Rounding up the Usual Suspects: Assessing Yorkie, AP-1, and Stat Coactivation in Tumorigenesis



Abstract
:1. Introduction
1.1. Studying Tumorigenesis in Drosophila is Fast, Cheap, and Effective
1.2. Cooperation between Different Signaling Pathways Is a Hallmark of Tumorigenesis
2. Background and Approach
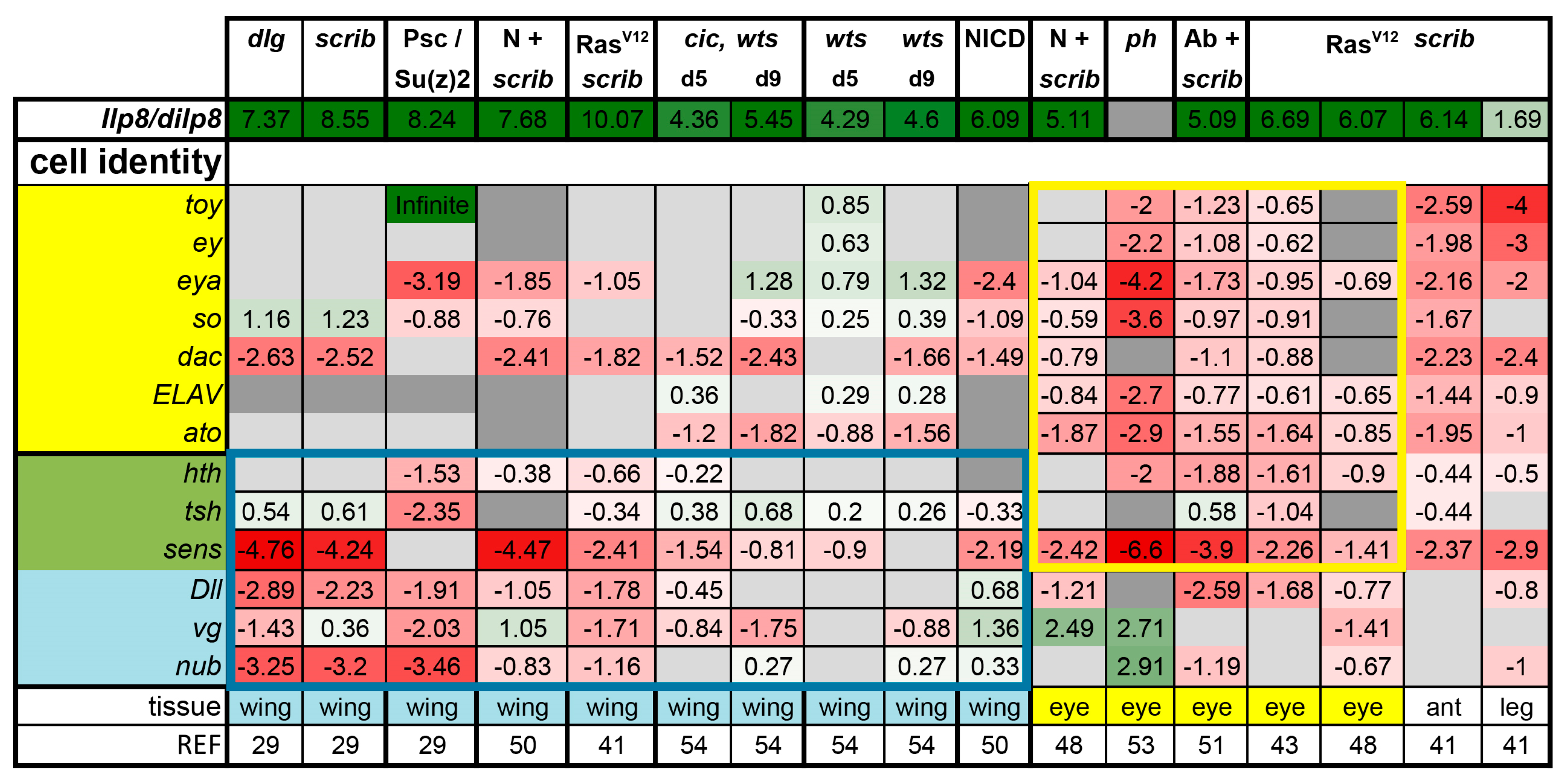
