Single Exon Skipping Can Address a Multi-Exon Duplication in the Dystrophin Gene

 and
and
Abstract
:1. Introduction
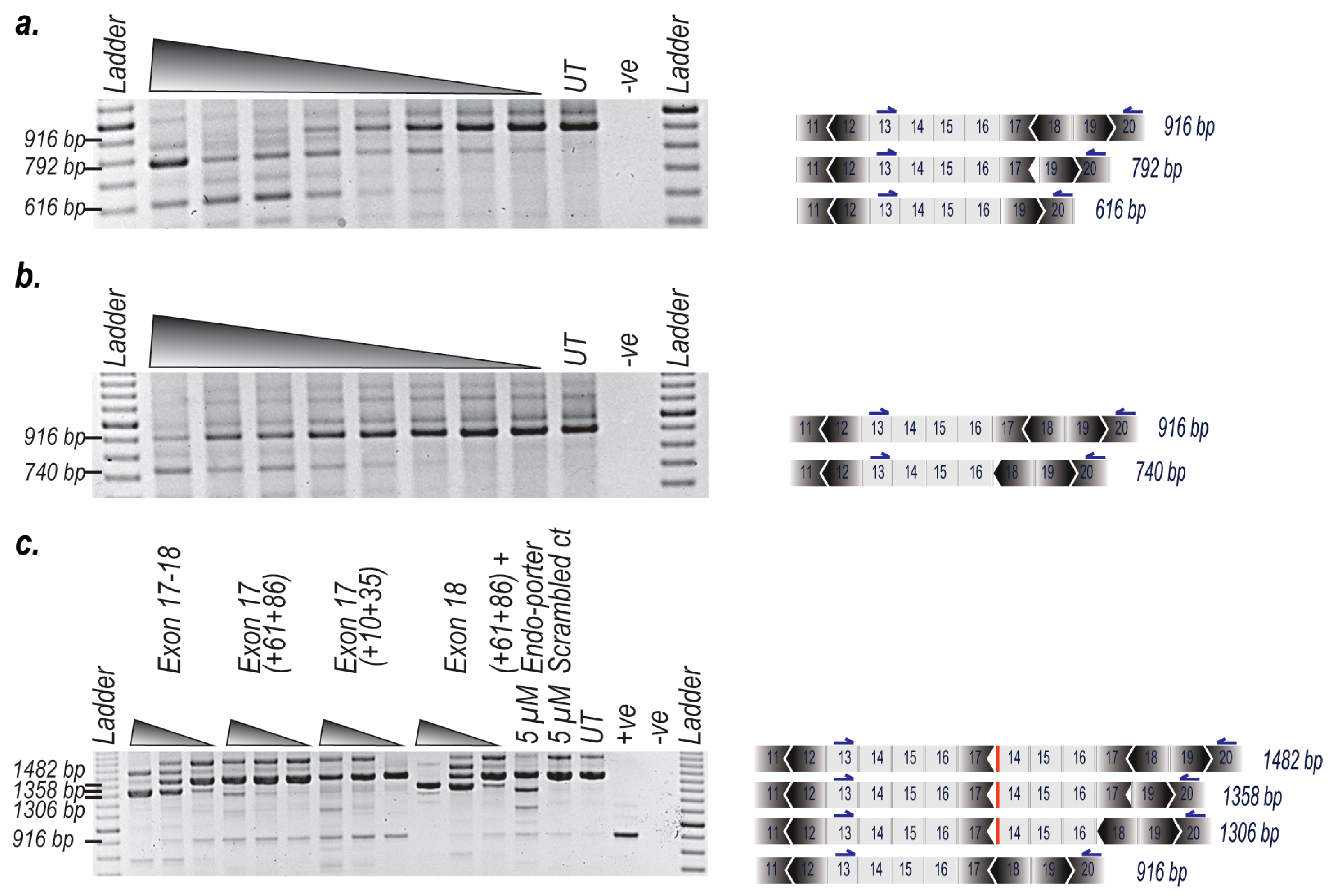
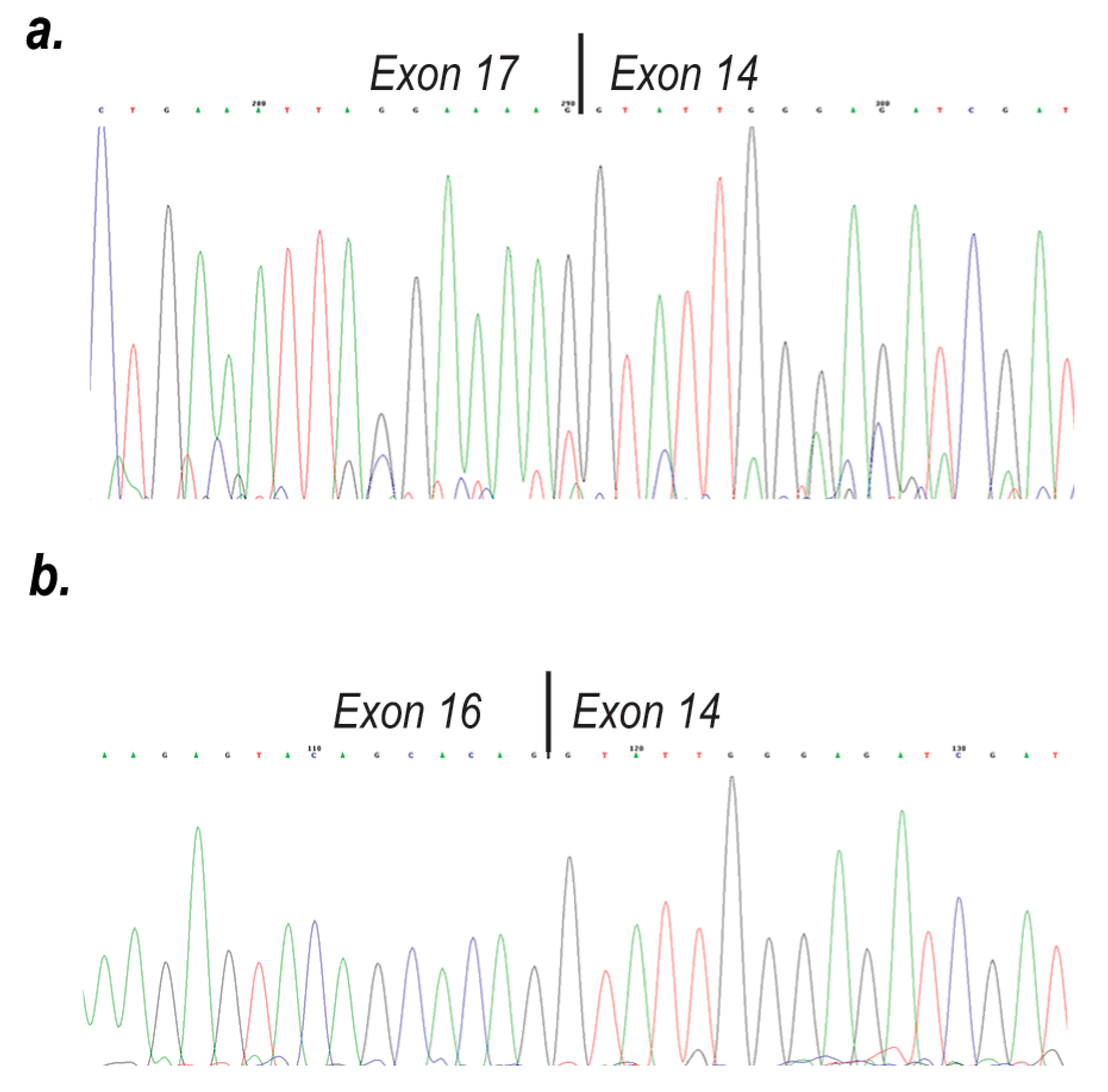
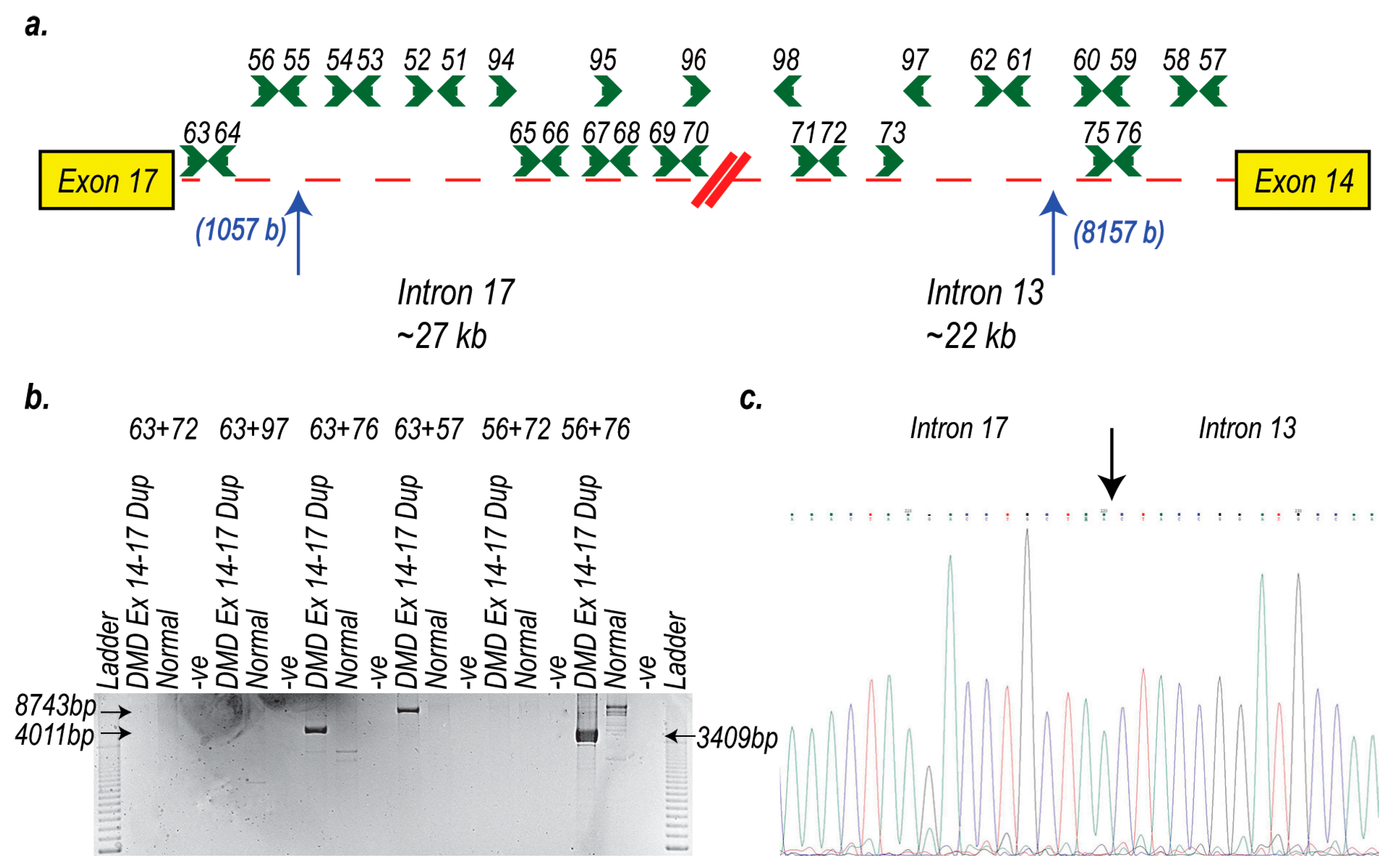
2. Results
3. Discussion
4. Materials and Methods
4.1. AO Design and Synthesis
4.2. Cell Propagation
4.3. Transfection
4.4. RNA Extraction and RT-PCR Assays
4.5. Gel Analysis and Imaging
4.6. Western Blotting
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DMD | Duchenne muscular dystrophy |
| BMD | Becker muscular dystrophy |
| AO | Antisense oligomer |
| PMO | Phosphorodiamidate morpholino oligomer |
| PPMO | Peptide conjugated phosphorodiamidate morpholino oligomer |
References
- Mendell, J.R.; Shilling, C.; Leslie, N.D.; Flanigan, K.M.; al-Dahhak, R.; Gastier-Foster, J.; Kneile, K.; Dunn, D.M.; Duval, B.; Aoyagi, A.; et al. Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann. Neurol. 2012, 71, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, E.P.; Brown, R.H., Jr.; Kunkel, L.M. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef]
- Hoffman, E.P.; Fischbeck, K.H.; Brown, R.H.; Johnson, M.; Medori, R.; Loike, J.D.; Harris, J.B.; Waterston, R.; Brooke, M.; Specht, L.; et al. Characterization of dystrophin in muscle-biopsy specimens from patients with Duchenne‘s or Becker‘s muscular dystrophy. N. Engl. J. Med. 1988, 318, 1363–1368. [Google Scholar] [CrossRef] [PubMed]
- Moser, H. Duchenne muscular dystrophy: Pathogenetic aspects and genetic prevention. Hum. Genet. 1984, 66, 17–40. [Google Scholar] [CrossRef] [PubMed]
