magness, R.R. Maternal cardiovascular and other physiologic responses to the endocrinology of pregnancy. In Endocrinology of Pregnancy; Springer: Berlin/Heidelberg, Germany, 1998; pp. 507–539. [Google Scholar]
- Pepe, G.J.; Albrecht, E.D. Actions of placental and fetal adrenal steroid hormones in primate pregnancy. Endocr. Rev. 1995, 16, 608–648. [Google Scholar] [CrossRef]
- Reynolds, L.P.; Redmer, D.A. Utero-placental vascular development and placental function. J. Anim. Sci. 1995, 73, 1839–1851. [Google Scholar] [CrossRef]
- Magness, R.R.; Rosenfeld, C.R. Local and systemic estradiol-17 beta: Effects on uterine and systemic vasodilation. Am. J. Physiol. 1989, 256, E536–E542. [Google Scholar] [CrossRef]
- Reynolds, L.P.; Kirsch, J.D.; Kraft, K.C.; Knutson, D.L.; McClaflin, W.J.; Redmer, D.A. Time-course of the uterine response to estradiol-17beta in ovariectomized ewes: Uterine growth and microvascular development. Biol. Reprod. 1998, 59, 606–612. [Google Scholar] [CrossRef]
- Magness, R.; Ford, S. Maternal cardiovascular adaptation and uterine circulation-physiology and pathophysiology. In Stress and Developmental Programming of Health and Disease: Beyond Phenomenology; Nova Science Publishers: Hauppauge, NY, USA, 2014; pp. 341–374. [Google Scholar]
- Rosenfeld, C.R. Distribution of cardiac output in ovine pregnancy. Am. J. Physiol. 1977, 232, H231–H235. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, C.R. The Uterine Circulation; Perinatology Press: Ithaca, NY, USA, 1989. [Google Scholar]
- Rosenfeld, C.R.; Morriss, F.H., Jr.; Makowski, E.L.; Meschia, G.; Battaglia, F.C. Circulatory changes in the reproductive tissues of ewes during pregnancy. Gynecol. Investig. 1974, 5, 252–268. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, E.D.; Pepe, G.J. Placental steroid hormone biosynthesis in primate pregnancy. Endocr. Rev. 1990, 11, 124–150. [Google Scholar] [CrossRef]
- Carnegie, J.A.; Robertson, H.A. Conjugated and unconjugated estrogens in fetal and maternal fluids of the pregnant ewe: A possible role for estrone sulfate during early pregnancy. Biol. Reprod. 1978, 19, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Ford, S.P. Control of uterine and ovarian blood flow throughout the estrous cycle and pregnancy of ewes, sows and cows. J. Anim. Sci. 1982, 55 (Suppl. 2), 32–42. [Google Scholar] [CrossRef]
- Markee, J. Rhythmic uterine vascular changes. Am. J. Physiol. 1932, 100, 32–39. [Google Scholar] [CrossRef]
- Magness, R.R.; Rosenfeld, C.R. Systemic and uterine responses to alpha-adrenergic stimulation in pregnant and nonpregnant ewes. Am. J. Obstet. Gynecol. 1986, 155, 897–904. [Google Scholar] [CrossRef]
- Magness, R.R.; Parker, C.R., Jr.; Rosenfeld, C.R. Systemic and uterine responses to chronic infusion of estradiol-17 beta. Am. J. Physiol. 1993, 265, E690–E698. [Google Scholar] [CrossRef]
- Magness, R.R.; Phernetton, T.M.; Zheng, J. Systemic and uterine blood flow distribution during prolonged infusion of 17beta-estradiol. Am. J. Physiol. 1998, 275, H731–H743. [Google Scholar] [CrossRef]
- Rosenfeld, C.R.; Chen, C.; Roy, T.; Liu, X. Estrogen selectively up-regulates eNOS and nNOS in reproductive arteries by transcriptional mechanisms. J. Soc. Gynecol. Investig. 2003, 10, 205–215. [Google Scholar] [CrossRef]
- Naden, R.P.; Rosenfeld, C.R. Systemic and uterine responsiveness to angiotensin II and norepinephrine in estrogen-treated nonpregnant sheep. Am. J. Obstet. Gynecol. 1985, 153, 417–425. [Google Scholar] [CrossRef]
- Killam, A.P.; Rosenfeld, C.R.; Battaglia, F.C.; Makowski, E.L.; Meschia, G. Effect of estrogens on the uterine blood flow of oophorectomized ewes. Am. J. Obstet. Gynecol. 1973, 115, 1045–1052. [Google Scholar] [CrossRef]
- Magness, R.R.; Phernetton, T.M.; Gibson, T.C.; Chen, D.B. Uterine blood flow responses to ICI 182 780 in ovariectomized oestradiol-17beta-treated, intact follicular and pregnant sheep. J. Physiol. 2005, 565, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Gibson, T.C.; Phernetton, T.M.; Wiltbank, M.C.; Magness, R.R. Development and use of an ovarian synchronization model to study the effects of endogenous estrogen and nitric oxide on uterine blood flow during ovarian cycles in sheep. Biol. Reprod. 2004, 70, 1886–1894. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, P.; Boyne, P.; Flett, P.; Beilby, J.; James, I. Longitudinal assessment of changes in reproductive hormones during normal pregnancy. Clin. Chem. 1991, 37, 667–672. [Google Scholar] [CrossRef] [PubMed]
- Lang, U.; Baker, R.S.; Braems, G.; Zygmunt, M.; Kunzel, W.; Clark, K.E. Uterine blood flow—A determinant of fetal growth. Eur. J. Obstet. Gynecol. Reprod. Biol. 2003, 110 (Suppl. 1), S55–S61. [Google Scholar] [CrossRef]
- Herrera-Garcia, G.; Contag, S. Maternal preeclampsia and risk for cardiovascular disease in offspring. Curr. Hypertens. Rep. 2014, 16, 475. [Google Scholar] [CrossRef]
- Barker, D.J.; Bull, A.R.; Osmond, C.; Simmonds, S.J. Fetal and placental size and risk of hypertension in adult life. BMJ 1990, 301, 259–262. [Google Scholar] [CrossRef]
- Terragno, N.A.; Terragno, D.A.; Pacholczyk, D.; McGiff, J.C. Prostaglandins and the regulation of uterine blood flow in pregnancy. Nature 1974, 249, 57–58. [Google Scholar] [CrossRef]
- Van Buren, G.A.; Yang, D.S.; Clark, K.E. Estrogen-induced uterine vasodilatation is antagonized by L-nitroarginine methyl ester, an inhibitor of nitric oxide synthesis. Am. J. Obstet. Gynecol. 1992, 167, 828–833. [Google Scholar] [CrossRef]
- Tschugguel, W.; Stonek, F.; Zhegu, Z.; Dietrich, W.; Schneeberger, C.; Stimpfl, T.; Waldhoer, T.; Vycudilik, W.; Huber, J.C. Estrogen increases endothelial carbon monoxide, heme oxygenase 2, and carbon monoxide-derived cGMP by a receptor-mediated system. J. Clin. Endocrinol. Metab. 2001, 86, 3833–3839. [Google Scholar] [CrossRef] [PubMed]
- Burger, N.Z.; Kuzina, O.Y.; Osol, G.; Gokina, N.I. Estrogen replacement enhances EDHF-mediated vasodilation of mesenteric and uterine resistance arteries: Role of endothelial cell Ca2+. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E503–E512. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, C.R.; Cox, B.E.; Roy, T.; Magness, R.R. Nitric oxide contributes to estrogen-induced vasodilation of the ovine uterine circulation. J. Clin. Investig. 1996, 98, 2158–2166. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.J.; Vallance, P.J.; Neild, G.H.; Spencer, J.A.; Imms, F.J. Nitric oxide-mediated vasodilation in human pregnancy. Am. J. Physiol. 1997, 272, H748–H752. [Google Scholar] [CrossRef]
- Sheibani, L.; Lechuga, T.J.; Zhang, H.; Hameed, A.; Wing, D.A.; Kumar, S.; Rosenfeld, C.R.; Chen, D.B. Augmented H2S production via cystathionine-beta-synthase upregulation plays a role in pregnancy-associated uterine vasodilation. Biol. Reprod. 2017, 96, 664–672. [Google Scholar] [CrossRef]
- Pfaffl, M.W.; Lange, I.G.; Daxenberger, A.; Meyer, H.H. Tissue-specific expression pattern of estrogen receptors (ER): Quantification of ER alpha and ER beta mRNA with real-time RT-PCR. APMIS 2001, 109, 345–355. [Google Scholar] [CrossRef]
- Byers, M.J.; Zangl, A.; Phernetton, T.M.; Lopez, G.; Chen, D.B.; Magness, R.R. Endothelial vasodilator production by ovine uterine and systemic arteries: Ovarian steroid and pregnancy control of ERalpha and ERbeta levels. J. Physiol. 2005, 565, 85–99. [Google Scholar] [CrossRef]
- Lemmen, J.G.; Broekhof, J.L.; Kuiper, G.G.; Gustafsson, J.A.; van der Saag, P.T.; van der Burg, B. Expression of estrogen receptor alpha and beta during mouse embryogenesis. Mech. Dev. 1999, 81, 163–167. [Google Scholar] [CrossRef]
- Shughrue, P.J.; Lane, M.V.; Scrimo, P.J.; Merchenthaler, I. Comparative distribution of estrogen receptor-alpha (ER-alpha) and beta (ER-beta) mRNA in the rat pituitary, gonad, and reproductive tract. Steroids 1998, 63, 498–504. [Google Scholar] [CrossRef]
- Maxwell, B.L.; McDonnell, D.P.; Conneely, O.M.; Schulz, T.Z.; Greene, G.L.; O’Malley, B.W. Structural organization and regulation of the chicken estrogen receptor. Mol. Endocrinol. 1987, 1, 25–35. [Google Scholar] [CrossRef] [Green Version]
- Weiler, I.J.; Lew, D.; Shapiro, D.J. The Xenopus laevis estrogen receptor: Sequence homology with human and avian receptors and identification of multiple estrogen receptor messenger ribonucleic acids. Mol. Endocrinol. 1987, 1, 355–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, E.R.; Habibi, H.R. Estrogen receptor function and regulation in fish and other vertebrates. Gen. Comp. Endocrinol. 2013, 192, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Enmark, E.; Pelto-Huikko, M.; Grandien, K.; Lagercrantz, S.; Lagercrantz, J.; Fried, G.; Nordenskjold, M.; Gustafsson, J.A. Human estrogen receptor beta-gene structure, chromosomal localization, and expression pattern. J. Clin. Endocrinol. Metab. 1997, 82, 4258–4265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gosden, J.R.; Middleton, P.G.; Rout, D. Localization of the human oestrogen receptor gene to chromosome 6q24→q27 by in situ hybridization. Cytogenet. Genome Res. 1986, 43, 218–220. [Google Scholar] [CrossRef] [PubMed]
- Heldring, N.; Pike, A.; Andersson, S.; Matthews, J.; Cheng, G.; Hartman, J.; Tujague, M.; Strom, A.; Treuter, E.; Warner, M.; et al. Estrogen receptors: How do they signal and what are their targets. Physiol. Rev. 2007, 87, 905–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuiper, G.G.; Carlsson, B.; Grandien, K.; Enmark, E.; Haggblad, J.; Nilsson, S.; Gustafsson, J.A. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology 1997, 138, 863–870. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, X.; Shen, P.; Loggie, B.W.; Chang, Y.; Deuel, T.F. A variant of estrogen receptor-{alpha}, hER-{alpha}36: Transduction of estrogen- and antiestrogen-dependent membrane-initiated mitogenic signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 9063–9068. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Ding, L.; Coleman, M.; Wang, Z. Estrogen receptor-alpha (ER-alpha) suppresses expression of its variant ER-alpha 36. FEBS Lett. 2009, 583, 1368–1374. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.L.; Yan, L.Y.; Liang, X.W.; Wang, Z.B.; Wang, Z.Y.; Qiao, J.; Schatten, H.; Sun, Q.Y. A novel variant of ER-alpha, ER-alpha36 mediates testosterone-stimulated ERK and Akt activation in endometrial cancer Hec1A cells. Reprod. Biol. Endocrinol. 2009, 7, 102. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Haynes, M.P.; Bender, J.R. Plasma membrane localization and function of the estrogen receptor alpha variant (ER46) in human endothelial cells. Proc. Natl. Acad. Sci. USA 2003, 100, 4807–4812. [Google Scholar] [CrossRef] [Green Version]
- Anamthathmakula, P.; Kyathanahalli, C.; Ingles, J.; Hassan, S.S.; Condon, J.C.; Jeyasuria, P. Estrogen receptor alpha isoform ERdelta7 in myometrium modulates uterine quiescence during pregnancy. EBioMedicine 2019, 39, 520–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stricker, R.; Eberhart, R.; Chevailler, M.C.; Quinn, F.A.; Bischof, P.; Stricker, R. Establishment of detailed reference values for luteinizing hormone, follicle stimulating hormone, estradiol, and progesterone during different phases of the menstrual cycle on the Abbott ARCHITECT analyzer. Clin. Chem. Lab. Med. 2006, 44, 883–887. [Google Scholar] [CrossRef] [PubMed]
- Kang, L.; Zhang, X.; Xie, Y.; Tu, Y.; Wang, D.; Liu, Z.; Wang, Z.Y. Involvement of estrogen receptor variant ER-alpha36, not GPR30, in nongenomic estrogen signaling. Mol. Endocrinol. 2010, 24, 709–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkins, M.B.; Thornton, J.W.; Crews, D.; Skipper, J.K.; Dotte, A.; Thomas, P. Identification of a third distinct estrogen receptor and reclassification of estrogen receptors in teleosts. Proc. Natl. Acad. Sci. USA 2000, 97, 10751–10756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legler, J.; Zeinstra, L.M.; Schuitemaker, F.; Lanser, P.H.; Bogerd, J.; Brouwer, A.; Vethaak, A.D.; De Voogt, P.; Murk, A.J.; Van der Burg, B. Comparison of in vivo and in vitro reporter gene assays for short-term screening of estrogenic activity. Environ. Sci. Technol. 2002, 36, 4410–4415. [Google Scholar] [CrossRef] [Green Version]
- Sabo-Attwood, T.; Kroll, K.J.; Denslow, N.D. Differential expression of largemouth bass (Micropterus salmoides) estrogen receptor isotypes alpha, beta, and gamma by estradiol. Mol. Cell. Endocrinol. 2004, 218, 107–118. [Google Scholar] [CrossRef]
- Toran-Allerand, C.D. Minireview: A plethora of estrogen receptors in the brain: Where will it end? Endocrinology 2004, 145, 1069–1074. [Google Scholar] [CrossRef] [Green Version]
- Rao, B.R. Isolation and characterization of an estrogen binding protein which may integrate the plethora of estrogenic actions in non-reproductive organs. J. Steroid Biochem. Mol. Biol. 1998, 65, 3–41. [Google Scholar] [CrossRef]
- Karas, R.H.; Patterson, B.L.; Mendelsohn, M.E. Human vascular smooth muscle cells contain functional estrogen receptor. Circulation 1994, 89, 1943–1950. [Google Scholar] [CrossRef] [Green Version]
- Orimo, A.; Inoue, S.; Ouchi, Y.; Orimo, H. Vascular smooth muscle cells possess estrogen receptor and respond to estrogen. Ann. N. Y. Acad. Sci. 1995, 748, 592–594. [Google Scholar] [CrossRef]
- Register, T.C.; Adams, M.R. Coronary artery and cultured aortic smooth muscle cells express mRNA for both the classical estrogen receptor and the newly described estrogen receptor beta. J. Steroid Biochem. Mol. Biol. 1998, 64, 187–191. [Google Scholar] [CrossRef]