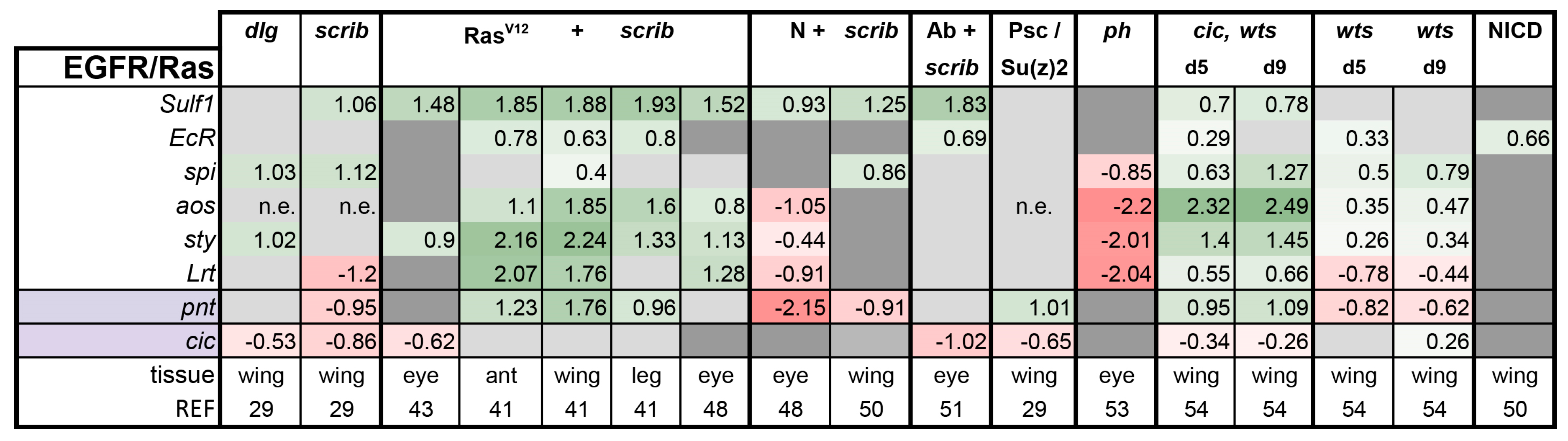
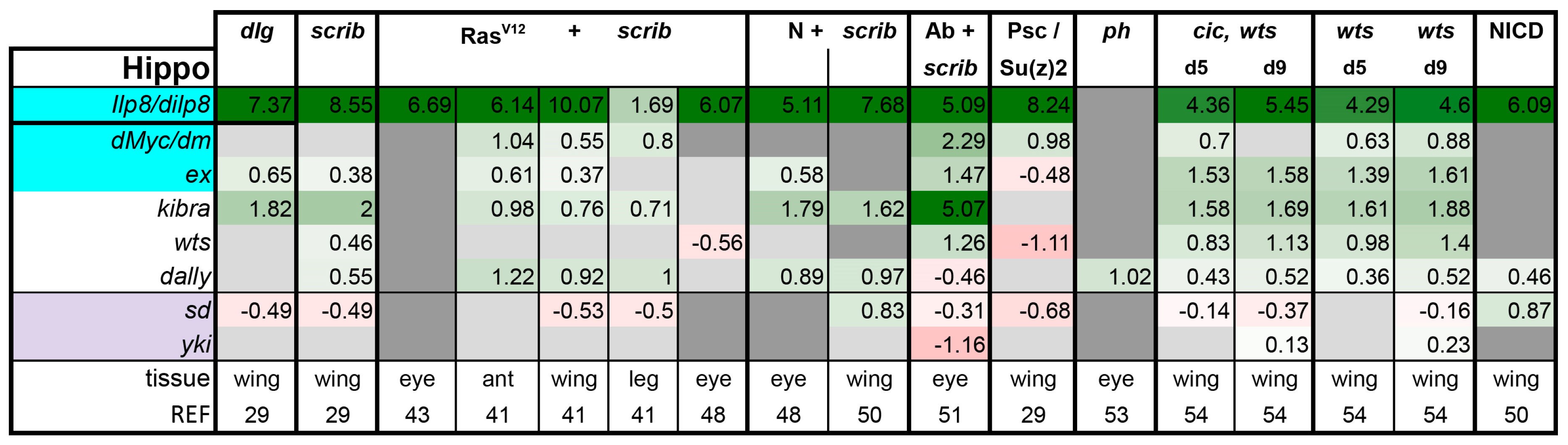
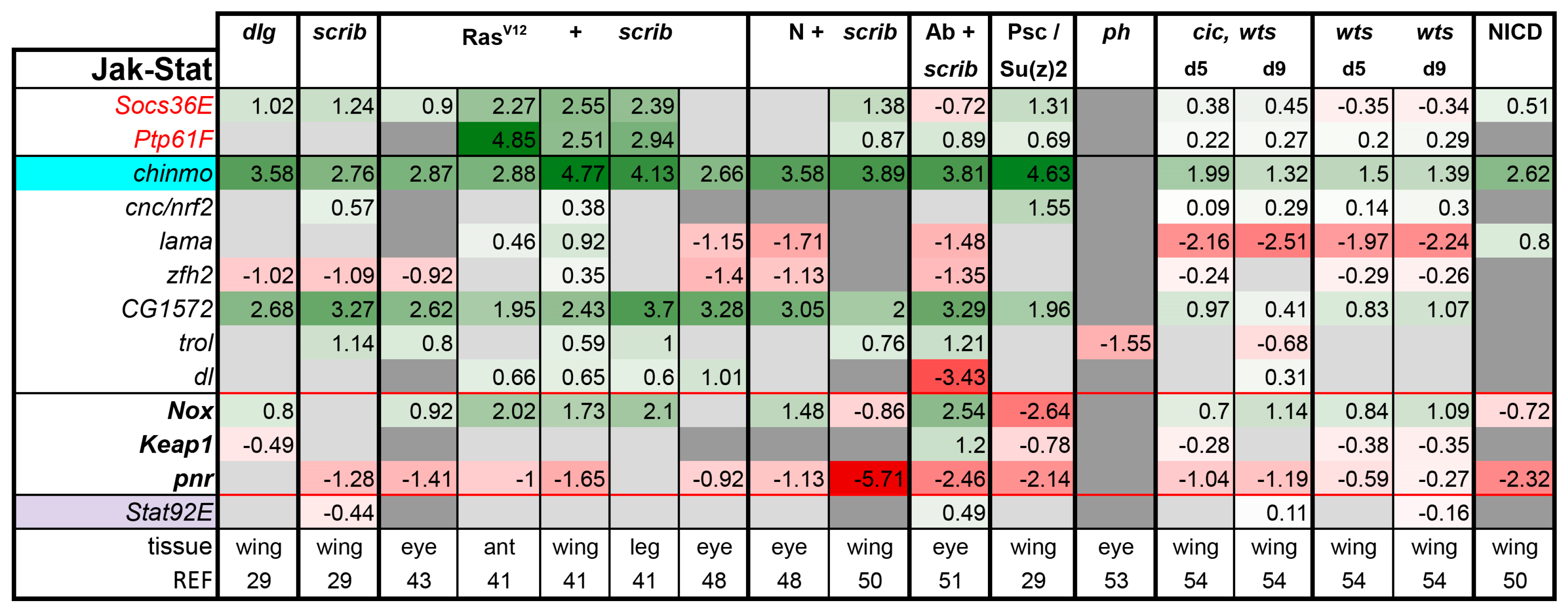
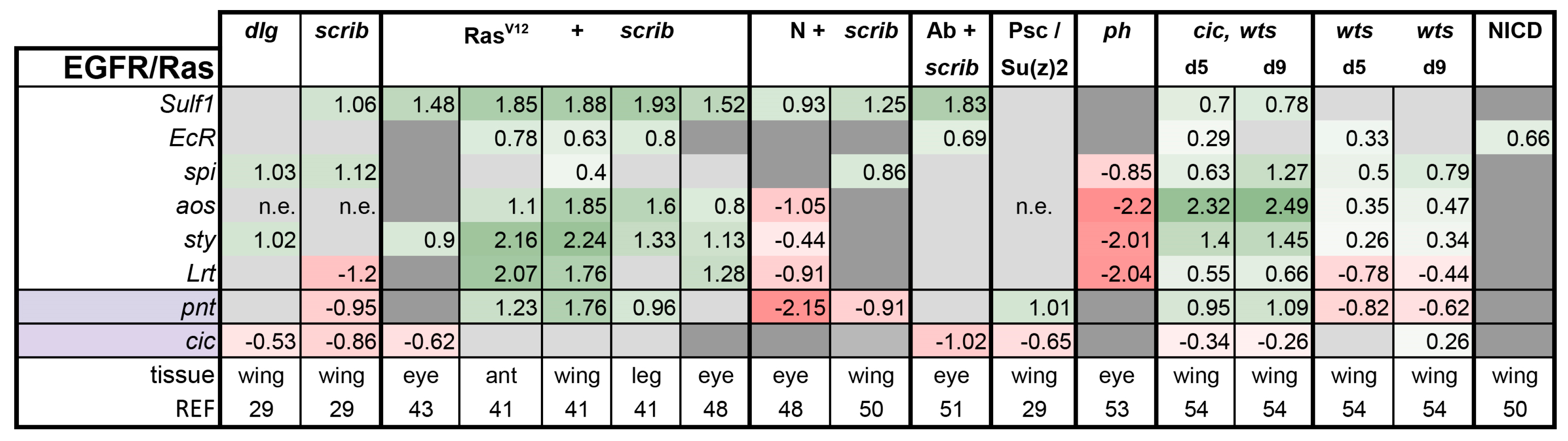
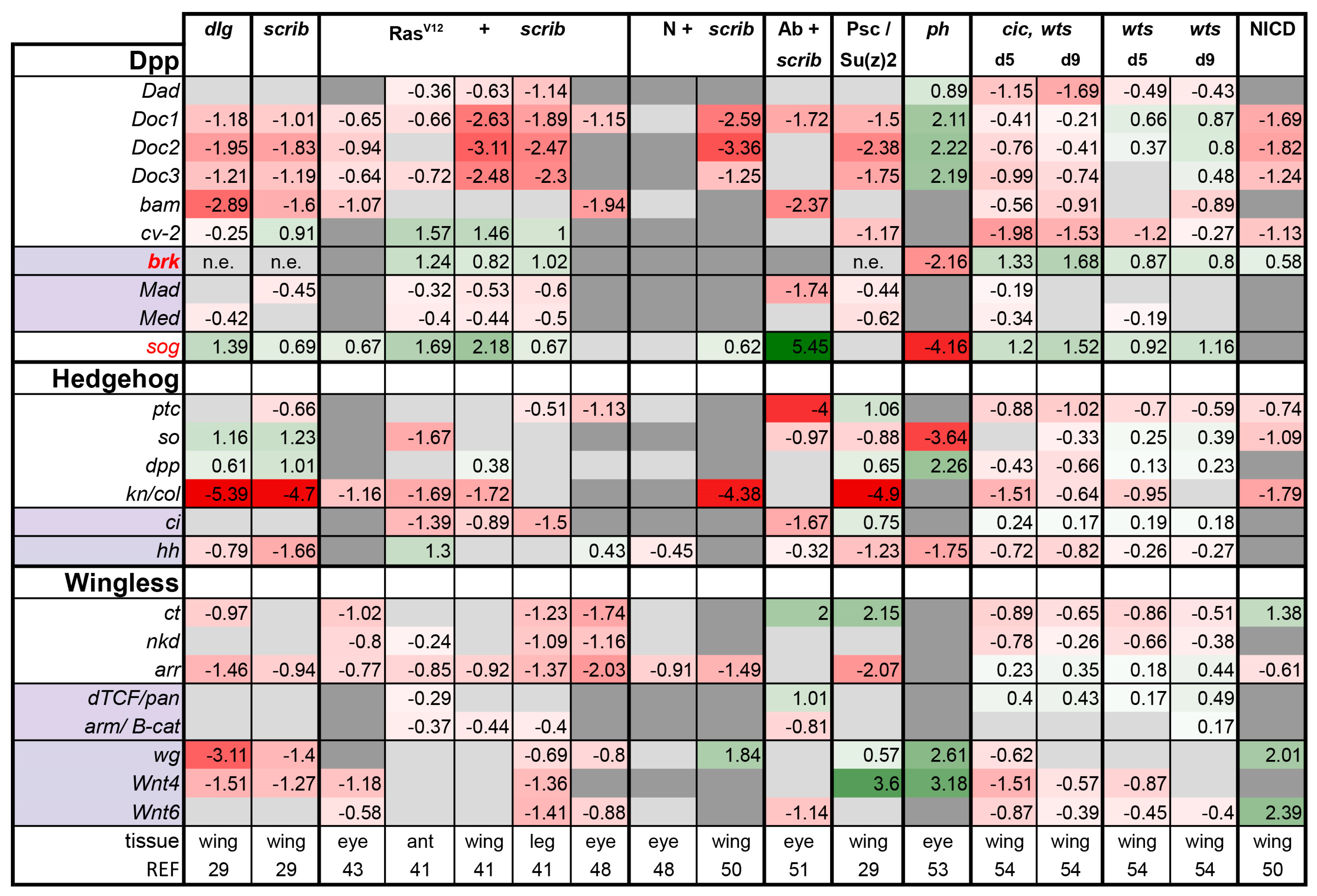
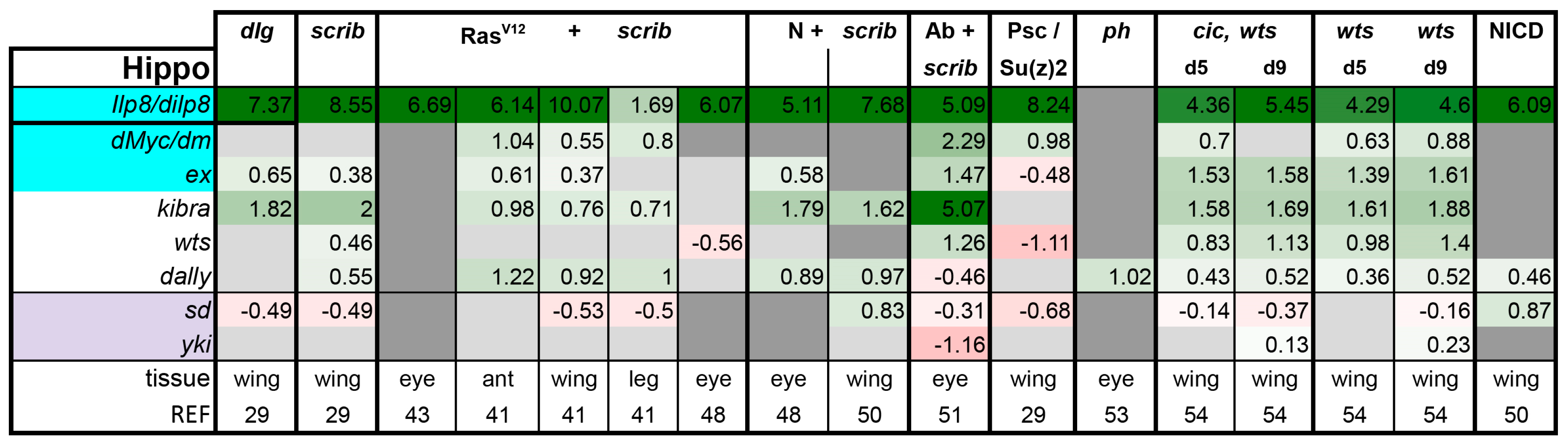
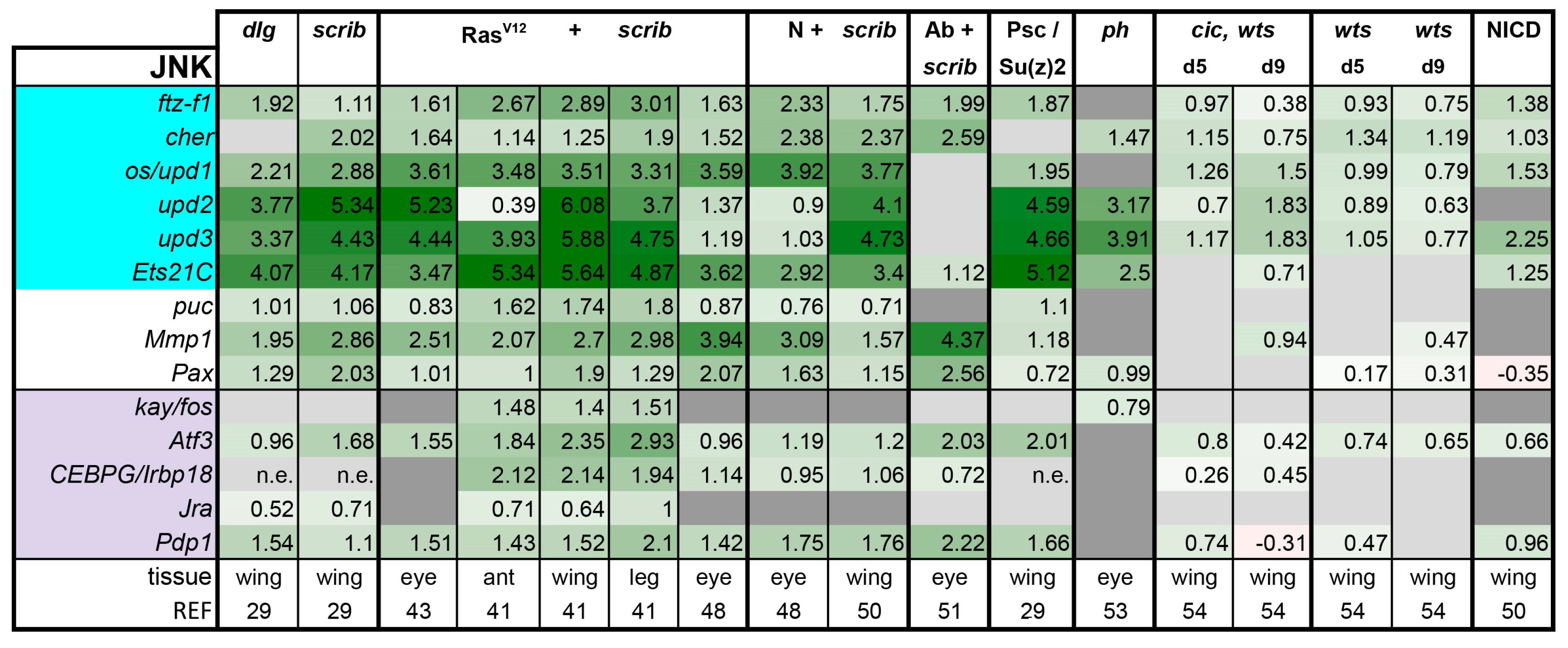
Is There a Common Signaling Signature of Tumorigenesis?
