Small-Angle Scattering and Multifractal Analysis of DNA Sequences


Abstract
:1. Introduction
2. Theoretical Background
2.1. Iterated Function Systems and Chaos Game Representation of DNA Sequences
2.2. Fractals and Multifractals
2.3. Small-Angle Scattering
3. Results and Discussion
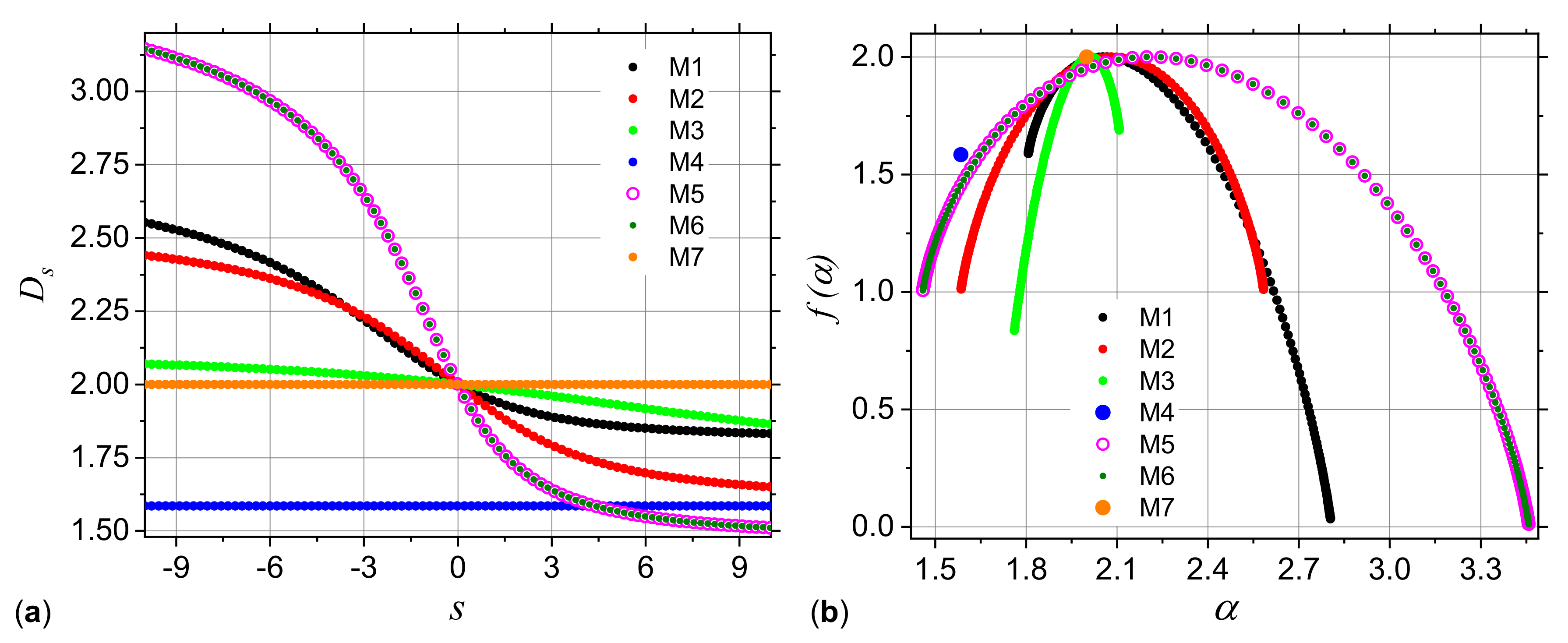
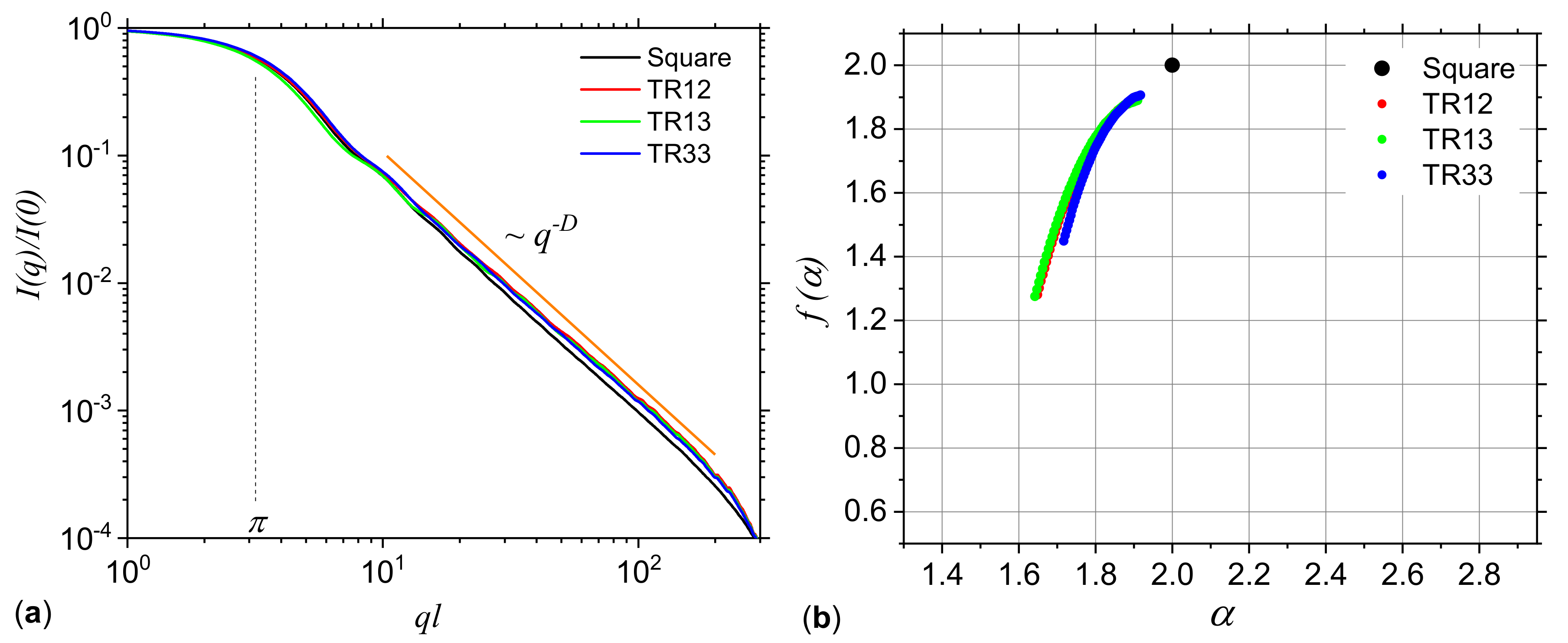
3.1. Analysis of Theoretical Models
3.1.1. Multiplicative Deterministic Cascades
3.1.2. Missing Sequences Models
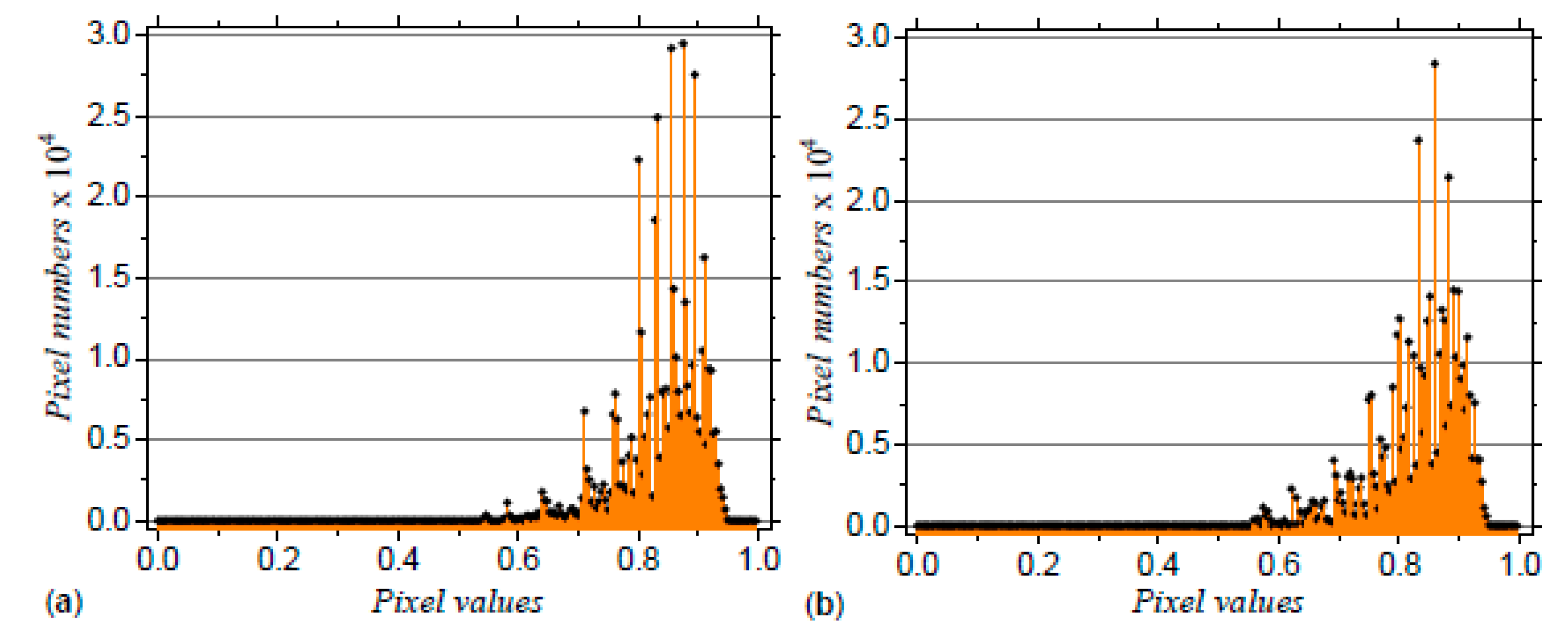
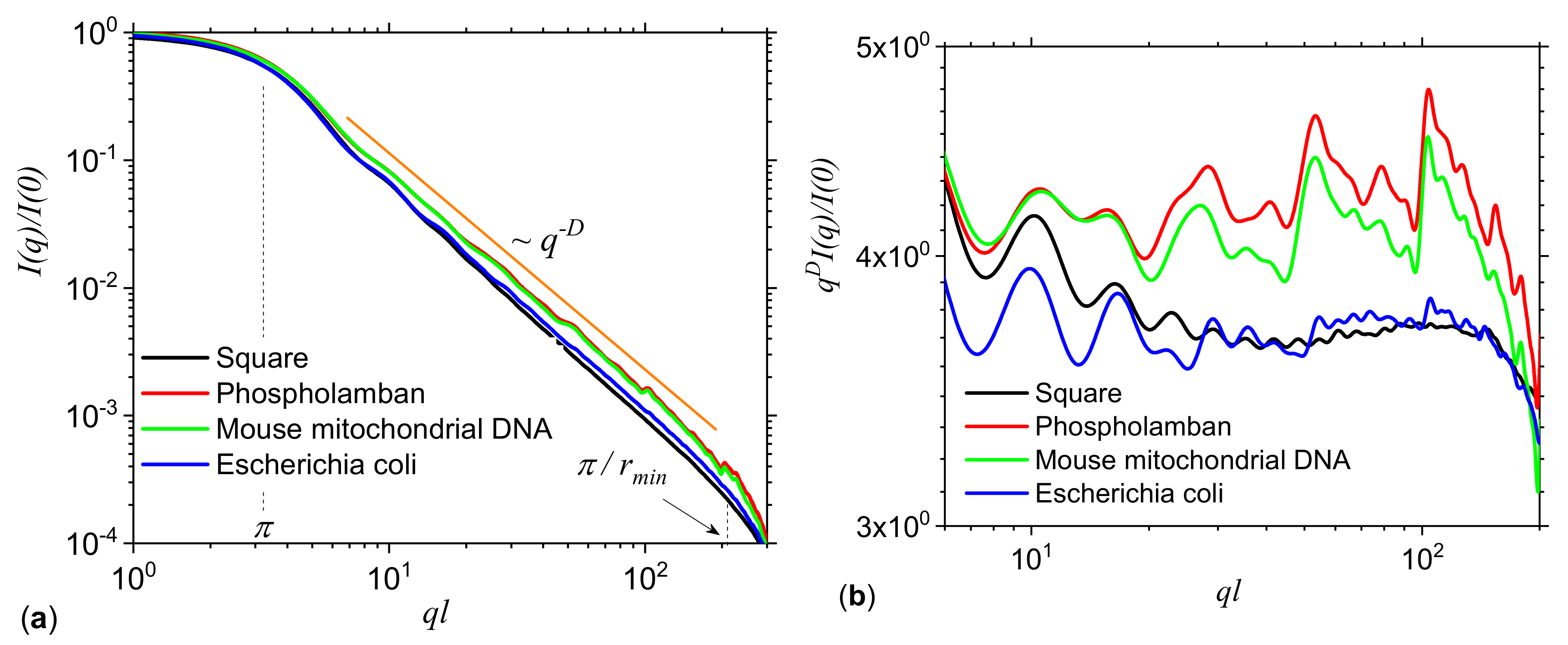
3.2. Application to DNA Sequences: Phospholamban, Mouse mitochondrion and Escherichia coli
4. Conclusions
Funding
Conflicts of Interest
