Impaired ECM Remodeling and Macrophage Activity Define Necrosis and Regeneration Following Damage in Aged Skeletal Muscle

Abstract
:1. Introduction
2. Results
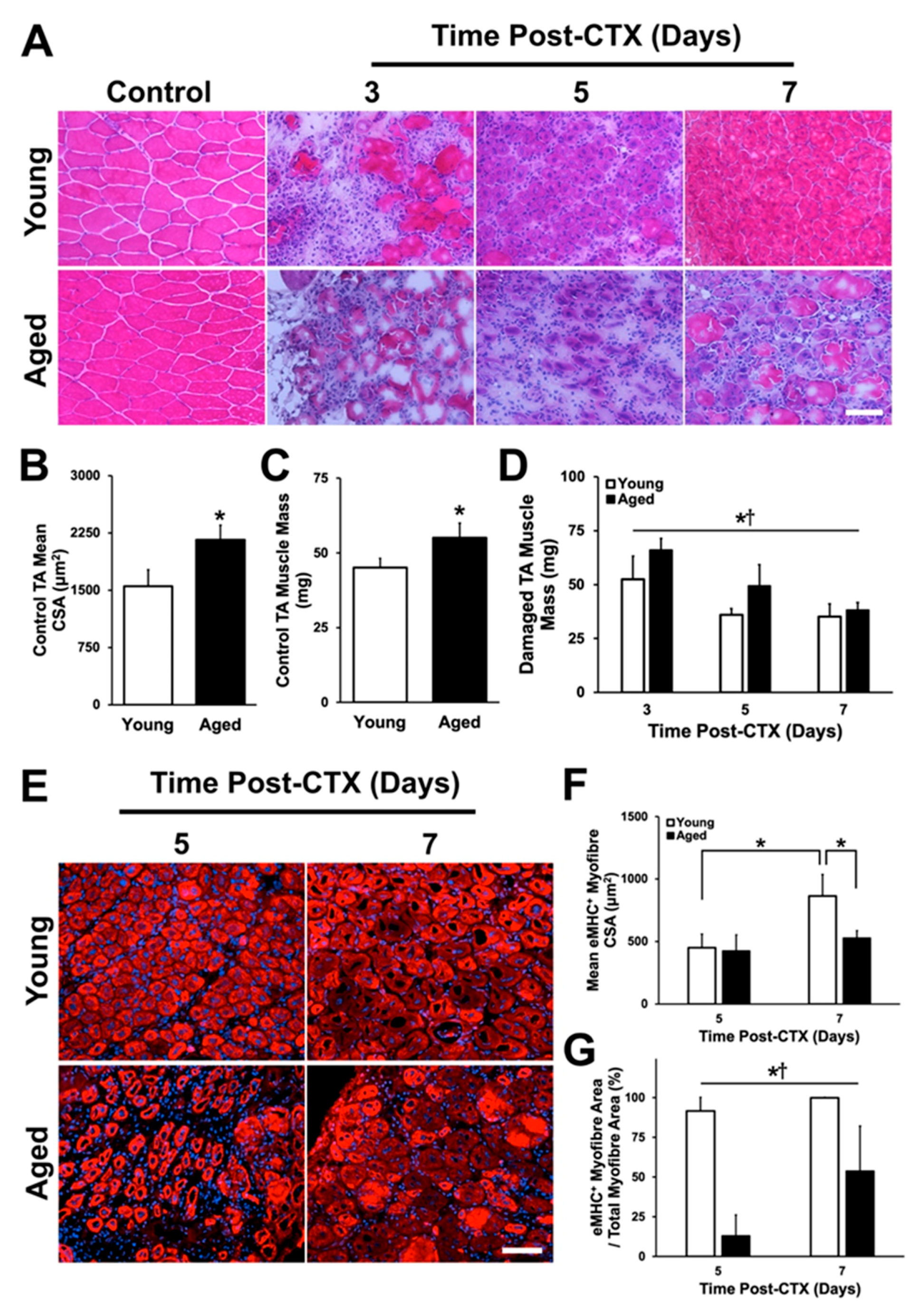
2.1. Body Mass, Muscle Mass, and Myofiber Cross-Sectional Area
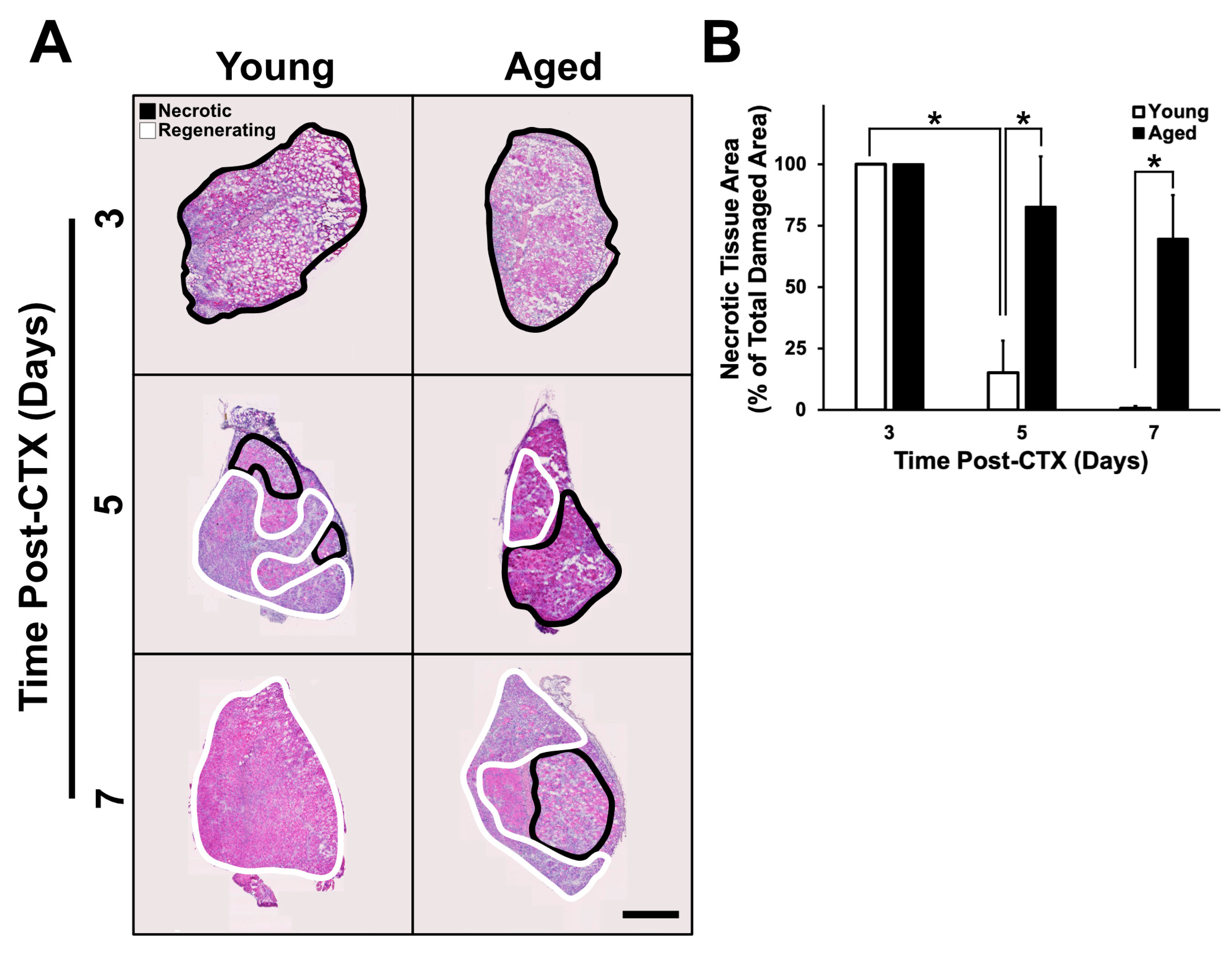
2.2. Assessment of Regenerative Capacity
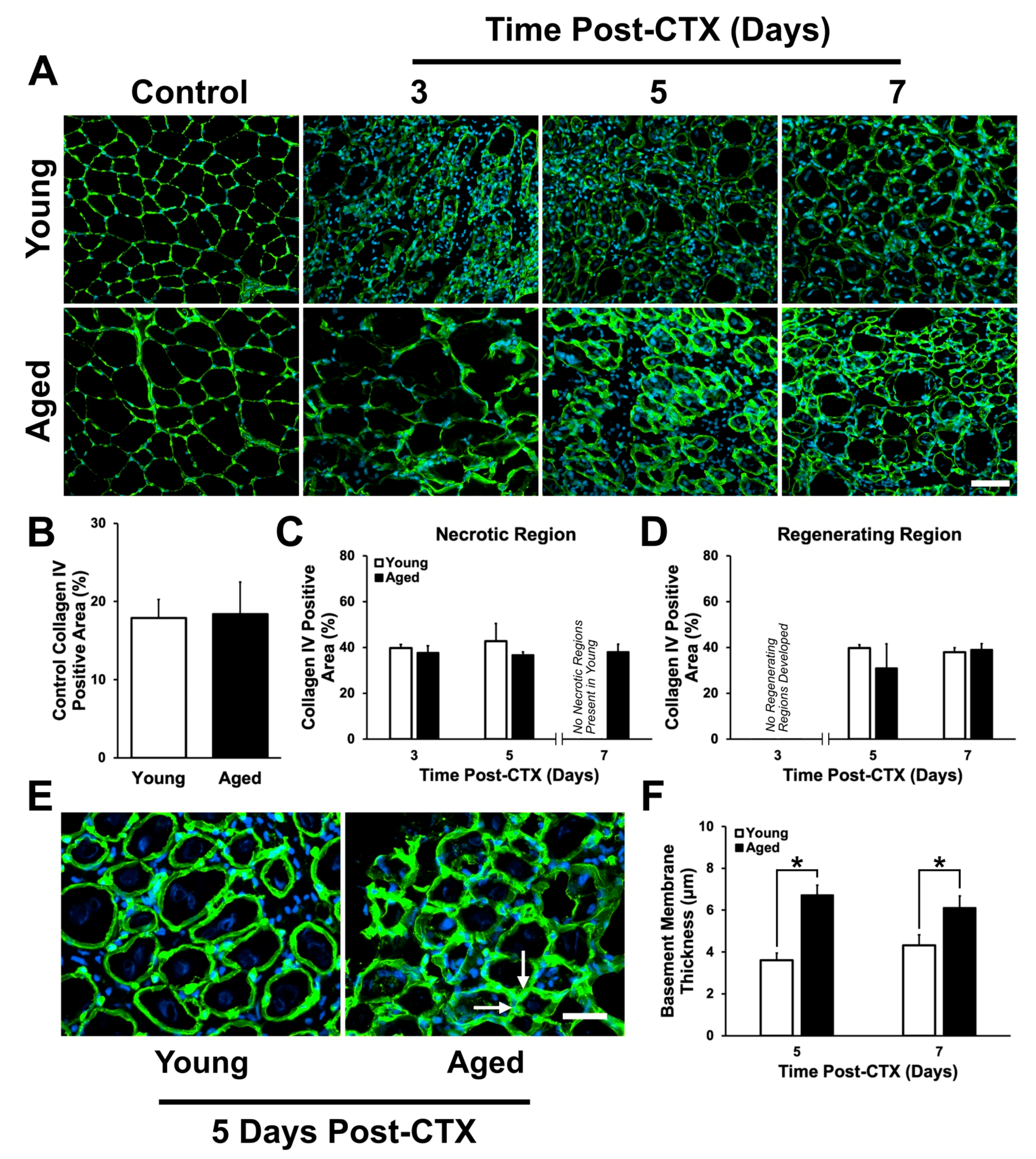
2.3. Assessment of ECM Remodeling
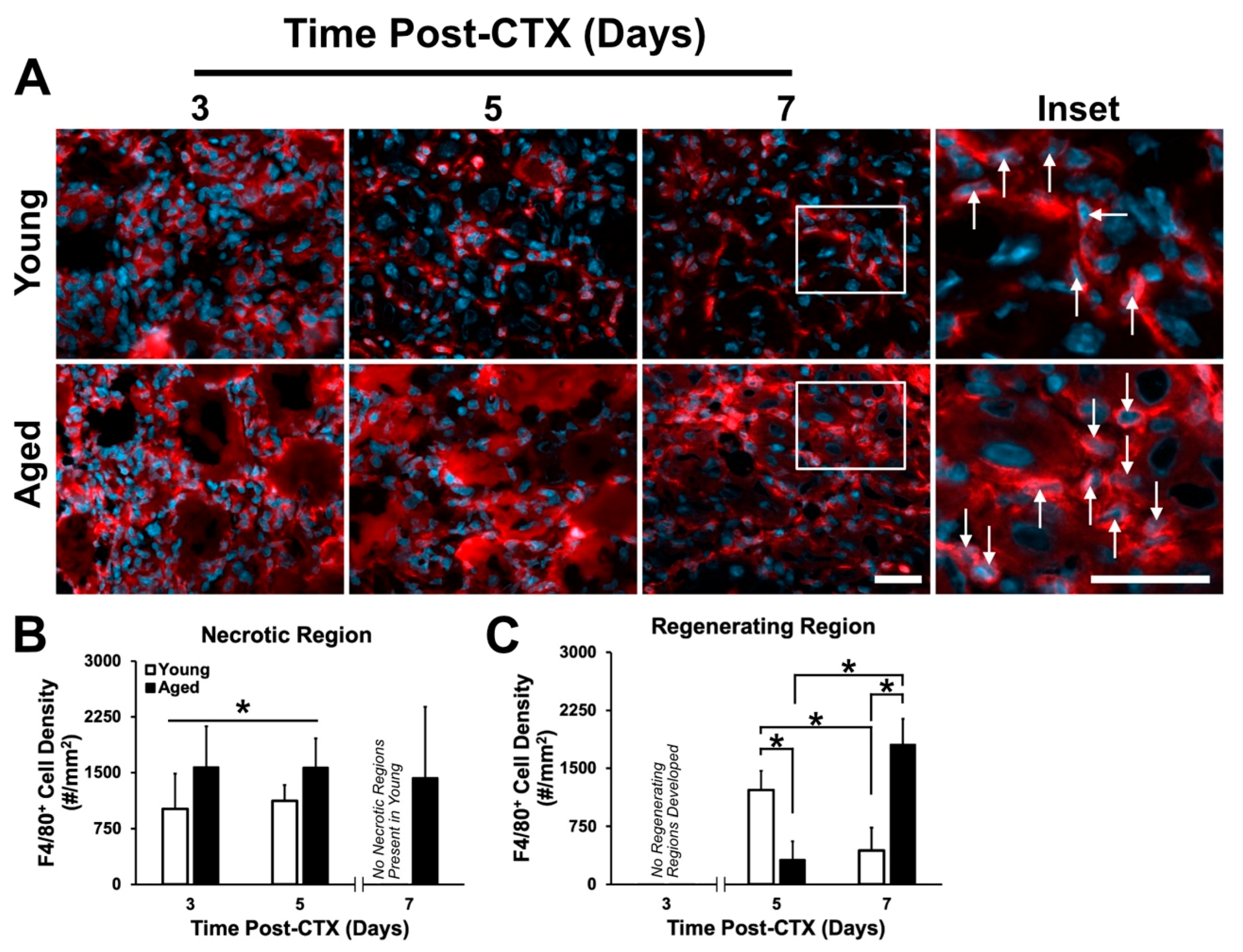
2.4. Macrophage Density Following Damage
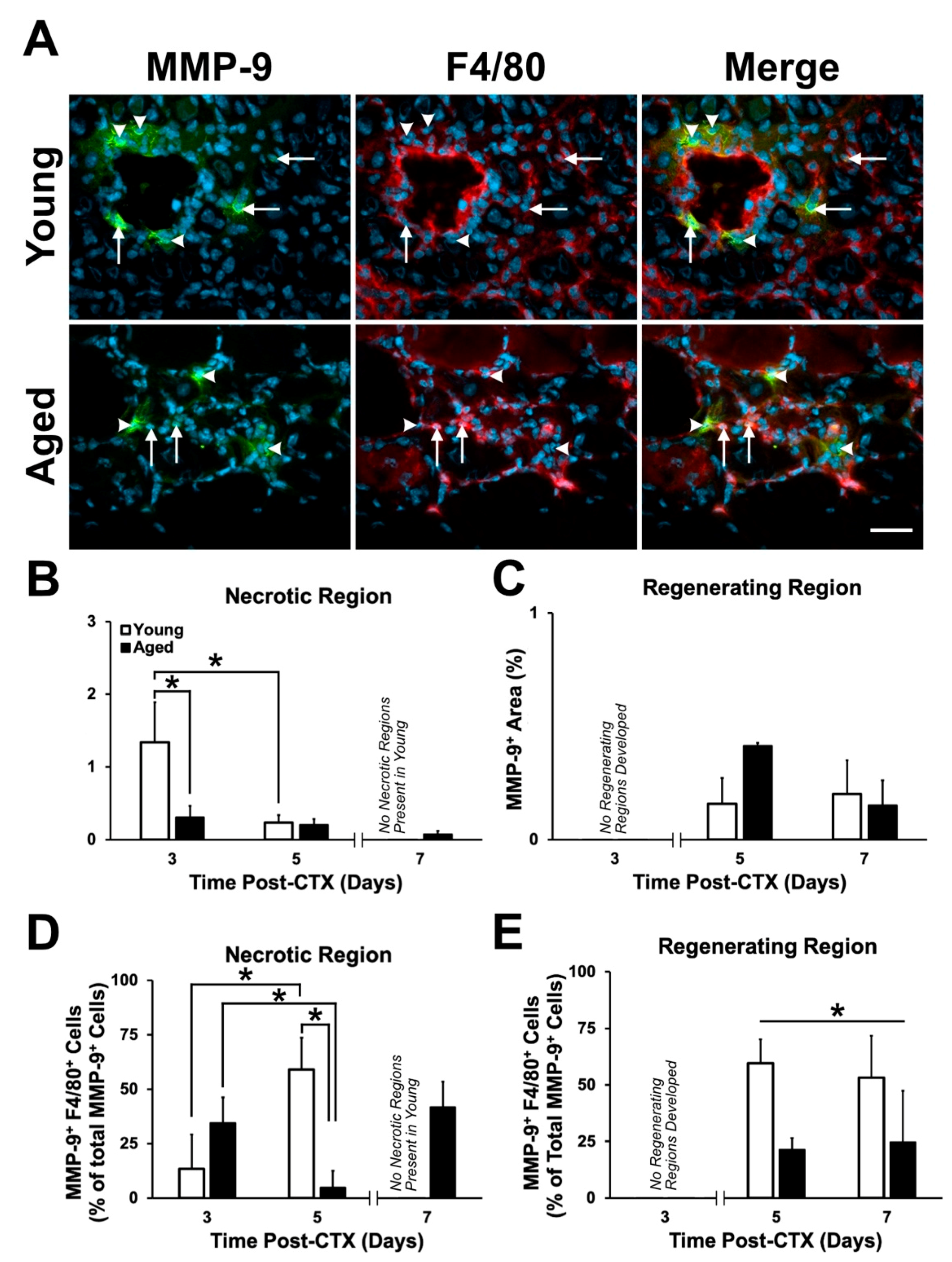
2.5. Expression Pattern and Localization of MMP-9
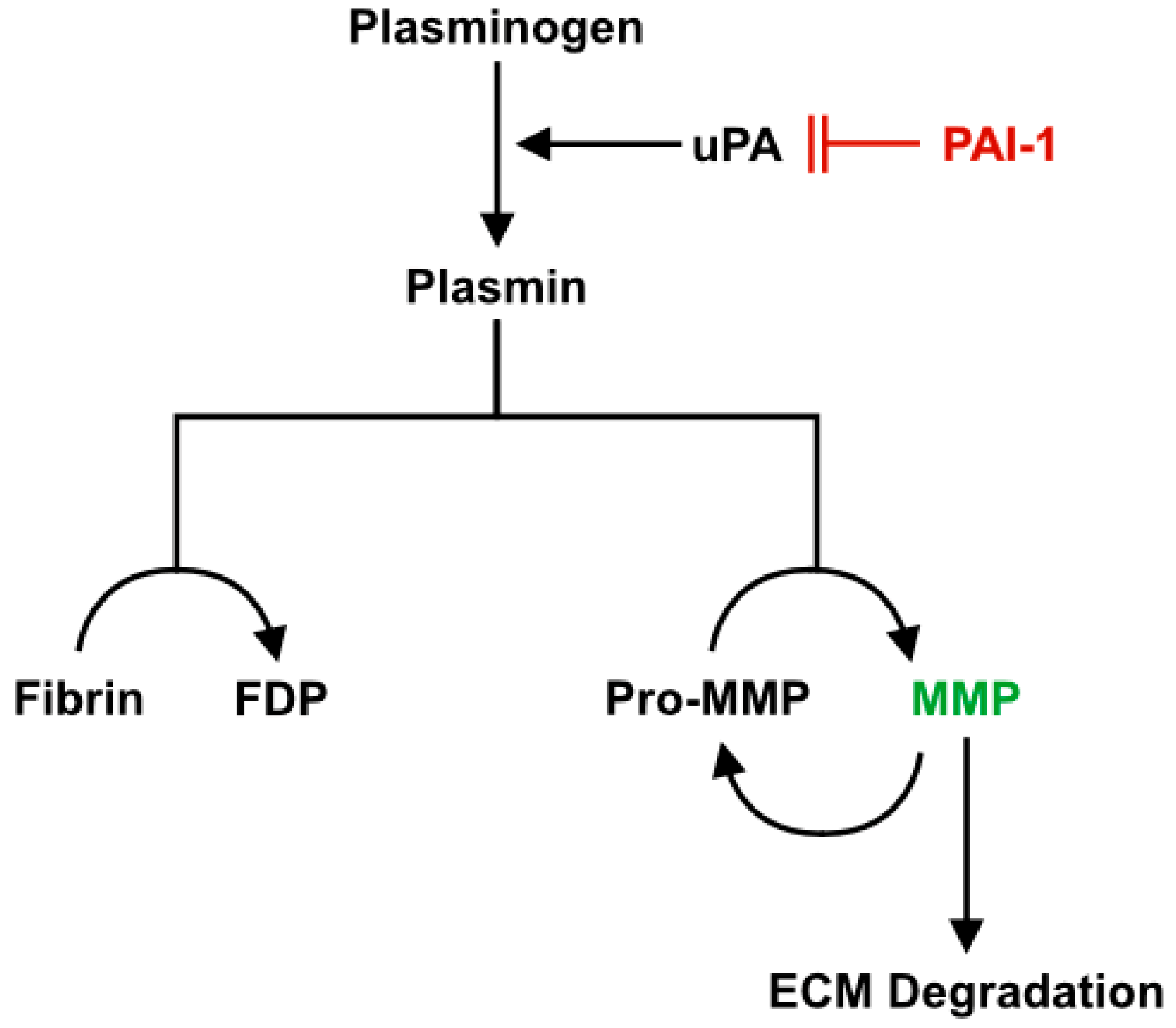
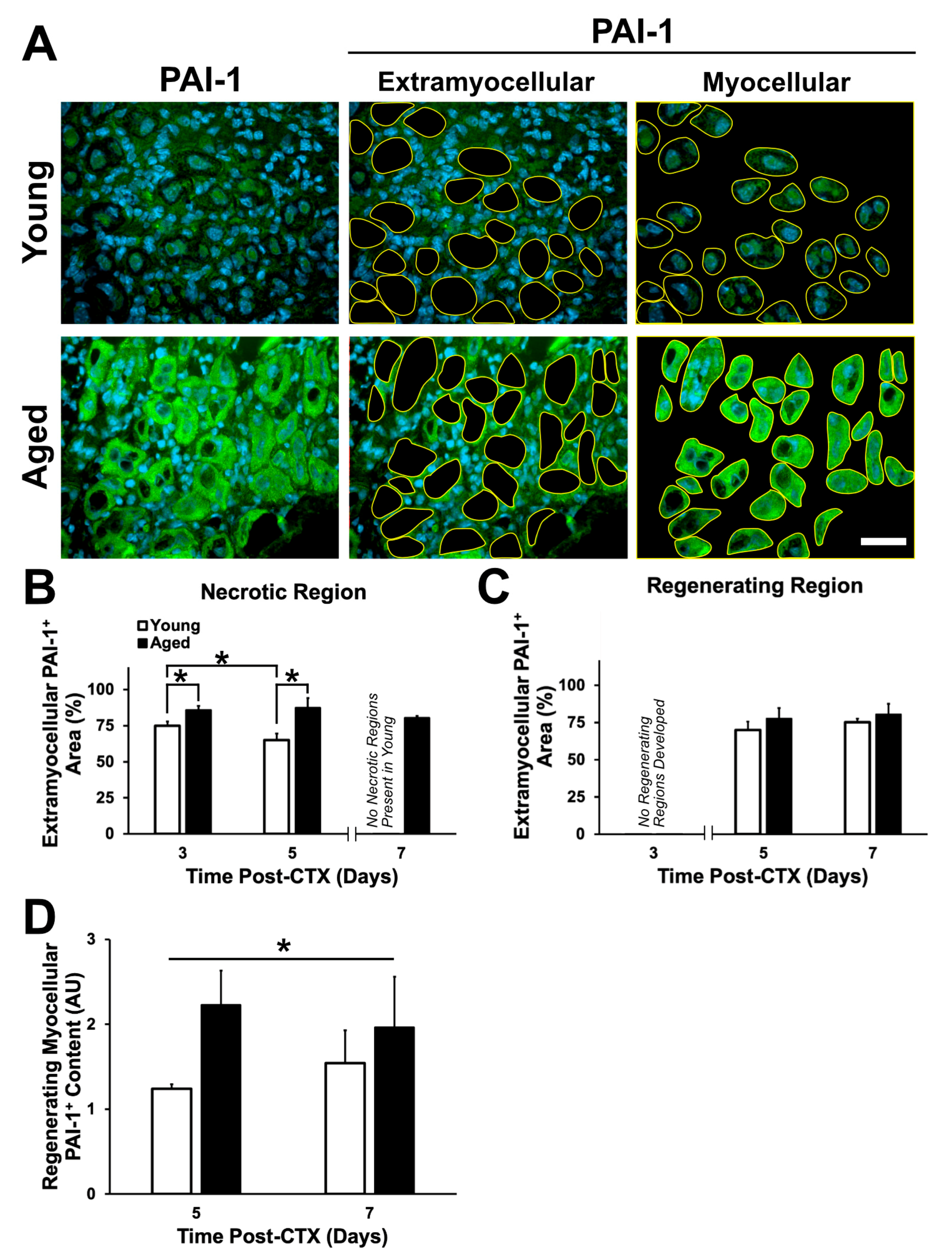
2.6. Expression Pattern and Localization of PAI-1
3. Discussion
3.1. Role of PAI-1 in Impaired Muscle Regeneration with Aging
3.2. Alterations to the ECM During Regeneration in Aged Skeletal Muscle
3.3. Study Limitations
4. Materials and Methods
4.1. Animal Care
4.2. Skeletal Muscle Damage and Tissue Collection
4.3. Histological Analyses
4.4. Image Analysis
4.5. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| CSA | Cross-sectional Area |
| CTX | Cardiotoxin |
| DAPI | 4,6-diamidino-2-phenylindole |
| ECM | Extracellular Matrix |
| eMHC | Embryonic Myosin Heavy Chain |
| MMP-9 | Matrix Metalloproteinase-9 |
| PAI-1 | Plasminogen Activator Inhibitor-1 |
| TA | Tibialis Anterior |
| TGF-β | Transforming Growth Factor-β |
| uPA | Urokinase Plasminogen Activator |
References
- Heymsfield, S.B.; Adamek, M.; Gonzalez, M.C.; Jia, G.; Thomas, D.M. Assessing skeletal muscle mass: Historical overview and state of the art. J. Cachexia Sarcopenia Muscle 2014, 5, 9–18. [Google Scholar] [CrossRef]
- White, H.K.; Petrie, C.D.; Landschulz, W.; MacLean, D.; Taylor, A.; Lyles, K.; Wei, J.Y.; Hoffman, A.R.; Salvatori, R.; Ettinger, M.P.; et al. Effects of an oral growth hormone secretagogue in older adults. J. Clin. Endocrinol. Metab. 2009, 94, 1198–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beasley, J.M.; Shikany, J.M.; Thomson, C.A. The role of dietary protein intake in the prevention of sarcopenia of aging. Nutr. Clin. Pract. 2013, 28, 684–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deer, R.R.; Volpi, E. Protein intake and muscle function in older adults. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 248–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neto, W.K.; Gama, E.F.; Rocha, L.Y.; Ramos, C.C.; Taets, W.; Scapini, K.B.; Ferreira, J.B.; Rodrigues, B.; Caperuto, É. Effects of testosterone on lean mass gain in elderly men: Systematic review with meta-analysis of controlled and randomized studies. Age 2015, 37, 9742. [Google Scholar] [CrossRef]
- Sharples, A.P.; Hughes, D.C.; Deane, C.S.; Saini, A.; Selman, C.; Stewart, C.E. Longevity and skeletal muscle mass: The role of IGF signalling, the sirtuins, dietary restriction and protein intake. Aging Cell 2015, 14, 511–523. [Google Scholar] [CrossRef]
- Hawke, T.J.; Garry, D.J. Myogenic satellite cells: Physiology to molecular biology. J. Appl. Physiol. 2001, 91, 534–551. [Google Scholar] [CrossRef]
- Dumont, N.A.; Bentzinger, C.F.; Sincennes, M.-C.; Rudnicki, M.A. Satellite Cells and Skeletal Muscle Regeneration. Compr. Physiol. 2015, 5, 1027–1059. [Google Scholar] [CrossRef]