- (1)
- (2)
- scrib mutant wing discs, data from [29]. Dlg and Scrib act together in a complex and scrib mutant wing discs phenocopy dlg mutants, about 70% of all genes that are differentially expressed in dlg mutant wing discs are also differentially expressed in scrib discs; 311 upregulated and 263 downregulated genes [29].
- (3)
- (4)
- N + scrib-: A microarray analysis was performed on N + scrib- tumors revealing common as well as unique changes compared with RasV12 + scrib- tumors in the eye [43]. These tumors express NICD in scrib- mitotic clones. Transcriptomes of wing discs with N + scrib- tumors were also published recently [50]. N + scrib- tumors are neoplastic in nature.
- (5)
- Abrupt + scrib-: The zinc finger transcription factor Abrupt was identified as a scrib cooperating oncogene in a screen [51]. Ectopic Abrupt expression has no discernable phenotypes on differentiation and gives the cells a slight growth advantage, whereas Abrupt overexpression in scrib- cells maintains cells in a progenitor-like state and prevents the formation of photoreceptors. Eye discs with such cells are severely overgrown and neoplastic [51,52].
- (6)
- (7)
- polyhomeotic (ph) mutants (ph-p and ph-d double), member of the PRC1, which often display a loss of polarity along with overgrowth [33]. Clones can be invasive, and display cooperative tumorigenesis with RasV12 [33]. A small proportion of animals with ph clones in the eye can reach adulthood and display overgrown eyes [33,53].
- (8)
- capicua, warts (cic, wts) double mutants: Cic is the transcriptional repressor of EGFR/Ras signaling [54]. The Ras/Raf/MAPK Kinase (MEK) cascade culminates in activation of Mitogen-Activated Protein Kinase (MAPK), which targets Cic for degradation, allowing target gene induction [55]. Wts kinase acts in the Hippo pathway [49,56,57,58,59,60]. In wts mutants, Yki is stabilized and can accumulate in the nucleus and help induce expression of genes that drive cell growth (such as Myc), proliferation (e.g., Cyclin E), as well as conferring apoptotic resistance via induction of Diap1 [7,8,61]. Hippo signaling controls the transcriptional output of the Ras pathway and their mutual disruption, as in cic,wts mutants, causes synergistic overgrowth in larval discs [54]. Such discs stay hyperplastic and lose apical-basal polarity only at the very late stages [54]. We have data for day 5 (prior to overgrowth) and with overgrown, heavily folded day 9 discs [54].
- (9)
- wts mutants at day 5 and day 9, data from [54]. These discs are hyperplastic owing to overactivation of Yki.
- (10)
- NICD-overexpressing wing discs display hyperplastic overgrowth, data from [50].
3. Analysis/Results
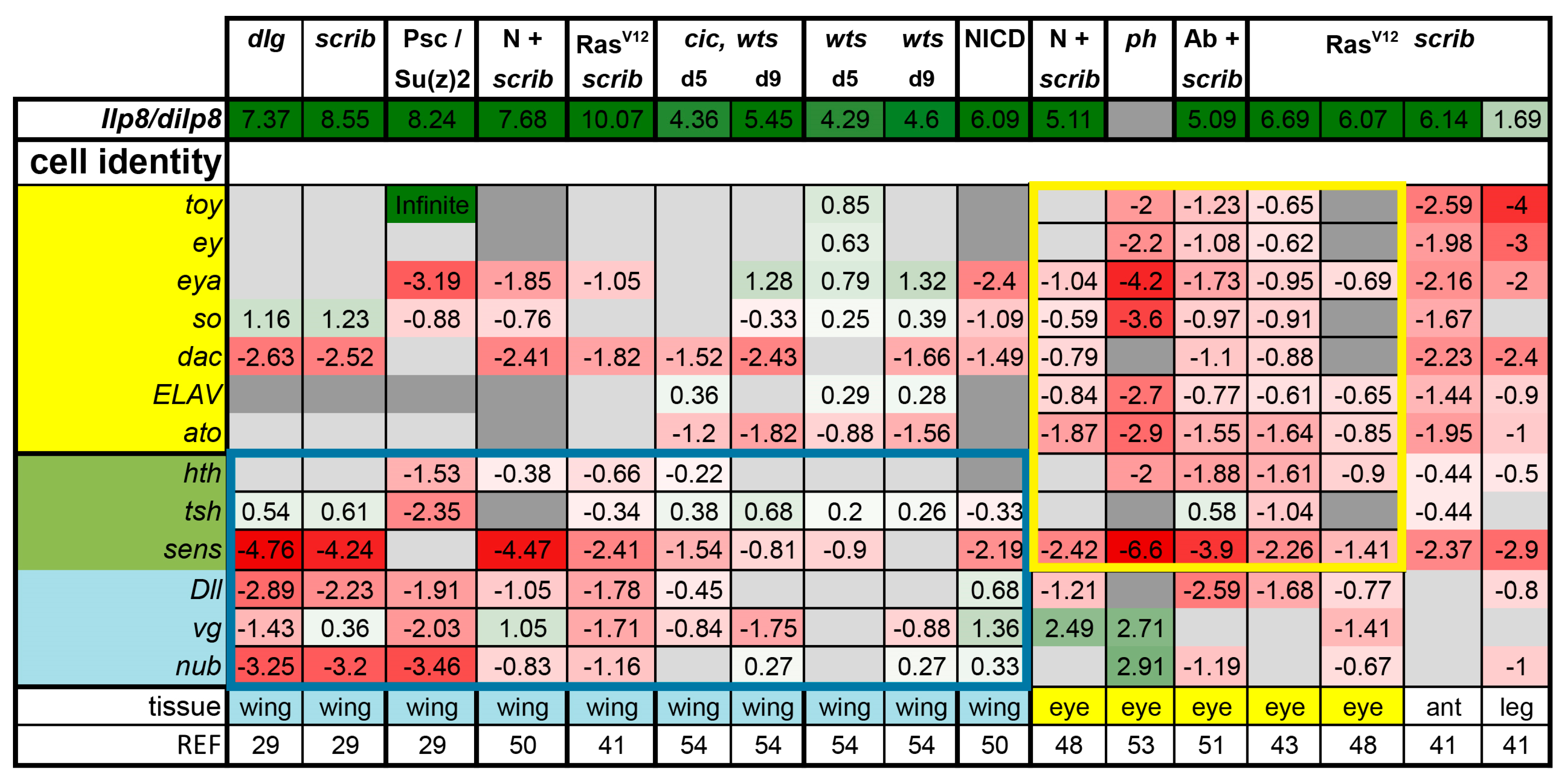
3.1. Tumors Cause Delayed Pupariation and Loss of Cell Fate Specification
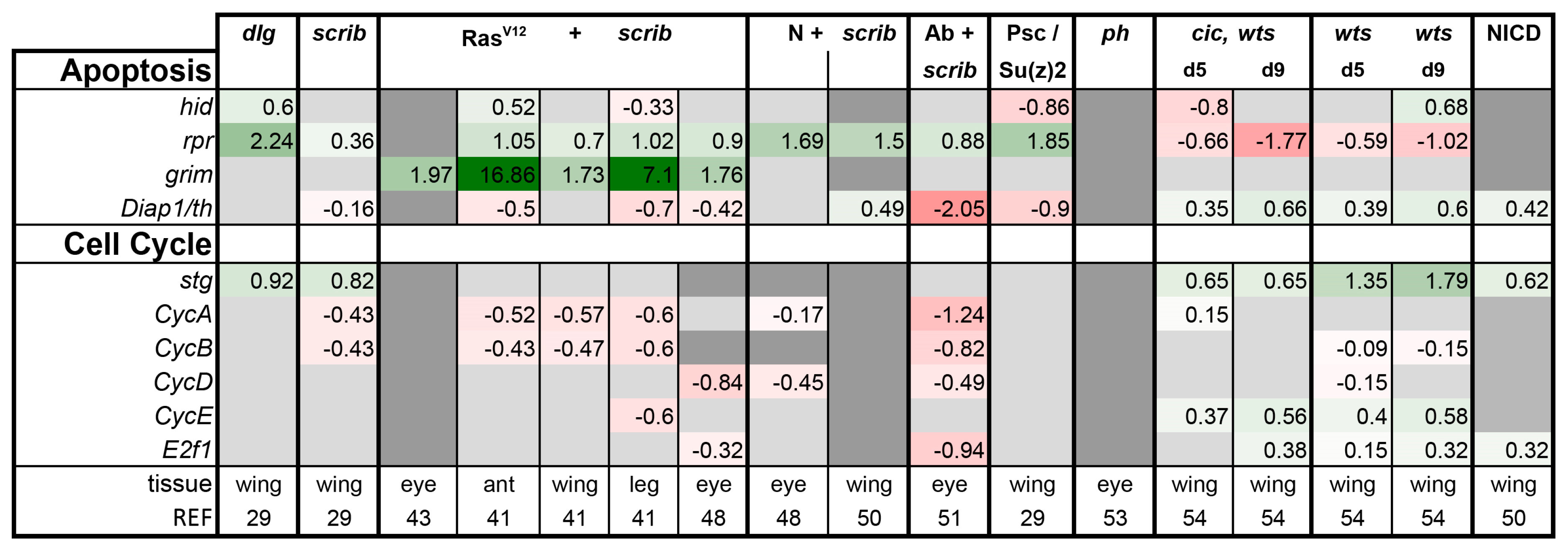
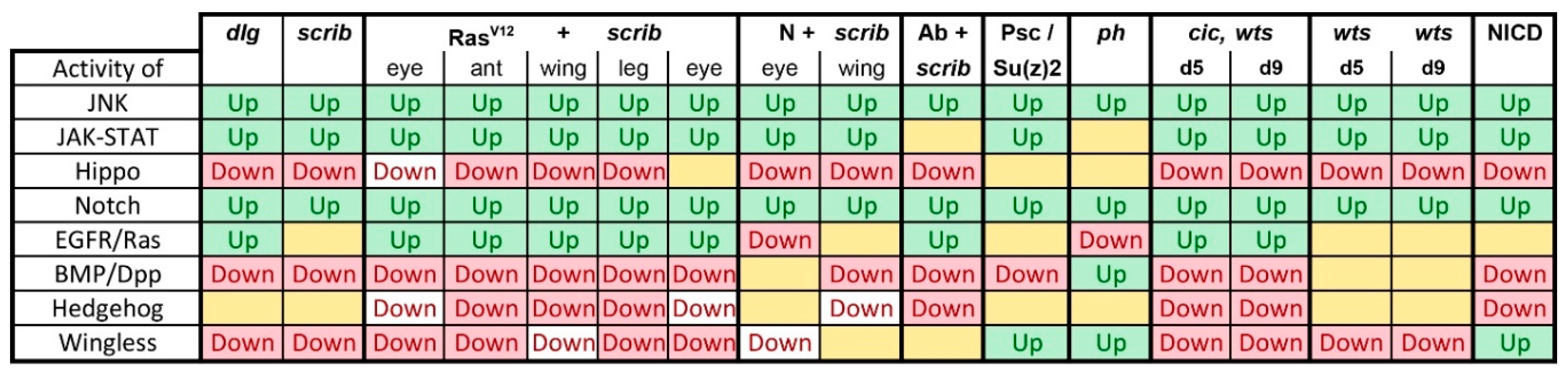
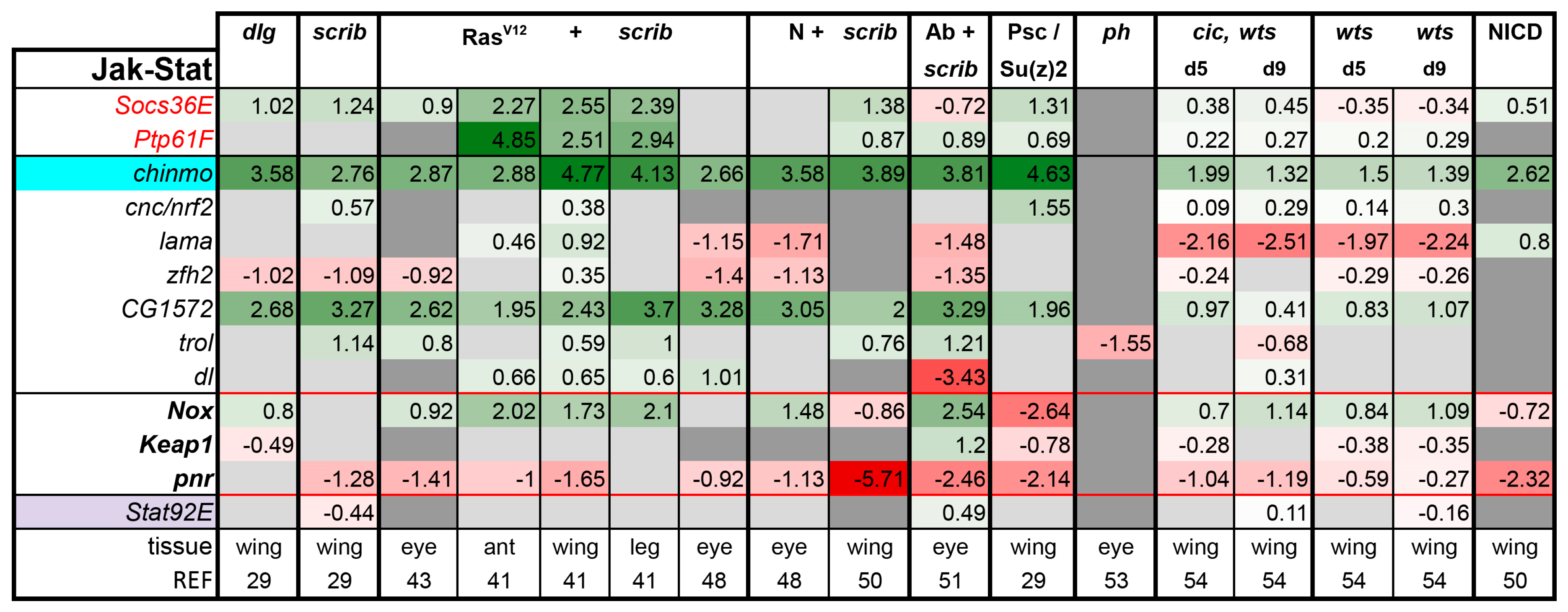
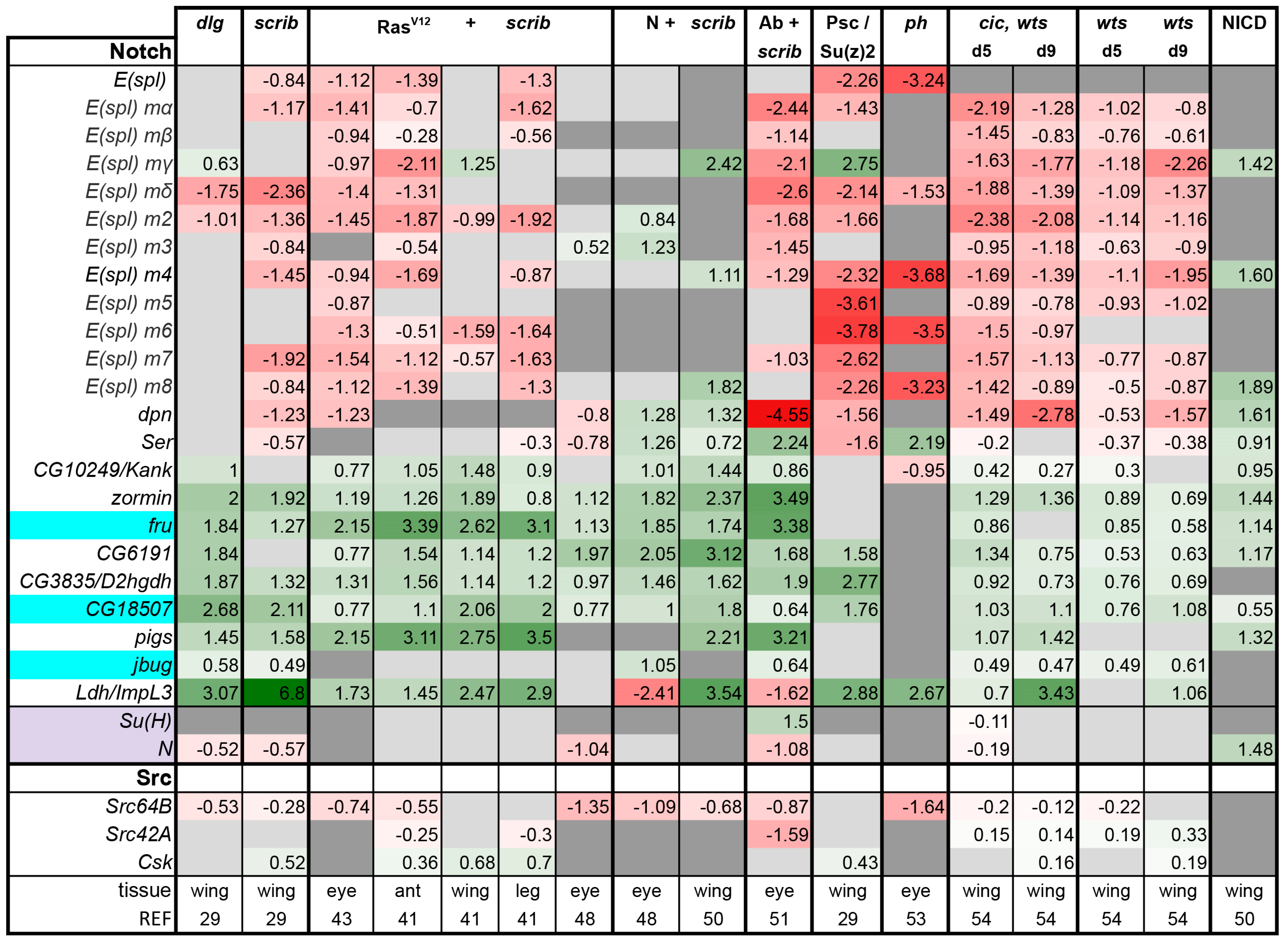
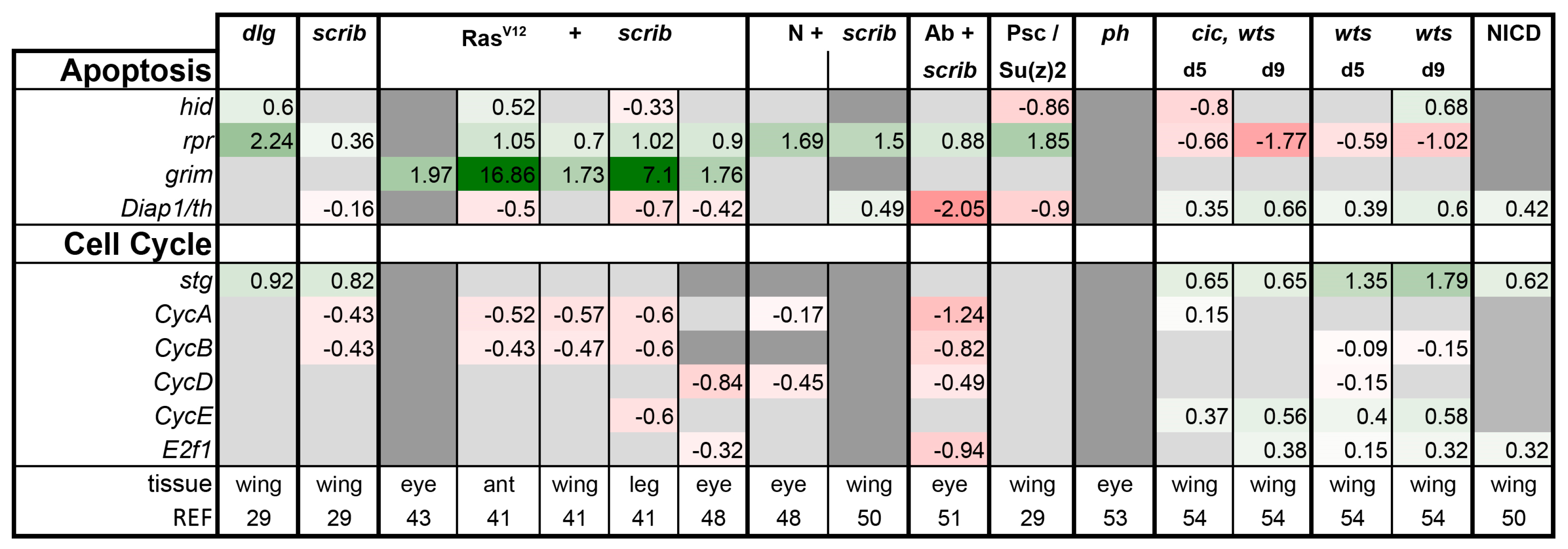
3.2. Inactivation of Hippo, Dpp, Hh, Wg, and Activation of JNK, JAK/STAT, and Notch Are Commonly Seen in Tumors
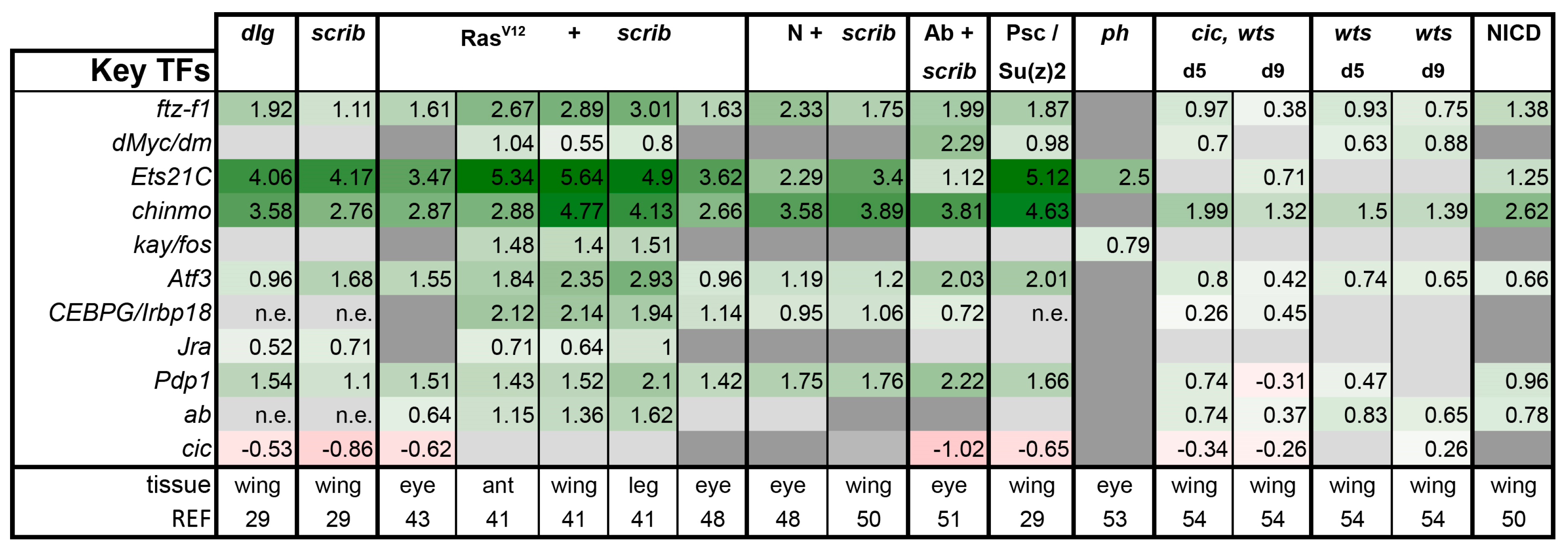
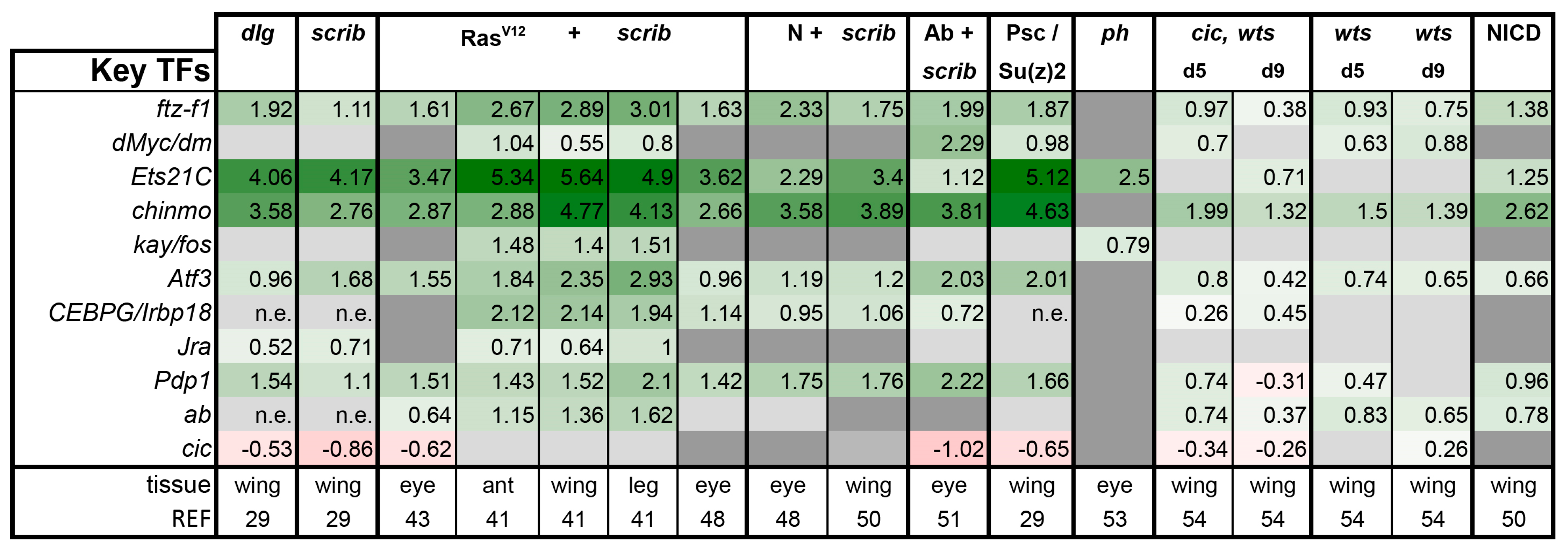
3.3. A Set of Transcription Factors Is Commonly Upregulated in Tumors
4. Discussion and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sonoshita, M.; Cagan, R.L. Modeling Human Cancers in Drosophila. Curr. Top. Dev. Biol. 2017, 121, 287–309. [Google Scholar]
- Richardson, H.E.; Portela, M. Modelling Cooperative Tumorigenesis in Drosophila. Biomed. Res. Int. 2018, 2018, 4258387. [Google Scholar] [CrossRef] [Green Version]
- Herranz, H.; Eichenlaub, T.; Cohen, S.M. Cancer in Drosophila: Imaginal Discs as a Model for Epithelial Tumor Formation. Curr. Top. Dev. Biol. 2016, 116, 181–199. [Google Scholar] [PubMed]
- Bangi, E.; Ang, C.; Smibert, P.; Uzilov, A.V.; Teague, A.G.; Antipin, Y.; Chen, R.; Hecht, C.; Gruszczynski, N.; Yon, W.J.; et al. A personalized platform identifies trametinib plus zoledronate for a patient with KRAS-mutant metastatic colorectal cancer. Sci. Adv. 2019, 5, eaav6528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonoshita, M.; Scopton, A.P.; Ung, P.M.U.; Murray, M.A.; Silber, L.; Maldonado, A.Y.; Real, A.; Schlessinger, A.; Cagan, R.L.; Dar, A.C. A whole-animal platform to advance a clinical kinase inhibitor into new disease space. Nat. Methods 2018, 14, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Bangi, E.; Murgia, C.; Teague, A.G.; Sansom, O.J.; Cagan, R.L. Functional exploration of colorectal cancer genomes using Drosophila. Nat. Commun. 2016, 7, 13615. [Google Scholar] [CrossRef] [PubMed]
- Harvey, K.F.; Tapon, N. The Salvador–Warts–Hippo pathway—An emerging tumour-suppressor network. Nat. Rev. Cancer 2007, 7, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Pan, D. Hippo signaling in organ size control. Genes Dev. 2007, 21, 886–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halder, G.; Johnson, R.L. Hippo signaling: Growth control and beyond. Development 2011, 138, 9–22. [Google Scholar] [CrossRef] [Green Version]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef]
