Benchmarking Data Sets from PubChem BioAssay Data: Current Scenario and Room for Improvement


Abstract
:
1. Introduction
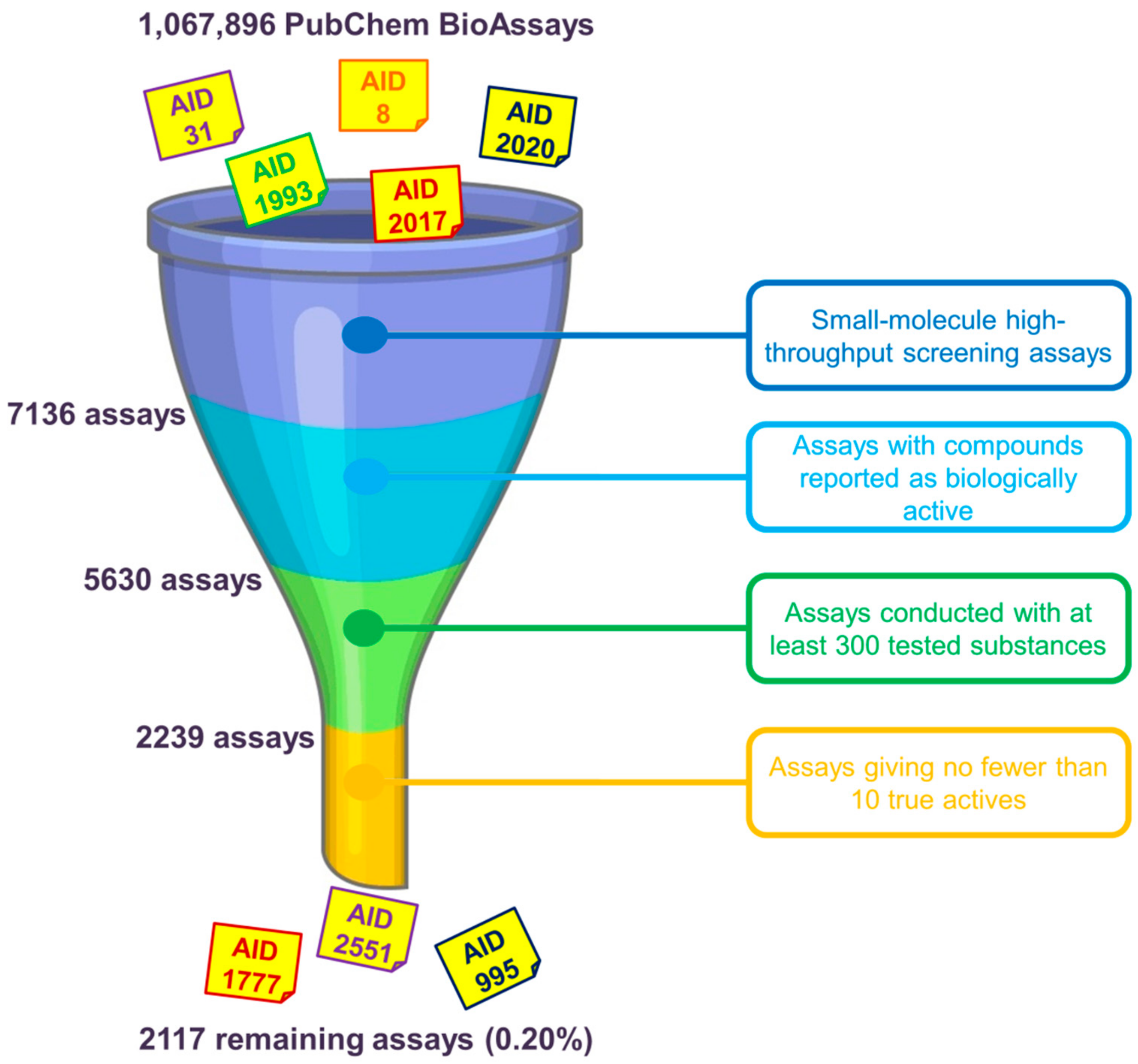
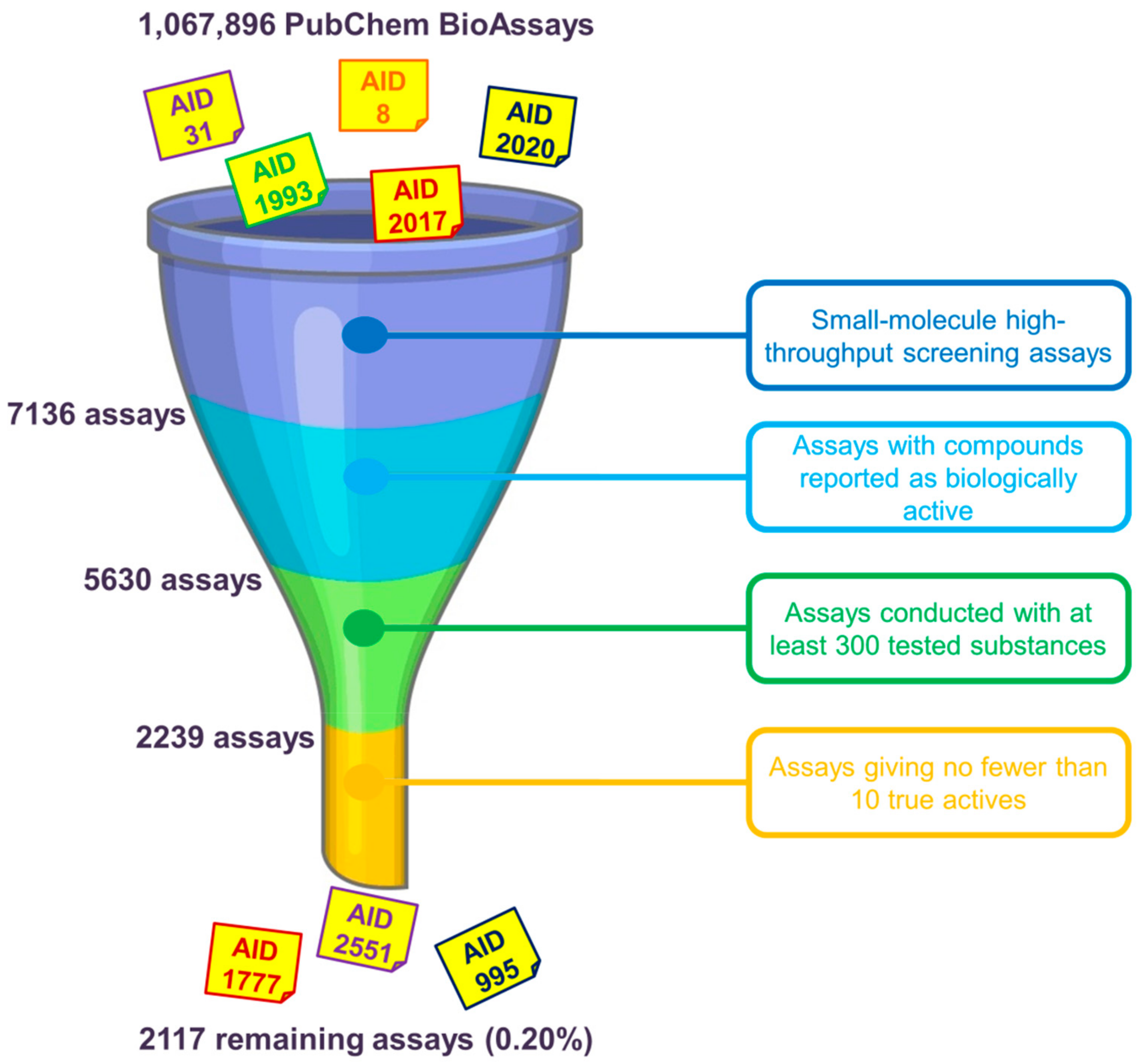
2. PubChem BioAssay Statistics: Assays and Compounds
3. What We Can Do with PubChem BioAssay Data: From the Data Set Construction Point of View
3.1. The MUV Data Sets
3.2. The UCI Repository
3.3. The Butkiewicz et al. Data Collection
3.4. The Lindh et al. Data Collection
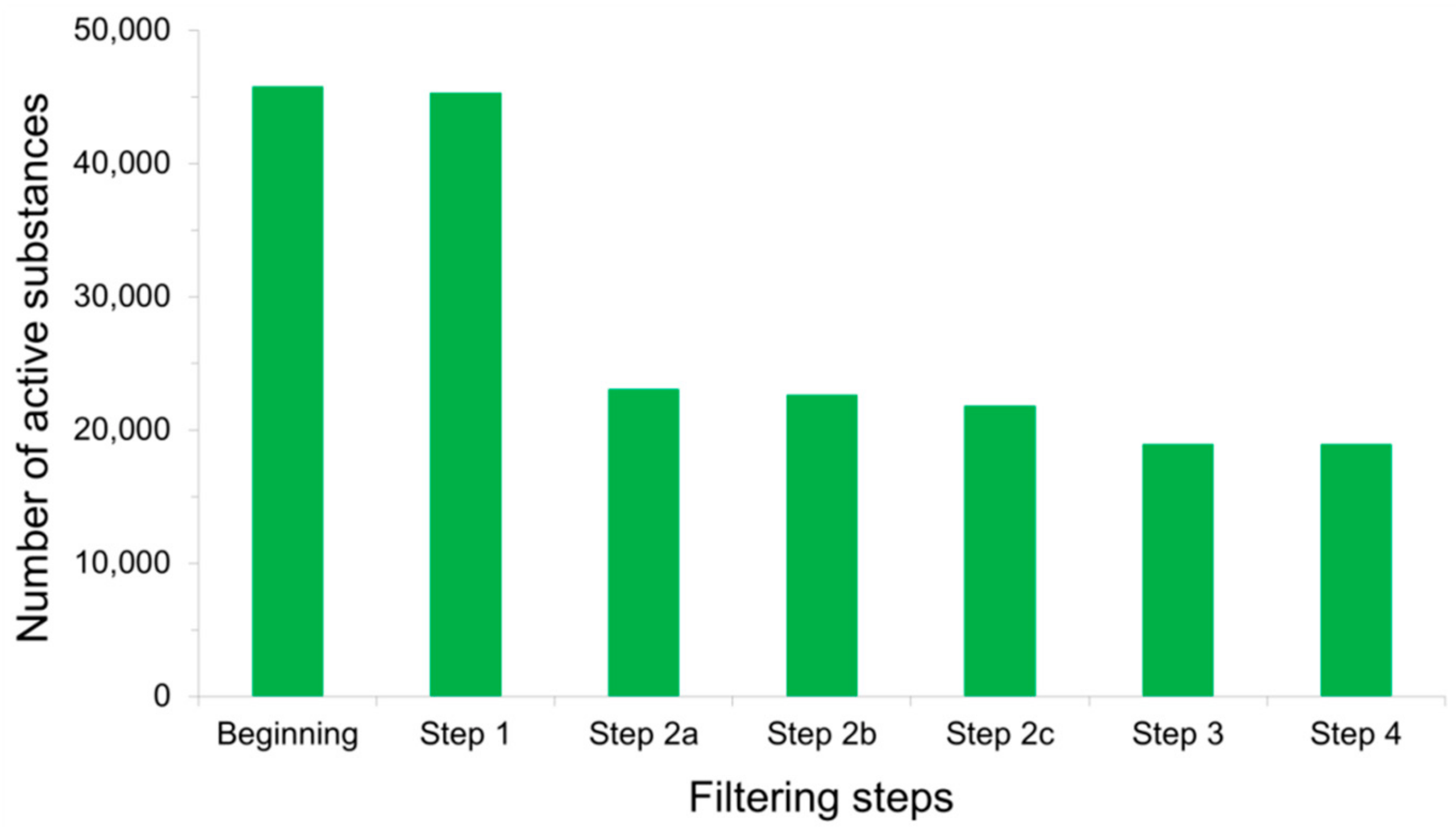
3.5. The LIT-PCBA Data Sets
4. Noteworthy Issues with Using Data from PubChem BioAssay for Constructing Benchmarking Data Sets
4.1. Assay Selection for Evaluating Virtual Screening Methods
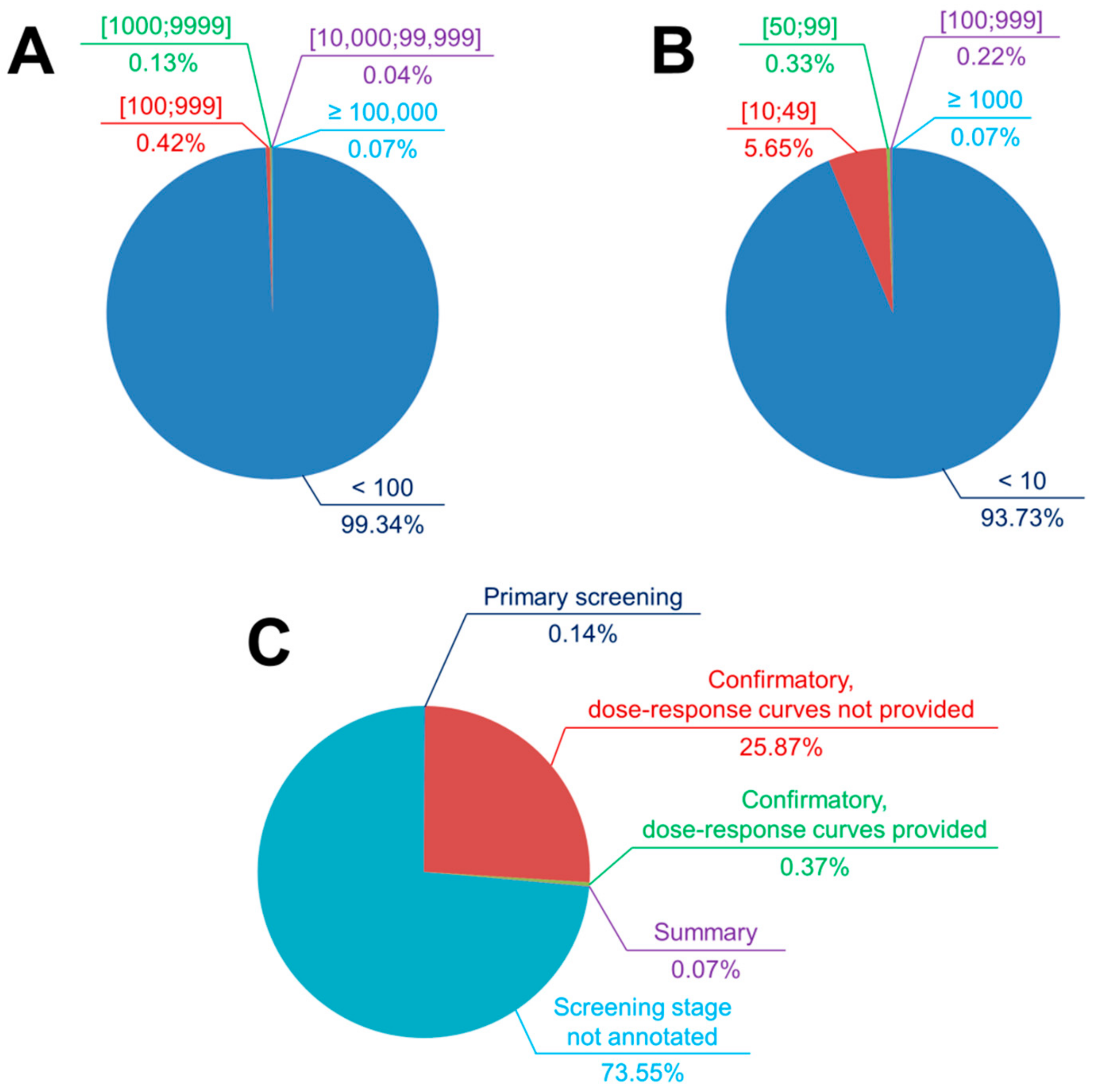
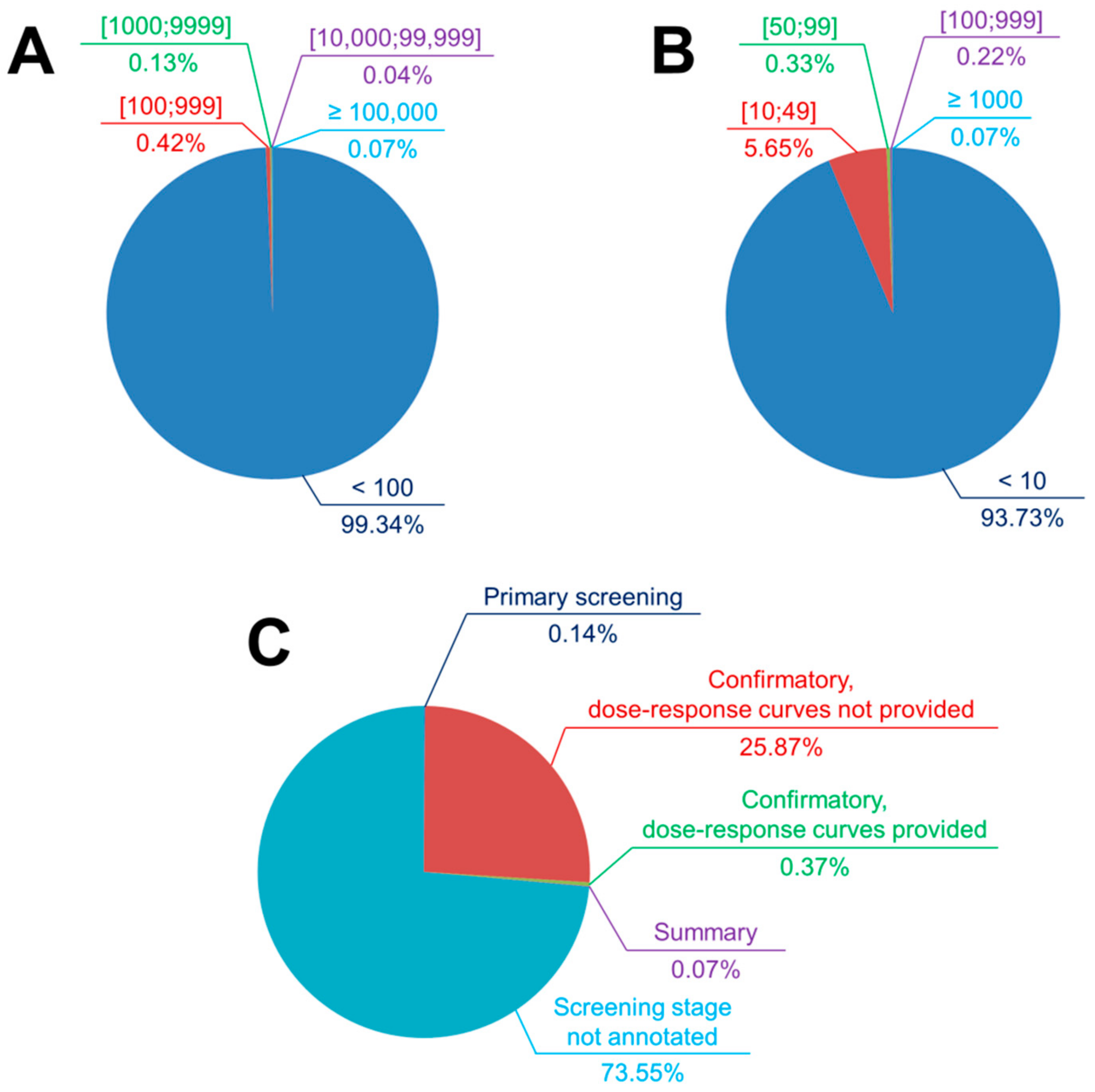
4.1.1. Assay Selection as Regards the Data Size and Hit Rates