- Thomas, P.; Pang, Y.; Filardo, E.J.; Dong, J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology 2005, 146, 624–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batra, S.; Iosif, S. Nuclear estrogen receptors in human uterine arteries. Gynecol. Obstet. Investig. 1987, 24, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.X.; Ma, X.H.; Smith, G.C.; Nathanielsz, P.W. Differential distribution of ERalpha and ERbeta mRNA in intrauterine tissues of the pregnant rhesus monkey. Am. J. Physiol. Cell Physiol. 2000, 278, C190–C198. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.; Lubo, Z. Review article: Steroid hormones and uterine vascular adaptation to pregnancy. Reprod. Sci. 2008, 15, 336–348. [Google Scholar] [CrossRef] [Green Version]
- Reslan, O.M.; Yin, Z.; do Nascimento, G.R.; Khalil, R.A. Subtype-specific estrogen receptor-mediated vasodilator activity in the cephalic, thoracic, and abdominal vasculature of female rat. J. Cardiovasc. Pharmacol. 2013, 62, 26–40. [Google Scholar] [CrossRef]
- Mata, K.M.; Li, W.; Reslan, O.M.; Siddiqui, W.T.; Opsasnick, L.A.; Khalil, R.A. Adaptive increases in expression and vasodilator activity of estrogen receptor subtypes in a blood vessel-specific pattern during pregnancy. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1679–H1696. [Google Scholar] [CrossRef] [Green Version]
- Lechuga, T.J.; Bilg, A.K.; Patel, B.A.; Nguyen, N.A.; Qi, Q.R.; Chen, D.B. Estradiol-17beta stimulates H2 S biosynthesis by ER-dependent CBS and CSE transcription in uterine artery smooth muscle cells in vitro. J. Cell. Physiol. 2019, 234, 9264–9273. [Google Scholar] [CrossRef]
- Liao, W.X.; Magness, R.R.; Chen, D.B. Expression of estrogen receptors-alpha and -beta in the pregnant ovine uterine artery endothelial cells in vivo and in vitro. Biol. Reprod. 2005, 72, 530–537. [Google Scholar] [CrossRef]
- Scott, P.A.; Tremblay, A.; Brochu, M.; St-Louis, J. Vasorelaxant action of 17-estradiol in rat uterine arteries: Role of nitric oxide synthases and estrogen receptors. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H3713–H3719. [Google Scholar] [CrossRef]
- Hilgers, R.H.; Oparil, S.; Wouters, W.; Coelingh Bennink, H.J. Vasorelaxing effects of estetrol in rat arteries. J. Endocrinol. 2012, 215, 97–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandala, M. Influence of Estrogens on Uterine Vascular Adaptation in Normal and Preeclamptic Pregnancies. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubanyi, G.M.; Freay, A.D.; Kauser, K.; Sukovich, D.; Burton, G.; Lubahn, D.B.; Couse, J.F.; Curtis, S.W.; Korach, K.S. Vascular estrogen receptors and endothelium-derived nitric oxide production in the mouse aorta. Gender difference and effect of estrogen receptor gene disruption. J. Clin. Investig. 1997, 99, 2429–2437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupont, S.; Krust, A.; Gansmuller, A.; Dierich, A.; Chambon, P.; Mark, M. Effect of single and compound knockouts of estrogen receptors alpha (ERalpha) and beta (ERbeta) on mouse reproductive phenotypes. Development 2000, 127, 4277–4291. [Google Scholar] [PubMed]
- Zhu, Y.; Bian, Z.; Lu, P.; Karas, R.H.; Bao, L.; Cox, D.; Hodgin, J.; Shaul, P.W.; Thoren, P.; Smithies, O.; et al. Abnormal vascular function and hypertension in mice deficient in estrogen receptor beta. Science 2002, 295, 505–508. [Google Scholar] [CrossRef]
- Stauffer, S.R.; Coletta, C.J.; Tedesco, R.; Nishiguchi, G.; Carlson, K.; Sun, J.; Katzenellenbogen, B.S.; Katzenellenbogen, J.A. Pyrazole ligands: Structure-affinity/activity relationships and estrogen receptor-alpha-selective agonists. J. Med. Chem. 2000, 43, 4934–4947. [Google Scholar] [CrossRef]
- Meyers, M.J.; Sun, J.; Carlson, K.E.; Marriner, G.A.; Katzenellenbogen, B.S.; Katzenellenbogen, J.A. Estrogen receptor-beta potency-selective ligands: Structure-activity relationship studies of diarylpropionitriles and their acetylene and polar analogues. J. Med. Chem. 2001, 44, 4230–4251. [Google Scholar] [CrossRef]
- Sun, J.; Huang, Y.R.; Harrington, W.R.; Sheng, S.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S. Antagonists selective for estrogen receptor alpha. Endocrinology 2002, 143, 941–947. [Google Scholar] [CrossRef]
- Compton, D.R.; Sheng, S.; Carlson, K.E.; Rebacz, N.A.; Lee, I.Y.; Katzenellenbogen, B.S.; Katzenellenbogen, J.A. Pyrazolo[1,5-a]pyrimidines: Estrogen receptor ligands possessing estrogen receptor beta antagonist activity. J. Med. Chem. 2004, 47, 5872–5893. [Google Scholar] [CrossRef]
- Corcoran, J.J.; Nicholson, C.; Sweeney, M.; Charnock, J.C.; Robson, S.C.; Westwood, M.; Taggart, M.J. Human uterine and placental arteries exhibit tissue-specific acute responses to 17beta-estradiol and estrogen-receptor-specific agonists. Mol. Hum. Reprod. 2014, 20, 433–441. [Google Scholar] [CrossRef]
- Nicholson, C.J.; Sweeney, M.; Robson, S.C.; Taggart, M.J. Estrogenic vascular effects are diminished by chronological aging. Sci. Rep. 2017, 7, 12153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Resnik, R.; Killam, A.P.; Battaglia, F.C.; Makowski, E.L.; Meschia, G. The stimulation of uterine blood flow by various estrogens. Endocrinology 1974, 94, 1192–1196. [Google Scholar] [CrossRef] [PubMed]
- Resnik, R.; Brink, G.W.; Plumer, M.H. The effect of progesterone on estrogen-induced uterine blood flow. Am. J. Obstet. Gynecol. 1977, 128, 251–254. [Google Scholar] [CrossRef]
- Huckabee, W.E.; Crenshaw, C.; Curet, L.B.; Mann, L.; Barron, D.H. The effect of exogenous oestrogen on the blood flow and oxygen consumption of the uterus of the non-pregnant ewe. Q. J. Exp. Physiol. Cogn. Med. Sci. 1970, 55, 16–24. [Google Scholar] [CrossRef] [Green Version]
- Simkiss, K. Influence of large doses of oestrogens on the structure of the bones of some reptiles. Nature 1961, 190, 1217–1218. [Google Scholar] [CrossRef]
- Flandroy, L.; Galand, P. Changes in cGMP and cAMP content in the estrogen-stimulated rat uterus: Temporal relationship with other parameters of hormonal stimulation. J. Cyclic Nucleotide Res. 1978, 4, 145–158. [Google Scholar]
- Aronica, S.M.; Kraus, W.L.; Katzenellenbogen, B.S. Estrogen action via the cAMP signaling pathway: Stimulation of adenylate cyclase and cAMP-regulated gene transcription. Proc. Natl. Acad. Sci. USA 1994, 91, 8517–8521. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.B.; Bird, I.M.; Zheng, J.; Magness, R.R. Membrane estrogen receptor-dependent extracellular signal-regulated kinase pathway mediates acute activation of endothelial nitric oxide synthase by estrogen in uterine artery endothelial cells. Endocrinology 2004, 145, 113–125. [Google Scholar] [CrossRef]
- Gu, Q.; Korach, K.S.; Moss, R.L. Rapid action of 17beta-estradiol on kainate-induced currents in hippocampal neurons lacking intracellular estrogen receptors. Endocrinology 1999, 140, 660–666. [Google Scholar] [CrossRef]
- Chambliss, K.L.; Yuhanna, I.S.; Mineo, C.; Liu, P.; German, Z.; Sherman, T.S.; Mendelsohn, M.E.; Anderson, R.G.; Shaul, P.W. Estrogen receptor alpha and endothelial nitric oxide synthase are organized into a functional signaling module in caveolae. Circ. Res. 2000, 87, E44–E52. [Google Scholar] [CrossRef] [Green Version]
- Pastore, M.B.; Landeros, R.V.; Chen, D.B.; Magness, R.R. Structural analysis of estrogen receptors: Interaction between estrogen receptors and cav-1 within the caveolae. Biol. Reprod. 2019, 100, 495–504. [Google Scholar] [CrossRef]
- Pastore, M.B.; Talwar, S.; Conley, M.R.; Magness, R.R. Identification of Differential ER-Alpha Versus ER-Beta Mediated Activation of eNOS in Ovine Uterine Artery Endothelial Cells. Biol. Reprod. 2016, 94, 139. [Google Scholar] [CrossRef] [PubMed]
- Carmeci, C.; Thompson, D.A.; Ring, H.Z.; Francke, U.; Weigel, R.J. Identification of a gene (GPR30) with homology to the G-protein-coupled receptor superfamily associated with estrogen receptor expression in breast cancer. Genomics 1997, 45, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Mizukami, Y. In vivo functions of GPR30/GPER-1, a membrane receptor for estrogen: From discovery to functions in vivo. Endocr. J. 2010, 57, 101–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Revankar, C.M.; Cimino, D.F.; Sklar, L.A.; Arterburn, J.B.; Prossnitz, E.R. A transmembrane intracellular estrogen receptor mediates rapid cell signaling. Science 2005, 307, 1625–1630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filardo, E.J.; Quinn, J.A.; Bland, K.I.; Frackelton, A.R., Jr. Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol. Endocrinol. 2000, 14, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Levin, E.R. G protein-coupled receptor 30: Estrogen receptor or collaborator? Endocrinology 2009, 150, 1563–1565. [Google Scholar] [CrossRef] [Green Version]
- Alexander, S.P.; Mathie, A.; Peters, J.A. Guide to Receptors and Channels (GRAC), 3rd ed. Br. J. Pharmacol. 2008, 153 (Suppl. 2), S1–S209. [Google Scholar] [CrossRef] [Green Version]
- O’Dowd, B.F.; Nguyen, T.; Marchese, A.; Cheng, R.; Lynch, K.R.; Heng, H.H.; Kolakowski, L.F., Jr.; George, S.R. Discovery of three novel G-protein-coupled receptor genes. Genomics 1998, 47, 310–313. [Google Scholar] [CrossRef]
- Owman, C.; Blay, P.; Nilsson, C.; Lolait, S.J. Cloning of human cDNA encoding a novel heptahelix receptor expressed in Burkitt’s lymphoma and widely distributed in brain and peripheral tissues. Biochem. Biophys. Res. Commun. 1996, 228, 285–292. [Google Scholar] [CrossRef]
- Kolkova, Z.; Noskova, V.; Ehinger, A.; Hansson, S.; Casslen, B. G protein-coupled estrogen receptor 1 (GPER, GPR 30) in normal human endometrium and early pregnancy decidua. Mol. Hum. Reprod. 2010, 16, 743–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ylikomi, T.; Vienonen, A.; Ahola, T.M. G protein-coupled receptor 30 down-regulates cofactor expression and interferes with the transcriptional activity of glucocorticoid. Eur. J. Biochem. 2004, 271, 4159–4168. [Google Scholar] [CrossRef] [PubMed]
- Isensee, J.; Meoli, L.; Zazzu, V.; Nabzdyk, C.; Witt, H.; Soewarto, D.; Effertz, K.; Fuchs, H.; Gailus-Durner, V.; Busch, D.; et al. Expression pattern of G protein-coupled receptor 30 in LacZ reporter mice. Endocrinology 2009, 150, 1722–1730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broughton, B.R.; Miller, A.A.; Sobey, C.G. Endothelium-dependent relaxation by G protein-coupled receptor 30 agonists in rat carotid arteries. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H1055–H1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindsey, S.H.; Carver, K.A.; Prossnitz, E.R.; Chappell, M.C. Vasodilation in response to the GPR30 agonist G-1 is not different from estradiol in the mRen2.Lewis female rat. J. Cardiovasc. Pharmacol. 2011, 57, 598–603. [Google Scholar] [CrossRef] [Green Version]
- Tropea, T.; De Francesco, E.M.; Rigiracciolo, D.; Maggiolini, M.; Wareing, M.; Osol, G.; Mandala, M. Pregnancy Augments G Protein Estrogen Receptor (GPER) Induced Vasodilation in Rat Uterine Arteries via the Nitric Oxide—cGMP Signaling Pathway. PLoS ONE 2015, 10, e0141997. [Google Scholar] [CrossRef] [Green Version]
- Haas, E.; Meyer, M.R.; Schurr, U.; Bhattacharya, I.; Minotti, R.; Nguyen, H.H.; Heigl, A.; Lachat, M.; Genoni, M.; Barton, M. Differential effects of 17beta-estradiol on function and expression of estrogen receptor alpha, estrogen receptor beta, and GPR30 in arteries and veins of patients with atherosclerosis. Hypertension 2007, 49, 1358–1363. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Dehghani, B.; Magrisso, I.J.; Rick, E.A.; Bonhomme, E.; Cody, D.B.; Elenich, L.A.; Subramanian, S.; Murphy, S.J.; Kelly, M.J.; et al. GPR30 contributes to estrogen-induced thymic atrophy. Mol. Endocrinol. 2008, 22, 636–648. [Google Scholar] [CrossRef] [Green Version]
- Otto, C.; Fuchs, I.; Kauselmann, G.; Kern, H.; Zevnik, B.; Andreasen, P.; Schwarz, G.; Altmann, H.; Klewer, M.; Schoor, M.; et al. GPR30 does not mediate estrogenic responses in reproductive organs in mice. Biol. Reprod. 2009, 80, 34–41. [Google Scholar] [CrossRef] [Green Version]
- Martensson, U.E.; Salehi, S.A.; Windahl, S.; Gomez, M.F.; Sward, K.; Daszkiewicz-Nilsson, J.; Wendt, A.; Andersson, N.; Hellstrand, P.; Grande, P.O.; et al. Deletion of the G protein-coupled receptor 30 impairs glucose tolerance, reduces bone growth, increases blood pressure, and eliminates estradiol-stimulated insulin release in female mice. Endocrinology 2009, 150, 687–698. [Google Scholar] [CrossRef]
- Meyer, M.R.; Prossnitz, E.R.; Barton, M. The G protein-coupled estrogen receptor GPER/GPR30 as a regulator of cardiovascular function. Vascul. Pharmacol. 2011, 55, 17–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bologa, C.G.; Revankar, C.M.; Young, S.M.; Edwards, B.S.; Arterburn, J.B.; Kiselyov, A.S.; Parker, M.A.; Tkachenko, S.E.; Savchuck, N.P.; Sklar, L.A.; et al. Virtual and biomolecular screening converge on a selective agonist for GPR30. Nat. Chem. Biol. 2006, 2, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Dennis, M.K.; Field, A.S.; Burai, R.; Ramesh, C.; Petrie, W.K.; Bologa, C.G.; Oprea, T.I.; Yamaguchi, Y.; Hayashi, S.; Sklar, L.A.; et al. Identification of a GPER/GPR30 antagonist with improved estrogen receptor counterselectivity. J. Steroid Biochem. Mol. Biol. 2011, 127, 358–366. [Google Scholar] [CrossRef] [Green Version]
- Dennis, M.K.; Burai, R.; Ramesh, C.; Petrie, W.K.; Alcon, S.N.; Nayak, T.K.; Bologa, C.G.; Leitao, A.; Brailoiu, E.; Deliu, E.; et al. In vivo effects of a GPR30 antagonist. Nat. Chem. Biol. 2009, 5, 421–427. [Google Scholar] [CrossRef] [Green Version]