- Harvey, K.F.; Zhang, X.; Thomas, D.M. The Hippo pathway and human cancer. Nat. Rev. Cancer 2013, 13, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Jenny, F.H.; Basler, K. Powerful Drosophila screens that paved the wingless pathway. Fly (Austin) 2014, 8, 218–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamaratoglu, F.; Affolter, M.; Pyrowolakis, G. Dpp/BMP signaling in flies: From molecules to biology. Semin. Cell Dev. Biol. 2014, 32, 128–136. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, S.; Schulze, K.L.; Bellen, H.J. Introduction to Notch signaling. Methods Mol. Biol. 2014, 1187, 1–14. [Google Scholar] [PubMed]
- Casci, T.; Freeman, M. Control of EGF receptor signalling: Lessons from fruitflies. Cancer Metastasis Rev. 1999, 18, 181–201. [Google Scholar] [CrossRef] [PubMed]
- Oldham, S.; Hafen, E. Insulin/IGF and target of rapamycin signaling: A TOR de force in growth control. Trends Cell Biol. 2003, 13, 79–85. [Google Scholar] [CrossRef]
- Davis, R.J. Signal transduction by the JNK group of MAP kinases. Cell 2000, 103, 239–252. [Google Scholar] [CrossRef] [Green Version]
- Weitzman, J.B. Quick guide. Jnk. Curr. Biol. 2000, 10, R290. [Google Scholar] [CrossRef] [Green Version]
- Pinal, N.; Calleja, M.; Morata, G. Pro-apoptotic and pro-proliferation functions of the JNK pathway of Drosophila: Roles in cell competition, tumorigenesis and regeneration. Open Biol. 2019, 9, 180256. [Google Scholar] [CrossRef] [Green Version]
- Herrera, S.C.; Bach, E.A. JAK/STAT signaling in stem cells and regeneration: From. Development 2019, 146, dev167643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Read, R.D.; Goodfellow, P.J.; Mardis, E.R.; Novak, N.; Armstrong, J.R.; Cagan, R.L. A Drosophila model of multiple endocrine neoplasia type 2. Genetics 2005, 171, 1057–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thornton, K.; Kim, G.; Maher, V.E.; Chattopadhyay, S.; Tang, S.; Moon, Y.J.; Song, P.; Marathe, A.; Balakrishnan, S.; Zhu, H.; et al. Vandetanib for the Treatment of Symptomatic or Progressive Medullary Thyroid Cancer in Patients with Unresectable Locally Advanced or Metastatic Disease: U.S. Food and Drug Administration Drug Approval Summary. Clin. Cancer Res. 2012, 18, 3722–3730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dar, A.C.; Das, T.K.; Shokat, K.; Cagan, R.L. Chemical genetic discovery of targets and anti-targets for cancer polypharmacology. Nature 2012, 486, 80–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasai, Y.; Cagan, R. Drosophila as a tool for personalized medicine: A primer. Per Med. 2010, 7, 621–632. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337.e10. [Google Scholar] [CrossRef] [Green Version]
- Bryant, P.J.; Schmidt, O. The genetic control of cell proliferation in Drosophila imaginal discs. J. Cell Sci. Suppl. 1990, 13, 169–189. [Google Scholar] [CrossRef] [Green Version]
- Humbert, P.; Russell, S.; Richardson, H. Dlg, Scribble and Lgl in cell polarity, cell proliferation and cancer. Bioessays 2003, 25, 542–553. [Google Scholar] [CrossRef]
- Bunker, B.D.; Nellimoottil, T.T.; Boileau, R.M.; Classen, A.-K.; Bilder, D. The transcriptional response to tumorigenic polarity loss in Drosophila. eLife 2015, 4, 4. [Google Scholar] [CrossRef]
- Classen, A.K.; Bunker, B.D.; Harvey, K.F.; Vaccari, T.; Bilder, D. A tumor suppressor activity of Drosophila Polycomb genes mediated by JAK-STAT signaling. Nat. Genet. 2009, 41, 1150–1155. [Google Scholar] [CrossRef] [Green Version]
- Hodgson, J.W.; Cheng, N.N.; Sinclair, D.A.; Kyba, M.; Randsholt, N.B.; Brock, H.W. The polyhomeotic locus of Drosophila melanogaster is transcriptionally and post-transcriptionally regulated during embryogenesis. Mech. Dev. 1997, 66, 69–81. [Google Scholar] [CrossRef]
- Dura, J.-M.; Randsholt, N.B.; Deatrick, J.; Erk, I.; Santamaria, P.; Freeman, J.; Freeman, S.J.; Weddell, D.; Brock, H.W. A complex genetic locus, polyhomeotic, is required for segmental specification and epidermal development in D. melanogaster. Cell 1987, 51, 829–839. [Google Scholar] [CrossRef]
- Martinez, A.M.; Schuettengruber, B.; Sakr, S.; Janic, A.; Gonzalez, C.; Cavalli, G. Polyhomeotic has a tumor suppressor activity mediated by repression of Notch signaling. Nat. Genet. 2009, 41, 1076–1082. [Google Scholar] [CrossRef] [PubMed]
- Beira, J.V.; Torres, J.; Paro, R. Signalling crosstalk during early tumorigenesis in the absence of Polycomb silencing. PLoS Genet. 2018, 14, e1007187. [Google Scholar] [CrossRef]
- Brumby, A.M.; Richardson, H.E. Scribble mutants cooperate with oncogenic Ras or Notch to cause neoplastic overgrowth in Drosophila. EMBO J. 2003, 22, 5769–5779. [Google Scholar] [CrossRef]
- Pagliarini, R.A.; Xu, T. A genetic screen in Drosophila for metastatic behavior. Science 2003, 302, 1227–1231. [Google Scholar] [CrossRef]
- Fahey-Lozano, N.; La Marca, J.E.; Portela, M.; Richardson, H.E. Drosophila Models of Cell Polarity and Cell Competition in Tumourigenesis. Adv. Exp. Med. Biol. 2019, 1167, 37–64. [Google Scholar]