- Koenig, M.; Beggs, A.H.; Moyer, M.; Scherpf, S.; Heindrich, K.; Bettecken, T.; Meng, G.; Muller, C.R.; Lindlof, M.; Kaariainen, H.; et al. The molecular basis for Duchenne versus Becker muscular dystrophy: Correlation of severity with type of deletion. Am. J. Hum. Genet. 1989, 45, 498–506. [Google Scholar] [PubMed]
- Koenig, M.; Monaco, A.P.; Kunkel, L.M. The complete sequence of dystrophin predicts a rod-shaped cytoskeletal protein. Cell 1988, 53, 219–228. [Google Scholar] [CrossRef]
- Monaco, A.P.; Bertelson, C.J.; Liechti-Gallati, S.; Moser, H.; Kunkel, L.M. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics 1988, 2, 90–95. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Janson, A.A.; van Ommen, G.J.; van Deutekom, J.C. Antisense-induced exon skipping for duplications in Duchenne muscular dystrophy. BMC Med. Genet. 2007, 8, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Mann, C.J.; Honeyman, K.; McClorey, G.; Fletcher, S.; Wilton, S.D. Improved antisense oligonucleotide induced exon skipping in the mdx mouse model of muscular dystrophy. J. Gene Med. 2002, 4, 644–654. [Google Scholar] [CrossRef]
- Mitrpant, C.; Fletcher, S.; Iversen, P.L.; Wilton, S.D. By-passing the nonsense mutation in the 4 CV mouse model of muscular dystrophy by induced exon skipping. J. Gene Med. 2009, 11, 46–56. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; De Winter, C.L.; Janson, A.A.; Kaman, W.E.; Van Ommen, G.J.; Den Dunnen, J.T.; Van Deutekom, J.C. Functional analysis of 114 exon-internal AONs for targeted DMD exon skipping: Indication for steric hindrance of SR protein binding sites. Oligonucleotides 2005, 15, 284–297. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Janson, A.A.; Kaman, W.E.; Bremmer-Bout, M.; den Dunnen, J.T.; Baas, F.; van Ommen, G.J.; van Deutekom, J.C. Therapeutic antisense-induced exon skipping in cultured muscle cells from six different DMD patients. Hum. Mol. Genet. 2003, 12, 907–914. [Google Scholar] [CrossRef] [PubMed]
