A Peptidic Thymidylate-Synthase Inhibitor Loaded on Pegylated Liposomes Enhances the Antitumour Effect of Chemotherapy Drugs in Human Ovarian Cancer Cells

 , ,
, ,  , and
, and
Abstract
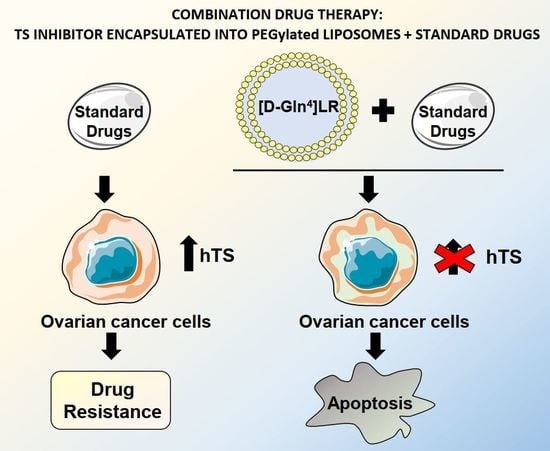
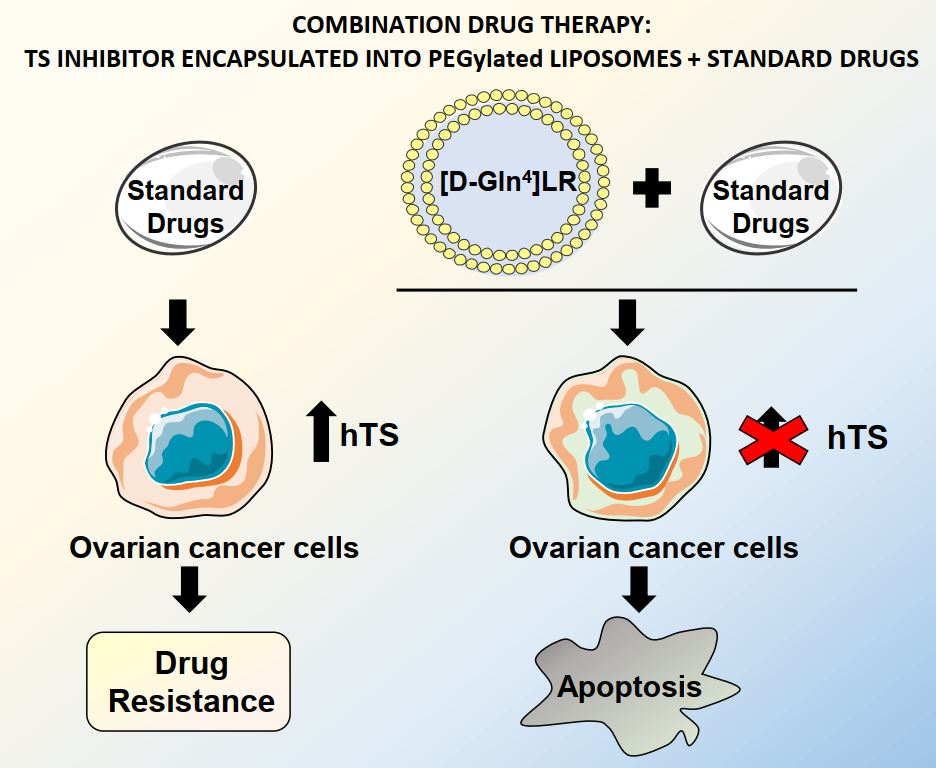
:
1. Introduction
2. Results
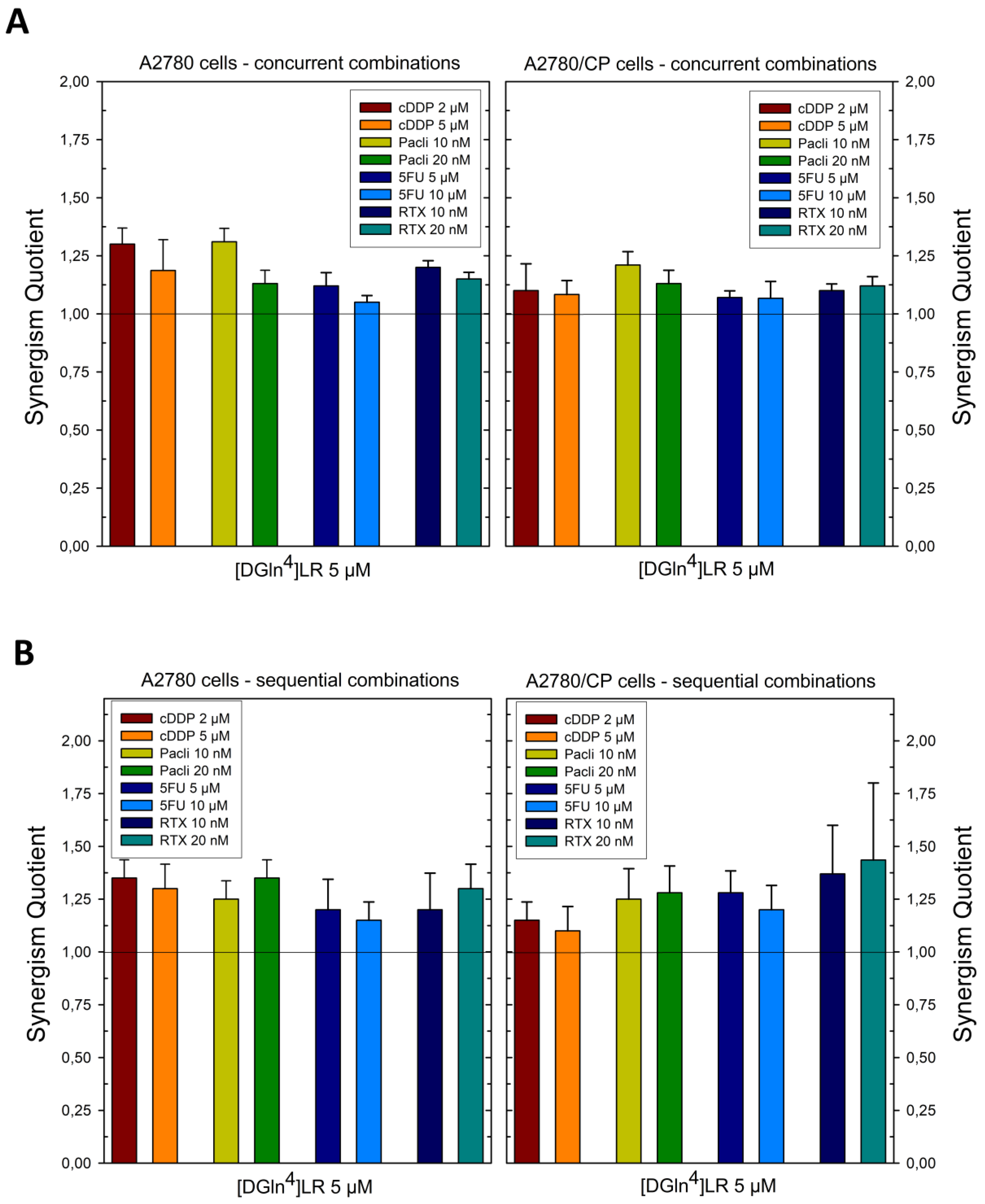
2.1. Sequence-Dependent Synergistic Antiproliferative Effects of [DGln4]LR/Drug Combinations
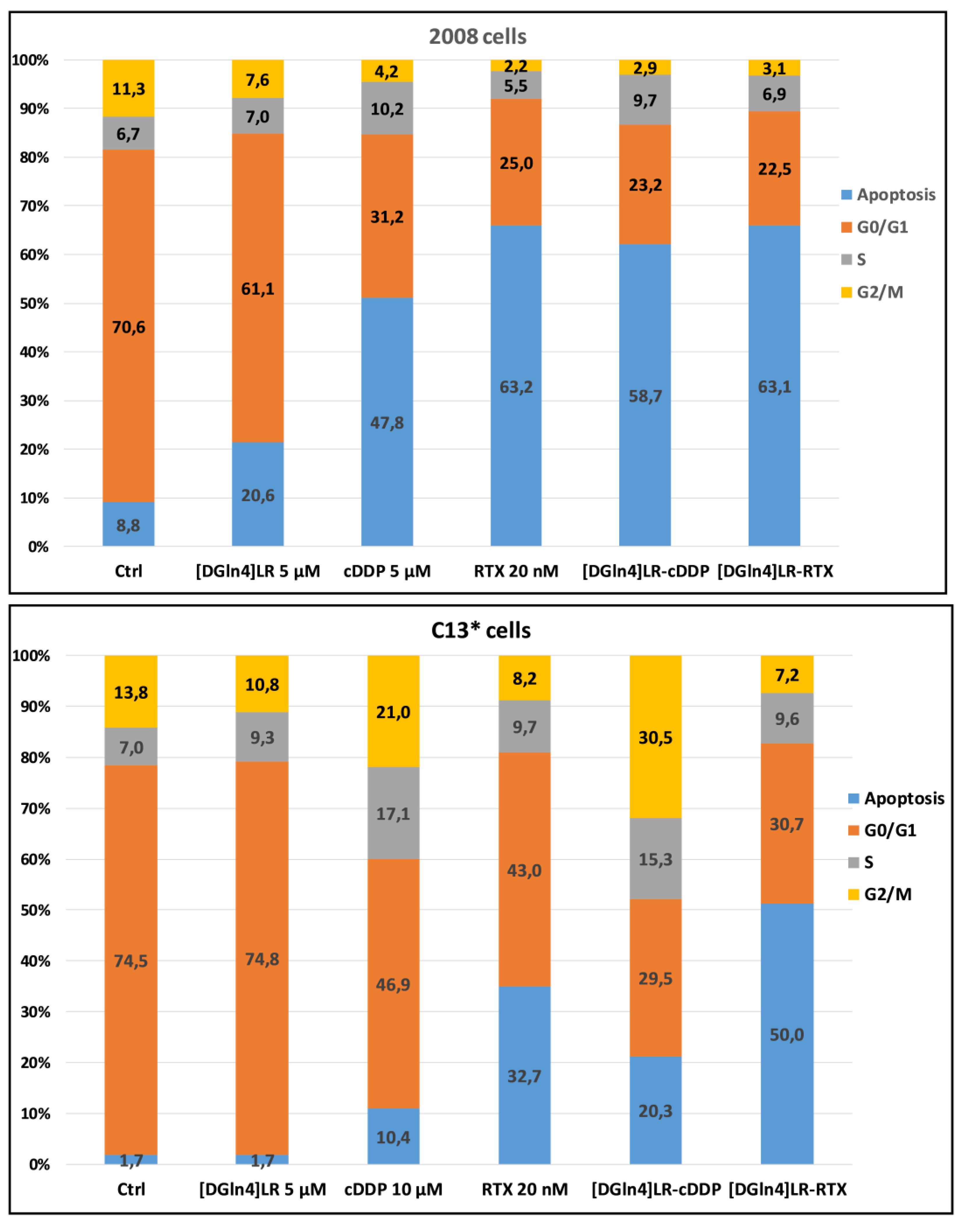
2.2. [DGln4]LR Combination with Chemotherapy Drugs Cause Great Perturbation of Cell Cycle and Promotes Apoptosis
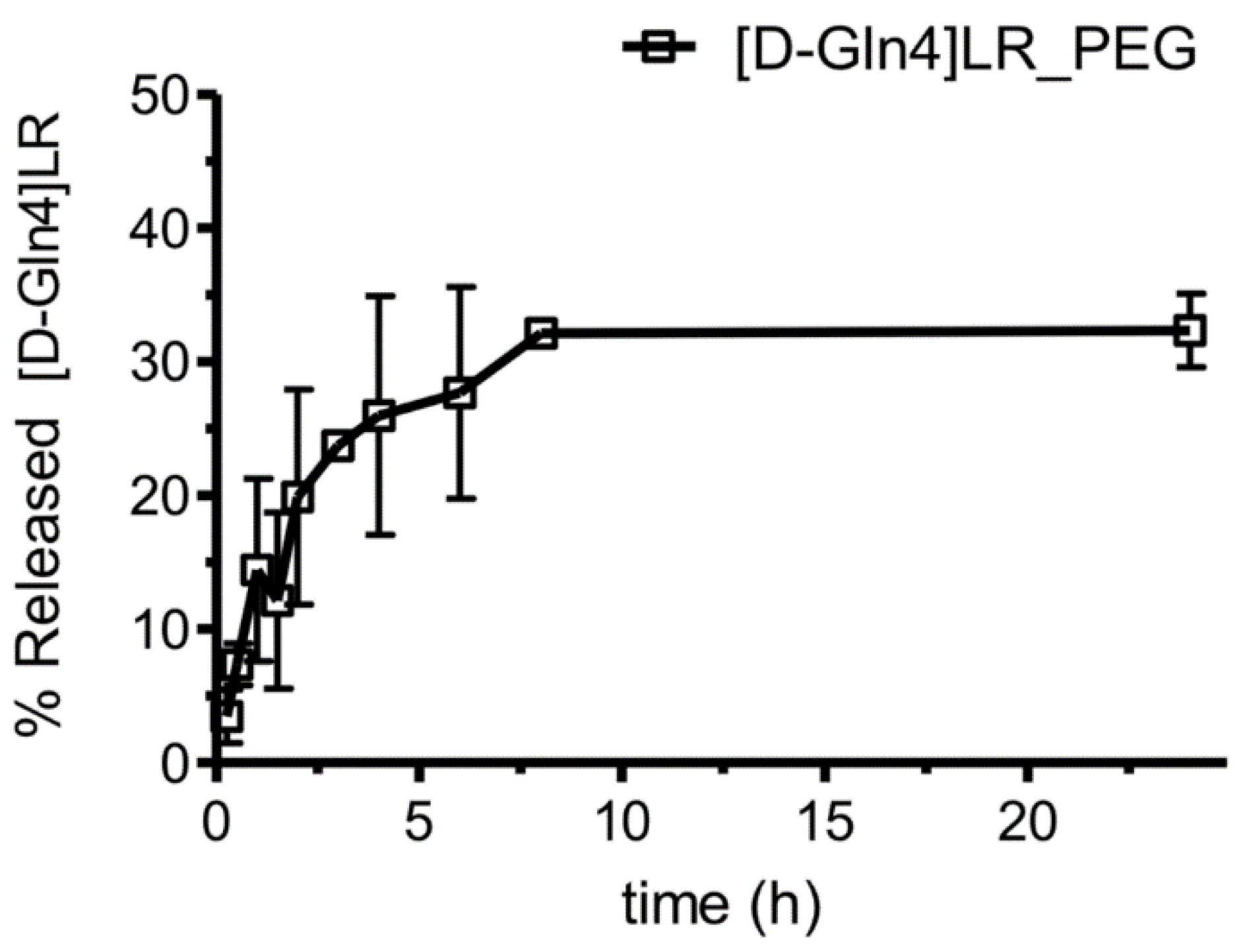
2.3. Liposome Characterization
2.4. Cytotoxicity of Peptide-Loaded Liposomes
2.5. Sequence-Dependent Synergistic Antiproliferative Effect of Peptide-Loaded Liposomes in Combination with RTX or cDDP
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Lines
4.3. Cell Transfection with [D-Gln4]LR Peptide by Means of the Delivery System SAINT-PhD
4.4. Liposome Characterization
4.5. Cytotoxicity of Liposome-Drug Combinations by MTT Assay
4.6. Synergy Analysis
4.7. Flow Cytometric Analysis of Cell Cycle
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bowtell, D.D.; Böhm, S.; Ahmed, A.A.; Aspuria, P.J.; Bast, R.C.; Beral, V.; Berek, J.S.; Birrer, M.J.; Blagden, S.; Bookman, M.A.; et al. Rethinking ovarian cancer II: Reducing mortality from high-grade serous ovarian cancer. Nat. Rev. Cancer 2015, 15, 668–679. [Google Scholar] [CrossRef]
- Lai, G.M.; Ozols, R.F.; Smyth, J.F.; Young, R.C.; Hamilton, T.C. Enhanced DNA repair and resistance to cisplatin in human ovarian cancer. Biochem. Pharmacol. 1988, 37, 4597–4600. [Google Scholar] [CrossRef]
- Scanlon, K.J.; Kashani-Sabet, M. Elevated expression of thymidylate synthase cycle genes in cisplatin-resistant human ovarian carcinoma A2780 cells. Proc. Natl. Acad. Sci. USA 1988, 85, 650–653. [Google Scholar] [CrossRef] [Green Version]
- Costi, M.P.; Ferrari, S. Update on antifolate drugs targets. Curr. Drug Targets 2001, 2, 135–166. [Google Scholar] [CrossRef]
- Boyer, Q.; Li, C.; Lee, J.Y.; Shepard, H.M. A novel approach to thymidylate synthase as a target for cancer chemotherapy. Mol. Pharmacol. 2001, 59, 446–452. [Google Scholar]
- Ackland, S.P.; Clarke, S.J.; Beale, P.; Peters, G.J. Thymidylate synthase inhibitors. Cancer Chemother. Biol. Response Modif. 2002, 20, 1–36. [Google Scholar]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef]
- Jackman, A.L.; Taylor, G.A.; Gibson, W.; Kimbell, R.; Brown, M.; Calvert, A.H.; Judson, I.R.; Hughes, L.R. ICI D1694, a quinazoline antifolate thymidylate synthase inhibitor that is a potent inhibitor of L1210 tumor cell growth in vitro and in vivo: A new agent for clinical study. Cancer Res. 1991, 51, 5579–5586. [Google Scholar]