4.1.2. Assay Selection as Regards the Nature of Virtual Screening
4.1.3. Assay Selection as Regards the Screening Stage
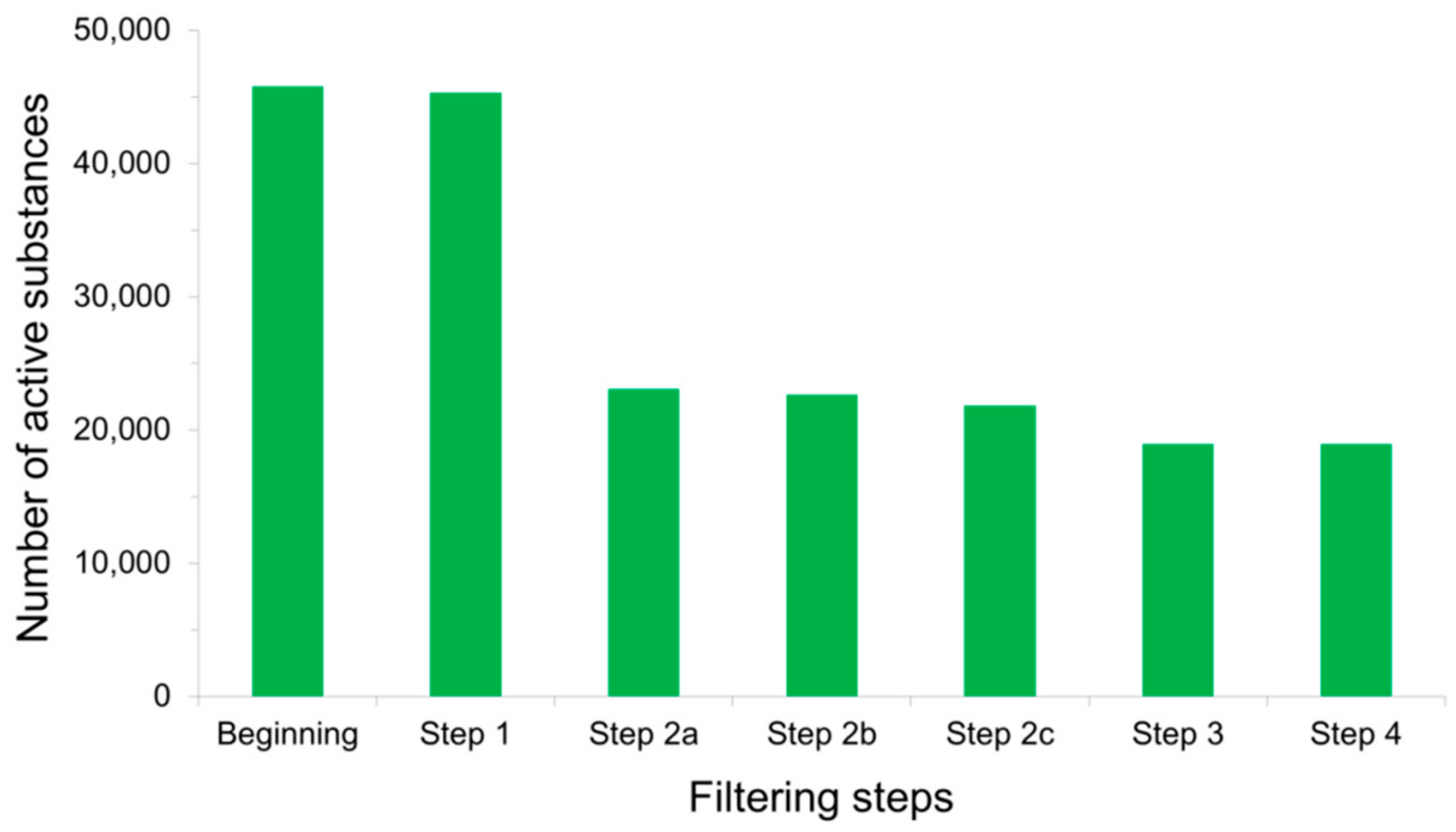
4.2. Detecting False Positives among Active Substances
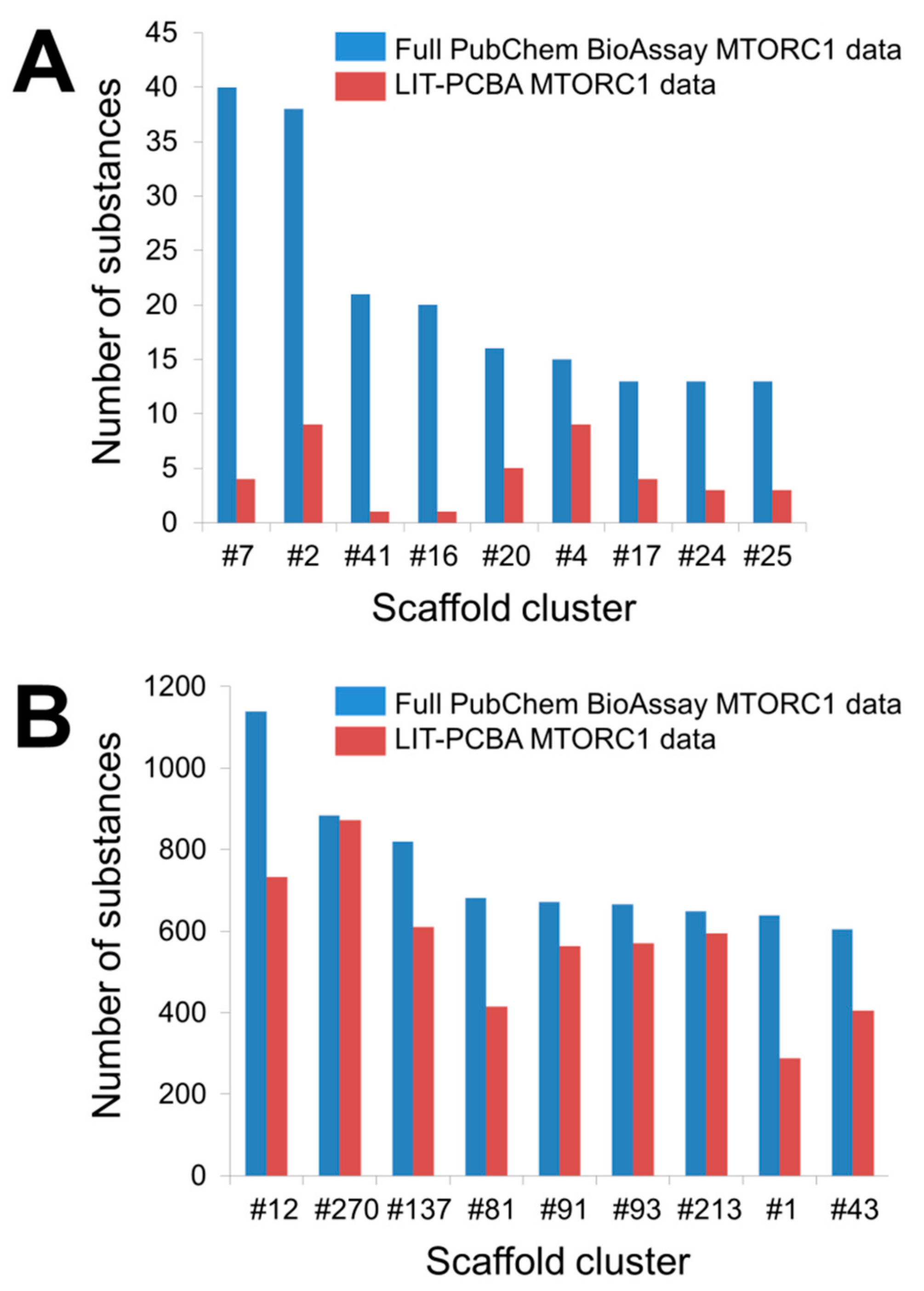
4.3. Possible Chemical Bias in Assembling Active and Inactive Substances
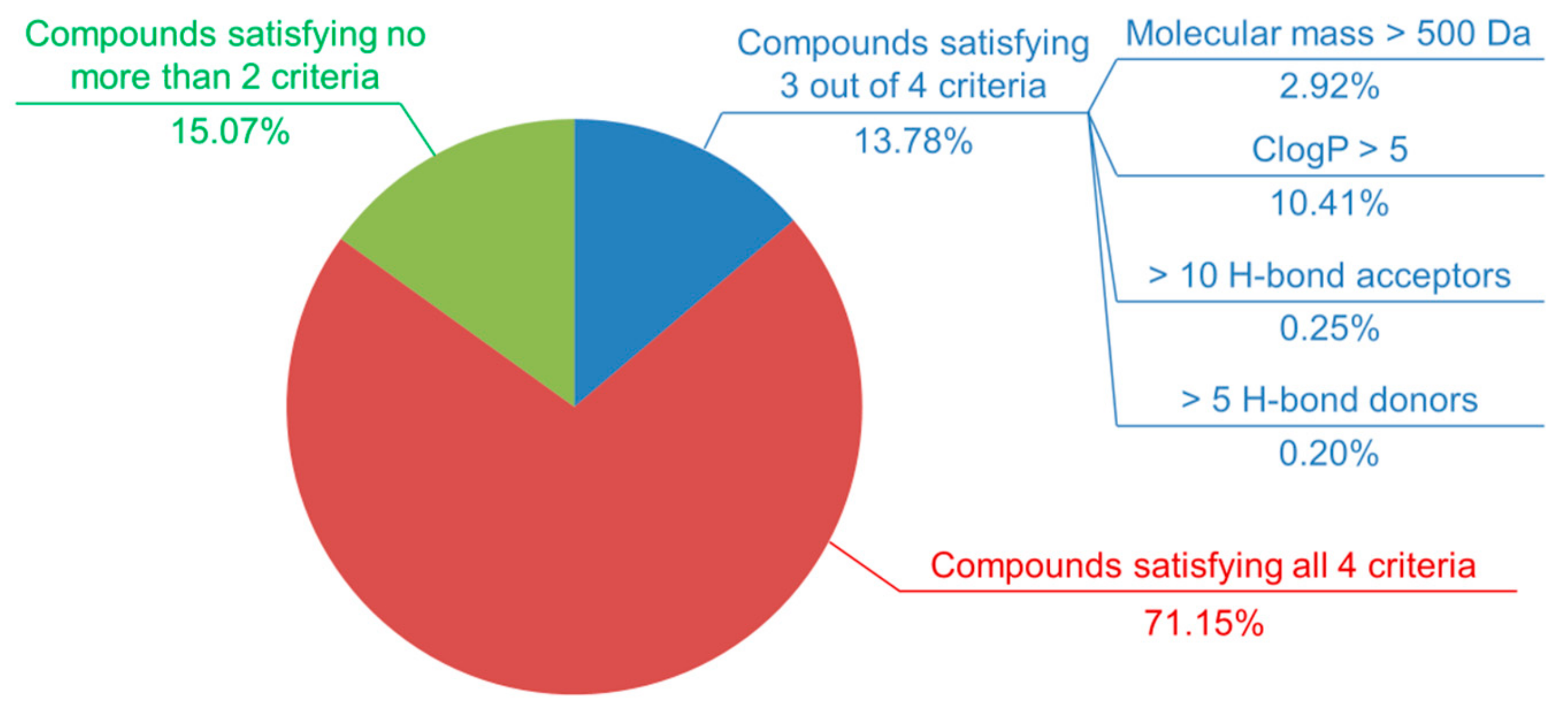
4.4. Potency Bias in the Composition of Active Ligand Sets
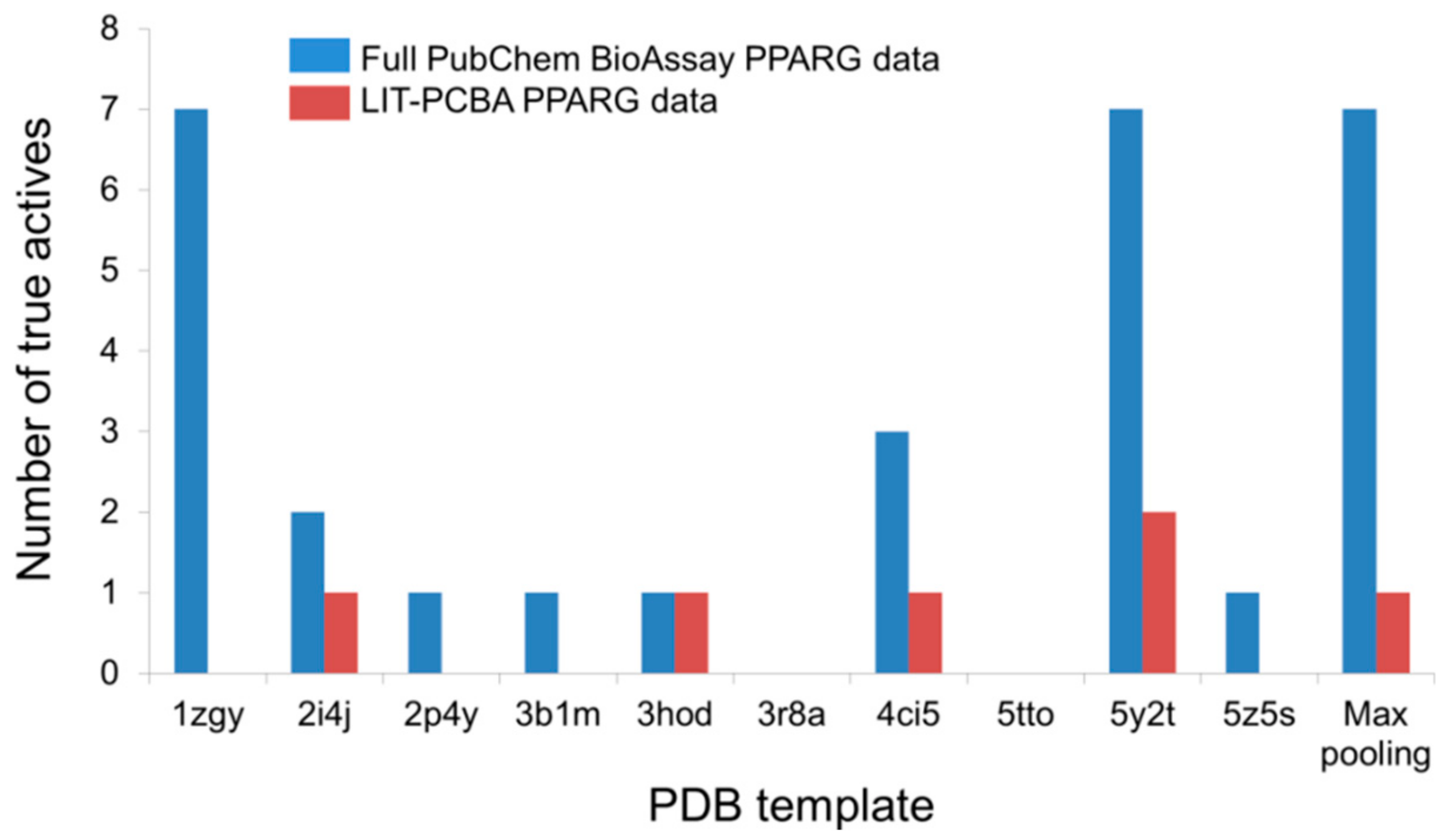
4.5. Processing Input Structures Prior to Virtual Screening
5. Conclusions
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| HTS | High-throughput screening |
| AID | Assay identifier |
| MUV | Maximum unbiased validation |
| AVE | Asymmetric validation embedding |
| PAINS | Pan-assay interference compounds |
| EF1% | Enrichment in true actives at a constant 1% false positive rate over random picking |