- Greer, K.L.; Lochmuller, H.; Flanigan, K.; Fletcher, S.; Wilton, S.D. Targeted exon skipping to correct exon duplications in the dystrophin gene. Mol. Nucleic Acids 2014, 3, e155. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Van Deutekom, J.C.; Fokkema, I.F.; Van Ommen, G.J.; Den Dunnen, J.T. Entries in the Leiden Duchenne muscular dystrophy mutation database: An overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve 2006, 34, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Forrest, S.; Meloni, P.L.; Muntoni, F.; Kim, J.; Fletcher, S.; Wilton, S.D. Personalized exon skipping strategies to address clustered non-deletion dystrophin mutations. Neuromuscul Disord 2010, 20, 810–816. [Google Scholar] [CrossRef] [Green Version]
- Wilton, S.D.; Fall, A.M.; Harding, P.L.; McClorey, G.; Coleman, C.; Fletcher, S. Antisense oligonucleotide-induced exon skipping across the human dystrophin gene transcript. Mol. Ther. 2007, 15, 1288–1296. [Google Scholar] [CrossRef]
- Moulton, H.M.; Moulton, J.D. Morpholinos and their peptide conjugates: Therapeutic promise and challenge for Duchenne muscular dystrophy. Biochim. Biophys. Acta 2010, 1798, 2296–2303. [Google Scholar] [CrossRef] [Green Version]
- Amantana, A.; Moulton, H.M.; Cate, M.L.; Reddy, M.T.; Whitehead, T.; Hassinger, J.N.; Youngblood, D.S.; Iversen, P.L. Pharmacokinetics, biodistribution, stability and toxicity of a cell-penetrating peptide-morpholino oligomer conjugate. Bioconjug. Chem. 2007, 18, 1325–1331. [Google Scholar] [CrossRef]