- Ohsawa, S. Elimination of oncogenic cells that regulate epithelial homeostasis in Drosophila. Dev. Growth Differ. 2019, 61, 337–342. [Google Scholar] [CrossRef] [Green Version]
- Merino, M.M.; Levayer, R.; Moreno, E. Survival of the Fittest: Essential Roles of Cell Competition in Development, Aging, and Cancer. Trends Cell Biol. 2016, 26, 776–788. [Google Scholar] [CrossRef]
- Morata, G.; Calleja, M. Cell competition and tumorigenesis in the imaginal discs of Drosophila. Semin. Cancer Biol. 2020, 63, 19–26. [Google Scholar] [CrossRef]
- Atkins, M.; Potier, D.; Romanelli, L.; Jacobs, J.; Mach, J.; Hamaratoglu, F.; Aerts, S.; Halder, G. An Ectopic Network of Transcription Factors Regulated by Hippo Signaling Drives Growth and Invasion of a Malignant Tumor Model. Curr. Biol. 2016, 26, 2101–2113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davie, K.; Jacobs, J.; Atkins, M.; Potier, D.; Christiaens, V.; Halder, G.; Aerts, S. Discovery of Transcription Factors and Regulatory Regions Driving In Vivo Tumor Development by ATAC-seq and FAIRE-seq Open Chromatin Profiling. PLoS Genet. 2015, 11, e1004994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Külshammer, E.; Mundorf, J.; Kilinc, M.; Frommolt, P.; Wagle, P.; Uhlirova, M. Interplay among Drosophila transcription factors Ets21c, Fos and Ftz-F1 drives JNK-mediated tumor malignancy. Dis. Model Mech. 2015, 8, 1279–1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhlirova, M.; Bohmann, D. JNK- and Fos-regulated Mmp1 expression cooperates with Ras to induce invasive tumors in Drosophila. EMBO J. 2006, 25, 5294–5304. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Pastor-Pareja, J.C.; Xu, T. Interaction between Ras(V12) and scribbled clones induces tumour growth and invasion. Nature 2010, 463, 545–548. [Google Scholar] [CrossRef] [Green Version]
- Igaki, T.; Pagliarini, R.A.; Xu, T. Loss of cell polarity drives tumor growth and invasion through JNK activation in Drosophila. Curr. Biol. 2006, 16, 1139–1146. [Google Scholar] [CrossRef] [Green Version]
- Bilder, D.; Li, M.; Perrimon, N. Cooperative regulation of cell polarity and growth by Drosophila tumor suppressors. Science 2000, 289, 113–116. [Google Scholar] [CrossRef] [Green Version]
- Doggett, K.; Turkel, N.; Willoughby, L.F.; Ellul, J.; Murray, M.J.; Richardson, H.E.; Brumby, A.M. BTB-Zinc Finger Oncogenes Are Required for Ras and Notch-Driven Tumorigenesis in Drosophila. PLoS ONE 2015, 10, e0132987. [Google Scholar] [CrossRef] [Green Version]
- Pantalacci, S.; Tapon, N.; Leopold, P. The Salvador partner Hippo promotes apoptosis and cell-cycle exit in Drosophila. Nat. Cell Biol. 2003, 5, 921–927. [Google Scholar] [CrossRef]
- Logeay, R.; Géminard, C.; Lassus, P.; Héron-Milhavet, L.; Profile, V.O.; Fischer, B.; Bray, S.J.; Colinge, J.; Djiane, A. Loss of epithelial polarity redirects Notch signaling and triggers a Xrp1 response during neoplastic growth in Drosophila. BioRxiv 2020. [Google Scholar]
- Turkel, N.; Sahota, V.K.; Bolden, J.E.; Goulding, K.R.; Doggett, K.; Willoughby, L.F.; Blanco, E.; Martin-Blanco, E.; Corominas, M.; Ellul, J.; et al. The BTB-zinc finger transcription factor abrupt acts as an epithelial oncogene in Drosophila melanogaster through maintaining a progenitor-like cell state. PLoS Genet. 2013, 9, e1003627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turkel, N.; Portela, M.; Poon, C.; Li, J.; Brumby, A.M.; Richardson, H.E. Cooperation of the BTB-Zinc finger protein, Abrupt, with cytoskeletal regulators in Drosophila epithelial tumorigenesis. Biol. Open 2015, 4, 1024–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres, J.; Monti, R.; Moore, A.L.; Seimiya, M.; Jiang, Y.; Beerenwinkel, N.; Beisel, C.; Beira, J.V.; Paro, R. A switch in transcription and cell fate governs the onset of an epigenetically-deregulated tumor in Drosophila. eLife 2018, 7, e32697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascual, J.; Jacobs, J.; Sansores-Garcia, L.; Natarajan, M.; Zeitlinger, J.; Aerts, S.; Halder, G.; Hamaratoglu, F. Hippo Reprograms the Transcriptional Response to Ras Signaling. Dev. Cell 2017, 42, 667–680. [Google Scholar] [CrossRef] [Green Version]
- Jiménez, G.; Shvartsman, S.Y.; Paroush, Z. The Capicua repressor—A general sensor of RTK signaling in development and disease. J. Cell Sci. 2012, 125, 1383–1391. [Google Scholar] [CrossRef] [Green Version]
- Udan, R.S.; Kango-Singh, M.; Nolo, R.; Tao, C.; Halder, G. Hippo promotes proliferation arrest and apoptosis in the Salvador/Warts pathway. Nat. Cell Biol. 2003, 5, 914–920. [Google Scholar] [CrossRef]
- Wu, S.; Huang, J.; Dong, J.; Pan, D. hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts. Cell 2003, 114, 445–456. [Google Scholar] [CrossRef] [Green Version]
- Tapon, N.; Harvey, K.F.; Bell, D.W.; Wahrer, K.C.; Schiripo, T.A.; Haber, D.A.; Hariharan, I.K. salvador Promotes Both Cell Cycle Exit and Apoptosis in Drosophila and Is Mutated in Human Cancer Cell Lines. Cell 2002, 110, 467. [Google Scholar] [CrossRef] [Green Version]