- Grounds, M.D. Towards Understanding Skeletal Muscle Regeneration. Pathol. Res. Pract. 1991, 187, 1–22. [Google Scholar] [CrossRef]
- Mann, C.J.; Perdiguero, E.; Kharraz, Y.; Aguilar, S.; Pessina, P.; Serrano, A.L.; Muñoz-Cánoves, P. Aberrant repair and fibrosis development in skeletal muscle. Skelet. Muscle 2011, 1, 21. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wehling-Henricks, M.; Samengo, G.; Tidball, J.G. Increases of M2a macrophages and fibrosis in aging muscle are influenced by bone marrow aging and negatively regulated by muscle-derived nitric oxide. Aging Cell 2015, 14, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Blau, H.M.; Cosgrove, B.D.; Ho, A.T.V. The central role of muscle stem cells in regenerative failure with aging. Nat. Med. 2015, 21, 854–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grounds, M.D. Age-associated changes in the response of skeletal muscle cells to exercise and regeneration. Ann. NY Acad. Sci. 1998, 854, 78–91. [Google Scholar] [CrossRef]
- Brack, A.S.; Rando, T.A. Intrinsic changes and extrinsic influences of myogenic stem cell function during aging. Stem Cell Rev. 2007, 3, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Stearns-Reider, K.M.; D’Amore, A.; Beezhold, K.; Rothrauff, B.; Cavalli, L.; Wagner, W.R.; Vorp, D.A.; Tsamis, A.; Shinde, S.; Zhang, C.; et al. Aging of the skeletal muscle extracellular matrix drives a stem cell fibrogenic conversion. Aging Cell 2017, 16, 518–528. [Google Scholar] [CrossRef] [Green Version]
- Sadeh, M. Effects of aging on skeletal muscle regeneration. J. Neurol. Sci. 1988, 87, 67–74. [Google Scholar] [CrossRef]
- Carlson, B.M.; Faulkner, J.A. Muscle transplantation between young and old rats: Age of host determines recovery. Am. J. Physiol. Cell Physiol. 1989, 256, C1262–C1266. [Google Scholar] [CrossRef] [PubMed]
- Grounds, M.D. Phagocytosis of necrotic muscle in muscle isografts is influenced by the strain, age, and sex of host mice. J. Pathol. 1987, 153, 71–82. [Google Scholar] [CrossRef]
- Gürlek, A.; Bayraktar, M.; Kirazli, S. Increased plasminogen activator inhibitor-1 activity in offspring of type 2 diabetic patients: Lack of association with plasma insulin levels. Diabetes Care 2000, 23, 88–92. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef]
- Ma, L.-J.; Fogo, A.B. PAI-1 and kidney fibrosis. Front. Biosci. 2009, 14, 2028–2041. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Kobayashi, A.; Yamazaki, N.; Sugawara, Y.; Takada, Y.; Takada, A. Relationship between age and plasma t-PA, PA-inhibitor, and PA activity. Thromb. Res. 1987, 46, 625–633. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Takeshita, K.; Saito, H. Plasminogen activator inhibitor-1 in aging. Semin. Thromb. Hemost. 2014, 40, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Takeshita, K.; Kojima, T.; Takamatsu, J.; Saito, H. Aging and plasminogen activator inhibitor-1 (PAI-1) regulation: Implication in the pathogenesis of thrombotic disorders in the elderly. Cardiovasc. Res. 2005, 66, 276–285. [Google Scholar] [CrossRef] [Green Version]
- Collen, D. The Plasminogen (Fibrinolytic) System. Thromb. Haemost. 1999, 82, 259–270. [Google Scholar] [CrossRef]