- Greco, S.; Cardinali, B.; Falcone, G.; Martelli, F. Circular RNAs in Muscle Function and Disease. Int. J. Mol. Sci. 2018, 19, 3454. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.; Aoki, Y.; Kameyama, T.; Saito, T.; Masuda, S.; Tanihata, J.; Nagata, T.; Mayeda, A.; Takeda, S.; Tsukahara, T. Endogenous Multiple Exon Skipping and Back-Splicing at the DMD Mutation Hotspot. Int. J. Mol. Sci. 2016, 17, 1722. [Google Scholar] [CrossRef]
- Vulin, A.; Wein, N.; Simmons, T.R.; Rutherford, A.M.; Findlay, A.R.; Yurkoski, J.A.; Kaminoh, Y.; Flanigan, K.M. The first exon duplication mouse model of Duchenne muscular dystrophy: A tool for therapeutic development. Neuromuscul. Disord. 2015, 25, 827–834. [Google Scholar] [CrossRef]
- Greer, K.; Fletcher, S.; Wilton, S.D. Skipping of Duplicated Dystrophin Exons: In Vitro Induction and Assessment. Methods Mol. Biol. 2018, 1828, 219–228. [Google Scholar] [PubMed]
- Greer, K.; Mizzi, K.; Rice, E.; Kuster, L.; Barrero, R.A.; Bellgard, M.I.; Lynch, B.J.; Foley, A.R.; E., O.R.; Wilton, S.D.; et al. Pseudoexon activation increases phenotype severity in a Becker muscular dystrophy patient. Mol. Genet. Genom. Med. 2015, 3, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Kunkel, T.A.; Bebenek, K. DNA replication fidelity. Annu Rev. Biochem 2000, 69, 497–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panda, A.C.; Gorospe, M. Detection and Analysis of Circular RNAs by RT-PCR. Bio Protoc 2018, 8, e2775. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.; Zuo, Y.; Wang, J.; Zhang, M.Q.; Malhotra, A.; Mayeda, A. Characterization of RNase R-digested cellular RNA source that consists of lariat and circular RNAs from pre-mRNA splicing. Nucleic Acids Res. 2006, 34, e63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cartegni, L.; Wang, J.; Zhu, Z.; Zhang, M.Q.; Krainer, A.R. ESEfinder: A web resource to identify exonic splicing enhancers. Nucleic Acids Res. 2003, 31, 3568–3571. [Google Scholar] [CrossRef] [Green Version]