- Cocconi, G.; Cunningham, D.; Van Cutsem, E.; Francois, E.; Gustavsson, B.; Van Hazel, G.; Kerr, D.; Possinger, K.; Hietschold, S.M. Open, randomized, multicenter trial of raltitrexed versus fluorouracil plus high-dose leucovorin in patients with advanced colorectal cancer. Tomudex Colorectal Cancer Study Group. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1998, 16, 2943–2952. [Google Scholar] [CrossRef] [PubMed]
- Van Triest, B.; Pinedo, H.M.; Van Hensbergen, Y.; Smid, K.; Telleman, F.; Schoenmakers, P.S.; Van der Wilt, C.L.; Van Laar, J.A.; Noordhuis, P.; Jansen, G.; et al. Thymidylate synthase level as the main predictive parameter for sensitivity to 5-fluorouracil, but not for folate-based thymidylate synthase inhibitors, in 13 nonselected colon cancer cell lines. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 1999, 5, 643–654. [Google Scholar]
- Farrugia, D.C.; Ford, H.E.R.; Cunningham, D.; Danenberg, K.D.; Danenberg, P.V.; Brabender, J.; McVicar, A.D.; Aherne, G.W.; Hardcastle, A.; McCarthy, K.; et al. Thymidylate synthase expression in advanced colorectal cancer predicts for response to raltitrexed. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2003, 9, 792–801. [Google Scholar]
- Welsh, S.J.; Titley, J.; Brunton, L.; Valenti, M.; Monaghan, P.; Jackman, A.L.; Aherne, G.W. Comparison of thymidylate synthase (TS) protein up-regulation after exposure to TS inhibitors in normal and tumor cell lines and tissues. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2000, 6, 2538–2546. [Google Scholar]
- Peters, G.J.; Backus, H.H.J.; Freemantle, S.; Van Triest, B.; Codacci-Pisanelli, G.; Van der Wilt, C.L.; Smid, K.; Lunec, J.; Calvert, A.H.; Marsh, S.; et al. Induction of thymidylate synthase as a 5-fluorouracil resistance mechanism. Biochim. Biophys. Acta 2002, 1587, 194–205. [Google Scholar] [CrossRef] [Green Version]
- Rahman, L.; Voeller, D.; Rahman, M.; Lipkowitz, S.; Allegra, C.; Barrett, J.C.; Kaye, F.J.; Zajac-Kaye, M. Thymidylate synthase as an oncogene: A novel role for an essential DNA synthesis enzyme. Cancer Cell 2004, 5, 341–351. [Google Scholar] [CrossRef] [Green Version]
- Chu, E.; Takechi, T.; Jones, K.L.; Voeller, D.M.; Copur, S.M.; Maley, G.F.; Maley, F.; Segal, S.; Allegra, C.J. Thymidylate synthase binds to c-myc RNA in human colon cancer cells and in vitro. Mol. Cell. Biol. 1995, 15, 179–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, E.; Copur, S.M.; Ju, J.; Chen, T.M.; Khleif, S.; Voeller, D.M.; Mizunuma, N.; Patel, M.; Maley, G.F.; Maley, F.; et al. Thymidylate synthase protein and p53 mRNA form an in vivo ribonucleoprotein complex. Mol. Cell. Biol. 1999, 19, 1582–1594. [Google Scholar] [CrossRef] [Green Version]