- Plow, E.F.; Herren, T.; Redlitz, A.; Miles, L.A.; Hoover-Plow, J.L. The cell biology of the plasminogen system. FASEB J. 1995, 9, 939–945. [Google Scholar] [CrossRef]
- Irigoyen, J.P.; Muñoz-Cánoves, P.; Montero, L.; Koziczak, M.; Nagamine, Y. The plasminogen activator system: Biology and regulation. Cell. Mol. Life Sci. 1999, 56, 104–132. [Google Scholar] [CrossRef]
- Garg, K.; Boppart, M.D. Influence of exercise and aging on extracellular matrix composition in the skeletal muscle stem cell niche. J. Appl. Physiol. 2016, 121, 1053–1058. [Google Scholar] [CrossRef] [Green Version]
- Krause, M.P.; Moradi, J.; Nissar, A.A.; Riddell, M.C.; Hawke, T.J. Inhibition of plasminogen activator inhibitor-1 restores skeletal muscle regeneration in untreated type 1 diabetic mice. Diabetes 2011, 60, 1964–1972. [Google Scholar] [CrossRef] [Green Version]
- Francis, R.M.; Romeyn, C.L.; Coughlin, A.M.; Nagelkirk, P.R.; Womack, C.J.; Lemmer, J.T. Age and aerobic training status effects on plasma and skeletal muscle tPA and PAI-1. Eur. J. Appl. Physiol. 2014, 114, 1229–1238. [Google Scholar] [CrossRef]
- Koh, T.J.; Bryer, S.C.; Pucci, A.M.; Sisson, T.H. Mice deficient in plasminogen activator inhibitor-1 have improved skeletal muscle regeneration. Am. J. Physiol. Cell Physiol. 2005, 289, C217–C223. [Google Scholar] [CrossRef] [PubMed]
- Higazi, A.A.-R. Fibrinolysis|Overview. In Encyclopedia of Respiratory Medicine; Laurent, G.J., Shapiro, S.D., Eds.; Academic Press: Oxford, UK, 2006; pp. 201–205. ISBN 978-0-12-370879-3. [Google Scholar]
- Krause, M.P.; Al-Sajee, D.; D’Souza, D.M.; Rebalka, I.A.; Moradi, J.; Riddell, M.C.; Hawke, T.J. Impaired Macrophage and Satellite Cell Infiltration Occurs in a Muscle-Specific Fashion Following Injury in Diabetic Skeletal Muscle. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, Y.; Kita, S.; Nishizawa, H.; Fukuda, S.; Fujishima, Y.; Obata, Y.; Nagao, H.; Masuda, S.; Nakamura, Y.; Shimizu, Y.; et al. Adiponectin promotes muscle regeneration through binding to T-cadherin. Sci. Rep. 2019, 9, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldspink, G.; Fernandes, K.; Williams, P.E.; Wells, D.J. Age-related changes in collagen gene expression in the muscles of mdx dystrophic and normal mice. Neuromuscul. Disord. 1994, 4, 183–191. [Google Scholar] [CrossRef]
- Wood, L.K.; Kayupov, E.; Gumucio, J.P.; Mendias, C.L.; Claflin, D.R.; Brooks, S.V. Intrinsic stiffness of extracellular matrix increases with age in skeletal muscles of mice. J. Appl. Physiol. 2014, 117, 363–369. [Google Scholar] [CrossRef] [Green Version]
- Brack, A.S.; Conboy, M.J.; Roy, S.; Lee, M.; Kuo, C.J.; Keller, C.; Rando, T.A. Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Science 2007, 317, 807–810. [Google Scholar] [CrossRef]
- Lacraz, G.; Rouleau, A.-J.; Couture, V.; Söllrald, T.; Drouin, G.; Veillette, N.; Grandbois, M.; Grenier, G. Increased Stiffness in Aged Skeletal Muscle Impairs Muscle Progenitor Cell Proliferative Activity. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [Green Version]
- Kjær, M. Role of Extracellular Matrix in Adaptation of Tendon and Skeletal Muscle to Mechanical Loading. Physiol. Rev. 2004, 84, 649–698. [Google Scholar] [CrossRef]