- Meyer, M.R.; Fredette, N.C.; Howard, T.A.; Hu, C.; Ramesh, C.; Daniel, C.; Amann, K.; Arterburn, J.B.; Barton, M.; Prossnitz, E.R. G protein-coupled estrogen receptor protects from atherosclerosis. Sci. Rep. 2014, 4, 7564. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Sun, X.; Chou, J.; Lin, M.; Ferrario, C.M.; Zapata-Sudo, G.; Groban, L. Cardiomyocyte-specific deletion of the G protein-coupled estrogen receptor (GPER) leads to left ventricular dysfunction and adverse remodeling: A sex-specific gene profiling analysis. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1870–1882. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, S.H.; Cohen, J.A.; Brosnihan, K.B.; Gallagher, P.E.; Chappell, M.C. Chronic treatment with the G protein-coupled receptor 30 agonist G-1 decreases blood pressure in ovariectomized mRen2.Lewis rats. Endocrinology 2009, 150, 3753–3758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, E.; Bhattacharya, I.; Brailoiu, E.; Damjanovic, M.; Brailoiu, G.C.; Gao, X.; Mueller-Guerre, L.; Marjon, N.A.; Gut, A.; Minotti, R.; et al. Regulatory role of G protein-coupled estrogen receptor for vascular function and obesity. Circ. Res. 2009, 104, 288–291. [Google Scholar] [CrossRef] [Green Version]
- Mazzuca, M.Q.; Mata, K.M.; Li, W.; Rangan, S.S.; Khalil, R.A. Estrogen receptor subtypes mediate distinct microvascular dilation and reduction in [Ca2+]I in mesenteric microvessels of female rat. J. Pharmacol. Exp. Ther. 2015, 352, 291–304. [Google Scholar] [CrossRef] [Green Version]
- Chambliss, K.L.; Shaul, P.W. Rapid activation of endothelial NO synthase by estrogen: Evidence for a steroid receptor fast-action complex (SRFC) in caveolae. Steroids 2002, 67, 413–419. [Google Scholar] [CrossRef]
- Galluzzo, P.; Caiazza, F.; Moreno, S.; Marino, M. Role of ERbeta palmitoylation in the inhibition of human colon cancer cell proliferation. Endocr. Relat. Cancer 2007, 14, 153–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatia, M. Hydrogen sulfide as a vasodilator. IUBMB Life 2005, 57, 603–606. [Google Scholar] [CrossRef] [PubMed]
- Hurst, A.G.; Goad, D.W.; Mohan, M.; Malayer, J.R. Independent downstream gene expression profiles in the presence of estrogen receptor alpha or beta. Biol. Reprod. 2004, 71, 1252–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, R.X.; Barnes, C.J.; Zhang, Z.; Bao, Y.; Kumar, R.; Santen, R.J. The role of Shc and insulin-like growth factor 1 receptor in mediating the translocation of estrogen receptor alpha to the plasma membrane. Proc. Natl. Acad. Sci. USA 2004, 101, 2076–2081. [Google Scholar] [CrossRef] [Green Version]
- Marquez, D.C.; Lee, J.; Lin, T.; Pietras, R.J. Epidermal growth factor receptor and tyrosine phosphorylation of estrogen receptor. Endocrine 2001, 16, 73–81. [Google Scholar] [CrossRef]
- Pedram, A.; Razandi, M.; Levin, E.R. Nature of functional estrogen receptors at the plasma membrane. Mol. Endocrinol. 2006, 20, 1996–2009. [Google Scholar] [CrossRef]
- Pedram, A.; Razandi, M.; Aitkenhead, M.; Hughes, C.C.; Levin, E.R. Integration of the non-genomic and genomic actions of estrogen. Membrane-initiated signaling by steroid to transcription and cell biology. J. Biol. Chem. 2002, 277, 50768–50775. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Thompson, E.B. The structure of the nuclear hormone receptors. Steroids 1999, 64, 310–319. [Google Scholar] [CrossRef]
- Kumar, R.; Zakharov, M.N.; Khan, S.H.; Miki, R.; Jang, H.; Toraldo, G.; Singh, R.; Bhasin, S.; Jasuja, R. The dynamic structure of the estrogen receptor. J. Amino. Acids 2011, 2011, 812540. [Google Scholar] [CrossRef] [Green Version]
- Le Dily, F.; Beato, M. Signaling by Steroid Hormones in the 3D Nuclear Space. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef]
- Martini, P.G.; Katzenellenbogen, B.S. Modulation of estrogen receptor activity by selective coregulators. J. Steroid Biochem. Mol. Biol. 2003, 85, 117–122. [Google Scholar] [CrossRef]
- Driscoll, M.D.; Sathya, G.; Muyan, M.; Klinge, C.M.; Hilf, R.; Bambara, R.A. Sequence requirements for estrogen receptor binding to estrogen response elements. J. Biol. Chem. 1998, 273, 29321–29330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klinge, C.M. Estrogen receptor interaction with estrogen response elements. Nucleic Acids Res. 2001, 29, 2905–2919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Andersson, S.; Warner, M.; Gustafsson, J.A. Estrogen receptor (ER)beta knockout mice reveal a role for ERbeta in migration of cortical neurons in the developing brain. Proc. Natl. Acad. Sci. USA 2003, 100, 703–708. [Google Scholar] [CrossRef] [Green Version]
- Cowley, S.M.; Hoare, S.; Mosselman, S.; Parker, M.G. Estrogen receptors alpha and beta form heterodimers on DNA. J. Biol. Chem. 1997, 272, 19858–19862. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Chambon, P. The estrogen receptor binds tightly to its responsive element as a ligand-induced homodimer. Cell 1988, 55, 145–156. [Google Scholar] [CrossRef]
- Charn, T.H.; Liu, E.T.; Chang, E.C.; Lee, Y.K.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S. Genome-wide dynamics of chromatin binding of estrogen receptors alpha and beta: Mutual restriction and competitive site selection. Mol. Endocrinol. 2010, 24, 47–59. [Google Scholar] [CrossRef] [Green Version]
- Grober, O.M.; Mutarelli, M.; Giurato, G.; Ravo, M.; Cicatiello, L.; De Filippo, M.R.; Ferraro, L.; Nassa, G.; Papa, M.F.; Paris, O.; et al. Global analysis of estrogen receptor beta binding to breast cancer cell genome reveals an extensive interplay with estrogen receptor alpha for target gene regulation. BMC Genom. 2011, 12, 36. [Google Scholar] [CrossRef]
- Sausville, E.; Carney, D.; Battey, J. The human vasopressin gene is linked to the oxytocin gene and is selectively expressed in a cultured lung cancer cell line. J. Biol. Chem. 1985, 260, 10236–10241. [Google Scholar]
- Lechuga, T.J.; Qi, Q.R.; Kim, T.; Magness, R.R.; Chen, D.B. E2beta stimulates ovine uterine artery endothelial cell H2S production in vitro by estrogen receptor-dependent upregulation of cystathionine beta-synthase and cystathionine gamma-lyase expressiondagger. Biol. Reprod. 2019, 100, 514–522. [Google Scholar] [CrossRef]
- Greaves, E.; Collins, F.; Critchley, H.O.; Saunders, P.T. ERbeta-dependent effects on uterine endothelial cells are cell specific and mediated via Sp1. Hum. Reprod. 2013, 28, 2490–2501. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Mayer, J.A.; Mazumdar, A.; Fertuck, K.; Kim, H.; Brown, M.; Brown, P.H. Estrogen induces c-myc gene expression via an upstream enhancer activated by the estrogen receptor and the AP-1 transcription factor. Mol. Endocrinol. 2011, 25, 1527–1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, S.R.; Lu, B.; Scott, G.K.; Kushner, P.J.; Smith, I.E.; Dowsett, M.; Benz, C.C. Increased activator protein-1 DNA binding and c-Jun NH2-terminal kinase activity in human breast tumors with acquired tamoxifen resistance. Clin. Cancer Res. 1999, 5, 251–256. [Google Scholar] [PubMed]
- Webb, P.; Nguyen, P.; Valentine, C.; Lopez, G.N.; Kwok, G.R.; McInerney, E.; Katzenellenbogen, B.S.; Enmark, E.; Gustafsson, J.A.; Nilsson, S.; et al. The estrogen receptor enhances AP-1 activity by two distinct mechanisms with different requirements for receptor transactivation functions. Mol. Endocrinol. 1999, 13, 1672–1685. [Google Scholar] [CrossRef] [PubMed]
- Bajic, V.B.; Tan, S.L.; Chong, A.; Tang, S.; Strom, A.; Gustafsson, J.A.; Lin, C.Y.; Liu, E.T. Dragon ERE Finder version 2: A tool for accurate detection and analysis of estrogen response elements in vertebrate genomes. Nucleic Acids Res. 2003, 31, 3605–3607. [Google Scholar] [CrossRef] [Green Version]
- Shaul, P.W.; North, A.J.; Wu, L.C.; Wells, L.B.; Brannon, T.S.; Lau, K.S.; Michel, T.; Margraf, L.R.; Star, R.A. Endothelial nitric oxide synthase is expressed in cultured human bronchiolar epithelium. J. Clin. Investig. 1994, 94, 2231–2236. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Tang, Z.P.; Zhao, W.; Cong, B.H.; Lu, J.Q.; Tang, X.L.; Li, X.H.; Zhu, X.Y.; Ni, X. MiR-22/Sp-1 Links Estrogens With the Up-Regulation of Cystathionine gamma-Lyase in Myocardium, Which Contributes to Estrogenic Cardioprotection Against Oxidative Stress. Endocrinology 2015, 156, 2124–2137. [Google Scholar] [CrossRef]
- O’Lone, R.; Frith, M.C.; Karlsson, E.K.; Hansen, U. Genomic targets of nuclear estrogen receptors. Mol. Endocrinol. 2004, 18, 1859–1875. [Google Scholar] [CrossRef]
- Fujimoto, N.; Kitamura, S. Effects of environmental estrogenic chemicals on AP1 mediated transcription with estrogen receptors alpha and beta. J. Steroid Biochem. Mol. Biol. 2004, 88, 53–59. [Google Scholar] [CrossRef]
- Lupien, M.; Eeckhoute, J.; Meyer, C.A.; Krum, S.A.; Rhodes, D.R.; Liu, X.S.; Brown, M. Coactivator function defines the active estrogen receptor alpha cistrome. Mol. Cell. Biol. 2009, 29, 3413–3423. [Google Scholar] [CrossRef] [Green Version]
- Zaret, K.S.; Carroll, J.S. Pioneer transcription factors: Establishing competence for gene expression. Genes Dev. 2011, 25, 2227–2241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filardo, E.J.; Quinn, J.A.; Frackelton, A.R., Jr.; Bland, K.I. Estrogen action via the G protein-coupled receptor, GPR30: Stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signaling axis. Mol. Endocrinol. 2002, 16, 70–84. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Gros, R.; Limbird, L.E.; Chorazyczewski, J.; Feldman, R.D. Estradiol-mediated ERK phosphorylation and apoptosis in vascular smooth muscle cells requires GPR 30. Am. J. Physiol. Cell Physiol. 2009, 297, C1178–C1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acconcia, F.; Bocedi, A.; Ascenzi, P.; Marino, M. Does palmitoylation target estrogen receptors to plasma membrane caveolae? IUBMB Life 2003, 55, 33–35. [Google Scholar] [CrossRef] [PubMed]
- Acconcia, F.; Ascenzi, P.; Bocedi, A.; Spisni, E.; Tomasi, V.; Trentalance, A.; Visca, P.; Marino, M. Palmitoylation-dependent estrogen receptor alpha membrane localization: Regulation by 17beta-estradiol. Mol. Biol. Cell 2005, 16, 231–237. [Google Scholar] [CrossRef]
- Kim, H.P.; Lee, J.Y.; Jeong, J.K.; Bae, S.W.; Lee, H.K.; Jo, I. Nongenomic stimulation of nitric oxide release by estrogen is mediated by estrogen receptor alpha localized in caveolae. Biochem. Biophys. Res. Commun. 1999, 263, 257–262. [Google Scholar] [CrossRef]
- Chambliss, K.L.; Yuhanna, I.S.; Anderson, R.G.; Mendelsohn, M.E.; Shaul, P.W. ERbeta has nongenomic action in caveolae. Mol. Endocrinol. 2002, 16, 938–946. [Google Scholar] [CrossRef] [Green Version]
- Hisamoto, K.; Ohmichi, M.; Kurachi, H.; Hayakawa, J.; Kanda, Y.; Nishio, Y.; Adachi, K.; Tasaka, K.; Miyoshi, E.; Fujiwara, N.; et al. Estrogen induces the Akt-dependent activation of endothelial nitric-oxide synthase in vascular endothelial cells. J. Biol. Chem. 2001, 276, 3459–3467. [Google Scholar] [CrossRef] [Green Version]
- Bird, I.M.; Sullivan, J.A.; Di, T.; Cale, J.M.; Zhang, L.; Zheng, J.; Magness, R.R. Pregnancy-dependent changes in cell signaling underlie changes in differential control of vasodilator production in uterine artery endothelial cells. Endocrinology 2000, 141, 1107–1117. [Google Scholar] [CrossRef]
- Bernier, S.G.; Haldar, S.; Michel, T. Bradykinin-regulated interactions of the mitogen-activated protein kinase pathway with the endothelial nitric-oxide synthase. J. Biol. Chem. 2000, 275, 30707–30715. [Google Scholar] [CrossRef] [Green Version]
- Wyatt, A.W.; Steinert, J.R.; Wheeler-Jones, C.P.; Morgan, A.J.; Sugden, D.; Pearson, J.D.; Sobrevia, L.; Mann, G.E. Early activation of the p42/p44MAPK pathway mediates adenosine-induced nitric oxide production in human endothelial cells: A novel calcium-insensitive mechanism. FASEB J. 2002, 16, 1584–1594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di, T.; Sullivan, J.A.; Magness, R.R.; Zhang, L.; Bird, I.M. Pregnancy-specific enhancement of agonist-stimulated ERK-1/2 signaling in uterine artery endothelial cells increases Ca2+ sensitivity of endothelial nitric oxide synthase as well as cytosolic phospholipase A(2). Endocrinology 2001, 142, 3014–3026. [Google Scholar] [CrossRef]
- Xu, X.; Yan, Q.; Liu, X.; Li, P.; Li, X.; Chen, Y.; Simoncini, T.; Liu, J.; Zhu, D.; Fu, X. 17beta-Estradiol nongenomically induces vascular endothelial H2S release by promoting phosphorylation of cystathionine gamma-lyase. J. Biol. Chem. 2019, 294, 15577–15592. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Moriarty, K.; Bender, J.R. Vascular cell signaling by membrane estrogen receptors. Steroids 2008, 73, 864–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marino, M.; Ascenzi, P. Membrane association of estrogen receptor alpha and beta influences 17beta-estradiol-mediated cancer cell proliferation. Steroids 2008, 73, 853–858. [Google Scholar] [CrossRef]
- Kato, S.; Endoh, H.; Masuhiro, Y.; Kitamoto, T.; Uchiyama, S.; Sasaki, H.; Masushige, S.; Gotoh, Y.; Nishida, E.; Kawashima, H.; et al. Activation of the estrogen receptor through phosphorylation by mitogen-activated protein kinase. Science 1995, 270, 1491–1494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razandi, M.; Pedram, A.; Park, S.T.; Levin, E.R. Proximal events in signaling by plasma membrane estrogen receptors. J. Biol. Chem. 2003, 278, 2701–2712. [Google Scholar] [CrossRef] [Green Version]
- Wade, C.B.; Robinson, S.; Shapiro, R.A.; Dorsa, D.M. Estrogen receptor (ER)alpha and ERbeta exhibit unique pharmacologic properties when coupled to activation of the mitogen-activated protein kinase pathway. Endocrinology 2001, 142, 2336–2342. [Google Scholar] [CrossRef]
- Ma, Y.; Qiao, X.; Falone, A.E.; Reslan, O.M.; Sheppard, S.J.; Khalil, R.A. Gender-specific reduction in contraction is associated with increased estrogen receptor expression in single vascular smooth muscle cells of female rat. Cell. Physiol. Biochem. 2010, 26, 457–470. [Google Scholar] [CrossRef]
- Lindsey, S.H.; Liu, L.; Chappell, M.C. Vasodilation by GPER in mesenteric arteries involves both endothelial nitric oxide and smooth muscle cAMP signaling. Steroids 2014, 81, 99–102. [Google Scholar] [CrossRef] [Green Version]