- Benhattar, J.; Cerottini, J.P.; Saraga, E.; Metthez, G.; Givel, J.C. p53 mutations as a possible predictor of response to chemotherapy in metastatic colorectal carcinomas. Int. J. Cancer 1996, 69, 190–192. [Google Scholar] [CrossRef]
- Giovannetti, E.; Backus, H.H.J.; Wouters, D.; Ferreira, C.G.; Van Houten, V.M.M.; Brakenhoff, R.H.; Poupon, M.F.; Azzarello, A.; Pinedo, H.M.; Peters, G.J. Changes in the status of p53 affect drug sensitivity to thymidylate synthase (TS) inhibitors by altering TS levels. Br. J. Cancer 2007, 96, 769–775. [Google Scholar] [CrossRef] [Green Version]
- Cardinale, D.; Guaitoli, G.; Tondi, D.; Luciani, R.; Henrich, S.; Salo-Ahen, O.M.H.; Ferrari, S.; Marverti, G.; Guerrieri, D.; Ligabue, A.; et al. Protein-protein interface-binding peptides inhibit the cancer therapy target human thymidylate synthase. Proc. Natl. Acad. Sci. USA 2011, 108, E542–E549. [Google Scholar] [CrossRef] [Green Version]
- Ponterini, G.; Martello, A.; Pavesi, G.; Lauriola, A.; Luciani, R.; Santucci, M.; Pelà, M.; Gozzi, G.; Pacifico, S.; Guerrini, R.; et al. Intracellular quantitative detection of human thymidylate synthase engagement with an unconventional inhibitor using tetracysteine-diarsenical-probe technology. Sci. Rep. 2016, 6, 27198. [Google Scholar] [CrossRef] [Green Version]
- Pelà, M.; Saxena, P.; Luciani, R.; Santucci, M.; Ferrari, S.; Marverti, G.; Marraccini, C.; Martello, A.; Pirondi, S.; Genovese, F.; et al. Optimization of peptides that target human thymidylate synthase to inhibit ovarian cancer cell growth. J. Med. Chem. 2014, 57, 1355–1367. [Google Scholar] [CrossRef]
- Genovese, F.; Gualandi, A.; Taddia, L.; Marverti, G.; Pirondi, S.; Marraccini, C.; Perco, P.; Pelà, M.; Guerrini, R.; Amoroso, M.R.; et al. Mass spectrometric/bioinformatic identification of a protein subset that characterizes the cellular activity of anticancer peptides. J. Proteome Res. 2014, 13, 5250–5261. [Google Scholar] [CrossRef]
- Miller, D.S.; Blessing, J.A.; Krasner, C.N.; Mannel, R.S.; Hanjani, P.; Pearl, M.L.; Waggoner, S.E.; Boardman, C.H. Phase II evaluation of pemetrexed in the treatment of recurrent or persistent platinum-resistant ovarian or primary peritoneal carcinoma: A study of the Gynecologic Oncology Group. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 2686–2691. [Google Scholar] [CrossRef] [Green Version]
- Sacchetti, F.; Marraccini, C.; D’Arca, D.; Pelà, M.; Pinetti, D.; Maretti, E.; Hanuskova, M.; Iannuccelli, V.; Costi, M.P.; Leo, E. Enhanced anti-hyperproliferative activity of human thymidylate synthase inhibitor peptide by solid lipid nanoparticle delivery. Colloids Surf. B Biointerfaces 2015, 136, 346–354. [Google Scholar] [CrossRef]