- Thomas, K.; Engler, A.J.; Meyer, G.A. Extracellular matrix regulation in the muscle satellite cell niche. Connect. Tissue Res. 2015, 56, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Kühl, U.; Ocalan, M.; Timpl, R.; Mayne, R.; Hay, E.; von der Mark, K. Role of muscle fibroblasts in the deposition of type-IV collagen in the basal lamina of myotubes. Differentiation 1984, 28, 164–172. [Google Scholar] [CrossRef]
- Gulati, A.K.; Reddi, A.H.; Zalewski, A.A. Changes in the basement membrane zone components during skeletal muscle fiber degeneration and regeneration. J. Cell. Biol. 1983, 97, 957–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bentzinger, C.F.; Wang, Y.X.; von Maltzahn, J.; Soleimani, V.D.; Yin, H.; Rudnicki, M.A. Fibronectin regulates Wnt7a signaling and satellite cell expansion. Cell Stem Cell 2013, 12, 75–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calve, S.; Odelberg, S.J.; Simon, H.-G. A Transitional Extracellular Matrix Instructs Cell Behavior During Muscle Regeneration. Dev. Biol. 2010, 344, 259–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kherif, S.; Lafuma, C.; Dehaupas, M.; Lachkar, S.; Fournier, J.G.; Verdière-Sahuqué, M.; Fardeau, M.; Alameddine, H.S. Expression of matrix metalloproteinases 2 and 9 in regenerating skeletal muscle: A study in experimentally injured and mdx muscles. Dev. Biol. 1999, 205, 158–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmeli, E.; Moas, M.; Reznick, A.Z.; Coleman, R. Matrix metalloproteinases and skeletal muscle: A brief review. Muscle Nerve 2004, 29, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, K.; Nakamura, A.; Ueda, H.; Yuasa, K.; Yoshida, K.; Takeda, S.; Ikeda, S. Activation and localization of matrix metalloproteinase-2 and -9 in the skeletal muscle of the muscular dystrophy dog (CXMDJ). BMC Musculoskelet. Disord. 2007, 8, 54. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Li, Y. Role of matrix metalloproteinases in skeletal muscle. Cell Adhes. Migr. 2009, 3, 337–341. [Google Scholar] [CrossRef]
- Eren, M.; Boe, A.E.; Klyachko, E.A.; Vaughan, D.E. Role of plasminogen activator inhibitor-1 in senescence and aging. Semin. Thromb. Hemost. 2014, 40, 645–651. [Google Scholar] [CrossRef]
- Naderi, J.; Bernreuther, C.; Grabinski, N.; Putman, C.T.; Henkel, B.; Bell, G.; Glatzel, M.; Sultan, K.R. Plasminogen Activator Inhibitor Type 1 Up-Regulation Is Associated with Skeletal Muscle Atrophy and Associated Fibrosis. Am. J. Pathol. 2009, 175, 763–771. [Google Scholar] [CrossRef] [Green Version]
- Cesari, M.; Pahor, M.; Incalzi, R.A. Plasminogen Activator Inhibitor-1 (PAI-1): A Key Factor Linking Fibrinolysis and Age-Related Subclinical and Clinical Conditions. Cardiovasc. Ther. 2010, 28, e72–e91. [Google Scholar] [CrossRef] [Green Version]
- Ali, S.; Garcia, J.M. Sarcopenia, cachexia and aging: Diagnosis, mechanisms and therapeutic options-A mini-review. Gerontology 2014, 60, 294–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muscaritoli, M.; Anker, S.D.; Argilés, J.; Aversa, Z.; Bauer, J.M.; Biolo, G.; Boirie, Y.; Bosaeus, I.; Cederholm, T.; Costelli, P.; et al. Consensus definition of sarcopenia, cachexia and pre-cachexia: Joint document elaborated by Special Interest Groups (SIG) “cachexia-anorexia in chronic wasting diseases” and “nutrition in geriatrics”. Clin. Nutr. 2010, 29, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Xia, J.; Zhang, X.I.; Gathirua-Mwangi, W.G.; Guo, J.; Li, Y.; McKenzie, S.; Song, Y. Associations of Muscle Mass and Strength with All-Cause Mortality among US Older Adults. Med. Sci. Sports Exerc. 