| ABC | ATP-binding cassette |
| PDB | Protein Data Bank |
| SMILES | Simplified molecular-input line-entry system |
| SDF | Spatial data file |
| GPCRs | G protein-coupled receptors |
References
- Wang, Y.; Xiao, J.; Suzek, T.O.; Zhang, J.; Wang, J.; Bryant, S.H. PubChem: A public information system for analyzing bioactivities of small molecules. Nucleic Acids Res. 2009, 37, W623–W633. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Cheng, T.; Wang, Y.; Bryant, S.H. PubChem as a public resource for drug discovery. Drug Discov. Today 2010, 15, 1052–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Bolton, E.; Dracheva, S.; Karapetyan, K.; Shoemaker, B.A.; Suzek, T.O.; Wang, J.; Xiao, J.; Zhang, J.; Bryant, S.H. An overview of the PubChem BioAssay resource. Nucleic Acids Res. 2010, 38, D255–D266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Xiao, J.; Suzek, T.O.; Zhang, J.; Wang, J.; Zhou, Z.; Han, L.; Karapetyan, K.; Dracheva, S.; Shoemaker, B.A.; et al. PubChem’s BioAssay database. Nucleic Acids Res. 2012, 40, D400–D412. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Cheng, T.; Bryant, S.H. PubChem BioAssay: A decade’s development toward open high-throughput screening data sharing. SLAS Discov. 2017, 22, 655–666. [Google Scholar] [CrossRef] [Green Version]
- Austin, C.P.; Brady, L.S.; Insel, T.R.; Collins, F.S. NIH molecular libraries initiative. Science 2004, 306, 1138–1139. [Google Scholar] [CrossRef]
- Cheng, T.; Pan, Y.; Hao, M.; Wang, Y.; Bryant, S.H. PubChem applications in drug discovery: A bibliometric analysis. Drug Discov. Today 2014, 19, 1751–1756. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem substance and compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef]
- PubChem Data Sources. Available online: https://pubchem.ncbi.nlm.nih.gov/sources/ (accessed on 30 April 2020).
- PubChem Classification Browser. Available online: https://pubchem.ncbi.nlm.nih.gov/classification/#hid=80/ (accessed on 30 April 2020).
- PubChem Data Counts. Available online: https://pubchemdocs.ncbi.nlm.nih.gov/statistics/ (accessed on 30 April 2020).
- Kim, S.; Thiessen, P.A.; Cheng, T.; Yu, B.; Shoemaker, B.A.; Wang, J.; Bolton, E.E.; Wang, Y.; Bryant, S.H. Literature information in PubChem: Associations between PubChem records and scientific articles. J. Cheminform 2016, 8, 32. [Google Scholar] [CrossRef] [Green Version]
- PubChem BioAssay. Available online: https://www.ncbi.nlm.nih.gov/pcassay/ (accessed on 30 April 2020).
- Entrez Programming Utilities Help. Available online: https://www.ncbi.nlm.nih.gov/books/NBK25501/ (accessed on 30 April 2020).