- Ge, X.; Guo, R.; Qiao, Y.; Zhang, Y.; Lei, J.; Wang, X.; Li, L.; Hu, D. The G protein-coupled receptor GPR30 mediates the nontranscriptional effect of estrogen on the activation of PI3K/Akt pathway in endometrial cancer cells. Int. J. Gynecol. Cancer 2013, 23, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Vivacqua, A.; Bonofiglio, D.; Recchia, A.G.; Musti, A.M.; Picard, D.; Ando, S.; Maggiolini, M. The G protein-coupled receptor GPR30 mediates the proliferative effects induced by 17beta-estradiol and hydroxytamoxifen in endometrial cancer cells. Mol. Endocrinol. 2006, 20, 631–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filice, E.; Recchia, A.G.; Pellegrino, D.; Angelone, T.; Maggiolini, M.; Cerra, M.C. A new membrane G protein-coupled receptor (GPR30) is involved in the cardiac effects of 17beta-estradiol in the male rat. J. Physiol. Pharmacol. 2009, 60, 3–10. [Google Scholar]
- Tian, R.; Wang, Z.; Shi, Z.; Li, D.; Wang, Y.; Zhu, Y.; Lin, W.; Gui, Y.; Zheng, X.L. Differential expression of G-protein-coupled estrogen receptor-30 in human myometrial and uterine leiomyoma smooth muscle. Fertil. Steril. 2013, 99, 256–263. [Google Scholar] [CrossRef] [PubMed]
- Levin, E.R. Rapid signaling by steroid receptors. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, R1425–R1430. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, S.; Davidge, S.T. G-protein coupled receptor 30 (GPR30): A novel regulator of endothelial inflammation. PLoS ONE 2012, 7, e52357. [Google Scholar] [CrossRef] [Green Version]
- Gao, F.; Ma, X.; Ostmann, A.B.; Das, S.K. GPR30 activation opposes estrogen-dependent uterine growth via inhibition of stromal ERK1/2 and estrogen receptor alpha (ERalpha) phosphorylation signals. Endocrinology 2011, 152, 1434–1447. [Google Scholar] [CrossRef] [Green Version]
- Richards, R.G.; DiAugustine, R.P.; Petrusz, P.; Clark, G.C.; Sebastian, J. Estradiol stimulates tyrosine phosphorylation of the insulin-like growth factor-1 receptor and insulin receptor substrate-1 in the uterus. Proc. Natl. Acad. Sci. USA 1996, 93, 12002–12007. [Google Scholar] [CrossRef] [Green Version]
- Richards, R.G.; Walker, M.P.; Sebastian, J.; DiAugustine, R.P. Insulin-like growth factor-1 (IGF-1) receptor-insulin receptor substrate complexes in the uterus. Altered signaling response to estradiol in the IGF-1(m/m) mouse. J. Biol. Chem. 1998, 273, 11962–11969. [Google Scholar] [CrossRef] [Green Version]
- Sukocheva, O.; Wadham, C.; Holmes, A.; Albanese, N.; Verrier, E.; Feng, F.; Bernal, A.; Derian, C.K.; Ullrich, A.; Vadas, M.A.; et al. Estrogen transactivates EGFR via the sphingosine 1-phosphate receptor Edg-3: The role of sphingosine kinase-1. J. Cell Biol. 2006, 173, 301–310. [Google Scholar] [CrossRef]
- Joel, P.B.; Smith, J.; Sturgill, T.W.; Fisher, T.L.; Blenis, J.; Lannigan, D.A. pp90rsk1 regulates estrogen receptor-mediated transcription through phosphorylation of Ser-167. Mol. Cell. Biol. 1998, 18, 1978–1984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, M.; Paciga, J.E.; Feldman, R.I.; Yuan, Z.; Coppola, D.; Lu, Y.Y.; Shelley, S.A.; Nicosia, S.V.; Cheng, J.Q. Phosphatidylinositol-3-OH Kinase (PI3K)/AKT2, activated in breast cancer, regulates and is induced by estrogen receptor alpha (ERalpha) via interaction between ERalpha and PI3K. Cancer Res. 2001, 61, 5985–5991. [Google Scholar] [PubMed]
- Arnold, S.F.; Melamed, M.; Vorojeikina, D.P.; Notides, A.C.; Sasson, S. Estradiol-binding mechanism and binding capacity of the human estrogen receptor is regulated by tyrosine phosphorylation. Mol. Endocrinol. 1997, 11, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Schnitzler, G.R.; Vallaster, C.S.; Ueda, K.; Erdkamp, S.; Briggs, C.E.; Iyer, L.K.; Jaffe, I.Z.; Karas, R.H. Unliganded estrogen receptor alpha regulates vascular cell function and gene expression. Mol. Cell. Endocrinol. 2017, 442, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, S.C.; Winuthayanon, W.; Korach, K.S. What’s new in estrogen receptor action in the female reproductive tract. J. Mol. Endocrinol. 2016, 56, R55–R71. [Google Scholar] [CrossRef] [Green Version]
- Karas, R.H.; Gauer, E.A.; Bieber, H.E.; Baur, W.E.; Mendelsohn, M.E. Growth factor activation of the estrogen receptor in vascular cells occurs via a mitogen-activated protein kinase-independent pathway. J. Clin. Investig. 1998, 101, 2851–2861. [Google Scholar] [CrossRef] [Green Version]
- Mendelsohn, M.E.; Karas, R.H. Rapid progress for non-nuclear estrogen receptor signaling. J. Clin. Investig. 2010, 120, 2277–2279. [Google Scholar] [CrossRef]
- Mendelsohn, M.E.; Karas, R.H. The protective effects of estrogen on the cardiovascular system. N. Engl. J. Med. 1999, 340, 1801–1811. [Google Scholar] [CrossRef]
- Sentis, S.; Le Romancer, M.; Bianchin, C.; Rostan, M.C.; Corbo, L. Sumoylation of the estrogen receptor alpha hinge region regulates its transcriptional activity. Mol. Endocrinol. 2005, 19, 2671–2684. [Google Scholar] [CrossRef]
- Nirmala, P.B.; Thampan, R.V. Ubiquitination of the rat uterine estrogen receptor: Dependence on estradiol. Biochem. Biophys. Res. Commun. 1995, 213, 24–31. [Google Scholar] [CrossRef]
- Wang, C.; Fu, M.; Angeletti, R.H.; Siconolfi-Baez, L.; Reutens, A.T.; Albanese, C.; Lisanti, M.P.; Katzenellenbogen, B.S.; Kato, S.; Hopp, T.; et al. Direct acetylation of the estrogen receptor alpha hinge region by p300 regulates transactivation and hormone sensitivity. J. Biol. Chem. 2001, 276, 18375–18383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, M.; Wang, C.; Zhang, X.; Pestell, R. Nuclear receptor modifications and endocrine cell proliferation. J. Steroid Biochem. Mol. Biol. 2003, 85, 133–138. [Google Scholar] [CrossRef]
- Fuqua, S.A.; Wiltschke, C.; Zhang, Q.X.; Borg, A.; Castles, C.G.; Friedrichs, W.E.; Hopp, T.; Hilsenbeck, S.; Mohsin, S.; O’Connell, P.; et al. A hypersensitive estrogen receptor-alpha mutation in premalignant breast lesions. Cancer Res. 2000, 60, 4026–4029. [Google Scholar] [PubMed]
- Picard, N.; Caron, V.; Bilodeau, S.; Sanchez, M.; Mascle, X.; Aubry, M.; Tremblay, A. Identification of estrogen receptor beta as a SUMO-1 target reveals a novel phosphorylated sumoylation motif and regulation by glycogen synthase kinase 3beta. Mol. Cell. Biol. 2012, 32, 2709–2721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, M.; Kotcherguina, L.; Dharia, A.; Fujimoto, S.; Dahiya, R. Cytosine-phosphoguanine methylation of estrogen receptors in endometrial cancer. Cancer Res. 2001, 61, 3262–3266. [Google Scholar] [PubMed]
- Nothnick, W.B.; Healy, C. Estrogen induces distinct patterns of microRNA expression within the mouse uterus. Reprod. Sci. 2010, 17, 987–994. [Google Scholar] [CrossRef]
- Hou, L.; Lu, Y.; Li, Y.; Li, L. MiRNA-451 is a potential biomarker for estrogenicity in mouse uterus. Front. Environ. Sci. Eng. 2014, 8, 99–105. [Google Scholar] [CrossRef]
- Perez-Cremades, D.; Mompeon, A.; Vidal-Gomez, X.; Hermenegildo, C.; Novella, S. Role of miRNA in the Regulatory Mechanisms of Estrogens in Cardiovascular Ageing. Oxid. Med. Cell. Longev. 2018, 2018, 6082387. [Google Scholar] [CrossRef]
- Williams, K.C.; Renthal, N.E.; Gerard, R.D.; Mendelson, C.R. The microRNA (miR)-199a/214 cluster mediates opposing effects of progesterone and estrogen on uterine contractility during pregnancy and labor. Mol. Endocrinol. 2012, 26, 1857–1867. [Google Scholar] [CrossRef]
- Zierau, O.; Helle, J.; Schadyew, S.; Morgenroth, Y.; Bentler, M.; Hennig, A.; Chittur, S.; Tenniswood, M.; Kretzschmar, G. Role of miR-203 in estrogen receptor-mediated signaling in the rat uterus and endometrial carcinoma. J. Cell. Biochem. 2018, 119, 5359–5372. [Google Scholar] [CrossRef]
- Hu, X.Q.; Dasgupta, C.; Xiao, D.; Huang, X.; Yang, S.; Zhang, L. MicroRNA-210 Targets Ten-Eleven Translocation Methylcytosine Dioxygenase 1 and Suppresses Pregnancy-Mediated Adaptation of Large Conductance Ca(2+)-Activated (K+) Channel Expression and Function in Ovine Uterine Arteries. Hypertension 2017, 70, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Pan, Q.; Luo, X.; Toloubeydokhti, T.; Chegini, N. The expression profile of micro-RNA in endometrium and endometriosis and the influence of ovarian steroids on their expression. Mol. Hum. Reprod. 2007, 13, 797–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidal-Gomez, X.; Perez-Cremades, D.; Mompeon, A.; Dantas, A.P.; Novella, S.; Hermenegildo, C. MicroRNA as Crucial Regulators of Gene Expression in Estradiol-Treated Human Endothelial Cells. Cell. Physiol. Biochem. 2018, 45, 1878–1892. [Google Scholar] [CrossRef]
- Dasgupta, C.; Chen, M.; Zhang, H.; Yang, S.; Zhang, L. Chronic hypoxia during gestation causes epigenetic repression of the estrogen receptor-alpha gene in ovine uterine arteries via heightened promoter methylation. Hypertension 2012, 60, 697–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Xiao, D.; Hu, X.Q.; Dasgupta, C.; Yang, S.; Zhang, L. Hypoxia Represses ER-alpha Expression and Inhibits Estrogen-Induced Regulation of Ca2+-Activated K+ Channel Activity and Myogenic Tone in Ovine Uterine Arteries: Causal Role of DNA Methylation. Hypertension 2015, 66, 44–51. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Dasgupta, C.; Xiong, F.; Zhang, L. Epigenetic upregulation of large-conductance Ca2+-activated K+ channel expression in uterine vascular adaptation to pregnancy. Hypertension 2014, 64, 610–618. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.Q.; Dasgupta, C.; Chen, M.; Xiao, D.; Huang, X.; Han, L.; Yang, S.; Xu, Z.; Zhang, L. Pregnancy Reprograms Large-Conductance Ca(2+)-Activated K(+) Channel in Uterine Arteries: Roles of Ten-Eleven Translocation Methylcytosine Dioxygenase 1-Mediated Active Demethylation. Hypertension 2017, 69, 1181–1191. [Google Scholar] [CrossRef]
- Graham, A.; Falcone, T.; Nothnick, W.B. The expression of microRNA-451 in human endometriotic lesions is inversely related to that of macrophage migration inhibitory factor (MIF) and regulates MIF expression and modulation of epithelial cell survival. Hum. Reprod. 2015, 30, 642–652. [Google Scholar] [CrossRef] [Green Version]
- Brkic, J.; Dunk, C.; O’Brien, J.; Fu, G.; Nadeem, L.; Wang, Y.L.; Rosman, D.; Salem, M.; Shynlova, O.; Yougbare, I.; et al. MicroRNA-218-5p Promotes Endovascular Trophoblast Differentiation and Spiral Artery Remodeling. Mol. Ther. 2018, 26, 2189–2205. [Google Scholar] [CrossRef] [Green Version]
- Myatt, S.S.; Lam, E.W. The emerging roles of forkhead box (Fox) proteins in cancer. Nat. Rev. Cancer 2007, 7, 847–859. [Google Scholar] [CrossRef]
- Zhao, W.; Shen, W.W.; Cao, X.M.; Ding, W.Y.; Yan, L.P.; Gao, L.J.; Li, X.L.; Zhong, T.Y. Novel mechanism of miRNA-365-regulated trophoblast apoptosis in recurrent miscarriage. J. Cell. Mol. Med. 2017, 21, 2412–2425. [Google Scholar] [CrossRef] [Green Version]
- Fu, G.; Ye, G.; Nadeem, L.; Ji, L.; Manchanda, T.; Wang, Y.; Zhao, Y.; Qiao, J.; Wang, Y.L.; Lye, S.; et al. MicroRNA-376c impairs transforming growth factor-beta and nodal signaling to promote trophoblast cell proliferation and invasion. Hypertension 2013, 61, 864–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Feng, L.; Zhang, H.; Hachy, S.; Satohisa, S.; Laurent, L.C.; Parast, M.; Zheng, J.; Chen, D.B. Preeclampsia up-regulates angiogenesis-associated microRNA (i.e., miR-17, -20a, and -20b) that target ephrin-B2 and EPHB4 in human placenta. J. Clin. Endocrinol. Metab. 2012, 97, E1051–E1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penney, L.L.; Frederick, R.J.; Parker, G.W. 17 beta-estradiol stimulation of uterine blood flow in oophorectomized rabbits with complete inhibition of uterine ribonucleic acid synthesis. Endocrinology 1981, 109, 1672–1676. [Google Scholar] [CrossRef] [PubMed]
- Keyes, L.E.; Majack, R.; Dempsey, E.C.; Moore, L.G. Pregnancy stimulation of DNA synthesis and uterine blood flow in the guinea pig. Pediatric Res. 1997, 41, 708–715. [Google Scholar] [CrossRef] [Green Version]
- Dickson, W.M.; Bosc, M.J.; Locatelli, A. Effect of estrogen and progesterone on uterine blood flow of castrate sows. Am. J. Physiol. 1969, 217, 1431–1434. [Google Scholar] [CrossRef]
- Resnik, R.; Battaglia, F.C.; Makowski, E.L.; Meschia, G. The effect of actinomycin D on estrogen-induced uterine blood flow. Am. J. Obstet. Gynecol. 1975, 122, 273–277. [Google Scholar] [CrossRef]
- Rosenfeld, C.R.; Roy, T.; Cox, B.E. Mechanisms modulating estrogen-induced uterine vasodilation. Vascul. Pharmacol. 2002, 38, 115–125. [Google Scholar] [CrossRef]
- Furchgott, R.F.; Zawadzki, J.V. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 1980, 288, 373–376. [Google Scholar] [CrossRef]
- Ignarro, L.J.; Buga, G.M.; Wood, K.S.; Byrns, R.E.; Chaudhuri, G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc. Natl. Acad. Sci. USA 1987, 84, 9265–9269. [Google Scholar] [CrossRef] [Green Version]
- Knowles, R.G.; Moncada, S. Nitric oxide synthases in mammals. Biochem. J. 1994, 298 Pt 2, 249–258. [Google Scholar] [CrossRef]
- Koshland, D.E., Jr. The molecule of the year. Science 1992, 258, 1861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, R.M.; Ferrige, A.G.; Moncada, S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 1987, 327, 524–526. [Google Scholar] [CrossRef] [PubMed]
- Gross, S.S.; Lane, P. Physiological reactions of nitric oxide and hemoglobin: A radical rethink. Proc. Natl. Acad. Sci. USA 1999, 96, 9967–9969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blomberg, L.M.; Blomberg, M.R.; Siegbahn, P.E. A theoretical study of myoglobin working as a nitric oxide scavenger. J. Biol. Inorg. Chem. 2004, 9, 923–935. [Google Scholar] [CrossRef]
- Satoh, K.; Ikeda, Y.; Shioda, S.; Tobe, T.; Yoshikawa, T. Edarabone scavenges nitric oxide. Redox Rep. 2002, 7, 219–222. [Google Scholar] [CrossRef]