2018, 50, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Sisson, T.H.; Nguyen, M.-H.; Yu, B.; Novak, M.L.; Simon, R.H.; Koh, T.J. Urokinase-type plasminogen activator increases hepatocyte growth factor activity required for skeletal muscle regeneration. Blood 2009, 114, 5052–5061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, L.; Kawao, N.; Tamura, Y.; Okumoto, K.; Okada, K.; Yano, M.; Matsuo, O.; Kaji, H. Plasminogen Activator Inhibitor-1 Is Involved in Impaired Bone Repair Associated with Diabetes in Female Mice. PLoS ONE 2014, 9, e92686. [Google Scholar] [CrossRef] [Green Version]
- Nicholas, S.B.; Aguiniga, E.; Ren, Y.; Kim, J.; Wong, J.; Govindarajan, N.; Noda, M.; Wang, W.; Kawano, Y.; Collins, A.; et al. Plasminogen activator inhibitor-1 deficiency retards diabetic nephropathy. Kidney Int. 2005, 67, 1297–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyon, C.J.; Hsueh, W.A. Effect of plasminogen activator inhibitor-1 in diabetes mellitus and cardiovascular disease. Am. J. Med. 2003, 115, 62S–68S. [Google Scholar] [CrossRef]
- Goldberg, R.B. Cytokine and Cytokine-Like Inflammation Markers, Endothelial Dysfunction, and Imbalanced Coagulation in Development of Diabetes and Its Complications. J. Clin. Endocrinol. Metab. 2009, 94, 3171–3182. [Google Scholar] [CrossRef] [Green Version]
- Baar, M.P.; Perdiguero, E.; Muñoz-Cánoves, P.; de Keizer, P.L. Musculoskeletal senescence: A moving target ready to be eliminated. Curr. Opin. Pharmacol. 2018, 40, 147–155. [Google Scholar] [CrossRef]
- Kortlever, R.M.; Nijwening, J.H.; Bernards, R. Transforming Growth Factor-β Requires Its Target Plasminogen Activator Inhibitor-1 for Cytostatic Activity. J. Biol. Chem. 2008, 283, 24308–24313. [Google Scholar] [CrossRef] [Green Version]
- Kortlever, R.M.; Higgins, P.J.; Bernards, R. Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat. Cell Biol. 2006, 8, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Bigg, H.F.; Rowan, A.D.; Barker, M.D.; Cawston, T.E. Activity of matrix metalloproteinase-9 against native collagen types I and III. FEBS J. 2007, 274, 1246–1255. [Google Scholar] [CrossRef] [PubMed]
- Novak, M.L.; Bryer, S.C.; Cheng, M.; Nguyen, M.-H.; Conley, K.L.; Cunningham, A.K.; Xue, B.; Sisson, T.H.; You, J.-S.; Hornberger, T.A.; et al. Macrophage-specific expression of urokinase-type plasminogen activator promotes skeletal muscle regeneration. J. Immunol. 2011, 187, 1448–1457. [Google Scholar] [CrossRef] [PubMed]
- DiPasquale, D.M.; Cheng, M.; Billich, W.; Huang, S.A.; van Rooijen, N.; Hornberger, T.A.; Koh, T.J. Urokinase-type plasminogen activator and macrophages are required for skeletal muscle hypertrophy in mice. Am. J. Physiol. Cell Physiol. 2007, 293, C1278–C1285. [Google Scholar] [CrossRef] [PubMed]
- Fibbi, G.; Barletta, E.; Dini, G.; Del Rosso, A.; Pucci, M.; Cerletti, M.; Del Rosso, M. Cell invasion is affected by differential expression of the urokinase plasminogen activator/urokinase plasminogen activator receptor system in muscle satellite cells from normal and dystrophic patients. Lab. Invest. 2001, 81, 27–39. [Google Scholar] [CrossRef] [Green Version]