- PubMed Central. Available online: https://www.ncbi.nlm.nih.gov/pmc/ (accessed on 1 May 2020).
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Suzek, T.; Zhang, J.; Wang, J.; He, S.; Cheng, T.; Shoemaker, B.A.; Gindulyte, A.; Bryant, S.H. PubChem BioAssay: 2014 Update. Nucleic Acids Res. 2014, 42, D1075–D1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Bryant, S.H.; Cheng, T.; Wang, J.; Gindulyte, A.; Shoemaker, B.A.; Thiessen, P.A.; He, S.; Zhang, J. PubChem BioAssay: 2017 Update. Nucleic Acids Res. 2017, 45, D955–D963. [Google Scholar] [CrossRef]
- Kim, S.; Thiessen, P.A.; Cheng, T.; Yu, B.; Bolton, E.E. An update on PUG-REST: RESTful interface for programmatic access to PubChem. Nucleic Acids Res. 2018, 46, W563–W570. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- About PubChem. Available online: https://pubchemdocs.ncbi.nlm.nih.gov/about/ (accessed on 30 April 2020).
- Tran-Nguyen, V.K.; Jacquemard, C.; Rognan, D. LIT-PCBA: An unbiased data set for machine learning and virtual screening. J. Chem. Inf. Model. 2020. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Pyka, A.; Babuska, M.; Zachariasz, M. A Comparison of Theoretical methods of calculation of partition coefficients for selected drugs. Acta. Pol. Pharm. 2006, 63, 159–167. [Google Scholar]
- Huang, N.; Shoichet, B.K.; Irwin, J.J. Benchmarking sets for molecular docking. J. Med. Chem. 2006, 49, 6789–6801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef] [PubMed]
- Bauer, M.R.; Ibrahim, T.M.; Vogel, S.M.; Boeckler, F.M. Evaluation and optimization of virtual screening workflows with DEKOIS 2.0—A public library of challenging docking benchmark sets. J. Chem. Inf. Model. 2013, 53, 1447–1462. [Google Scholar] [CrossRef] [PubMed]
- Wassermann, A.M.; Lounkine, E.; Hoepfner, D.; Le Goff, G.; King, F.J.; Studer, C.; Peltier, J.M.; Grippo, M.L.; Prindle, V.; Tao, J.; et al. Dark chemical matter as a promising starting point for drug lead discovery. Nat. Chem. Biol. 2015, 11, 958–966. [Google Scholar] [CrossRef]
- PubChem BioAssay “Limits” Search. Available online: https://www.ncbi.nlm.nih.gov/pcassay/limits (accessed on 30 April 2020).
- PubChem BioAssay “Advanced” Search. Available online: https://www.ncbi.nlm.nih.gov/pcassay/advanced (accessed on 30 April 2020).
- PubChem Power User Gateway (PUG) Help. Available online: https://pubchemdocs.ncbi.nlm.nih.gov/power-user-gateway (accessed on 30 April 2020).
- PubChem PUG SOAP. Available online: https://pubchemdocs.ncbi.nlm.nih.gov/pug-soap (accessed on 30 April 2020).
- PubChem PUG REST. Available online: https://pubchemdocs.ncbi.nlm.nih.gov/pug-rest (accessed on 30 April 2020).
- PubChem PUG View. Available online: https://pubchemdocs.ncbi.nlm.nih.gov/pug-view (accessed on 30 April 2020).
- PubChemRDF. Available online: https://pubchemdocs.ncbi.nlm.nih.gov/rdf (accessed on 30 April 2020).
- ScrubChem by Jason Bret Harris. Available online: http://scrubchem.org/ (accessed on 1 May 2020).
- Kim, S. Getting the most out of PubChem for virtual screening. Expert Opin. Drug Discov. 2016, 11, 843–855. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Shoemaker, B.A.; Bolton, E.E.; Bryant, S.H. Finding potential multitarget ligands using PubChem. Methods Mol. Biol. 2018, 1825, 63–91. [Google Scholar] [CrossRef]
- Li, Q.Y.; Jorgensen, F.S.; Oprea, T.; Brunak, S.; Taboureau, O. hERG classification model based on a combination of support vector machine method and GRIND descriptors. Mol. Pharm. 2008, 5, 117–127. [Google Scholar] [CrossRef]
- Su, B.H.; Shen, M.Y.; Esposito, E.X.; Hopfinger, A.J.; Tseng, Y.J. In silico binary classification QSAR models based on 4D-fingerprints and MOE descriptors for prediction of hERG blockage. J. Chem. Inf. Model. 2010, 50, 1304–1318. [Google Scholar] [CrossRef]
- Wang, S.; Li, Y.; Wang, J.; Chen, L.; Zhang, L.; Yu, H.; Hou, T. ADMET evaluation in drug discovery. 12. development of binary classification models for prediction of hERG potassium channel blockage. Mol. Pharm. 2012, 9, 996–1010. [Google Scholar] [CrossRef]
- Shen, M.Y.; Su, B.H.; Esposito, E.X.; Hopfinger, A.J.; Tseng, Y.J. A comprehensive support vector machine binary hERG classification model based on extensive but biased end point hERG data sets. Chem. Res. Toxicol. 2011, 24, 934–949. [Google Scholar] [CrossRef]
- Cheng, F.; Yu, Y.; Shen, J.; Yang, L.; Li, W.; Liu, G.; Lee, P.W.; Tang, Y. Classification of cytochrome P450 inhibitors and noninhibitors using combined classifiers. J. Chem. Inf. Model. 2011, 51, 996–1011. [Google Scholar] [CrossRef]
- Su, B.H.; Tu, Y.S.; Lin, C.; Shao, C.Y.; Lin, O.A.; Tseng, Y.J. Rule-based prediction models of cytochrome P450 inhibition. J. Chem. Inf. Model. 2015, 55, 1426–1434. [Google Scholar] [CrossRef] [PubMed]
- Didziapetris, R.; Dapkunas, J.; Sazonovas, A.; Japertas, P. Trainable structure-activity relationship model for virtual screening of CYP3A4 inhibition. J. Comput. Aided Mol. Des. 2010, 24, 891–906. [Google Scholar] [CrossRef] [PubMed]
- Novotarskyi, S.; Sushko, I.; Korner, R.; Pandey, A.K.; Tetko, I.V. A comparison of different QSAR approaches to modeling CYP450 1A2 inhibition. J. Chem. Inf. Model. 2011, 51, 1271–1280. [Google Scholar] [CrossRef] [PubMed]
- Buchwald, P. Activity-limiting role of molecular size: Size-dependency of maximum activity for P450 inhibition as revealed by qHTS data. Drug Metab. Dispos. 2014, 42, 1785–1790. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Rusyn, I.; Richard, A.; Tropsha, A. Use of cell viability assay data improves the prediction accuracy of conventional quantitative structure-activity relationship models of animal carcinogenicity. Environ. Health Perspect. 2008, 116, 506–513. [Google Scholar] [CrossRef] [Green Version]
- Guha, R.; Schurer, S.C. Utilizing high throughput screening data for predictive toxicology models: Protocols and application to MLSCN assays. J. Comput. Aided Mol. Des. 2008, 22, 367–384. [Google Scholar] [CrossRef]
- Zhang, J.; Hsieh, J.H.; Zhu, H. Profiling animal toxicants by automatically mining public bioassay data: A big data approach for computational toxicology. PLoS ONE 2014, 9, 11. [Google Scholar] [CrossRef] [PubMed]
- Sedykh, A.; Zhu, H.; Tang, H.; Zhang, L.; Richard, A.; Rusyn, I.; Tropsha, A. Use of in vitro HTS-derived concentration-response data as biological descriptors improves the accuracy of QSAR models of in vivo toxicity. Environ. Health Perspect. 2011, 119, 364–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.T.; Huang, R.; Sedykh, A.; Wang, W.; Xia, M.; Zhu, H. Mechanism profiling of hepatotoxicity caused by oxidative stress using antioxidant response element reporter gene assay models and big data. Environ. Health Perspect. 2016, 124, 634–641. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Zhang, J.; Kim, M.T.; Boison, A.; Sedykh, A.; Moran, K. Big data in chemical toxicity research: The use of high-throughput screening assays to identify potential toxicants. Chem. Res. Toxicol. 2014, 27, 1643–1651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.; Wild, D.; Guha, R. PubChem as a source of polypharmacology. J. Chem. Inf. Model. 2009, 49, 2044–2055. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Han, B.; Wei, X.; Tan, C.; Chen, Y.; Jiang, Y. A two-step target binding and selectivity support vector machines approach for virtual screening of dopamine receptor subtype-selective ligands. PLoS ONE 2012, 7, e39076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swamidass, S.J.; Schillebeeckx, C.N.; Matlock, M.; Hurle, M.R.; Agarwal, P. Combined analysis of phenotypic and target-based screening in assay networks. J. Biomol. Screen. 2014, 19, 782–790. [Google Scholar] [CrossRef] [Green Version]