- Stamler, J.S. S-nitrosothiols and the bioregulatory actions of nitrogen oxides through reactions with thiol groups. Curr. Top. Microbiol. Immunol. 1995, 196, 19–36. [Google Scholar] [CrossRef]
- Beckman, J.S.; Beckman, T.W.; Chen, J.; Marshall, P.A.; Freeman, B.A. Apparent hydroxyl radical production by peroxynitrite: Implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. USA 1990, 87, 1620–1624. [Google Scholar] [CrossRef] [Green Version]
- Tsikas, D.; Denker, K.; Frolich, J.C. Artifactual-free analysis of S-nitrosoglutathione and S-nitroglutathione by neutral-pH, anion-pairing, high-performance liquid chromatography. Study on peroxynitrite-mediated S-nitration of glutathione to S-nitroglutathione under physiological conditions. J. Chromatogr. A 2001, 915, 107–116. [Google Scholar] [CrossRef]
- Mayer, B.; Schmidt, K.; Humbert, P.; Bohme, E. Biosynthesis of endothelium-derived relaxing factor: A cytosolic enzyme in porcine aortic endothelial cells Ca2+-dependently converts L-arginine into an activator of soluble guanylyl cyclase. Biochem. Biophys. Res. Commun. 1989, 164, 678–685. [Google Scholar] [CrossRef]
- Burstyn, J.N.; Yu, A.E.; Dierks, E.A.; Hawkins, B.K.; Dawson, J.H. Studies of the heme coordination and ligand binding properties of soluble guanylyl cyclase (sGC): Characterization of Fe(II)sGC and Fe(II)sGC(CO) by electronic absorption and magnetic circular dichroism spectroscopies and failure of CO to activate the enzyme. Biochemistry 1995, 34, 5896–5903. [Google Scholar] [CrossRef] [PubMed]
- Francis, S.H.; Busch, J.L.; Corbin, J.D.; Sibley, D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol. Rev. 2010, 62, 525–563. [Google Scholar] [CrossRef] [PubMed]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Goulopoulou, S.; Hannan, J.L.; Matsumoto, T.; Ergul, A.; Webb, R.C. Augmented dilation to nitric oxide in uterine arteries from rats with type 2 diabetes: Implications for vascular adaptations to pregnancy. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H610–H618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magness, R.R.; Shaw, C.E.; Phernetton, T.M.; Zheng, J.; Bird, I.M. Endothelial vasodilator production by uterine and systemic arteries. II. Pregnancy effects on NO synthase expression. Am. J. Physiol. 1997, 272, H1730–H1740. [Google Scholar] [CrossRef] [PubMed]
- Vagnoni, K.E.; Shaw, C.E.; Phernetton, T.M.; Meglin, B.M.; Bird, I.M.; Magness, R.R. Endothelial vasodilator production by uterine and systemic arteries. III. Ovarian and estrogen effects on NO synthase. Am. J. Physiol. 1998, 275, H1845–H1856. [Google Scholar] [CrossRef]
- Weiner, C.P.; Lizasoain, I.; Baylis, S.A.; Knowles, R.G.; Charles, I.G.; Moncada, S. Induction of calcium-dependent nitric oxide synthases by sex hormones. Proc. Natl. Acad. Sci. USA 1994, 91, 5212–5216. [Google Scholar] [CrossRef] [Green Version]
- Magness, R.R.; Rosenfeld, C.R.; Hassan, A.; Shaul, P.W. Endothelial vasodilator production by uterine and systemic arteries. I. Effects of ANG II on PGI2 and NO in pregnancy. Am. J. Physiol. 1996, 270, H1914–H1923. [Google Scholar] [CrossRef]
- Nelson, S.H.; Steinsland, O.S.; Wang, Y.; Yallampalli, C.; Dong, Y.L.; Sanchez, J.M. Increased nitric oxide synthase activity and expression in the human uterine artery during pregnancy. Circ. Res. 2000, 87, 406–411. [Google Scholar] [CrossRef] [Green Version]
- Van der Heijden, O.W.; Essers, Y.P.; Fazzi, G.; Peeters, L.L.; De Mey, J.G.; van Eys, G.J. Uterine artery remodeling and reproductive performance are impaired in endothelial nitric oxide synthase-deficient mice. Biol. Reprod. 2005, 72, 1161–1168. [Google Scholar] [CrossRef]
- Cadnapaphornchai, M.A.; Ohara, M.; Morris, K.G., Jr.; Knotek, M.; Rogachev, B.; Ladtkow, T.; Carter, E.P.; Schrier, R.W. Chronic NOS inhibition reverses systemic vasodilation and glomerular hyperfiltration in pregnancy. Am. J. Physiol. Renal. Physiol. 2001, 280, F592–F598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamura, T.; Ghoneim, H.A.; Ayajiki, K.; Ammar, E.S.; Toda, N. Mechanisms underlying contraction and relaxation induced by nerve stimulation in monkey uterine arteries. Pharmacology 2000, 61, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Kublickiene, K.R.; Cockell, A.P.; Nisell, H.; Poston, L. Role of nitric oxide in the regulation of vascular tone in pressurized and perfused resistance myometrial arteries from term pregnant women. Am. J. Obstet. Gynecol. 1997, 177, 1263–1269. [Google Scholar] [CrossRef]
- Yi, F.X.; Magness, R.R.; Bird, I.M. Simultaneous imaging of [Ca2+]i and intracellular NO production in freshly isolated uterine artery endothelial cells: Effects of ovarian cycle and pregnancy. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R140–R148. [Google Scholar] [CrossRef]
- Honnens, A.; Weisser, S.; Welter, H.; Einspanier, R.; Bollwein, H. Relationships between uterine blood flow, peripheral sex steroids, expression of endometrial estrogen receptors and nitric oxide synthases during the estrous cycle in mares. J. Reprod. Dev. 2011, 57, 43–48. [Google Scholar] [CrossRef] [Green Version]
- Arroyo, J.A.; Anthony, R.V.; Parker, T.A.; Galan, H.L. Differential expression of placental and vascular endothelial nitric oxide synthase in an ovine model of fetal growth restriction. Am. J. Obstet. Gynecol. 2006, 195, 771–777. [Google Scholar] [CrossRef]
- Magness, R.R.; Sullivan, J.A.; Li, Y.; Phernetton, T.M.; Bird, I.M. Endothelial vasodilator production by uterine and systemic arteries. VI. Ovarian and pregnancy effects on eNOS and NO(x). Am. J. Physiol. Heart Circ. Physiol. 2001, 280, H1692–H1698. [Google Scholar] [CrossRef]
- Zhang, H.H.; Chen, J.C.; Sheibani, L.; Lechuga, T.J.; Chen, D.B. Pregnancy Augments VEGF-Stimulated In Vitro Angiogenesis and Vasodilator (NO and H2S) Production in Human Uterine Artery Endothelial Cells. J. Clin. Endocrinol. Metab. 2017, 102, 2382–2393. [Google Scholar] [CrossRef]
- Zheng, J.; Bird, I.M.; Chen, D.B.; Magness, R.R. Angiotensin II regulation of ovine fetoplacental artery endothelial functions: Interactions with nitric oxide. J. Physiol. 2005, 565, 59–69. [Google Scholar] [CrossRef]
- Bird, I.M.; Zhang, L.; Magness, R.R. Possible mechanisms underlying pregnancy-induced changes in uterine artery endothelial function. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 284, R245–R258. [Google Scholar] [CrossRef] [Green Version]
- Bird, I.M. Endothelial nitric oxide synthase activation and nitric oxide function: New light through old windows. J. Endocrinol 2011, 210, 239–241. [Google Scholar] [CrossRef] [PubMed]
- Sladek, S.M.; Magness, R.R.; Conrad, K.P. Nitric oxide and pregnancy. Am. J. Physiol. 1997, 272, R441–R463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.H.; Feng, L.; Wang, W.; Magness, R.R.; Chen, D.B. Estrogen-responsive nitroso-proteome in uterine artery endothelial cells: Role of endothelial nitric oxide synthase and estrogen receptor-beta. J. Cell. Physiol. 2012, 227, 146–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Yuhanna, I.S.; Galcheva-Gargova, Z.; Karas, R.H.; Mendelsohn, M.E.; Shaul, P.W. Estrogen receptor alpha mediates the nongenomic activation of endothelial nitric oxide synthase by estrogen. J. Clin. Investig. 1999, 103, 401–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hohmann, N.; Xia, N.; Steinkamp-Fenske, K.; Forstermann, U.; Li, H. Estrogen Receptor Signaling and the PI3K/Akt Pathway Are Involved in Betulinic Acid-Induced eNOS Activation. Molecules 2016, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastore, M.B.; Jobe, S.O.; Ramadoss, J.; Magness, R.R. Estrogen receptor-alpha and estrogen receptor-beta in the uterine vascular endothelium during pregnancy: Functional implications for regulating uterine blood flow. Semin. Reprod. Med. 2012, 30, 46–61. [Google Scholar] [CrossRef] [Green Version]
- Palmer, S.K.; Zamudio, S.; Coffin, C.; Parker, S.; Stamm, E.; Moore, L.G. Quantitative estimation of human uterine artery blood flow and pelvic blood flow redistribution in pregnancy. Obstet. Gynecol. 1992, 80, 1000–1006. [Google Scholar] [PubMed]
- Nelson, S.H.; Steinsland, O.S.; Suresh, M.S.; Lee, N.M. Pregnancy augments nitric oxide-dependent dilator response to acetylcholine in the human uterine artery. Hum. Reprod. 1998, 13, 1361–1367. [Google Scholar] [CrossRef] [Green Version]
- Magness, R.R.; Shideman, C.R.; Habermehl, D.A.; Sullivan, J.A.; Bird, I.M. Endothelial vasodilator production by uterine and systemic arteries. V. Effects of ovariectomy, the ovarian cycle, and pregnancy on prostacyclin synthase expression. Prostaglandins Other Lipid Mediat. 2000, 60, 103–118. [Google Scholar] [CrossRef]
- Chen, D.B.; Jia, S.; King, A.G.; Barker, A.; Li, S.M.; Mata-Greenwood, E.; Zheng, J.; Magness, R.R. Global protein expression profiling underlines reciprocal regulation of caveolin 1 and endothelial nitric oxide synthase expression in ovariectomized sheep uterine artery by estrogen/progesterone replacement therapy. Biol. Reprod. 2006, 74, 832–838. [Google Scholar] [CrossRef] [Green Version]
- Chambliss, K.L.; Shaul, P.W. Estrogen modulation of endothelial nitric oxide synthase. Endocr. Rev. 2002, 23, 665–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumi, D.; Ignarro, L.J. Estrogen-related receptor alpha 1 up-regulates endothelial nitric oxide synthase expression. Proc. Natl. Acad. Sci. USA 2003, 100, 14451–14456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, X.X.; Mata-Greenwood, E.; Liao, W.X.; Zhang, H.; Zheng, J.; Chen, D.B. Transcriptional regulation of endothelial nitric oxide synthase expression in uterine artery endothelial cells by c-Jun/AP-1. Mol. Cell. Endocrinol. 2007, 279, 39–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukao, M.; Mason, H.S.; Britton, F.C.; Kenyon, J.L.; Horowitz, B.; Keef, K.D. Cyclic GMP-dependent protein kinase activates cloned BKCa channels expressed in mammalian cells by direct phosphorylation at serine 1072. J. Biol. Chem. 1999, 274, 10927–10935. [Google Scholar] [CrossRef] [Green Version]
- Rosenfeld, C.R.; Cornfield, D.N.; Roy, T. Ca(2+)-activated K(+) channels modulate basal and E(2)beta-induced rises in uterine blood flow in ovine pregnancy. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H422–H431. [Google Scholar] [CrossRef]
- Rosenfeld, C.R.; Roy, T. Large conductance Ca2+-activated and voltage-activated K+ channels contribute to the rise and maintenance of estrogen-induced uterine vasodilation and maintenance of blood pressure. Endocrinology 2012, 153, 6012–6020. [Google Scholar] [CrossRef]
- Gokina, N.I.; Bonev, A.D.; Phillips, J.; Gokin, A.P.; Veilleux, K.; Oppenheimer, K.; Goloman, G. Impairment of IKCa channels contributes to uteroplacental endothelial dysfunction in rat diabetic pregnancy. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H592–H604. [Google Scholar] [CrossRef] [Green Version]
- Osol, G.; Mandala, M. Maternal uterine vascular remodeling during pregnancy. Physiology (Bethesda) 2009, 24, 58–71. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.H.; Feng, L.; Livnat, I.; Hoh, J.K.; Shim, J.Y.; Liao, W.X.; Chen, D.B. Estradiol-17beta stimulates specific receptor and endogenous nitric oxide-dependent dynamic endothelial protein S-nitrosylation: Analysis of endothelial nitrosyl-proteome. Endocrinology 2010, 151, 3874–3887. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, S.; Lekontseva, O.; Peters, A.; Davidge, S.T. 17beta-Estradiol induces protein S-nitrosylation in the endothelium. Cardiovasc. Res. 2010, 85, 796–805. [Google Scholar] [CrossRef] [Green Version]
- Hess, D.T.; Matsumoto, A.; Kim, S.O.; Marshall, H.E.; Stamler, J.S. Protein S-nitrosylation: Purview and parameters. Nat. Rev. Mol. Cell. Biol. 2005, 6, 150–166. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.H.; Lechuga, T.J.; Tith, T.; Wang, W.; Wing, D.A.; Chen, D.B. S-nitrosylation of cofilin-1 mediates estradiol-17beta-stimulated endothelial cytoskeleton remodeling. Mol. Endocrinol. 2015, 29, 434–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.H.; Wang, W.; Feng, L.; Yang, Y.; Zheng, J.; Huang, L.; Chen, D.B. S-nitrosylation of Cofilin-1 Serves as a Novel Pathway for VEGF-Stimulated Endothelial Cell Migration. J. Cell Physiol. 2015, 230, 406–417. [Google Scholar] [CrossRef]
- Ghosh, M.; Song, X.; Mouneimne, G.; Sidani, M.; Lawrence, D.S.; Condeelis, J.S. Cofilin promotes actin polymerization and defines the direction of cell motility. Science 2004, 304, 743–746. [Google Scholar] [CrossRef] [Green Version]
- Hale, S.A.; Weger, L.; Mandala, M.; Osol, G. Reduced NO signaling during pregnancy attenuates outward uterine artery remodeling by altering MMP expression and collagen and elastin deposition. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1266–H1275. [Google Scholar] [CrossRef] [Green Version]
- Stirone, C.; Duckles, S.P.; Krause, D.N.; Procaccio, V. Estrogen increases mitochondrial efficiency and reduces oxidative stress in cerebral blood vessels. Mol. Pharmacol. 2005, 68, 959–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nazarewicz, R.R.; Zenebe, W.J.; Parihar, A.; Larson, S.K.; Alidema, E.; Choi, J.; Ghafourifar, P. Tamoxifen induces oxidative stress and mitochondrial apoptosis via stimulating mitochondrial nitric oxide synthase. Cancer Res. 2007, 67, 1282–1290. [Google Scholar] [CrossRef] [Green Version]
- Klinge, C.M. Estrogens regulate life and death in mitochondria. J. Bioenerg. Biomembr. 2017, 49, 307–324. [Google Scholar] [CrossRef]
- Klinge, C.M. Estrogenic control of mitochondrial function and biogenesis. J. Cell. Biochem. 2008, 105, 1342–1351. [Google Scholar] [CrossRef] [Green Version]