- Hama, S.; Itakura, S.; Nakai, M.; Nakayama, K.; Morimoto, S.; Suzuki, S.; Kogure, K. Overcoming the polyethylene glycol dilemma via pathological environment-sensitive change of the surface property of nanoparticles for cellular entry. J. Control. Release Off. J. Control. Release Soc. 2015, 206, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Sacchetti, F.; Marverti, G.; D’Arca, D.; Severi, L.; Maretti, E.; Iannuccelli, V.; Pacifico, S.; Ponterini, G.; Costi, M.P.; Leo, E. pH-promoted release of a novel anti-tumour peptide by “stealth” liposomes: Effect of nanocarriers on the drug activity in cis-platinum resistant cancer cells. Pharm. Res. 2018, 35, 206. [Google Scholar] [CrossRef]
- Righetti, S.C.; Perego, P.; Corna, E.; Pierotti, M.A.; Zunino, F. Emergence of p53 mutant cisplatin-resistant ovarian carcinoma cells following drug exposure: Spontaneously mutant selection. Cell Growth Differ. Mol. Biol. J. Am. Assoc. Cancer Res. 1999, 10, 473–478. [Google Scholar]
- Marverti, G.; Ligabue, A.; Paglietti, G.; Corona, P.; Piras, S.; Vitale, G.; Guerrieri, D.; Luciani, R.; Costi, M.P.; Frassineti, C.; et al. Collateral sensitivity to novel thymidylate synthase inhibitors correlates with folate cycle enzymes impairment in cisplatin-resistant human ovarian cancer cells. Eur. J. Pharmacol. 2009, 615, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Chu, E.; Koeller, D.M.; Johnston, P.G.; Zinn, S.; Allegra, C.J. Regulation of thymidylate synthase in human colon cancer cells treated with 5-fluorouracil and interferon-gamma. Mol. Pharmacol. 1993, 43, 527–533. [Google Scholar]
- Severi, L.; Losi, L.; Fonda, S.; Taddia, L.; Gozzi, G.; Marverti, G.; Magni, F.; Chinello, C.; Stella, M.; Sheouli, J.; et al. Proteomic and Bioinformatic Studies for the Characterization of Response to Pemetrexed in Platinum Drug Resistant Ovarian Cancer. Front. Pharmacol. 2018, 9, 454. [Google Scholar] [CrossRef] [Green Version]
- Carosati, E.; Tochowicz, A.; Marverti, G.; Guaitoli, G.; Benedetti, P.; Ferrari, S.; Stroud, R.M.; Finer-Moore, J.; Luciani, R.; Farina, D.; et al. Inhibitor of Ovarian Cancer Cells by Virtual Screening: A New Hydroxy-Thiazole Derivative Targeting Human Thymidylate Synthase. J. Med. Chem. 2012, 55, 10272–10276, (Brief Article). [Google Scholar] [CrossRef] [PubMed]
- Taddia, L.; D’Arca, D.; Ferrari, S.; Marraccini, C.; Severi, L.; Ponterini, G.; Assaraf, Y.G.; Marverti, G.; Costi, M.P. Inside the biochemical pathways of thymidylate synthase perturbed by anticancer drugs: Novel strategies to overcome cancer chemoresistance. Drug Resist. Updates 2015, 23, 20–54. [Google Scholar] [CrossRef] [PubMed]