- Harvey, K.F.; Pfleger, C.M.; Hariharan, I.K. The Drosophila Mst ortholog, hippo, restricts growth and cell proliferation and promotes apoptosis. Cell 2003, 114, 457–467. [Google Scholar] [CrossRef] [Green Version]
- Jia, J.; Zhang, W.; Wang, B.; Trinko, R.; Jiang, J. The Drosophila Ste20 family kinase dMST functions as a tumor suppressor by restricting cell proliferation and promoting apoptosis. Genes Dev. 2003, 17, 2514–2519. [Google Scholar] [CrossRef] [Green Version]
- Neto-Silva, R.M.; de Beco, S.; Johnston, L.A. Evidence for a growth-stabilizing regulatory feedback mechanism between Myc and Yorkie, the Drosophila homolog of Yap. Dev. Cell. 2010, 19, 507–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gontijo, A.M.; Garelli, A. The biology and evolution of the Dilp8-Lgr3 pathway: A relaxin-like pathway coupling tissue growth and developmental timing control. Mech. Dev. 2018, 154, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Colombani, J.; Andersen, D.S.; Léopold, P. Secreted peptide Dilp8 coordinates Drosophila tissue growth with developmental timing. Science 2012, 336, 582–585. [Google Scholar] [CrossRef] [PubMed]
- Boone, E.; Colombani, J.; Andersen, D.S.; Léopold, P. The Hippo signalling pathway coordinates organ growth and limits developmental variability by controlling dilp8 expression. Nat. Commun. 2016, 7, 13505. [Google Scholar] [CrossRef]
- Sitaram, P.; Lu, S.; Harsh, S.; Herrera, S.C.; Bach, E.A. Next-Generation Sequencing Reveals Increased Anti-oxidant Response and Ecdysone Signaling in STAT Supercompetitors in. G3 (Bethesda) 2019, 9, 2609–2622. [Google Scholar] [CrossRef] [Green Version]
- Muller, B.; Hartmann, B.; Pyrowolakis, G.; Affolter, M.; Basler, K. Conversion of an extracellular Dpp/BMP morphogen gradient into an inverse transcriptional gradient. Cell 2003, 113, 221–233. [Google Scholar] [CrossRef] [Green Version]
- Pyrowolakis, G.; Hartmann, B.; Muller, B.; Basler, K.; Affolter, M. A simple molecular complex mediates widespread BMP-induced repression during Drosophila development. Dev. Cell. 2004, 7, 229–240. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, E.L.; Anderson, K.V. Localized enhancement and repression of the activity of the TGF-beta family member, decapentaplegic, is necessary for dorsal-ventral pattern formation in the Drosophila embryo. Genes Dev. 1992, 114, 583–597. [Google Scholar]
- Francois, V.; Solloway, M.; O’Neill, J.W.; Emery, J.; Bier, E. Dorsal-ventral patterning of the Drosophila embryo depends on a putative negative growth factor encoded by the short gastrulation gene. Genes Dev. 1994, 8, 2602–2616. [Google Scholar] [CrossRef] [Green Version]
- Rulifson, E.J.; Blair, S.S. Notch regulates wingless expression and is not required for reception of the paracrine wingless signal during wing margin neurogenesis in Drosophila. Development 1995, 121, 2813–2824. [Google Scholar]
- Djiane, A.; Krejci, A.; Bernard, F.; Fexova, S.; Millen, K.; Bray, S.J. Dissecting the mechanisms of Notch induced hyperplasia. EMBO J. 2013, 32, 60–71. [Google Scholar] [CrossRef] [Green Version]
- Justice, R.W.; Zilian, O.; Woods, D.F.; Noll, M.; Bryant, P.J. The Drosophila tumor suppressor gene warts encodes a homolog of human myotonic dystrophy kinase and is required for the control of cell shape and proliferation. Genes Dev. 1995, 9, 534–546. [Google Scholar] [CrossRef] [Green Version]
- Xu, T.; Wang, W.; Zhang, S.; Stewart, R.A.; Yu, W. Identifying tumor suppressors in genetic mosaics: The Drosophila lats gene encodes a putative protein kinase. Development 1995, 121, 1053–1063. [Google Scholar] [PubMed]
- Kango-Singh, M.; Nolo, R.; Tao, C.; Verstreken, P.; Hiesinger, P.R.; Bellen, H.J.; Halder, G. Shar-pei mediates cell proliferation arrest during imaginal disc growth in Drosophila. Development 2002, 129, 5719–5730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, H.E.; Portela, M. Tissue growth and tumorigenesis in Drosophila: Cell polarity and the Hippo pathway. Curr. Opin. Cell Biol. 2017, 48, 1–9. [Google Scholar] [CrossRef]
- Fulford, A.; Tapon, N.; Ribeiro, P.S. Upstairs, downstairs: Spatial regulation of Hippo signalling. Curr. Opin. Cell Biol. 2018, 51, 22–32. [Google Scholar] [CrossRef]
- Huang, J.; Wu, S.; Barrera, J.; Matthews, K.; Pan, D. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell 2005, 122, 421–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamaratoglu, F.; Willecke, M.; Kango-Singh, M.; Nolo, R.; Hyun, E.; Tao, C.; Jafar-Nejad, H.; Halder, G. The tumour-suppressor genes NF2/Merlin and Expanded act through Hippo signalling to regulate cell proliferation and apoptosis. Nat. Cell Biol. 2006, 8, 27–36. [Google Scholar] [CrossRef]
- Nolo, R.; Morrison, C.M.; Tao, C.; Zhang, X.; Halder, G. The bantam microRNA is a target of the hippo tumor-suppressor pathway. Curr. Biol. 2006, 16, 1895–1904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, B.J.; Cohen, S.M. The Hippo pathway regulates the bantam microRNA to control cell proliferation and apoptosis in Drosophila. Cell 2006, 126, 767–774. [Google Scholar] [CrossRef] [Green Version]