- Satohisa, S.; Zhang, H.H.; Feng, L.; Yang, Y.Y.; Huang, L.; Chen, D.B. Endogenous NO upon estradiol-17beta stimulation and NO donor differentially regulate mitochondrial S-nitrosylation in endothelial cells. Endocrinology 2014, 155, 3005–3016. [Google Scholar] [CrossRef] [Green Version]
- Haldane, J. The Relation of the Action of Carbonic Oxide to Oxygen Tension. J. Physiol. 1895, 18, 201–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Burg, R. Toxicology update: Carbon monoxide. J. Appl. Toxicol. 1999, 19, 379–386. [Google Scholar] [CrossRef]
- Langston, P.; Gorman, D.; Runciman, W.; Upton, R. The effect of carbon monoxide on oxygen metabolism in the brains of awake sheep. Toxicology 1996, 114, 223–232. [Google Scholar] [CrossRef]
- Sjostrand, T. Endogenous formation of carbon monoxide; the CO concentration in the inspired and expired air of hospital patients. Acta Physiol. Scand. 1951, 22, 137–141. [Google Scholar] [CrossRef]
- Verma, A.; Hirsch, D.J.; Glatt, C.E.; Ronnett, G.V.; Snyder, S.H. Carbon monoxide: A putative neural messenger. Science 1993, 259, 381–384. [Google Scholar] [CrossRef]
- Suematsu, M.; Kashiwagi, S.; Sano, T.; Goda, N.; Shinoda, Y.; Ishimura, Y. Carbon monoxide as an endogenous modulator of hepatic vascular perfusion. Biochem. Biophys. Res. Commun. 1994, 205, 1333–1337. [Google Scholar] [CrossRef]
- Otterbein, L.E.; Bach, F.H.; Alam, J.; Soares, M.; Tao Lu, H.; Wysk, M.; Davis, R.J.; Flavell, R.A.; Choi, A.M. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat. Med. 2000, 6, 422–428. [Google Scholar] [CrossRef]
- Brouard, S.; Otterbein, L.E.; Anrather, J.; Tobiasch, E.; Bach, F.H.; Choi, A.M.; Soares, M.P. Carbon monoxide generated by heme oxygenase 1 suppresses endothelial cell apoptosis. J. Exp. Med. 2000, 192, 1015–1026. [Google Scholar] [CrossRef]
- Zhang, X.; Shan, P.; Otterbein, L.E.; Alam, J.; Flavell, R.A.; Davis, R.J.; Choi, A.M.; Lee, P.J. Carbon monoxide inhibition of apoptosis during ischemia-reperfusion lung injury is dependent on the p38 mitogen-activated protein kinase pathway and involves caspase 3. J. Biol. Chem. 2003, 278, 1248–1258. [Google Scholar] [CrossRef] [Green Version]
- Morita, T.; Mitsialis, S.A.; Koike, H.; Liu, Y.; Kourembanas, S. Carbon monoxide controls the proliferation of hypoxic vascular smooth muscle cells. J. Biol. Chem. 1997, 272, 32804–32809. [Google Scholar] [CrossRef] [Green Version]
- Maines, M.D. The heme oxygenase system: A regulator of second messenger gases. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 517–554. [Google Scholar] [CrossRef] [PubMed]
- Keyse, S.M.; Tyrrell, R.M. Heme oxygenase is the major 32-kDa stress protein induced in human skin fibroblasts by UVA radiation, hydrogen peroxide, and sodium arsenite. Proc. Natl. Acad. Sci. USA 1989, 86, 99–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durante, W.; Christodoulides, N.; Cheng, K.; Peyton, K.J.; Sunahara, R.K.; Schafer, A.I. cAMP induces heme oxygenase-1 gene expression and carbon monoxide production in vascular smooth muscle. Am. J. Physiol 1997, 273, H317–H323. [Google Scholar] [CrossRef] [PubMed]
- Kourembanas, S.; Morita, T.; Liu, Y.; Christou, H. Mechanisms by which oxygen regulates gene expression and cell-cell interaction in the vasculature. Kidney Int. 1997, 51, 438–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tenhunen, R.; Marver, H.S.; Schmid, R. Microsomal heme oxygenase. Characterization of the enzyme. J. Biol. Chem. 1969, 244, 6388–6394. [Google Scholar]
- Welch, W.J.; Feramisco, J.R. Purification of the major mammalian heat shock proteins. J. Biol. Chem. 1982, 257, 14949–14959. [Google Scholar]
- Acevedo, C.H.; Ahmed, A. Hemeoxygenase-1 inhibits human myometrial contractility via carbon monoxide and is upregulated by progesterone during pregnancy. J. Clin. Investig. 1998, 101, 949–955. [Google Scholar] [CrossRef]
- Zhao, H.; Wong, R.J.; Kalish, F.S.; Nayak, N.R.; Stevenson, D.K. Effect of heme oxygenase-1 deficiency on placental development. Placenta 2009, 30, 861–868. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Azuma, J.; Kalish, F.; Wong, R.J.; Stevenson, D.K. Maternal heme oxygenase 1 regulates placental vasculature development via angiogenic factors in mice. Biol. Reprod. 2011, 85, 1005–1012. [Google Scholar] [CrossRef] [Green Version]
- Meyer, N.; Langwisch, S.; Scharm, M.; Zenclussen, A.C. Using ultrasound to define the time point of intrauterine growth retardation in a mouse model of heme oxygenase-1 deficiency. Biol. Reprod. 2020. [Google Scholar] [CrossRef]
- Vile, G.F.; Basu-Modak, S.; Waltner, C.; Tyrrell, R.M. Heme oxygenase 1 mediates an adaptive response to oxidative stress in human skin fibroblasts. Proc. Natl. Acad. Sci. USA 1994, 91, 2607–2610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakhary, R.; Gaine, S.P.; Dinerman, J.L.; Ruat, M.; Flavahan, N.A.; Snyder, S.H. Heme oxygenase 2: Endothelial and neuronal localization and role in endothelium-dependent relaxation. Proc. Natl. Acad. Sci. USA 1996, 93, 795–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christodoulides, N.; Durante, W.; Kroll, M.H.; Schafer, A.I. Vascular smooth muscle cell heme oxygenases generate guanylyl cyclase-stimulatory carbon monoxide. Circulation 1995, 91, 2306–2309. [Google Scholar] [CrossRef] [PubMed]
- Vreman, H.J.; Wong, R.J.; Harmatz, P.; Fanaroff, A.A.; Berman, B.; Stevenson, D.K. Validation of the Natus CO-Stat End Tidal Breath Analyzer in children and adults. J. Clin. Monit. Comput. 1999, 15, 421–427. [Google Scholar] [CrossRef]
- Cook, M.N.; Nakatsu, K.; Marks, G.S.; McLaughlin, B.E.; Vreman, H.J.; Stevenson, D.K.; Brien, J.F. Heme oxygenase activity in the adult rat aorta and liver as measured by carbon monoxide formation. Can. J. Physiol. Pharmacol. 1995, 73, 515–518. [Google Scholar] [CrossRef]
- Lee, P.J.; Alam, J.; Sylvester, S.L.; Inamdar, N.; Otterbein, L.; Choi, A.M. Regulation of heme oxygenase-1 expression in vivo and in vitro in hyperoxic lung injury. Am. J. Respir. Cell Mol. Biol. 1996, 14, 556–568. [Google Scholar] [CrossRef]
- Otterbein, L.E.; Zuckerbraun, B.S.; Haga, M.; Liu, F.; Song, R.; Usheva, A.; Stachulak, C.; Bodyak, N.; Smith, R.N.; Csizmadia, E.; et al. Carbon monoxide suppresses arteriosclerotic lesions associated with chronic graft rejection and with balloon injury. Nat. Med. 2003, 9, 183–190. [Google Scholar] [CrossRef]
- Sato, K.; Balla, J.; Otterbein, L.; Smith, R.N.; Brouard, S.; Lin, Y.; Csizmadia, E.; Sevigny, J.; Robson, S.C.; Vercellotti, G.; et al. Carbon monoxide generated by heme oxygenase-1 suppresses the rejection of mouse-to-rat cardiac transplants. J. Immunol. 2001, 166, 4185–4194. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, K.; McDaid, J.; Ollinger, R.; Tsui, T.Y.; Berberat, P.O.; Usheva, A.; Csizmadia, E.; Smith, R.N.; Soares, M.P.; Bach, F.H. Biliverdin, a natural product of heme catabolism, induces tolerance to cardiac allografts. FASEB J. 2004, 18, 765–767. [Google Scholar] [CrossRef]
- Leffler, C.W.; Parfenova, H.; Jaggar, J.H. Carbon monoxide as an endogenous vascular modulator. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1–H11. [Google Scholar] [CrossRef] [Green Version]
- Leffler, C.W.; Nasjletti, A.; Yu, C.; Johnson, R.A.; Fedinec, A.L.; Walker, N. Carbon monoxide and cerebral microvascular tone in newborn pigs. Am. J. Physiol. 1999, 276, H1641–H1646. [Google Scholar] [CrossRef] [PubMed]
- Leffler, C.W.; Parfenova, H.; Jaggar, J.H.; Wang, R. Carbon monoxide and hydrogen sulfide: Gaseous messengers in cerebrovascular circulation. J. Appl. Physiol. 2006, 100, 1065–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adin, C.A.; Croker, B.P.; Agarwal, A. Protective effects of exogenous bilirubin on ischemia-reperfusion injury in the isolated, perfused rat kidney. Am. J. Physiol. Renal. Physiol. 2005, 288, F778–F784. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Liang, X.; Shi, L.; Wang, L.; Chen, J.; Kang, C.; Zhu, J.; Mi, M. Estrogen receptor and PI3K/Akt signaling pathway involvement in S-(-)equol-induced activation of Nrf2/ARE in endothelial cells. PLoS ONE 2013, 8, e79075. [Google Scholar] [CrossRef] [PubMed]
- Kreiser, D.; Baum, M.; Seidman, D.S.; Fanaroff, A.; Shah, D.; Hendler, I.; Stevenson, D.K.; Schiff, E.; Druzin, M.L. End tidal carbon monoxide levels are lower in women with gestational hypertension and pre-eclampsia. J. Perinatol. 2004, 24, 213–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramazzini, B. De morbis artificum diatriba [diseases of workers]. 1713. Am. J. Public Health 2001, 91, 1380–1382. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Kimura, H. The possible role of hydrogen sulfide as an endogenous neuromodulator. J. Neurosci. 1996, 16, 1066–1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, R. Physiological implications of hydrogen sulfide: A whiff exploration that blossomed. Physiol. Rev. 2012, 92, 791–896. [Google Scholar] [CrossRef] [Green Version]
- Hosoki, R.; Matsuki, N.; Kimura, H. The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem. Biophys. Res. Commun. 1997, 237, 527–531. [Google Scholar] [CrossRef]
- Stipanuk, M.H.; Beck, P.W. Characterization of the enzymic capacity for cysteine desulphhydration in liver and kidney of the rat. Biochem. J. 1982, 206, 267–277. [Google Scholar] [CrossRef] [Green Version]
- Swaroop, M.; Bradley, K.; Ohura, T.; Tahara, T.; Roper, M.D.; Rosenberg, L.E.; Kraus, J.P. Rat cystathionine beta-synthase. Gene organization and alternative splicing. J. Biol. Chem. 1992, 267, 11455–11461. [Google Scholar] [PubMed]
- Chiku, T.; Padovani, D.; Zhu, W.; Singh, S.; Vitvitsky, V.; Banerjee, R. H2S biogenesis by human cystathionine gamma-lyase leads to the novel sulfur metabolites lanthionine and homolanthionine and is responsive to the grade of hyperhomocysteinemia. J. Biol. Chem. 2009, 284, 11601–11612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.; Padovani, D.; Leslie, R.A.; Chiku, T.; Banerjee, R. Relative contributions of cystathionine beta-synthase and gamma-cystathionase to H2S biogenesis via alternative trans-sulfuration reactions. J. Biol. Chem. 2009, 284, 22457–22466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gadalla, M.M.; Snyder, S.H. Hydrogen sulfide as a gasotransmitter. J. Neurochem. 2010, 113, 14–26. [Google Scholar] [CrossRef] [Green Version]
- Mustafa, A.K.; Gadalla, M.M.; Snyder, S.H. Signaling by gasotransmitters. Sci. Signal. 2009, 2, re2. [Google Scholar] [CrossRef] [Green Version]
- Shibuya, N.; Mikami, Y.; Kimura, Y.; Nagahara, N.; Kimura, H. Vascular endothelium expresses 3-mercaptopyruvate sulfurtransferase and produces hydrogen sulfide. J. Biochem. 2009, 146, 623–626. [Google Scholar] [CrossRef]
- Lowicka, E.; Beltowski, J. Hydrogen sulfide (H2S)—The third gas of interest for pharmacologists. Pharmacol. Rep. 2007, 59, 4–24. [Google Scholar] [PubMed]
- Wang, R. Two’s company, three’s a crowd: Can H2S be the third endogenous gaseous transmitter? FASEB J. 2002, 16, 1792–1798. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Zhang, J.; Lu, Y.; Wang, R. The vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP) channel opener. EMBO J. 2001, 20, 6008–6016. [Google Scholar] [CrossRef] [Green Version]
- Kondo, K.; Bhushan, S.; King, A.L.; Prabhu, S.D.; Hamid, T.; Koenig, S.; Murohara, T.; Predmore, B.L.; Gojon, G., Sr.; Gojon, G., Jr.; et al. H(2)S protects against pressure overload-induced heart failure via upregulation of endothelial nitric oxide synthase. Circulation 2013, 127, 1116–1127. [Google Scholar] [CrossRef] [Green Version]
- Jackson, M.R.; Melideo, S.L.; Jorns, M.S. Human sulfide: Quinone oxidoreductase catalyzes the first step in hydrogen sulfide metabolism and produces a sulfane sulfur metabolite. Biochemistry 2012, 51, 6804–6815. [Google Scholar] [CrossRef] [PubMed]
- Olson, K.R. Hydrogen sulfide as an oxygen sensor. Clin. Chem. Lab. Med. 2013, 51, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Cuevasanta, E.; Zeida, A.; Carballal, S.; Wedmann, R.; Morzan, U.N.; Trujillo, M.; Radi, R.; Estrin, D.A.; Filipovic, M.R.; Alvarez, B. Insights into the mechanism of the reaction between hydrogen sulfide and peroxynitrite. Free Radic. Biol. Med. 2015, 80, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.H.; Krokowski, D.; Guan, B.J.; Bederman, I.; Majumder, M.; Parisien, M.; Diatchenko, L.; Kabil, O.; Willard, B.; Banerjee, R.; et al. Quantitative H2S-mediated protein sulfhydration reveals metabolic reprogramming during the integrated stress response. eLife 2015, 4, e10067. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, A.K.; Sikka, G.; Gazi, S.K.; Steppan, J.; Jung, S.M.; Bhunia, A.K.; Barodka, V.M.; Gazi, F.K.; Barrow, R.K.; Wang, R.; et al. Hydrogen sulfide as endothelium-derived hyperpolarizing factor sulfhydrates potassium channels. Circ. Res. 2011, 109, 1259–1268. [Google Scholar] [CrossRef]
- Wang, K.; Ahmad, S.; Cai, M.; Rennie, J.; Fujisawa, T.; Crispi, F.; Baily, J.; Miller, M.R.; Cudmore, M.; Hadoke, P.W.; et al. Dysregulation of hydrogen sulfide producing enzyme cystathionine gamma-lyase contributes to maternal hypertension and placental abnormalities in preeclampsia. Circulation 2013, 127, 2514–2522. [Google Scholar] [CrossRef] [Green Version]
- Badiei, A.; Chambers, S.T.; Gaddam, R.R.; Bhatia, M. Cystathionine-gamma-lyase gene silencing with siRNA in monocytes/macrophages attenuates inflammation in cecal ligation and puncture-induced sepsis in the mouse. J. Biosci. 2016, 41, 87–95. [Google Scholar] [CrossRef]
- Liu, Y.; Deng, Y.; Liu, H.; Yin, C.; Li, X.; Gong, Q. Hydrogen sulfide ameliorates learning memory impairment in APP/PS1 transgenic mice: A novel mechanism mediated by the activation of Nrf2. Pharmacol. Biochem. Behav. 2016, 150–151, 207–216. [Google Scholar] [CrossRef]