- Wilson, P.M.; LaBonte, M.J.; Lenz, H.J.; Mack, P.C.; Ladner, R.D. Inhibition of dUTPase induces synthetic lethality with thymidylate synthase-targeted therapies in non-small cell lung cancer. Mol. Cancer Ther. 2012, 11, 616–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackman, A.L.; Kelland, L.R.; Kimbell, R.; Brown, M.; Gibson, W.; Aherne, G.W.; Hardcastle, A.; Boyle, F.T. Mechanisms of acquired resistance to the quinazoline thymidylate synthase inhibitor ZD1694 (Tomudex) in one mouse and three human cell lines. Br. J. Cancer 1995, 71, 914–924. [Google Scholar] [CrossRef]
- Scanlon, K.J.; Newman, E.M.; Lu, Y.; Priest, D.G. Biochemical basis for cisplatin and 5-fluorouracil synergism in human ovarian carcinoma cells. Proc. Natl. Acad. Sci. USA 1986, 83, 8923–8925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacave, A.J.; Barón, F.J.; Antón, L.M.; Estrada, E.; De Sande, L.M.; Palacio, I.; Esteban, E.; Gracia, J.M.; Buesa, J.M.; Fernández, O.A. Combination chemotherapy with cisplatin and 5-fluorouracil 5-day infusion in the therapy of advanced gastric cancer: A phase II trial. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 1991, 2, 751–754. [Google Scholar] [CrossRef]
- Ahlgren, J.D.; Trocki, O.; Gullo, J.J.; Goldberg, R.; Muir, W.A.; Sisk, R.; Schacter, L. Protracted infusion of 5-FU with weekly low-dose cisplatin as second-line therapy in patients with metastatic colorectal cancer who have failed 5-FU monotherapy. Cancer Investig. 1991, 9, 27–33. [Google Scholar] [CrossRef]
- Chung, Y.S.; Yamashita, Y.; Inoue, T.; Matsuoka, T.; Nakata, B.; Onoda, N.; Maeda, K.; Sawada, T.; Kato, Y.; Shirasaka, T.; et al. Continuous infusion of 5-fluorouracil and low dose cisplatin infusion for the treatment of advanced and recurrent gastric adenocarcinoma. Cancer 1997, 80, 1–7. [Google Scholar] [CrossRef]
- Shirasaka, T.; Shimamoto, Y.; Ohshimo, H.; Saito, H.; Fukushima, M. Metabolic basis of the synergistic antitumor activities of 5-fluorouracil and cisplatin in rodent tumor models in vivo. Cancer Chemother. Pharmacol. 1993, 32, 167–172. [Google Scholar] [CrossRef]
- Nishiyama, M.; Yamamoto, W.; Park, J.S.; Okamoto, R.; Hanaoka, H.; Takano, H.; Saito, N.; Matsukawa, M.; Shirasaka, T.; Kurihara, M. Low-dose cisplatin and 5-fluorouracil in combination can repress increased gene expression of cellular resistance determinants to themselves. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 1999, 5, 2620–2628. [Google Scholar]
- Yeh, K.H.; Cheng, A.L.; Wan, J.P.; Lin, C.S.; Liu, C.C. Down-regulation of thymidylate synthase expression and its steady-state mRNA by oxaliplatin in colon cancer cells. Anticancer Drugs 2004, 15, 371–376. [Google Scholar] [CrossRef] [PubMed]
- Radivoyevitch, T. Folate system correlations in DNA microarray data. BMC Cancer 2005, 5, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.C.; Li, F.; Handler, J.; Huang, C.R.L.; Xiang, Y.; Neretti, N.; Sedivy, J.M.; Zeller, K.I.; Dang, C.V. Global regulation of nucleotide biosynthetic genes by c-Myc. PLoS ONE 2008, 3, e2722. [Google Scholar] [CrossRef] [PubMed]