- Rando, T.A.; Blau, H.M. Primary mouse myoblast purification, characterization, and transplantation for cell-mediated gene therapy. J. Cell Biol. 1994, 125, 1275–1287. [Google Scholar] [CrossRef] [Green Version]
- Harding, P.L.; Fall, A.M.; Honeyman, K.; Fletcher, S.; Wilton, S.D. The influence of antisense oligonucleotide length on dystrophin exon skipping. Mol 2007, 15, 157–166. [Google Scholar] [CrossRef]
- Adams, A.M.; Harding, P.L.; Iversen, P.L.; Coleman, C.; Fletcher, S.; Wilton, S.D. Antisense oligonucleotide induced exon skipping and the dystrophin gene transcript: cocktails and chemistries. Bmc Mol. Biol. 2007, 8, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Toh, Z.Y.; Thandar Aung-Htut, M.; Pinniger, G.; Adams, A.M.; Krishnaswarmy, S.; Wong, B.L.; Fletcher, S.; Wilton, S.D. Deletion of Dystrophin In-Frame Exon 5 Leads to a Severe Phenotype: Guidance for Exon Skipping Strategies. PLoS ONE 2016, 11, e0145620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, S.T.; Lo, H.P.; North, K.N. Single section Western blot: improving the molecular diagnosis of the muscular dystrophies. Neurology 2003, 61, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, L.V.; Davison, K.; Falkous, G.; Harwood, C.; O’Donnell, E.; Slater, C.R.; Harris, J.B. Dystrophin in skeletal muscle. I. Western blot analysis using a monoclonal antibody. J. Neurol. Sci. 1989, 94, 125–136. [Google Scholar] [CrossRef]
- Nicholson, L.V.; Johnson, M.A.; Davison, K.; O’Donnell, E.; Falkous, G.; Barron, M.; Harris, J.B. Dystrophin or a “related protein” in Duchenne muscular dystrophy? Acta Neurol. Scand. 1992, 86, 8–14. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nomenclature | Sequence (5′-3′) |