- Burgoyne, J.R.; Madhani, M.; Cuello, F.; Charles, R.L.; Brennan, J.P.; Schroder, E.; Browning, D.D.; Eaton, P. Cysteine redox sensor in PKGIa enables oxidant-induced activation. Science 2007, 317, 1393–1397. [Google Scholar] [CrossRef]
- Neo, B.H.; Kandhi, S.; Ahmad, M.; Wolin, M.S. Redox regulation of guanylate cyclase and protein kinase G in vascular responses to hypoxia. Respir. Physiol. Neurobiol. 2010, 174, 259–264. [Google Scholar] [CrossRef] [Green Version]
- Stubbert, D.; Prysyazhna, O.; Rudyk, O.; Scotcher, J.; Burgoyne, J.R.; Eaton, P. Protein kinase G Ialpha oxidation paradoxically underlies blood pressure lowering by the reductant hydrogen sulfide. Hypertension 2014, 64, 1344–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, K.; Ju, Y.; Li, S.; Altaany, Z.; Wang, R.; Yang, G. S-sulfhydration of MEK1 leads to PARP-1 activation and DNA damage repair. EMBO Rep. 2014, 15, 792–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukami, K.; Kawabata, A. Hydrogen sulfide and neuronal differentiation: Focus on Ca2+ channels. Nitric Oxide 2015, 46, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Ping, N.N.; Li, S.; Mi, Y.N.; Cao, L.; Cao, Y.X. Hydrogen sulphide induces vasoconstriction of rat coronary artery via activation of Ca(2+) influx. Acta Physiol. (Oxf.) 2015, 214, 88–96. [Google Scholar] [CrossRef]
- Zhang, W.; Xu, C.; Yang, G.; Wu, L.; Wang, R. Interaction of H2S with Calcium Permeable Channels and Transporters. Oxid. Med. Cell. Longev. 2015, 2015, 323269. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.W.; Cheng, Y.; Moore, P.K.; Bian, J.S. Hydrogen sulphide regulates intracellular pH in vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 2007, 358, 1142–1147. [Google Scholar] [CrossRef]
- Sun, Y.G.; Cao, Y.X.; Wang, W.W.; Ma, S.F.; Yao, T.; Zhu, Y.C. Hydrogen sulphide is an inhibitor of L-type calcium channels and mechanical contraction in rat cardiomyocytes. Cardiovasc. Res. 2008, 79, 632–641. [Google Scholar] [CrossRef] [Green Version]
- Malekova, L.; Krizanova, O.; Ondrias, K. H(2)S and HS(−) donor NaHS inhibits intracellular chloride channels. Gen. Physiol. Biophys. 2009, 28, 190–194. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Wu, L.; Jiang, B.; Yang, W.; Qi, J.; Cao, K.; Meng, Q.; Mustafa, A.K.; Mu, W.; Zhang, S.; et al. H2S as a physiologic vasorelaxant: Hypertension in mice with deletion of cystathionine gamma-lyase. Science 2008, 322, 587–590. [Google Scholar] [CrossRef] [Green Version]
- Jackson-Weaver, O.; Osmond, J.M.; Riddle, M.A.; Naik, J.S.; Gonzalez Bosc, L.V.; Walker, B.R.; Kanagy, N.L. Hydrogen sulfide dilates rat mesenteric arteries by activating endothelial large-conductance Ca(2+)-activated K(+) channels and smooth muscle Ca(2+) sparks. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1446–H1454. [Google Scholar] [CrossRef] [Green Version]
- Donovan, J.; Wong, P.S.; Roberts, R.E.; Garle, M.J.; Alexander, S.P.H.; Dunn, W.R.; Ralevic, V. A critical role for cystathionine-beta-synthase in hydrogen sulfide-mediated hypoxic relaxation of the coronary artery. Vascul. Pharmacol. 2017, 93–95, 20–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Ndisang, J.F.; Tang, G.; Cao, K.; Wang, R. Hydrogen sulfide-induced relaxation of resistance mesenteric artery beds of rats. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H2316–H2323. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Wu, L.; Liang, W.; Wang, R. Direct stimulation of K(ATP) channels by exogenous and endogenous hydrogen sulfide in vascular smooth muscle cells. Mol. Pharmacol. 2005, 68, 1757–1764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papapetropoulos, A.; Pyriochou, A.; Altaany, Z.; Yang, G.; Marazioti, A.; Zhou, Z.; Jeschke, M.G.; Branski, L.K.; Herndon, D.N.; Wang, R.; et al. Hydrogen sulfide is an endogenous stimulator of angiogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 21972–21977. [Google Scholar] [CrossRef] [Green Version]
- Naik, J.S.; Osmond, J.M.; Walker, B.R.; Kanagy, N.L. Hydrogen sulfide-induced vasodilation mediated by endothelial TRPV4 channels. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H1437–H1444. [Google Scholar] [CrossRef]
- Kang, M.; Hashimoto, A.; Gade, A.; Akbarali, H.I. Interaction between hydrogen sulfide-induced sulfhydration and tyrosine nitration in the KATP channel complex. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G532–G539. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.H.; Huang, X.; Meng, X.M.; Zhang, C.M.; Lu, H.L.; Kim, Y.C.; Xu, W.X. Exogenous H2S enhances mice gastric smooth muscle tension through S-sulfhydration of KV 4.3, mediating the inhibition of the voltage-dependent potassium current. Neurogastroenterol. Motil. 2014, 26, 1705–1716. [Google Scholar] [CrossRef]
- Itoh, H.; Bird, I.M.; Nakao, K.; Magness, R.R. Pregnancy increases soluble and particulate guanylate cyclases and decreases the clearance receptor of natriuretic peptides in ovine uterine, but not systemic, arteries. Endocrinology 1998, 139, 3329–3341. [Google Scholar] [CrossRef]
- Hu, X.Q.; Xiao, D.; Zhu, R.; Huang, X.; Yang, S.; Wilson, S.; Zhang, L. Pregnancy upregulates large-conductance Ca2+-activated K+ channel activity and attenuates myogenic tone in uterine arteries. Hypertension 2011, 58, 1132–1139. [Google Scholar] [CrossRef] [Green Version]
- Xiao, D.; Longo, L.D.; Zhang, L. Role of KATP and L-type Ca2+ channel activities in regulation of ovine uterine vascular contractility: Effect of pregnancy and chronic hypoxia. Am. J. Obstet. Gynecol. 2010, 203, 596.e6. [Google Scholar] [CrossRef] [Green Version]
- Khan, L.H.; Rosenfeld, C.R.; Liu, X.T.; Magness, R.R. Regulation of the cGMP-cPKG pathway and large-conductance Ca2+-activated K+ channels in uterine arteries during the ovine ovarian cycle. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E222–E228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.J.; Cai, W.J.; Li, N.; Ding, Y.J.; Chen, Y.; Zhu, Y.C. The hydrogen sulfide donor NaHS promotes angiogenesis in a rat model of hind limb ischemia. Antioxid. Redox Signal. 2010, 12, 1065–1077. [Google Scholar] [CrossRef] [PubMed]
- Szabo, C.; Papapetropoulos, A. Hydrogen sulphide and angiogenesis: Mechanisms and applications. Br. J. Pharmacol. 2011, 164, 853–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polhemus, D.J.; Kondo, K.; Bhushan, S.; Bir, S.C.; Kevil, C.G.; Murohara, T.; Lefer, D.J.; Calvert, J.W. Hydrogen sulfide attenuates cardiac dysfunction after heart failure via induction of angiogenesis. Circ. Heart Fail. 2013, 6, 1077–1086. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.B.; Feng, L.; Hodges, J.K.; Lechuga, T.J.; Zhang, H.H. Human trophoblast-derived hydrogen sulfide stimulates placental artery endothelial cell angiogenesis. Biol. Reprod. 2017, 97, 478–489. [Google Scholar] [CrossRef]
- Namba, T.; Koike, H.; Murakami, K.; Aoki, M.; Makino, H.; Hashiya, N.; Ogihara, T.; Kaneda, Y.; Kohno, M.; Morishita, R. Angiogenesis induced by endothelial nitric oxide synthase gene through vascular endothelial growth factor expression in a rat hindlimb ischemia model. Circulation 2003, 108, 2250–2257. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Sun, X.; Wang, R. Hydrogen sulfide-induced apoptosis of human aorta smooth muscle cells via the activation of mitogen-activated protein kinases and caspase-3. FASEB J. 2004, 18, 1782–1784. [Google Scholar] [CrossRef]
- Yang, G.; Wu, L.; Wang, R. Pro-apoptotic effect of endogenous H2S on human aorta smooth muscle cells. FASEB J. 2006, 20, 553–555. [Google Scholar] [CrossRef]
- Du, J.; Hui, Y.; Cheung, Y.; Bin, G.; Jiang, H.; Chen, X.; Tang, C. The possible role of hydrogen sulfide as a smooth muscle cell proliferation inhibitor in rat cultured cells. Heart Vessels 2004, 19, 75–80. [Google Scholar] [CrossRef]
- Lechuga, T.J.; Zhang, H.H.; Sheibani, L.; Karim, M.; Jia, J.; Magness, R.R.; Rosenfeld, C.R.; Chen, D.B. Estrogen Replacement Therapy in Ovariectomized Nonpregnant Ewes Stimulates Uterine Artery Hydrogen Sulfide Biosynthesis by Selectively Up-Regulating Cystathionine beta-Synthase Expression. Endocrinology 2015, 156, 2288–2298. [Google Scholar] [CrossRef]
- Lechuga, T.J.; Qi, Q.R.; Magness, R.R.; Chen, D.B. Ovine uterine artery hydrogen sulfide biosynthesis in vivo: Effects of ovarian cycle and pregnancydagger. Biol. Reprod. 2019, 100, 1630–1636. [Google Scholar] [CrossRef] [PubMed]
- Kraus, J.P.; Oliveriusova, J.; Sokolova, J.; Kraus, E.; Vlcek, C.; de Franchis, R.; Maclean, K.N.; Bao, L.; Bukovsk, G.; Patterson, D.; et al. The human cystathionine beta-synthase (CBS) gene: Complete sequence, alternative splicing, and polymorphisms. Genomics 1998, 52, 312–324. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Pei, Y.; Teng, H.; Cao, Q.; Wang, R. Specificity protein-1 as a critical regulator of human cystathionine gamma-lyase in smooth muscle cells. J. Biol. Chem. 2011, 286, 26450–26460. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Mani, S.; Cao, W.; Yang, G.; Lai, C.; Wu, L.; Wang, R. Interaction of hydrogen sulfide and estrogen on the proliferation of vascular smooth muscle cells. PLoS ONE 2012, 7, 1–10. [Google Scholar] [CrossRef]
- Mishra, J.S.; te Riele, G.M.; Qi, Q.R.; Lechuga, T.J.; Gopalakrishnan, K.; Chen, D.B.; Kumar, S. Estrogen receptor-β mediates estradiol-induced pregnancy-specific uterine artery endothelial cell angiotensin type-2 receptor expression. Hypertension 2019, in press. [Google Scholar] [CrossRef] [PubMed]
- D’Emmanuele di Villa Bianca, R.; Mitidieri, E.; Fusco, F.; Russo, A.; Pagliara, V.; Tramontano, T.; Donnarumma, E.; Mirone, V.; Cirino, G.; Russo, G.; et al. Urothelium muscarinic activation phosphorylates CBS(Ser227) via cGMP/PKG pathway causing human bladder relaxation through H2S production. Sci. Rep. 2016, 6, 31491. [Google Scholar] [CrossRef] [Green Version]
- Coletta, C.; Papapetropoulos, A.; Erdelyi, K.; Olah, G.; Modis, K.; Panopoulos, P.; Asimakopoulou, A.; Gero, D.; Sharina, I.; Martin, E.; et al. Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation. Proc. Natl. Acad. Sci. USA 2012, 109, 9161–9166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubo, S.; Doe, I.; Kurokawa, Y.; Nishikawa, H.; Kawabata, A. Direct inhibition of endothelial nitric oxide synthase by hydrogen sulfide: Contribution to dual modulation of vascular tension. Toxicology 2007, 232, 138–146. [Google Scholar] [CrossRef]
- Ali, M.Y.; Ping, C.Y.; Mok, Y.Y.; Ling, L.; Whiteman, M.; Bhatia, M.; Moore, P.K. Regulation of vascular nitric oxide in vitro and in vivo; a new role for endogenous hydrogen sulphide? Br. J. Pharmacol. 2006, 149, 625–634. [Google Scholar] [CrossRef] [Green Version]
- Filipovic, M.R.; Eberhardt, M.; Prokopovic, V.; Mijuskovic, A.; Orescanin-Dusic, Z.; Reeh, P.; Ivanovic-Burmazovic, I. Beyond H2S and NO interplay: Hydrogen sulfide and nitroprusside react directly to give nitroxyl (HNO). A new pharmacological source of HNO. J. Med. Chem. 2013, 56, 1499–1508. [Google Scholar] [CrossRef]
- Filipovic, M.R.; Miljkovic, J.; Nauser, T.; Royzen, M.; Klos, K.; Shubina, T.; Koppenol, W.H.; Lippard, S.J.; Ivanovic-Burmazovic, I. Chemical characterization of the smallest S-nitrosothiol, HSNO; cellular cross-talk of H2S and S-nitrosothiols. J. Am. Chem. Soc. 2012, 134, 12016–12027. [Google Scholar] [CrossRef] [PubMed]
- Cortese-Krott, M.M.; Fernandez, B.O.; Santos, J.L.; Mergia, E.; Grman, M.; Nagy, P.; Kelm, M.; Butler, A.; Feelisch, M. Nitrosopersulfide (SSNO(-)) accounts for sustained NO bioactivity of S-nitrosothiols following reaction with sulfide. Redox Biol. 2014, 2, 234–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berenyiova, A.; Grman, M.; Mijuskovic, A.; Stasko, A.; Misak, A.; Nagy, P.; Ondriasova, E.; Cacanyiova, S.; Brezova, V.; Feelisch, M.; et al. The reaction products of sulfide and S-nitrosoglutathione are potent vasorelaxants. Nitric. Oxide 2015, 46, 123–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foresti, R.; Motterlini, R. The heme oxygenase pathway and its interaction with nitric oxide in the control of cellular homeostasis. Free Radic. Res. 1999, 31, 459–475. [Google Scholar] [CrossRef]
- Thom, S.R.; Xu, Y.A.; Ischiropoulos, H. Vascular endothelial cells generate peroxynitrite in response to carbon monoxide exposure. Chem. Res. Toxicol. 1997, 10, 1023–1031. [Google Scholar] [CrossRef]
- Sun, M.W.; Zhong, M.F.; Gu, J.; Qian, F.L.; Gu, J.Z.; Chen, H. Effects of different levels of exercise volume on endothelium-dependent vasodilation: Roles of nitric oxide synthase and heme oxygenase. Hypertens Res. 2008, 31, 805–816. [Google Scholar] [CrossRef] [Green Version]
- Kajimura, M.; Shimoyama, M.; Tsuyama, S.; Suzuki, T.; Kozaki, S.; Takenaka, S.; Tsubota, K.; Oguchi, Y.; Suematsu, M. Visualization of gaseous monoxide reception by soluble guanylate cyclase in the rat retina. FASEB J. 2003, 17, 506–508. [Google Scholar] [CrossRef]
- Qingyou, Z.; Junbao, D.; Weijin, Z.; Hui, Y.; Chaoshu, T.; Chunyu, Z. Impact of hydrogen sulfide on carbon monoxide/heme oxygenase pathway in the pathogenesis of hypoxic pulmonary hypertension. Biochem. Biophys. Res. Commun. 2004, 317, 30–37. [Google Scholar] [CrossRef]
- Jin, H.F.; Du, J.B.; Li, X.H.; Wang, Y.F.; Liang, Y.F.; Tang, C.S. Interaction between hydrogen sulfide/cystathionine gamma-lyase and carbon monoxide/heme oxygenase pathways in aortic smooth muscle cells. Acta Pharmacol. Sin. 2006, 27, 1561–1566. [Google Scholar] [CrossRef]
- Polhemus, D.J.; Lefer, D.J. Emergence of hydrogen sulfide as an endogenous gaseous signaling molecule in cardiovascular disease. Circ. Res. 2014, 114, 730–737. [Google Scholar] [CrossRef]
- Cunningham, F.; Leveno, K.; Bloom, S.; Spong, C.Y.; Dashe, J. Williams Obstetrics, 24e; Mcgraw-Hill: New York, NY, USA, 2014. [Google Scholar]