- Biroccio, A.; Benassi, B.; Amodei, S.; Gabellini, C.; Del Bufalo, D.; Zupi, G. c-Myc down-regulation increases susceptibility to cisplatin through reactive oxygen species-mediated apoptosis in M14 human melanoma cells. Mol. Pharmacol. 2001, 60, 174–182. [Google Scholar] [CrossRef]
- Kugimiya, N.; Nishimoto, A.; Hosoyama, T.; Ueno, K.; Enoki, T.; Li, T.S.; Hamano, K. The c-MYC-ABCB5 axis plays a pivotal role in 5-fluorouracil resistance in human colon cancer cells. J. Cell. Mol. Med. 2015, 19, 1569–1581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longo, G.S.; Izzo, J.; Chang, Y.M.; Tong, W.P.; Zielinski, Z.; Gorlick, R.; Chou, T.C.; Bertino, J.R. Pretreatment of colon carcinoma cells with Tomudex enhances 5-fluorouracil cytotoxicity. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 1998, 4, 469–473. [Google Scholar]
- Kano, Y.; Akutsu, M.; Tsunoda, S.; Suzuki, K.; Yazawa, Y.; Furukawa, Y. Schedule-dependent synergism and antagonism between raltitrexed (“Tomudex”) and methotrexate in human colon cancer cell lines in vitro. Jpn. J. Cancer Res. GANN 2001, 92, 74–82. [Google Scholar] [CrossRef]
- Kano, Y.; Akutsu, M.; Suzuki, K.; Yazawa, Y.; Tsunoda, S. Schedule-dependent interactions between raltitrexed and cisplatin in human carcinoma cell lines in vitro. Jpn. J. Cancer Res. GANN 2000, 91, 424–432. [Google Scholar] [CrossRef]
- Wilson, P.M.; Danenberg, P.V.; Johnston, P.G.; Lenz, H.J.; Ladner, R.D. Standing the test of time: Targeting thymidylate biosynthesis in cancer therapy. Nat. Rev. Clin. Oncol. 2014, 1, 282–298. [Google Scholar] [CrossRef]
- Altieri, D.C.; Stein, G.S.; Lian, J.B.; Languino, L.R. TRAP-1, the mitochondrial Hsp90. Biochim. Biophys. Acta 2012, 1823, 767–773. [Google Scholar] [CrossRef] [Green Version]
- Banerji, U. Heat shock protein 90 as a drug target: Some like it hot. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 9–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, S.H.; Liu, Y.W.; Zhang, L.; Liu, B.; Li, L.; Shi, J.Z.; Li, L. Regulation of survival and chemoresistance by HSP90AA1 in ovarian cancer SKOV3 cells. Mol. Biol. Rep. 2013, 40, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Choi, I.J.; Kim, C.G.; Kim, H.S.; Oshima, A.; Michalowski, A.; Green, J.E. A gene expression signature of acquired chemoresistance to cisplatin and fluorouracil combination chemotherapy in gastric cancer patients. PLoS ONE 2011, 6, e16694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macleod, K.; Mullen, P.; Sewell, J.; Rabiasz, G.; Lawrie, S.; Miller, E.; Smyth, J.F.; Langdon, S.P. Altered ErbB receptor signaling and gene expression in cisplatin-resistant ovarian cancer. Cancer Res. 2005, 65, 