- Lucas, E.P.; Khanal, I.; Gaspar, P.; Fletcher, G.C.; Polesello, C.; Tapon, N.; Thompson, B.J. The Hippo pathway polarizes the actin cytoskeleton during collective migration of Drosophila border cells. J. Cell Biol. 2013, 201, 875–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sansores-Garcia, L.; Bossuyt, W.; Wada, K.; Yonemura, S.; Tao, C.; Sasaki, H.; Halder, G. Modulating F-actin organization induces organ growth by affecting the Hippo pathway. EMBO J. 2011, 30, 2325–2335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poon, C.L.C.; Brumby, A.M.; Richardson, H.E. Cooperates with Oncogenic. Int. J. Mol. Sci. 2018, 19. [Google Scholar]
- Rudrapatna, V.A.; Bangi, E.; Cagan, R.L. A Jnk-Rho-Actin remodeling positive feedback network directs Src-driven invasion. Oncogene 2014, 33, 2801–2806. [Google Scholar] [CrossRef] [Green Version]
- Pallavi, S.K.; Ho, D.M.; Hicks, C.; Miele, L.; Artavanis-Tsakonas, S. Notch and Mef2 synergize to promote proliferation and metastasis through JNK signal activation in Drosophila. EMBO J. 2012, 31, 2895–2907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, R.P.; Bajpai, A.; Sinha, P. Selector genes display tumor cooperation and inhibition in. Biol. Open 2017, 6, 1581–1591. [Google Scholar] [CrossRef] [Green Version]
- Moreno, E.; Yan, M.; Basler, K. Evolution of TNF signaling mechanisms: JNK-dependent apoptosis triggered by Eiger, the Drosophila homolog of the TNF superfamily. Curr. Biol. 2002, 12, 1263–1268. [Google Scholar] [CrossRef] [Green Version]
- Sluss, H.K.; Han, Z.; Barrett, T.; Davis, R.J.; Ip, Y.T. A JNK signal transduction pathway that mediates morphogenesis and an immune response in Drosophila. Genes Dev. 1996, 10, 2745–2758. [Google Scholar] [CrossRef] [Green Version]
- Igaki, T.; Kanda, H.; Yamamoto-Goto, Y.; Kanuka, H.; Kuranaga, E.; Aigaki, T.; Miura, M. Eiger, a TNF superfamily ligand that triggers the Drosophila JNK pathway. EMBO J. 2002, 21, 3009–3018. [Google Scholar] [CrossRef]
- Andersen, D.S.; Colombani, J.; Palmerini, V.; Chakrabandhu, K.; Boone, E.; Röthlisberger, M.; Toggweiler, J.; Basler, K.; Mapelli, M.; Hueber, A.-O.; et al. The Drosophila TNF receptor Grindelwald couples loss of cell polarity and neoplastic growth. Nature 2015, 522, 482–486. [Google Scholar] [CrossRef]
- Kanda, H.; Igaki, T.; Kanuka, H.; Yagi, T.; Miura, M. Wengen, a member of the Drosophila tumor necrosis factor receptor superfamily, is required for Eiger signaling. J. Biol. Chem. 2002, 277, 28372–28375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riesgo-Escovar, J.R.; Jenni, M.; Fritz, A.; Hafen, E. The Drosophila Jun-N-terminal kinase is required for cell morphogenesis but not for DJun-dependent cell fate specification in the eye. Genes Dev. 1996, 10, 2759–2768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donohoe, C.D.; Csordás, G.; Correia, A.; Jindra, M.; Klein, C.; Habermann, B.; Uhlirova, M. Atf3 links loss of epithelial polarity to defects in cell differentiation and cytoarchitecture. PLoS Genet. 2018, 14, e1007241. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Edgar, B.A.; Boutros, M. ATF3 acts as a rheostat to control JNK signalling during intestinal regeneration. Nat. Commun. 2017, 8, 14289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Marca, J.E.; Richardson, H.E. Two-Faced: Roles of JNK Signalling During Tumourigenesis in the. Front. Cell Dev. Biol. 2020, 8, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, E.; Alqadri, N.; Dodgson, L.; Mason, D.; Lyulcheva, E.; Messina, G.; Bennett, D. MRL proteins cooperate with activated Ras in glia to drive distinct oncogenic outcomes. Oncogene 2017, 36, 4311–4322. [Google Scholar] [CrossRef] [Green Version]
- Hirabayashi, S.; Baranski, T.J.; Cagan, R.L. Transformed Drosophila cells evade diet-mediated insulin resistance through wingless signaling. Cell 2013, 154, 664–675. [Google Scholar] [CrossRef] [Green Version]
- Ho, D.M.; Pallavi, S.K.; Artavanis-Tsakonas, S. The Notch-mediated hyperplasia circuitry in Drosophila reveals a Src-JNK signaling axis. Elife 2015, 4, e05996. [Google Scholar] [CrossRef]
- Bossuyt, W.; De Geest, N.; Aerts, S.; Leenaerts, I.; Marynen, P.; Hassan, B.A. The atonal proneural transcription factor links differentiation and tumor formation in Drosophila. PLoS Biol. 2009, 7, e40. [Google Scholar] [CrossRef]
- Gerlach, S.U.; Eichenlaub, T.; Herranz, H. Yorkie and JNK Control Tumorigenesis in Drosophila Cells with Cytokinesis Failure. Cell Rep. 2018, 23, 1491–1503. [Google Scholar] [CrossRef]
- Flaherty, M.S.; Salis, P.; Evans, C.J.; Ekas, L.A.; Marouf, A.; Zavadil, J.; Banerjee, U.; Bach, E.A. chinmo Is a Functional Effector of the JAK/STAT Pathway that Regulates Eye Development, Tumor Formation, and Stem Cell Self-Renewal in Drosophila. Dev. Cell 2010, 18, 556–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stec, W.; Vidal, O.; Zeidler, M.P. Drosophila SOCS36E negatively regulates JAK/STAT pathway signaling via two separable mechanisms. Mol. Biol. Cell. 2013, 24, 3000–3009. [Google Scholar] [CrossRef]