- Ardi, V.C.; Kupriyanova, T.A.; Deryugina, E.I.; Quigley, J.P. Human neutrophils uniquely release TIMP-free MMP-9 to provide a potent catalytic stimulator of angiogenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 20262–20267. [Google Scholar] [CrossRef] [Green Version]
- Bradley, L.M.; Douglass, M.F.; Chatterjee, D.; Akira, S.; Baaten, B.J.G. Matrix Metalloprotease 9 Mediates Neutrophil Migration into the Airways in Response to Influenza Virus-Induced Toll-Like Receptor Signaling. PLoS Pathog. 2012, 8, e1002641. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, S.; Zee, J.M.; Patel, K.D. Regulation of matrix metalloproteinase-9 (MMP-9) in TNF-stimulated neutrophils: Novel pathways for tertiary granule release. J. Leukoc. Biol. 2006, 79, 214–222. [Google Scholar] [CrossRef]
- Kobayashi, T.; Hattori, S.; Shinkai, H. Matrix metalloproteinases-2 and -9 are secreted from human fibroblasts. Acta Derm. Venereol. 2003, 83, 105–107. [Google Scholar] [CrossRef] [Green Version]
- Dayer, C.; Stamenkovic, I. Recruitment of Matrix Metalloproteinase-9 (MMP-9) to the Fibroblast Cell Surface by Lysyl Hydroxylase 3 (LH3) Triggers Transforming Growth Factor-β (TGF-β) Activation and Fibroblast Differentiation. J. Biol. Chem. 2015, 290, 13763–13778. [Google Scholar] [CrossRef] [Green Version]
- Lindner, D.; Zietsch, C.; Becher, P.M.; Schulze, K.; Schultheiss, H.-P.; Tschöpe, C.; Westermann, D. Differential expression of matrix metalloproteases in human fibroblasts with different origins. Biochem. Res. Int. 2012, 2012, 875742. [Google Scholar] [CrossRef] [Green Version]
- Guérin, C.W.; Holland, P.C. Synthesis and secretion of matrix-degrading metalloproteases by human skeletal muscle satellite cells. Dev. Dyn. 1995, 202, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Uezumi, A.; Fukada, S.; Yamamoto, N.; Takeda, S.; Tsuchida, K. Mesenchymal progenitors distinct from satellite cells contribute to ectopic fat cell formation in skeletal muscle. Nat. Cell Biol. 2010, 12, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Uezumi, A.; Ito, T.; Morikawa, D.; Shimizu, N.; Yoneda, T.; Segawa, M.; Yamaguchi, M.; Ogawa, R.; Matev, M.M.; Miyagoe-Suzuki, Y.; et al. Fibrosis and adipogenesis originate from a common mesenchymal progenitor in skeletal muscle. J. Cell. Sci. 2011, 124, 3654–3664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contreras, O.; Cruz-Soca, M.; Theret, M.; Soliman, H.; Tung, L.W.; Groppa, E.; Rossi, F.M.; Brandan, E. Cross-talk between TGF-β and PDGFRα signaling pathways regulates the fate of stromal fibro-adipogenic progenitors. J. Cell. Sci. 2019, 132. [Google Scholar] [CrossRef]
- Contreras, O.; Rebolledo, D.L.; Oyarzún, J.E.; Olguín, H.C.; Brandan, E. Connective tissue cells expressing fibro/adipogenic progenitor markers increase under chronic damage: Relevance in fibroblast-myofibroblast differentiation and skeletal muscle fibrosis. Cell Tissue Res. 2016, 364, 647–660. [Google Scholar] [CrossRef]
- Lemos, D.R.; Babaeijandaghi, F.; Low, M.; Chang, C.-K.; Lee, S.T.; Fiore, D.; Zhang, R.-H.; Natarajan, A.; Nedospasov, S.A.; Rossi, F.M.V. Nilotinib reduces muscle fibrosis in chronic muscle injury by promoting TNF-mediated apoptosis of fibro/adipogenic progenitors. Nat. Med. 2015, 21, 786–794. [Google Scholar] [CrossRef]