6789–6800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- French, J.B.; Zhao, H.; An, S.; Niessen, S.; Deng, Y.; Cravatt, B.F.; Benkovic, S.J. Hsp70/Hsp90 chaperone machinery is involved in the assembly of the purinosome. Proc. Natl. Acad. Sci. USA 2013, 110, 2528–2533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, S.; Chen, Y.X.; Qin, S.K.; Yang, A.Z.; Wang, L.; Xu, H.J.; Geng, H.Y. Raltitrexed induces mitochondrial-mediated apoptosis in SGC7901 human gastric cancer cells. Mol. Med. Rep. 2014, 10, 1927–1934. [Google Scholar] [CrossRef] [Green Version]
- Kang, G.H.; Kim, G.S.; Lee, H.R.; Yuh, Y.J.; Kim, S.R. A phase II trial of paclitaxel, 5-fluorouracil (5-FU) and cisplatin in patients with metastatic or recurrent gastric cancer. Cancer Res. Treat. Off. J. Korean Cancer Assoc. 2008, 40, 106–110. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Wang, Z.; Zhou, N.; An, X.; Xu, R.; He, Y.; Li, Y. Phase II study of biweekly paclitaxel plus infusional 5-fluorouracil and leucovorin as first-line chemotherapy in patients with advanced gastric cancer. Am. J. Clin. Oncol. 2011, 34, 401–405. [Google Scholar] [CrossRef]
- Cho, B.C.; Kim, J.H.; Kim, C.B.; Sohn, J.H.; Choi, H.J.; Lee, Y.C.; Ahn, J.B. Paclitaxel and leucovorin-modulated infusional 5-fluorouracil combination chemotherapy for metastatic gastric cancer. Oncol. Rep. 2006, 15, 621–627. [Google Scholar] [CrossRef] [Green Version]
- Andrews, P.A.; Murphy, M.P.; Howell, S.B. Differential potentiation of alkylating and platinating agent cytotoxicity in human ovarian carcinoma cells by glutathione depletion. Cancer Res. 1985, 45, 6250–6253. [Google Scholar]
- Korch, C.; Spillman, M.A.; Jackson, T.A.; Jacobsen, B.M.; Murphy, S.K.; Lessey, B.A.; Craig, J.V.; Bradford, A.P. DNA profiling analysis of endometrial and ovarian cell lines reveals misidentification, redundancy and contamination. Gynecol. Oncol. 2012, 127, 241–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, P.A.; Jones, J.A. Characterization of binding proteins from ovarian carcinoma and kidney tubule cells that are specific for cisplatin modified DNA. Cancer Commun. 1991, 3, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Bénard, J.; Da Silva, J.; De Blois, M.C.; Boyer, P.; Duvillard, P.; Chiric, E.; Riou, G. Characterization of a human ovarian adenocarcinoma line, IGROV1, in tissue culture and in nude mice. Cancer Res. 1985, 45, 4970–4979. [Google Scholar] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Marverti, G.; Ligabue, A.; Guerrieri, D.; Paglietti, G.; Piras, S.; Costi, M.P.; Farina, D.; Frassineti, C.; Monti, M.G.; Moruzzi, M.S. Spermidine/spermine N1-acetyltranferase modulation by novel folate cycle inhibitors in cisplatin-sensitive and -resistant human ovarian cancer cell lines. Gynecol. Oncol. 2010, 117, 202–210. [Google Scholar] [CrossRef]