- Osungbade, K.O.; Ige, O.K. Public health perspectives of preeclampsia in developing countries: Implication for health system strengthening. J. Pregnancy 2011, 2011, 481095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sibai, B.M. Diagnosis and management of gestational hypertension and preeclampsia. Obstet. Gynecol. 2003, 102, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Sibai, B.; Dekker, G.; Kupferminc, M. Pre-eclampsia. Lancet 2005, 365, 785–799. [Google Scholar] [CrossRef]
- Redman, C.W.; Sargent, I.L. Latest advances in understanding preeclampsia. Science 2005, 308, 1592–1594. [Google Scholar] [CrossRef]
- Karumanchi, S.A.; Bdolah, Y. Hypoxia and sFlt-1 in preeclampsia: The “chicken-and-egg” question. Endocrinology 2004, 145, 4835–4837. [Google Scholar] [CrossRef]
- Powe, C.E.; Levine, R.J.; Karumanchi, S.A. Preeclampsia, a disease of the maternal endothelium: The role of antiangiogenic factors and implications for later cardiovascular disease. Circulation 2011, 123, 2856–2869. [Google Scholar] [CrossRef]
- Stamilio, D.M.; Sehdev, H.M.; Morgan, M.A.; Propert, K.; Macones, G.A. Can antenatal clinical and biochemical markers predict the development of severe preeclampsia? Am. J. Obstet. Gynecol. 2000, 182, 589–594. [Google Scholar] [CrossRef]
- Gerretsen, G.; Huisjes, H.J.; Elema, J.D. Morphological changes of the spiral arteries in the placental bed in relation to pre-eclampsia and fetal growth retardation. Br. J. Obstet. Gynaecol. 1981, 88, 876–881. [Google Scholar] [CrossRef]
- Van Beck, E.; Peeters, L.L. Pathogenesis of preeclampsia: A comprehensive model. Obstet. Gynecol. Surv. 1998, 53, 233–239. [Google Scholar] [CrossRef]
- Magness, R.R.; Rosenfeld, C.R. The role of steroid hormones in the control of uterine blood flow. Uterine Circ. 1989, 10, 239–271. [Google Scholar]
- Jobe, S.O.; Tyler, C.T.; Magness, R.R. Aberrant synthesis, metabolism, and plasma accumulation of circulating estrogens and estrogen metabolites in preeclampsia implications for vascular dysfunction. Hypertension 2013, 61, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Kanasaki, K.; Palmsten, K.; Sugimoto, H.; Ahmad, S.; Hamano, Y.; Xie, L.; Parry, S.; Augustin, H.G.; Gattone, V.H.; Folkman, J.; et al. Deficiency in catechol-O-methyltransferase and 2-methoxyoestradiol is associated with pre-eclampsia. Nature 2008, 453, 1117–1121. [Google Scholar] [CrossRef] [PubMed]
- Nobunaga, T.; Tokugawa, Y.; Hashimoto, K.; Kimura, T.; Matsuzaki, N.; Nitta, Y.; Fujita, T.; Kidoguchi, K.I.; Azuma, C.; Saji, F. Plasma nitric oxide levels in pregnant patients with preeclampsia and essential hypertension. Gynecol. Obstet. Investig. 1996, 41, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Smarason, A.K.; Allman, K.G.; Young, D.; Redman, C.W. Elevated levels of serum nitrate, a stable end product of nitric oxide, in women with pre-eclampsia. Br. J. Obstet. Gynaecol. 1997, 104, 538–543. [Google Scholar] [CrossRef]
- Davidge, S.T.; Stranko, C.P.; Roberts, J.M. Urine but not plasma nitric oxide metabolites are decreased in women with preeclampsia. Am. J. Obstet. Gynecol. 1996, 174, 1008–1013. [Google Scholar] [CrossRef]
- Roberts, J.M.; Taylor, R.N.; Musci, T.J.; Rodgers, G.M.; Hubel, C.A.; McLaughlin, M.K. Preeclampsia: An endothelial cell disorder. Am. J. Obstet. Gynecol. 1989, 161, 1200–1204. [Google Scholar] [CrossRef]
- Mitchell, B.M.; Cook, L.G.; Danchuk, S.; Puschett, J.B. Uncoupled endothelial nitric oxide synthase and oxidative stress in a rat model of pregnancy-induced hypertension. Am. J. Hypertens. 2007, 20, 1297–1304. [Google Scholar] [CrossRef] [Green Version]
- Radi, R.; Beckman, J.S.; Bush, K.M.; Freeman, B.A. Peroxynitrite-induced membrane lipid peroxidation: The cytotoxic potential of superoxide and nitric oxide. Arch. Biochem. Biophys. 1991, 288, 481–487. [Google Scholar] [CrossRef]
- Kublickiene, K.R.; Lindblom, B.; Kruger, K.; Nisell, H. Preeclampsia: Evidence for impaired shear stress-mediated nitric oxide release in uterine circulation. Am. J. Obstet. Gynecol. 2000, 183, 160–166. [Google Scholar] [CrossRef]
- Lyall, F.; Barber, A.; Myatt, L.; Bulmer, J.N.; Robson, S.C. Hemeoxygenase expression in human placenta and placental bed implies a role in regulation of trophoblast invasion and placental function. FASEB J. 2000, 14, 208–219. [Google Scholar] [CrossRef]
- Lyall, F. Development of the utero-placental circulation: The role of carbon monoxide and nitric oxide in trophoblast invasion and spiral artery transformation. Microsc. Res. Technol. 2003, 60, 402–411. [Google Scholar] [CrossRef] [PubMed]
- Venditti, C.C.; Casselman, R.; Murphy, M.S.; Adamson, S.L.; Sled, J.G.; Smith, G.N. Chronic carbon monoxide inhalation during pregnancy augments uterine artery blood flow and uteroplacental vascular growth in mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 305, R939–R948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miles, E.W.; Kraus, J.P. Cystathionine beta-synthase: Structure, function, regulation, and location of homocystinuria-causing mutations. J. Biol. Chem. 2004, 279, 29871–29874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, T.; Wang, G.; Zhu, Z.; Huang, Y.; Gu, H.; Ni, X. Increased ADAM10 expression in preeclamptic placentas is associated with decreased expression of hydrogen sulfide production enzymes. Placenta 2015, 36, 947–950. [Google Scholar] [CrossRef] [PubMed]
- Holwerda, K.M.; Burke, S.D.; Faas, M.M.; Zsengeller, Z.; Stillman, I.E.; Kang, P.M.; van Goor, H.; McCurley, A.; Jaffe, I.Z.; Karumanchi, S.A.; et al. Hydrogen sulfide attenuates sFlt1-induced hypertension and renal damage by upregulating vascular endothelial growth factor. J. Am. Soc. Nephrol. 2014, 25, 717–725. [Google Scholar] [CrossRef] [Green Version]
- Cindrova-Davies, T.; Herrera, E.A.; Niu, Y.; Kingdom, J.; Giussani, D.A.; Burton, G.J. Reduced cystathionine gamma-lyase and increased miR-21 expression are associated with increased vascular resistance in growth-restricted pregnancies: Hydrogen sulfide as a placental vasodilator. Am. J. Pathol. 2013, 182, 1448–1458. [Google Scholar] [CrossRef] [Green Version]
- Babic, G.M.; Markovic, S.D.; Varjacic, M.; Djordjevic, N.Z.; Nikolic, T.; Stojic, I.; Jakovljevic, V. Estradiol decreases blood pressure in association with redox regulation in preeclampsia. Clin. Exp. Hypertens. 2018, 40, 281–286. [Google Scholar] [CrossRef]
- Djordjevic, N.Z.; Babic, G.M.; Markovic, S.D.; Ognjanovic, B.I.; Stajn, A.S.; Saicic, Z.S. The antioxidative effect of estradiol therapy on erythrocytes in women with preeclampsia. Reprod. Toxicol. 2010, 29, 231–236. [Google Scholar] [CrossRef]
- Yallampalli, C.; Garfield, R.E. Inhibition of nitric oxide synthesis in rats during pregnancy produces signs similar to those of preeclampsia. Am. J. Obstet. Gynecol. 1993, 169, 1316–1320. [Google Scholar] [CrossRef]
- Johal, T.; Lees, C.C.; Everett, T.R.; Wilkinson, I.B. The nitric oxide pathway and possible therapeutic options in pre-eclampsia. Br. J. Clin. Pharmacol. 2014, 78, 244–257. [Google Scholar] [CrossRef] [Green Version]
- De Belder, A.; Lees, C.; Martin, J.; Moncada, S.; Campbell, S. Treatment of HELLP syndrome with nitric oxide donor. Lancet 1995, 345, 124–125. [Google Scholar] [CrossRef]
- Lees, C.; Langford, E.; Brown, A.S.; de Belder, A.; Pickles, A.; Martin, J.F.; Campbell, S. The effects of S-nitrosoglutathione on platelet activation, hypertension, and uterine and fetal Doppler in severe preeclampsia. Obstet. Gynecol. 1996, 88, 14–19. [Google Scholar] [CrossRef]
- Vadillo-Ortega, F.; Perichart-Perera, O.; Espino, S.; Avila-Vergara, M.A.; Ibarra, I.; Ahued, R.; Godines, M.; Parry, S.; Macones, G.; Strauss, J.F. Effect of supplementation during pregnancy with L-arginine and antioxidant vitamins in medical food on pre-eclampsia in high risk population: Randomised controlled trial. BMJ 2011, 342, d2901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorniak-Wall, T.; Grivell, R.M.; Dekker, G.A.; Hague, W.; Dodd, J.M. The role of L-arginine in the prevention and treatment of pre-eclampsia: A systematic review of randomised trials. J. Hum. Hypertens. 2014, 28, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Trapani, A., Jr.; Goncalves, L.F.; Trapani, T.F.; Vieira, S.; Pires, M.; Pires, M.M. Perinatal and Hemodynamic Evaluation of Sildenafil Citrate for Preeclampsia Treatment: A Randomized Controlled Trial. Obstet. Gynecol. 2016, 128, 253–259. [Google Scholar] [CrossRef] [Green Version]
- Lombardi, N.; Crescioli, G.; Bettiol, A.; Ravaldi, C.; Vannacci, A. Perinatal deaths after sildenafil treatment of fetal growth restriction raise the issue of safety in randomised clinical trials. Pharmacoepidemiol. Drug Saf. 2019, 28, 437–438. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. The LDL receptor. Arterioscler Thromb. Vasc. Biol. 2009, 29, 431–438. [Google Scholar] [CrossRef] [Green Version]
- Ludman, A.; Venugopal, V.; Yellon, D.M.; Hausenloy, D.J. Statins and cardioprotection—More than just lipid lowering? Pharmacol. Ther. 2009, 122, 30–43. [Google Scholar] [CrossRef]
- Weis, M.; Heeschen, C.; Glassford, A.J.; Cooke, J.P. Statins have biphasic effects on angiogenesis. Circulation 2002, 105, 739–745. [Google Scholar] [CrossRef] [Green Version]
- Cudmore, M.; Ahmad, S.; Al-Ani, B.; Fujisawa, T.; Coxall, H.; Chudasama, K.; Devey, L.R.; Wigmore, S.J.; Abbas, A.; Hewett, P.W.; et al. Negative regulation of soluble Flt-1 and soluble endoglin release by heme oxygenase-1. Circulation 2007, 115, 1789–1797. [Google Scholar] [CrossRef] [Green Version]
- Muchova, L.; Wong, R.J.; Hsu, M.; Morioka, I.; Vitek, L.; Zelenka, J.; Schroder, H.; Stevenson, D.K. Statin treatment increases formation of carbon monoxide and bilirubin in mice: A novel mechanism of in vivo antioxidant protection. Can. J. Physiol. Pharmacol. 2007, 85, 800–810. [Google Scholar] [CrossRef] [PubMed]
- Maierean, S.M.; Mikhailidis, D.P.; Toth, P.P.; Grzesiak, M.; Mazidi, M.; Maciejewski, M.; Banach, M. The potential role of statins in preeclampsia and dyslipidemia during gestation: A narrative review. Expert Opin. Investig. Drugs 2018, 27, 427–435. [Google Scholar] [CrossRef]
- Banerjee, S.K.; Maulik, S.K. Effect of garlic on cardiovascular disorders: A review. Nutr. J. 2002, 1, 4. [Google Scholar] [CrossRef] [PubMed]
- Benavides, G.A.; Squadrito, G.L.; Mills, R.W.; Patel, H.D.; Isbell, T.S.; Patel, R.P.; Darley-Usmar, V.M.; Doeller, J.E.; Kraus, D.W. Hydrogen sulfide mediates the vasoactivity of garlic. Proc. Natl. Acad. Sci. USA 2007, 104, 17977–17982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
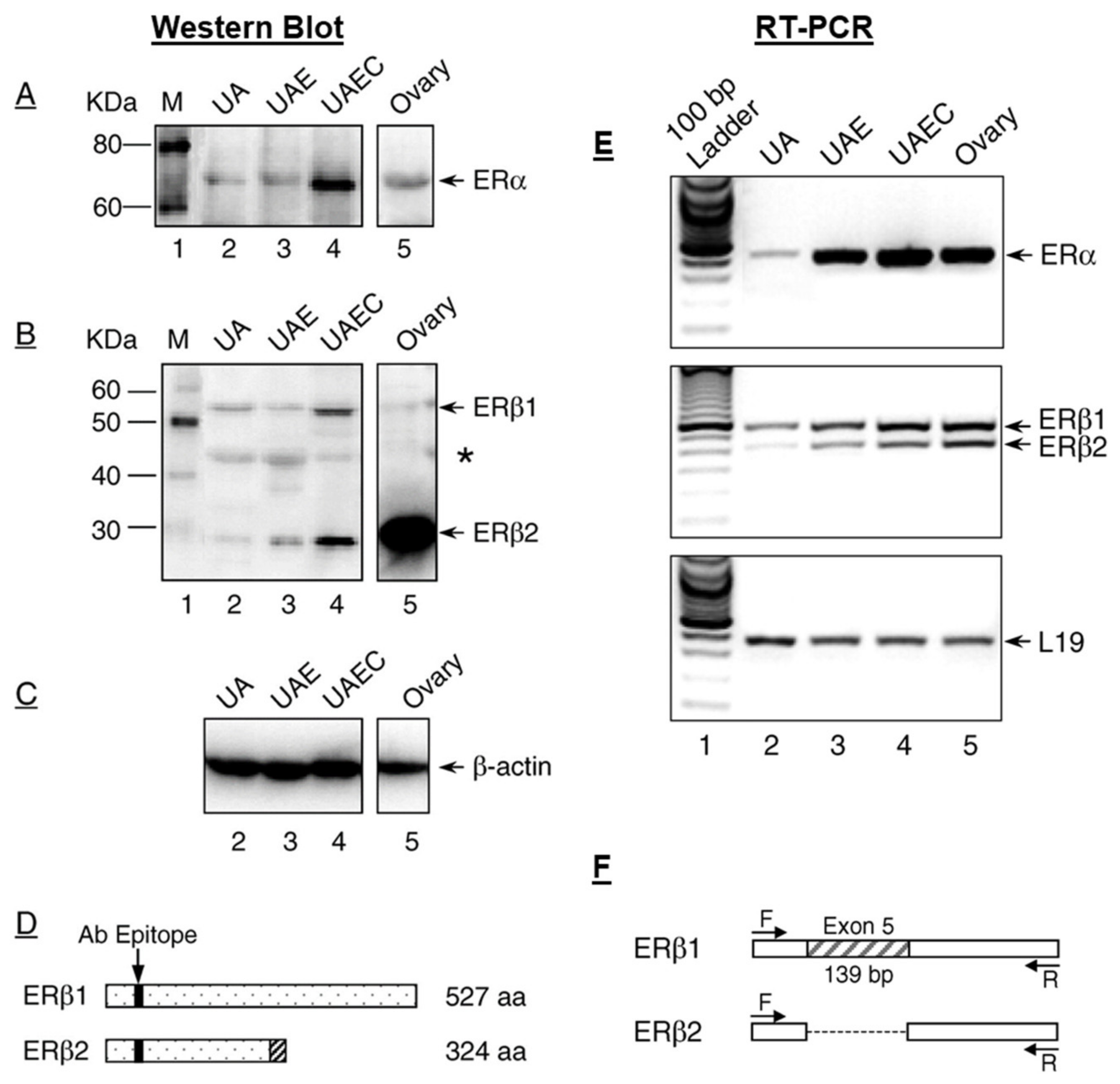
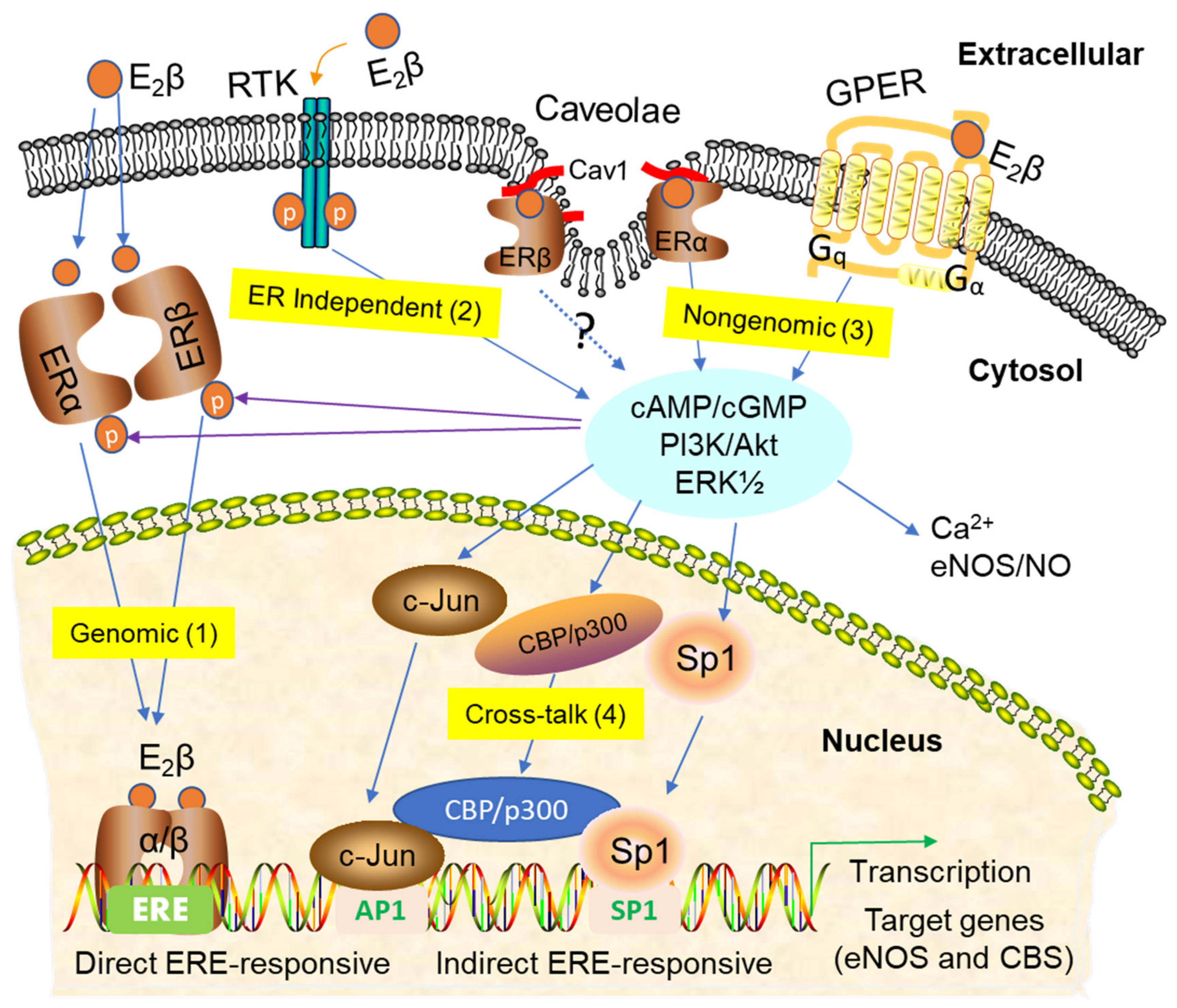

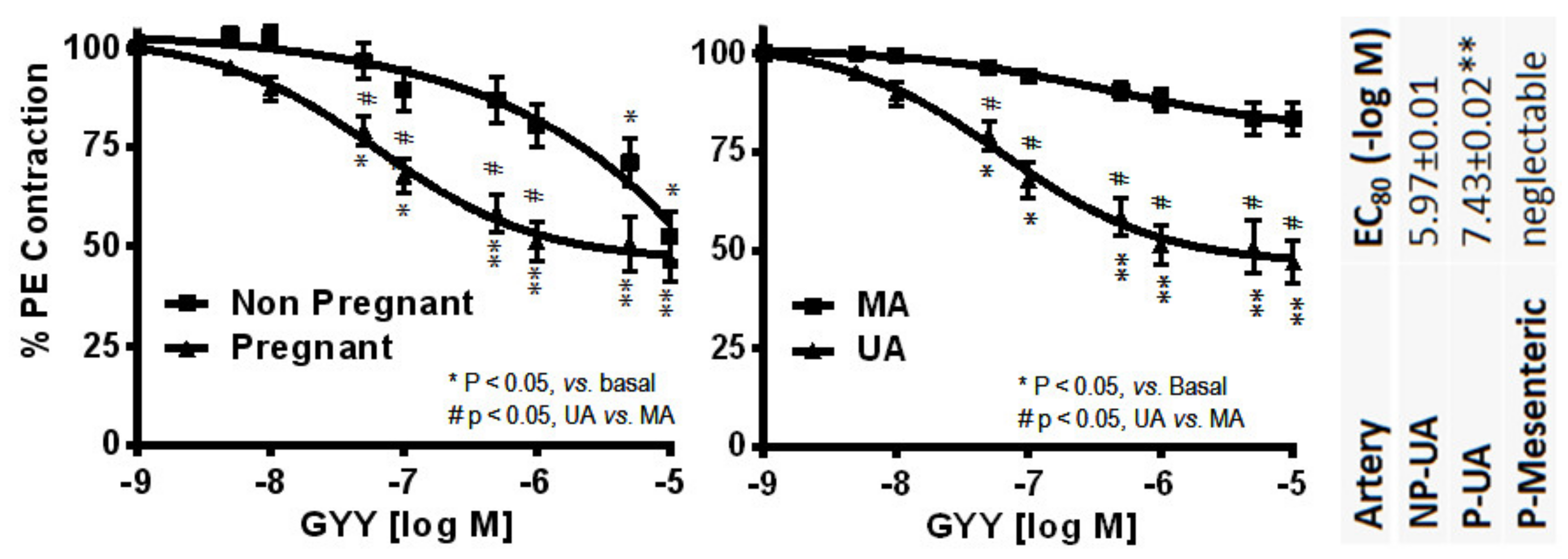

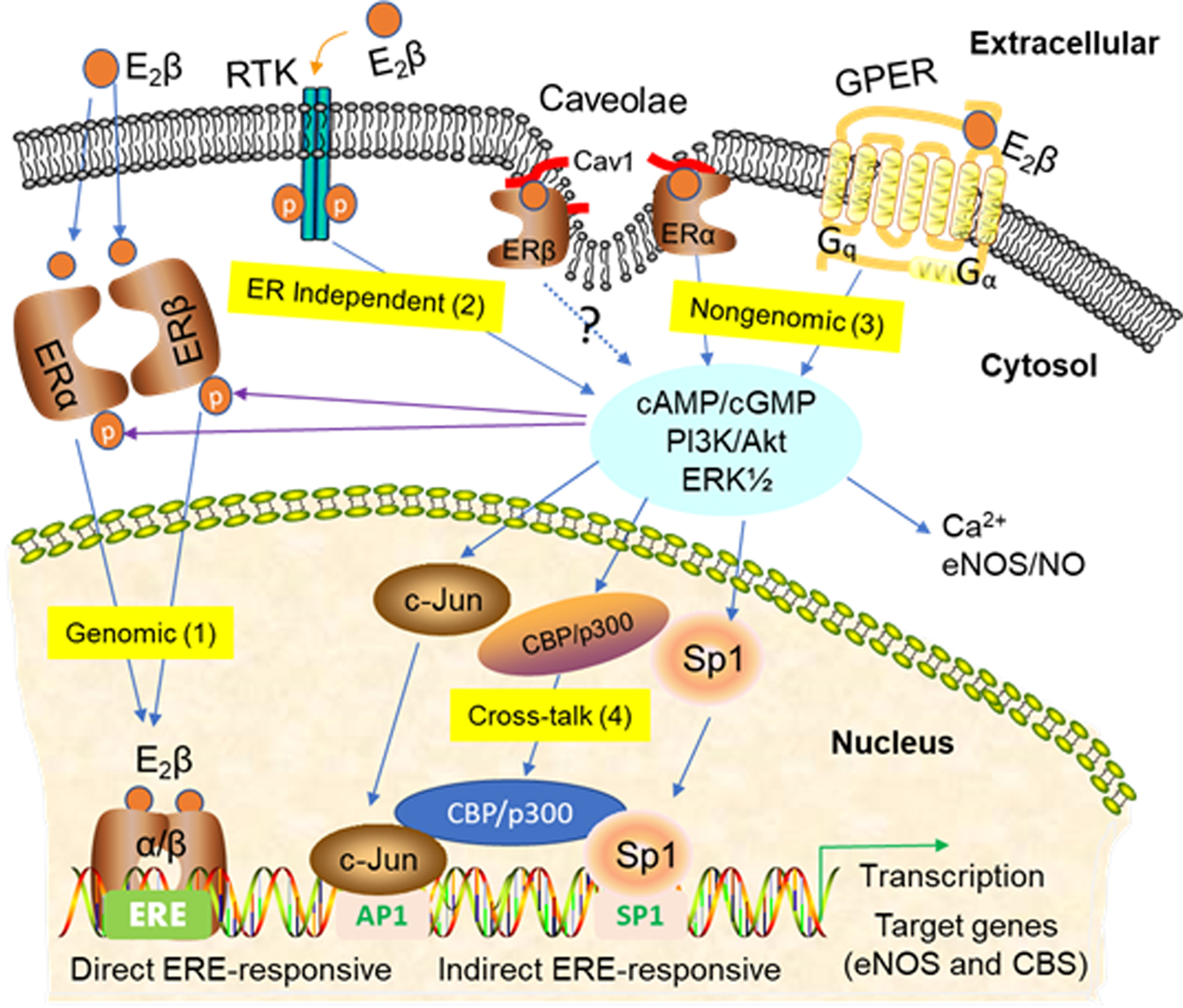{kind=link}
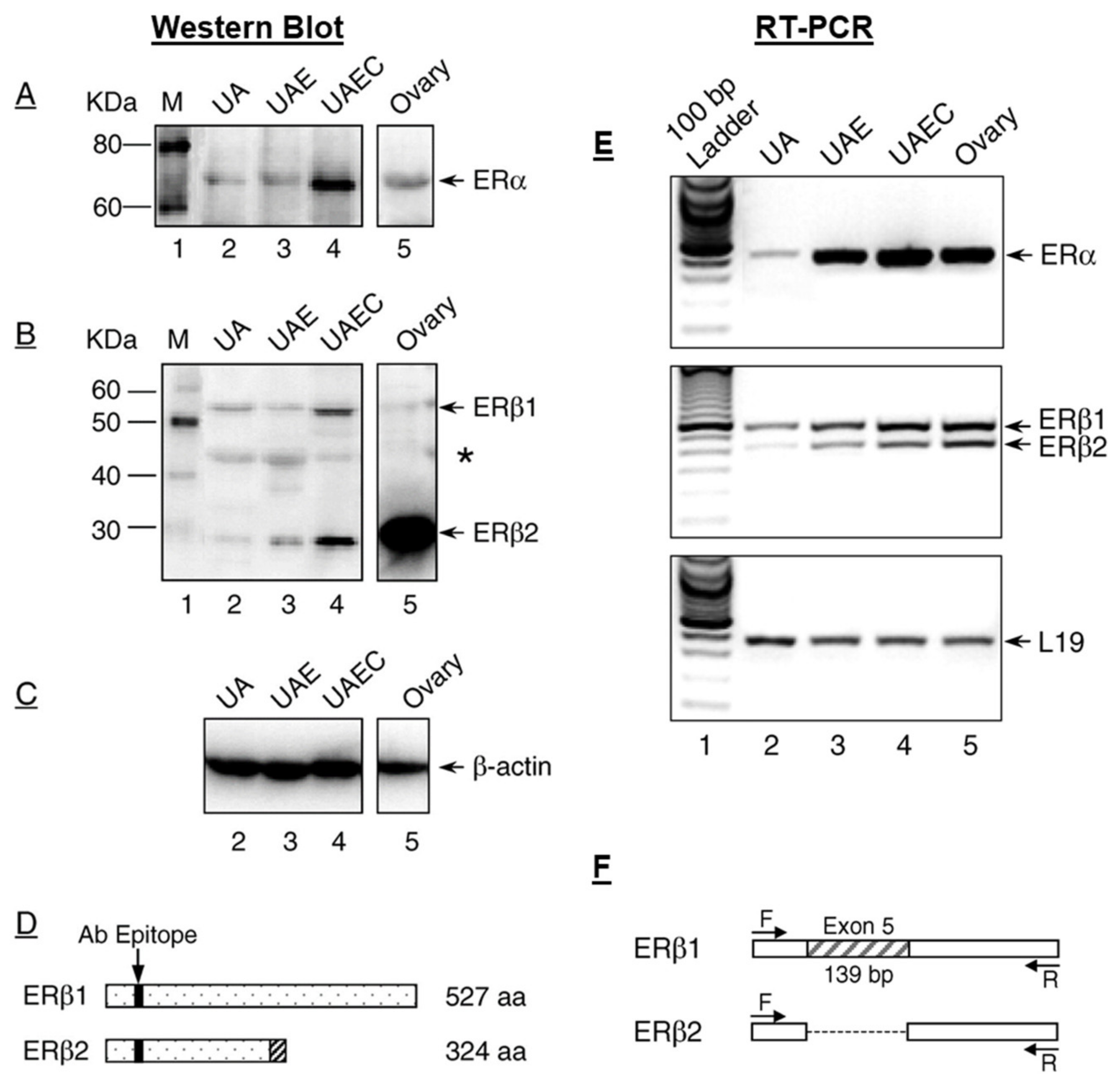{kind=link}
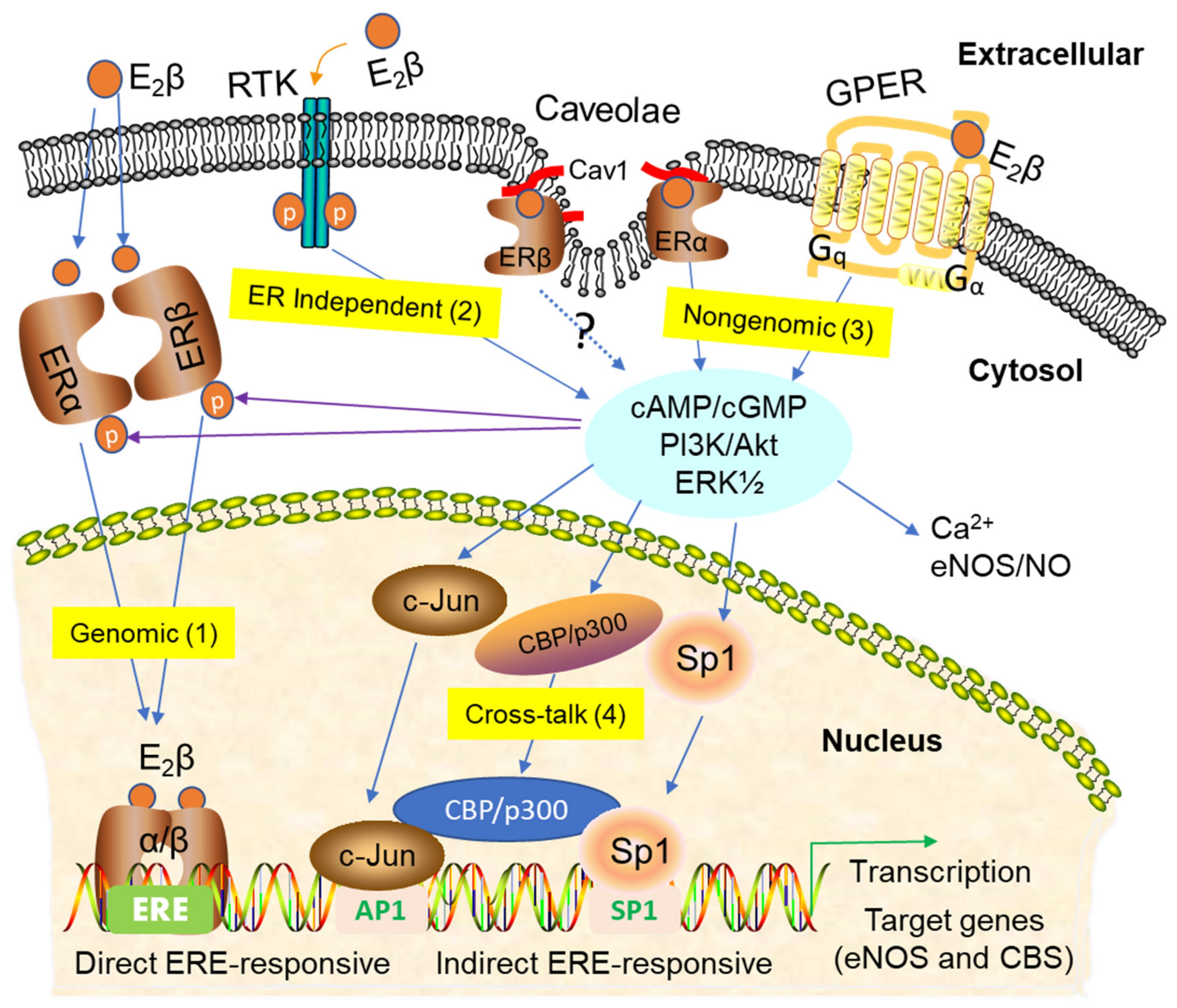{kind=link}
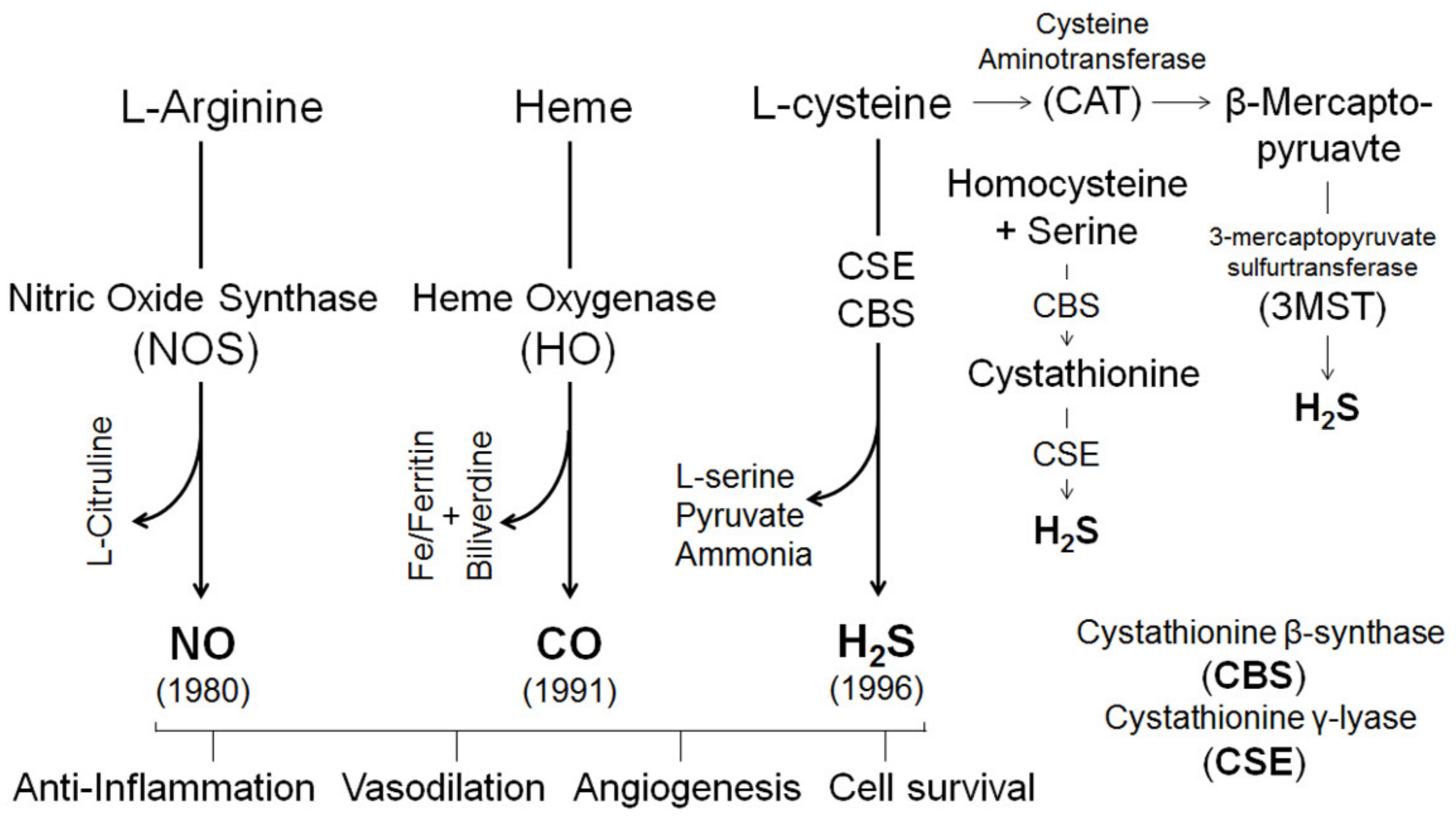{kind=link}
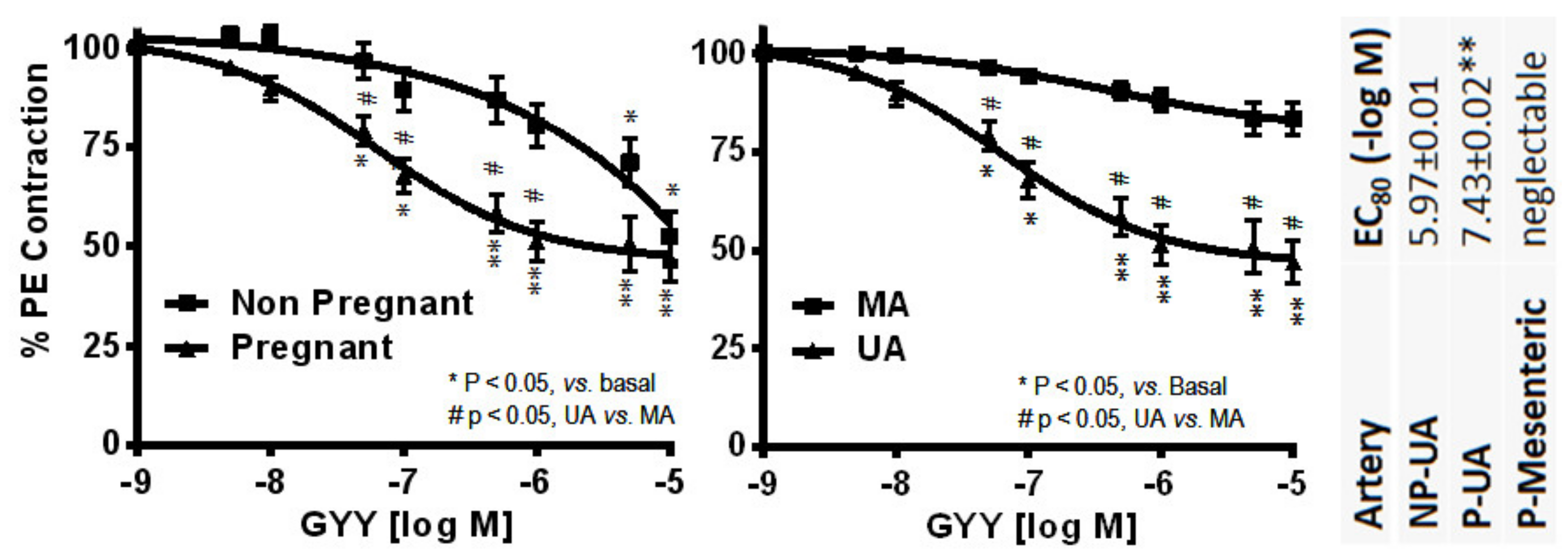{kind=link}
| miRNA | Regulation | Tissue/Cell | References |
|---|---|---|---|
| miR-451 | ↑ | Mouse uterus | [199] |
| miR-429 | ↑ | Mouse uterus | [198] |
| miR-155 | |||
| miR-181b | ↓ | Mouse uterus | [198] |
| miR-204 | |||
| miR-21 | ↓ | Human endometrial glandular epithelial cells | [204] |
| miR-20a | |||
| miR-21 | ↓ | Human leiomyoma SMC | [204] |
| miR-26a | |||
| miR-30b | ↑ | Human UVEC | [205] |
| miR-487a | |||
| miR-4710 | |||
| miR-501-3p | |||
| miR-378h | ↓ | Human UVEC | [205] |
| miR-1244 |
| miRNA | Tissue/Cell | Functions | Targets | References |
|---|---|---|---|---|
| miR-199a | human telomerase reverse transcriptase-immortalized human myometrial (hTERT-HM) cell line | Contractility | Cyclooxygenase-2 (COX-2) | [201] |
| miR-214 | ||||
| miR-203 | Rat uterus | Proliferation | Zinc finger and BTB domain containing 20 (Zbtb20), Alkaline Ceramidase 2 (Acer2) | [202] |
| Endometrial carcinoma | Migration | |||
| miR-451 | Human endometriotic lesions | Migration | Macrophage Migration Inhibitory Factor (MIF) | [210] |
| miR-218-5p | Extravillous cytotrophoblast (EVT) | Trophoblast differentiation | Transforming growth factor β 2 (TGFβ2) | [211] |
| Vessel remodeling | ||||
| miR-204 | Human endometrial cancer-1 (HEC-1A) cell line | Migration | Forkhead box C 1 (FOXC1) | [212] |
| Invasion | ||||
| miR-365 | Trophoblasts | Cell cycle | Mouse double minute 2 (MDM2), p53 | [213] |
| Apoptosis | ||||
| miR-376c | Trophoblasts | Proliferation | TGFβ2 | [214] |
| Invasion | ||||
| miR-17 | Placenta | Angiogenesis | Ephrin-B2 | [215] |
| miR-20a | Ephrin type-B receptor 4 (EPHB4) | |||
| miR-20b | ||||
| miR-210 | Ovine UA | Relaxation | Tet methylcytosine dioxygenase 1 (TET1) | [203] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bai, J.; Qi, Q.-R.; Li, Y.; Day, R.; Makhoul, J.; Magness, R.R.; Chen, D.-b. Estrogen Receptors and Estrogen-Induced Uterine Vasodilation in Pregnancy. Int. J. Mol. Sci. 2020, 21, 4349. https://doi.org/10.3390/ijms21124349
Bai J, Qi Q-R, Li Y, Day R, Makhoul J, Magness RR, Chen D-b. Estrogen Receptors and Estrogen-Induced Uterine Vasodilation in Pregnancy. International Journal of Molecular Sciences. 2020; 21(12):4349. https://doi.org/10.3390/ijms21124349
Chicago/Turabian StyleBai, Jin, Qian-Rong Qi, Yan Li, Robert Day, Josh Makhoul, Ronald R. Magness, and Dong-bao Chen. 2020. "Estrogen Receptors and Estrogen-Induced Uterine Vasodilation in Pregnancy" International Journal of Molecular Sciences 21, no. 12: 4349. https://doi.org/10.3390/ijms21124349
APA StyleBai, J., Qi, Q.-R., Li, Y., Day, R., Makhoul, J., Magness, R. R., & Chen, D.-b. (2020). Estrogen Receptors and Estrogen-Induced Uterine Vasodilation in Pregnancy. International Journal of Molecular Sciences, 21(12), 4349. https://doi.org/10.3390/ijms21124349