|---|---|
| H17A(+10+35) | AGU GAU GGC UGA GUG GUG GUG ACA GC |
| H17A(+61+86) | UGU UCC CUU GUG GUC ACC GUA GUU AC |
| H18A(+24+53) | CAG CUU CUG AGC GAG UAA UCC AGC UGU GAA |
| Scrambled Sequence | GGA UGU CCU GAG UCU AGA CCC UCC G |
| Primer Location | Primer Sequence |
|---|---|
| Exon 13 Forward | TGC TTC AAG AAG ATC TAG AAC AAG AAC |
| Exon 13 Forward | CAC GCA ACT GCT GCT TTG GAA G |
| Exon 16 Forward | GCA AAC TGT ATT CAC TCA AAC |
| Exon 15 Reverse | GTG AAT CTT GTT CAC TGC ATC |
| Exon 19 Reverse | ACG TTC CAC CAG GGC CTG AG |
| Exon 20 Reverse | GAT ACT CCA GCC AGT TAA GTC T |
| Exon 21 Reverse | GGC CAC AAA GTC TGC ATC CAG |
| Exon 25 Reverse | GTC TCA AGT CTC GAA GCA AAC |
| Intron 17 Forward | TGG TGT TTT CTG GGG TGA AA |
| Intron 17 Forward | CCA GGG TAG AGA AGT TTG AA |
| Intron 13 Reverse | ACC ACT ATA TAA AAT GGC TCC |
| Intron 13 Reverse | GAG GGA GGC AGA AAA CAA CC |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Greer, K.; Johnsen, R.; Nevo, Y.; Fellig, Y.; Fletcher, S.; Wilton, S.D. Single Exon Skipping Can Address a Multi-Exon Duplication in the Dystrophin Gene. Int. J. Mol. Sci. 2020, 21, 4511. https://doi.org/10.3390/ijms21124511
Greer K, Johnsen R, Nevo Y, Fellig Y, Fletcher S, Wilton SD. Single Exon Skipping Can Address a Multi-Exon Duplication in the Dystrophin Gene. International Journal of Molecular Sciences. 2020; 21(12):4511. https://doi.org/10.3390/ijms21124511
Chicago/Turabian StyleGreer, Kane, Russell Johnsen, Yoram Nevo, Yakov Fellig, Susan Fletcher, and Steve D. Wilton. 2020. "Single Exon Skipping Can Address a Multi-Exon Duplication in the Dystrophin Gene" International Journal of Molecular Sciences 21, no. 12: 4511. https://doi.org/10.3390/ijms21124511