- Cho, Y.S.; Cho-Chung, Y.S. Antisense protein kinase A RIalpha acts synergistically with hydroxycamptothecin to inhibit growth and induce apoptosis in human cancer cells: Molecular basis for combinatorial therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2003, 9, 1171–1178. [Google Scholar]
- Software R. Available online: https://cran.r-project.org/ (accessed on 6 March 2017).
- Bioconductor. Available online: https://www.bioconductor.org/ (accessed on 31 October 2017).
- Dolbeare, F.; Gratzner, H.; Pallavicini, M.G.; Gray, J.W. Flow cytometric measurement of total DNA content and incorporated bromodeoxyuridine. Proc. Natl. Acad. Sci. USA 1983, 80, 5573–5577. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Z-Average (nm) | PDI | Z-Potential (mV) | DL (µg/mg) | EE (%) |
|---|---|---|---|---|---|
| DOPE:CHEMS:DSPE_PEG (PpHL) | 198 ± 30 | 0.279 ± 0.077 | −14.54 ± 2.41 | - | - |
| [D-Gln4]LR_ DOPE:CHEMS:DSPE_PEG ([D-Gln4]LR-PpHL) | 209 ± 19 | 0.289 ± 0.072 | −14.28 ± 2.19 | 21.03 ± 1.66 | 42 ± 3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marverti, G.; Gozzi, G.; Maretti, E.; Lauriola, A.; Severi, L.; Sacchetti, F.; Losi, L.; Pacifico, S.; Ferrari, S.; Ponterini, G.; et al. A Peptidic Thymidylate-Synthase Inhibitor Loaded on Pegylated Liposomes Enhances the Antitumour Effect of Chemotherapy Drugs in Human Ovarian Cancer Cells. Int. J. Mol. Sci. 2020, 21, 4452. https://doi.org/10.3390/ijms21124452
Marverti G, Gozzi G, Maretti E, Lauriola A, Severi L, Sacchetti F, Losi L, Pacifico S, Ferrari S, Ponterini G, et al. A Peptidic Thymidylate-Synthase Inhibitor Loaded on Pegylated Liposomes Enhances the Antitumour Effect of Chemotherapy Drugs in Human Ovarian Cancer Cells. International Journal of Molecular Sciences. 2020; 21(12):4452. https://doi.org/10.3390/ijms21124452
Chicago/Turabian StyleMarverti, Gaetano, Gaia Gozzi, Eleonora Maretti, Angela Lauriola, Leda Severi, Francesca Sacchetti, Lorena Losi, Salvatore Pacifico, Stefania Ferrari, Glauco Ponterini, and et al. 2020. "A Peptidic Thymidylate-Synthase Inhibitor Loaded on Pegylated Liposomes Enhances the Antitumour Effect of Chemotherapy Drugs in Human Ovarian Cancer Cells" International Journal of Molecular Sciences 21, no. 12: 4452. https://doi.org/10.3390/ijms21124452
APA StyleMarverti, G., Gozzi, G., Maretti, E., Lauriola, A., Severi, L., Sacchetti, F., Losi, L., Pacifico, S., Ferrari, S., Ponterini, G., Leo, E., Costi, M. P., & D’Arca, D. (2020). A Peptidic Thymidylate-Synthase Inhibitor Loaded on Pegylated Liposomes Enhances the Antitumour Effect of Chemotherapy Drugs in Human Ovarian Cancer Cells. International Journal of Molecular Sciences, 21(12), 4452. https://doi.org/10.3390/ijms21124452