- Callus, B.A.; Mathey-Prevot, B. SOCS36E, a novel Drosophila SOCS protein, suppresses JAK/STAT and EGF-R signalling in the imaginal wing disc. Oncogene 2002, 21, 4812–4821. [Google Scholar] [CrossRef] [Green Version]
- Bina, S.; Wright, V.M.; Fisher, K.H.; Milo, M.; Zeidler, M.P. Transcriptional targets of Drosophila JAK/STAT pathway signalling as effectors of haematopoietic tumour formation. EMBO Rep. 2010, 11, 201–207. [Google Scholar] [CrossRef] [Green Version]
- Müller, P.; Kuttenkeuler, D.; Gesellchen, V.; Zeidler, M.P.; Boutros, M. Identification of JAK/STAT signalling components by genome-wide RNA interference. Nature 2005, 436, 871–875. [Google Scholar]
- Baeg, G.H.; Zhou, R.; Perrimon, N. Genome-wide RNAi analysis of JAK/STAT signaling components in Drosophila. Genes Dev. 2005, 19, 1861–1870. [Google Scholar] [CrossRef] [Green Version]
- Flaherty, M.S.; Zavadil, J.; Ekas, L.A.; Bach, E.A. Genome-wide expression profiling in the Drosophila eye reveals unexpected repression of notch signaling by the JAK/STAT pathway. Dev. Dyn. 2009, 238, 2235–2253. [Google Scholar] [CrossRef] [Green Version]
- Ayala-Camargo, A.; Ekas, L.A.; Flaherty, M.S.; Baeg, G.H.; Bach, E.A. The JAK/STAT pathway regulates proximo-distal patterning in Drosophila. Dev. Dyn. 2007, 236, 2721–2730. [Google Scholar] [CrossRef]
- Lopes, E.S.; Araujo, H.M. The maternal JAK/STAT pathway of Drosophila regulates embryonic dorsal-ventral patterning. Braz. J. Med. Biol. Res. 2004, 37, 1811–1818. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.L.; Schroeder, M.C.; Kango-Singh, M.; Tao, C.; Halder, G. Tumor suppression by cell competition through regulation of the Hippo pathway. Proc. Natl. Acad. Sci. USA 2012, 109, 484–489. [Google Scholar] [CrossRef] [Green Version]
- Artavanis-Tsakonas, S.; Rand, M.D.; Lake, R.J. Notch signaling: Cell fate control and signal integration in development. Science 1999, 284, 770–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krejcí, A.; Bernard, F.; Housden, B.E.; Collins, S.; Bray, S.J. Direct response to Notch activation: Signaling crosstalk and incoherent logic. Sci. Signal. 2009, 2, ra1. [Google Scholar] [CrossRef]
- Slaninova, V.; Krafcikova, M.; Perez-Gomez, R.; Steffal, P.; Trantirek, L.; Bray, S.J.; Krejci, A. Notch stimulates growth by direct regulation of genes involved in the control of glycolysis and the tricarboxylic acid cycle. Open Biol. 2016, 6, 150155. [Google Scholar] [CrossRef]
- Zhang, W.; Cohen, S.M. The Hippo pathway acts via p53 and microRNAs to control proliferation and proapoptotic gene expression during tissue growth. Biol. Open. 2013, 2, 822–828. [Google Scholar] [CrossRef] [Green Version]
- Perez, E.; Lindblad, J.L.; Bergmann, A. Tumor-promoting function of apoptotic caspases by an amplification loop involving ROS, macrophages and JNK in Drosophila. eLife 2017, 6, 6. [Google Scholar] [CrossRef]
- Letai, A. Functional precision cancer medicine-moving beyond pure genomics. Nat. Med. 2017, 23, 1028–1035. [Google Scholar] [CrossRef]
- Bangi, E. Strategies for Functional Interrogation of Big Cancer Data Using Drosophila Cancer Models. Int. J. Mol. Sci. 2020, 21, 3754. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway | EGFR/Ras | Notch | Dpp | Hh | Wg | JNK | Jak/Stat | Hippo |
|---|---|---|---|---|---|---|---|---|
| Ligand | Spi, Vn | D, Ser | Dpp | Hh | Wg | Egr | Upds | Ds |
| Receptor | EGFR | N | Tkv, Put | Ptc, Smo | Arr, Fz | Grnd, Wgn | Dome | Fat, Crb |
| Key Players | Ras, Raf, MAPK | Dsh, Sgg, Axin, APC | Hep, Bsk/JNK | Hop (Jak) | Hpo, Wts | |||
| Nuclear Factors | Cic, Pnt, Yan/Aop | N, Su(H), Mam | Mad, Med, Brk, Shn | Ci | Arm/βcat, dTCF | Kay, Jra, Atf3, Irbp18/ CEBPG, Pdp1 | Stat-92E | Yki Sd |
| Activity Assays | anti-dp-ERK anti-Cic pnt-lacZ aos-lacZ cic-GFP | E(spl)m8-lacZ NRE-EGFP Su(H)-lacZ anti-NICD | dad-GFP brk-GFP anti-P-Mad anti-Sal anti-Omb anti-Brk | anti-Ptc dpp-lacZ ptc-lacZ | anti-Cut nkd-lacZ arr-lacZ | puc-lacZ anti-Mmp1 anti-P-JNK TRE-dsRed | 10X STAT-GFP anti-P-STAT | ex-lacZ diap1-lacZ ban-GFP anti-Ex Yki::GFP |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamaratoglu, F.; Atkins, M. Rounding up the Usual Suspects: Assessing Yorkie, AP-1, and Stat Coactivation in Tumorigenesis. Int. J. Mol. Sci. 2020, 21, 4580. https://doi.org/10.3390/ijms21134580
Hamaratoglu F, Atkins M. Rounding up the Usual Suspects: Assessing Yorkie, AP-1, and Stat Coactivation in Tumorigenesis. International Journal of Molecular Sciences. 2020; 21(13):4580. https://doi.org/10.3390/ijms21134580
Chicago/Turabian StyleHamaratoglu, Fisun, and Mardelle Atkins. 2020. "Rounding up the Usual Suspects: Assessing Yorkie, AP-1, and Stat Coactivation in Tumorigenesis" International Journal of Molecular Sciences 21, no. 13: 4580. https://doi.org/10.3390/ijms21134580
APA StyleHamaratoglu, F., & Atkins, M. (2020). Rounding up the Usual Suspects: Assessing Yorkie, AP-1, and Stat Coactivation in Tumorigenesis. International Journal of Molecular Sciences, 21(13), 4580. https://doi.org/10.3390/ijms21134580