- Lukjanenko, L.; Karaz, S.; Stuelsatz, P.; Gurriaran-Rodriguez, U.; Michaud, J.; Dammone, G.; Sizzano, F.; Mashinchian, O.; Ancel, S.; Migliavacca, E.; et al. Aging Disrupts Muscle Stem Cell Function by Impairing Matricellular WISP1 Secretion from Fibro-Adipogenic Progenitors. Cell Stem Cell 2019, 24, 433–446. [Google Scholar] [CrossRef] [Green Version]
- Yahata, T.; Ibrahim, A.A.; Muguruma, Y.; Eren, M.; Shaffer, A.M.; Watanabe, N.; Kaneko, S.; Nakabayashi, T.; Dan, T.; Hirayama, N.; et al. TGF-β–induced intracellular PAI-1 is responsible for retaining hematopoietic stem cells in the niche. Blood 2017, 130, 2283–2294. [Google Scholar] [CrossRef] [Green Version]
- Cui, C.; Driscoll, R.K.; Piao, Y.; Chia, C.W.; Gorospe, M.; Ferrucci, L. Skewed macrophage polarization in aging skeletal muscle. Aging Cell 2019, 18. [Google Scholar] [CrossRef] [Green Version]
- Dadgar, S.; Wang, Z.; Johnston, H.; Kesari, A.; Nagaraju, K.; Chen, Y.-W.; Hill, D.A.; Partridge, T.A.; Giri, M.; Freishtat, R.J.; et al. Asynchronous remodeling is a driver of failed regeneration in Duchenne muscular dystrophy. J. Cell Biol. 2014, 207, 139–158. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Iezzi, S.; Carver, E.; Dressman, D.; Gridley, T.; Sartorelli, V.; Hoffman, E.P. Slug is a novel downstream target of MyoD. Temporal profiling in muscle regeneration. J. Biol. Chem. 2002, 277, 30091–30101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koensgen, D.; Stope, M.B.; Tuerbachova, I.; Bruennert, D.; Kohlmann, T.; Braicu, I.; Sehouli, J.; Denkert, C.; Darb-Esfahani, S.; Stickeler, E.; et al. Expression, Intracellular Localization, and Prognostic Value of Plasminogen Activator Inhibitor 1 and PAI-1 RNA-Binding Protein 1 in Primary and Recurrent Ovarian Cancer: A Study of the Tumor Bank Ovarian Cancer Network. Gynecol. Obstet. Invest. 2018, 83, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Eren, M.; Boe, A.E.; Murphy, S.B.; Place, A.T.; Nagpal, V.; Morales-Nebreda, L.; Urich, D.; Quaggin, S.E.; Budinger, G.R.S.; Mutlu, G.M.; et al. PAI-1–regulated extracellular proteolysis governs senescence and survival in Klotho mice. Proc. Natl. Acad. Sci. USA 2014, 111, 7090–7095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pöschl, E.; Schlötzer-Schrehardt, U.; Brachvogel, B.; Saito, K.; Ninomiya, Y.; Mayer, U. Collagen IV is essential for basement membrane stability but dispensable for initiation of its assembly during early development. Development 2004, 131, 1619–1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nederveen, J.P.; Joanisse, S.; Thomas, A.C.Q.; Snijders, T.; Manta, K.; Bell, K.E.; Phillips, S.M.; Kumbhare, D.; Parise, G. Age-related changes to the satellite cell niche are associated with reduced activation following exercise. FASEB J. 2020. [Google Scholar] [CrossRef]
- Gulati, A.K.; Reddi, A.H.; Zalewski, A.A. Distribution of fibronectin in normal and regenerating skeletal muscle. Anat. Rec. 1982, 204, 175–183. [Google Scholar] [CrossRef]
- Lukjanenko, L.; Jung, M.J.; Hegde, N.; Perruisseau-Carrier, C.; Migliavacca, E.; Rozo, M.; Karaz, S.; Jacot, G.; Schmidt, M.; Li, L.; et al. Loss of fibronectin from the aged stem cell niche affects the regenerative capacity of skeletal muscle in mice. Nat. Med. 2016, 22, 897–905. [Google Scholar] [CrossRef] [Green Version]