- Lounkine, E.; Nigsch, F.; Jenkins, J.L.; Glick, M. Activity-aware clustering of high throughput screening data and elucidation of orthogonal structure-activity relationships. J. Chem. Inf. Model. 2011, 51, 3158–3168. [Google Scholar] [CrossRef]
- Bissantz, C.; Folkers, G.; Rognan, D. Protein-based virtual screening of chemical databases. 1. Evaluation of different docking/scoring combinations. J. Med. Chem. 2000, 43, 4759–4767. [Google Scholar] [CrossRef]
- McGovern, S.L.; Shoichet, B.K. Information Decay in Molecular Docking Screens against holo, apo, and modeled conformations of enzymes. J. Med. Chem. 2003, 46, 2895–2907. [Google Scholar] [CrossRef]
- Diller, D.J.; Li, R. Kinases, homology models, and high throughput docking. J. Med. Chem. 2003, 46, 4638–4647. [Google Scholar] [CrossRef]
- Lorber, D.M.; Shoichet, B.K. Hierarchical docking of databases of multiple ligand conformations. Curr. Top. Med. Chem. 2005, 5, 739–749. [Google Scholar] [CrossRef]
- Irwin, J.J.; Raushel, F.M.; Shoichet, B.K. Virtual screening against metalloenzymes for inhibitors and substrates. Biochemistry 2005, 44, 12316–12328. [Google Scholar] [CrossRef]
- Miteva, M.A.; Lee, W.H.; Montes, M.O.; Villoutreix, B.O. Fast structure-based virtual ligand screening combining FRED, DOCK, and Surflex. J. Med. Chem. 2005, 48, 6012–6022. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.A.; Jain, A.N. Parameter estimation for scoring protein-ligand interactions using negative training data. J. Med. Chem. 2006, 49, 5856–5868. [Google Scholar] [CrossRef] [PubMed]
- Vogel, S.M.; Bauer, M.R.; Boeckler, F.M. DEKOIS: Demanding evaluation kits for objective in silico screening—A versatile tool for benchmarking docking programs and scoring functions. J. Chem. Inf. Model. 2011, 51, 2650–2665. [Google Scholar] [CrossRef] [PubMed]
- Gatica, E.A.; Cavasotto, C.N. Ligand and decoy sets for docking to g protein-coupled receptors. J. Chem. Inf. Model. 2012, 52, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Lagarde, N.; Ben Nasr, N.; Jeremie, A.; Guillemain, H.; Laville, V.; Labib, T.; Zagury, J.F.; Montes, M. NRLiSt BDB, the manually curated nuclear receptors ligands and structures benchmarking database. J. Med. Chem. 2014, 57, 3117–3125. [Google Scholar] [CrossRef]
- Xia, J.; Tilahun, E.L.; Kebede, E.H.; Reid, T.E.; Zhang, L.; Wang, X.S. Comparative modeling and benchmarking data sets for human histone deacetylases and sirtuin families. J. Chem. Inf. Model. 2015, 55, 374–388. [Google Scholar] [CrossRef] [Green Version]
- Chaput, L.; Martinez-Sanz, J.; Saettel, N.; Mouawad, L. Benchmark of four popular virtual screening programs: Construction of the active/decoy dataset remains a major determinant of measured performance. J. Cheminformatics 2016, 8, 56. [Google Scholar] [CrossRef] [Green Version]
- Wallach, I.; Heifets, A. most ligand-based classification benchmarks reward memorization rather than generalization. J. Chem. Inf. Model. 2018, 58, 916–932. [Google Scholar] [CrossRef]
- Chen, L.; Cruz, A.; Ramsey, S.; Dickson, C.J.; Duca, J.S.; Hornak, V.; Koes, D.R.; Kurtzman, T. Hidden bias in the DUD-E dataset leads to misleading performance of deep learning in structure-based virtual screening. PLoS ONE 2019, 14, e0220113. [Google Scholar] [CrossRef]
- Sieg, J.; Flachsenberg, F.; Rarey, M. In need of bias control: Evaluating chemical data for machine learning in structure-based virtual screening. J. Chem. Inf. Model. 2019, 59, 947–961. [Google Scholar] [CrossRef]
- Lagarde, N.; Zagury, J.F.; Montes, M. Benchmarking data sets for the evaluation of virtual ligand screening methods: Review and perspectives. J. Chem. Inf. Model. 2015, 55, 1297–1307. [Google Scholar] [CrossRef] [PubMed]
- BIOVIA Available Chemicals Directory (ACD). Available online: https://www.3dsbiovia.com/products/collaborative-science/databases/sourcing-databases/biovia-available-chemicals-directory.html (accessed on 1 May 2020).
- Irwin, J.J.; Shoichet, B.K. ZINC—A free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran-Nguyen, V.K.; Da Silva, F.; Bret, G.; Rognan, D. All in one: Cavity detection, druggability estimate, cavity-based pharmacophore perception, and virtual screening. J. Chem. Inf. Model. 2019, 59, 573–585. [Google Scholar] [CrossRef]
- Rohrer, S.G.; Baumann, K. Maximum unbiased validation (MUV) data sets for virtual screening based on PubChem bioactivity data. J. Chem. Inf. Model. 2009, 49, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Schierz, A.C. Virtual screening of bioassay data. J. Cheminformatics 2009, 1, 21. [Google Scholar] [CrossRef] [Green Version]
- Butkiewicz, M.; Lowe, E.W.; Mueller, R.; Mendenhall, J.L.; Teixeira, P.L.; Weaver, C.D.; Meiler, J. Benchmarking ligand-based virtual high-throughput screening with the PubChem database. Molecules 2013, 18, 735–756. [Google Scholar] [CrossRef] [Green Version]
- Lindh, M.; Svensson, F.; Schaal, W.; Zhang, J.; Sköld, C.; Brandt, P.; Karlen, A. Toward a benchmarking data set able to evaluate ligand- and structure-based virtual screening using public HTS data. J. Chem. Inf. Model. 2015, 55, 343–353. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [Green Version]
- Gilberg, E.; Jasial, S.; Stumpfe, D.; Dimova, D.; Bajorath, J. Highly promiscuous small molecules from biological screening assays include many pan-assay interference compounds but also candidates for polypharmacology. J. Med. Chem. 2016, 59, 10285–10290. [Google Scholar] [CrossRef]
- Baell, J.B. Feeling nature’s PAINS: Natural products, natural product drugs, and pan assay interference compounds (PAINS). J. Nat. Prod. 2016, 79, 616–628. [Google Scholar] [CrossRef]
- Capuzzi, S.J.; Muratov, E.N.; Tropsha, A. Phantom PAINS: Problems with the utility of alerts for pan-assay INterference CompoundS. J. Chem. Inf. Model. 2017, 57, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Kenny, P.W. Comment on the ecstasy and agony of assay interference compounds. J. Chem. Inf. Model. 2017, 57, 2640–2645. [Google Scholar] [CrossRef] [PubMed]
- Baell, J.B.; Nissink, J.W. Seven year itch: Pan-assay interference compounds (PAINS) in 2017—Utility and limitations. ACS Chem. Biol. 2018, 13, 36–44. [Google Scholar] [CrossRef] [Green Version]
- Nim, S.; Lobato, L.G.; Moreno, A.; Chaptal, V.; Rawal, M.K.; Falson, P.; Prasad, R. Atomic modelling and systematic mutagenesis identify residues in multiple drug binding sites that are essential for drug resistance in the major candida transporter Cdr1. Biochim. Biophys. Acta 2016, 1858, 2858–2870. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.P.; Rees, S.; Kalindjian, S.B.; Philpott, K.L. Principles of early drug discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, J.H. Accounting artifacts in high-throughput toxicity assays. Methods Mol. Biol. 2016, 1473, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Good, A.C.; Oprea, T.I. Optimization of CAMD techniques 3. Virtual screening enrichment studies: A help or hindrance in tool selection? J. Comput. Aided Mol. Des. 2008, 22, 169–178. [Google Scholar] [CrossRef]
- Bemis, G.W.; Murcko, M.A. The properties of known drugs. 1. Molecular frameworks. J. Med. Chem. 1996, 39, 2887–2893. [Google Scholar] [CrossRef]
- Dassault Systèmes, Biovia Corp. Available online: https://www.3dsbiovia.com/ (accessed on 1 April 2020).