- Tam, C.S.; Sparks, L.M.; Johannsen, D.L.; Covington, J.D.; Church, T.S.; Ravussin, E. Low macrophage accumulation in skeletal muscle of obese type 2 diabetics and elderly subjects. Obesity 2012, 20, 1530–1533. [Google Scholar] [CrossRef] [Green Version]
- Przybyla, B.; Gurley, C.; Harvey, J.F.; Bearden, E.; Kortebein, P.; Evans, W.J.; Sullivan, D.H.; Peterson, C.A.; Dennis, R.A. Aging alters macrophage properties in human skeletal muscle both at rest and in response to acute resistance exercise. Exp. Gerontol. 2006, 41, 320–327. [Google Scholar] [CrossRef]
- Villalta, S.A.; Nguyen, H.X.; Deng, B.; Gotoh, T.; Tidball, J.G. Shifts in macrophage phenotypes and macrophage competition for arginine metabolism affect the severity of muscle pathology in muscular dystrophy. Hum. Mol. Genet. 2009, 18, 482–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Mouse in Biomedical Research, 2nd ed. Volume 3. Available online: https://www.elsevier.com/books/the-mouse-in-biomedical-research/fox/978-0-12-369457-7 (accessed on 6 May 2020).
- Zhang, X. Hepatocyte growth factor system in the mouse uterus: Variation across the estrous cycle and regulation by 17-beta-estradiol and progesterone. Biol. Reprod. 2010, 82, 1037–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bibby, P. Simple Main Effects. In Encyclopedia of Research Design; SAGE Publications, Inc.: Thousand Oaks, CA, USA, 2010; ISBN 978-1-4129-6127-1. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Supplier | Catalogue | Host Species | Concentration |
|---|---|---|---|---|
| Collagen I | Abcam | ab34710 | Rabbit | 1:200 |
| Collagen IV | Abcam | ab6586 | Rabbit | 1:300 |
| Fibronectin | Abcam | ab23750 | Rabbit | 1:300 |
| PAI-1 | Abcam | ab66705 | Rabbit | 1:200 |
| MMP-9 | Abcam | ab38898 | Rabbit | 1:200 |
| eMHC | DSHB | F1.652 | Mouse | 1:1 |
| F4/80 | Abcam | ab90247 | Rat | 1:100 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, F.A.; Angus, S.A.; Stokes, K.; Karpowicz, P.; Krause, M.P. Impaired ECM Remodeling and Macrophage Activity Define Necrosis and Regeneration Following Damage in Aged Skeletal Muscle. Int. J. Mol. Sci. 2020, 21, 4575. https://doi.org/10.3390/ijms21134575
Rahman FA, Angus SA, Stokes K, Karpowicz P, Krause MP. Impaired ECM Remodeling and Macrophage Activity Define Necrosis and Regeneration Following Damage in Aged Skeletal Muscle. International Journal of Molecular Sciences. 2020; 21(13):4575. https://doi.org/10.3390/ijms21134575
Chicago/Turabian StyleRahman, Fasih Ahmad, Sarah Anne Angus, Kyle Stokes, Phillip Karpowicz, and Matthew Paul Krause. 2020. "Impaired ECM Remodeling and Macrophage Activity Define Necrosis and Regeneration Following Damage in Aged Skeletal Muscle" International Journal of Molecular Sciences 21, no. 13: 4575. https://doi.org/10.3390/ijms21134575
APA StyleRahman, F. A., Angus, S. A., Stokes, K., Karpowicz, P., & Krause, M. P. (2020). Impaired ECM Remodeling and Macrophage Activity Define Necrosis and Regeneration Following Damage in Aged Skeletal Muscle. International Journal of Molecular Sciences, 21(13), 4575. https://doi.org/10.3390/ijms21134575