- Schuffenhauer, A.; Ertl, P.; Roggo, S.; Wetzel, S.; Koch, M.A.; Waldmann, H. The scaffold tree, visualization of the scaffold universe by hierarchical scaffold classification. J. Chem. Inf. Model. 2007, 47, 47–58. [Google Scholar] [CrossRef]
- Rogers, D.; Hahn, M. Extended-connectivity fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef]
- Jain, A.N. Surflex-dock 2.1: Robust performance from ligand energetic modeling, ring flexibility, and knowledge-based search. J. Comput. Aided Mol. Des. 2007, 21, 281–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Dalby, A.; Nourse, J.G.; Hounshell, W.D.; Gushurst, A.K.; Grier, D.L.; Leland, B.A.; Laufer, J. Description of several chemical structure file formats used by computer programs developed at molecular design limited. J. Chem. Inf. Comput. Sci. 1992, 32, 244–255. [Google Scholar] [CrossRef]
- Cummings, M.D.; Gibbs, A.C.; DesJarlais, R.L. Processing of small molecule databases for automated docking. Med. Chem. 2007, 3, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Knox, A.J.; Meegan, M.J.; Carta, G.; Lloyd, D.G. Considerations in compound database preparatio—“hidden” impact on virtual screening results. J. Chem. Inf. Model. 2005, 45(6), 1908–1919. [Google Scholar] [CrossRef] [PubMed]
- Kellenberger, E.; Rodrigo, J.; Muller, P.; Rognan, D. Comparative evaluation of eight docking tools for docking and virtual screening accuracy. Proteins 2004, 57, 225–242. [Google Scholar] [CrossRef]
- Perola, E.; Charifson, P.S. Conformational analysis of drug-like molecules bound to proteins: An extensive study of ligand reorganization upon binding. J. Med. Chem. 2004, 47, 2499–2510. [Google Scholar] [CrossRef]
- Marcou, G.; Rognan, D. Optimizing fragment and scaffold docking by use of molecular interaction fingerprints. J. Chem. Inf. Model. 2007, 47, 195–207. [Google Scholar] [CrossRef]
- Desaphy, J.; Raimbaud, E.; Ducrot, P.; Rognan, D. Encoding protein-ligand interaction patterns in fingerprints and graphs. J. Chem. Inf. Model. 2013, 53, 623–637. [Google Scholar] [CrossRef]
- Polgar, T.; Keserue, G.M. Ensemble docking into flexible active sites. critical evaluation of FlexE against JNK-3 and β-secretase. J. Chem. Inf. Model. 2006, 46, 1795–1805. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkins, P.C.; Skillman, A.G.; Nicholls, A. Comparison of shape-matching and docking as virtual screening tools. J. Med. Chem. 2007, 50, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Bietz, S.; Urbaczek, S.; Schulz, B.; Rarey, M. Protoss: A holistic approach to predict tautomers and protonation states in proteinligand complexes. J. Cheminformatics 2014, 6, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molecular Networks Gmbh. Available online: https://www.mn-am.com/ (accessed on 30 April 2020).
- Molecular Operating Environment. Available online: https://www.chemcomp.com/Products.htm (accessed on 1 May 2020).
- Sybyl-X Molecular Modeling Software Packages, Version 2.0; TRIPOS Associates, Inc.: St. Louis, MO, USA, 2012.
- Daylight Chemical Information Systems. Available online: https://www.daylight.com/ (accessed on 1 May 2020).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Sets | Year | Number of Ligand Sets | Number of Molecules Per Ligand Set | Active-to-Inactive Ratio | Assay Data | Assay Artifacts Avoided | Chemical Bias Avoided | Virtual Screening Suitability | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Primary | Confirmatory | Ligand-Based | Structure-Based | |||||||
| MUV [80] | 2009 | 17 | 15,030 | 2 × 10−3 | ✓ | ✓ | ✓ | ✓ | ✓ a | ✓ |
| UCI [81] | 2009 | 21 | 69 to 59,795 | 2 × 10−4 to 0.33 | ✓ | ✓ | ✓ | |||
| Butkiewicz et al. [82] | 2013 | 9 | 61,849 to 344,769 | 5 × 10−4 to 7 × 10−3 | ✓ | ✓ | ||||
| Lindh et al. [83] | 2015 | 7 | 59,462 to 338,003 | 7 × 10−5 to 1 × 10−3 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
| LIT-PCBA [22] | 2020 | 15 | 4247 to 362,088 | 5 × 10−5 to 0.05 | ✓ | ✓ | ✓ b | ✓ | ✓ | |
| Data Sets | 2D ECFP4 Fingerprint Similarity Search | Molecular Docking | ||
|---|---|---|---|---|
| EF1% | Number of Retrieved Actives | EF1% | Number of Retrieved Actives | |
| Full PubChem data | 0.6 | 2 | 3.2 | 11 |
| LIT-PCBA MTORC1 data | 0.0 | 0 | 1.0 | 1 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tran-Nguyen, V.-K.; Rognan, D. Benchmarking Data Sets from PubChem BioAssay Data: Current Scenario and Room for Improvement. Int. J. Mol. Sci. 2020, 21, 4380. https://doi.org/10.3390/ijms21124380
Tran-Nguyen V-K, Rognan D. Benchmarking Data Sets from PubChem BioAssay Data: Current Scenario and Room for Improvement. International Journal of Molecular Sciences. 2020; 21(12):4380. https://doi.org/10.3390/ijms21124380
Chicago/Turabian StyleTran-Nguyen, Viet-Khoa, and Didier Rognan. 2020. "Benchmarking Data Sets from PubChem BioAssay Data: Current Scenario and Room for Improvement" International Journal of Molecular Sciences 21, no. 12: 4380. https://doi.org/10.3390/ijms21124380
APA StyleTran-Nguyen, V.-K., & Rognan, D. (2020). Benchmarking Data Sets from PubChem BioAssay Data: Current Scenario and Room for Improvement. International Journal of Molecular Sciences, 21(12), 4380. https://doi.org/10.3390/ijms21124380