References
- Arneodo, A.; Vaillant, C.; Audit, B.; Argoul, F.; d’Aubenton Carafa, Y.; Thermes, C. Multi-scale coding of genomic information: From DNA sequence to genome structure and function. Phys. Rep. 2011, 498, 45–188. [Google Scholar] [CrossRef]
- Felsenfeld, G.; Groudine, M. Controlling the double helix. Nature 2003, 421, 448–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albrecht-Buehler, G. Fractal genome sequences. Gene 2012, 498, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Albuquerque, E.L.; Fulco, U.L.; Freire, V.N.; Caetano, E.W.S.; Lyra, M.L.; de Moura, F.A.B.F. DNA-based nanobiostructured devices: The role of quasiperiodicity and correlation effects. Phys. Rep. 2014, 535, 139–209. [Google Scholar] [CrossRef]
- Niu, X.H.; Hu, X.H.; Shi, F.; Xia, J.B. Predicting DNA binding proteins using support vector machine with hybrid fractal features. J. Theor. Biol. 2014, 343, 186–192. [Google Scholar] [CrossRef]
- Lennon, F.E.; Cianci, G.C.; Cipriai, N.A.; Hensing, T.A.; Zhang, H.J.; Chen, C.T.; Murgu, S.T.; Vokes, E.E.; Vannier, M.W.; Salgia, R. Lung cancer—A fractal viewpoint. Nat. Rev. Clin. Oncol. 2015, 12, 664–675. [Google Scholar] [CrossRef] [Green Version]
- Babic, M.; Mihelic, J.; Calì, M. Complex Network Characterization Using Graph Theory and Fractal Geometry: The Case Study of Lung Cancer DNA Sequences. Appl. Sci. 2020, 10, 3037. [Google Scholar] [CrossRef]
- Li, W. Mutual information functions versus correlation functions. J. Stat. Phys. 1990, 60, 823–837. [Google Scholar] [CrossRef]
- Voss, R.F. Evolution of long-range fractal correlations and 1/f noise in DNA base sequences. Phys. Rev. Lett. 1992, 68, 3805–3808. [Google Scholar] [CrossRef]
- Havlin, S.; Buldyrev, S.V.; Goldberger, A.L.; Mantegna, R.N.; Peng, C.K.; Simons, M.; Stanley, H.E. Statistical and linguistic features of DNA sequences. Fractals 1995, 3, 269–284. [Google Scholar] [CrossRef]
- Herzel, H.; Große, I. Measuring correlations in symbol sequences. Phys. A Stat. Mech. Appl. 1995, 216, 518–542. [Google Scholar] [CrossRef]
- Bernaola-Galván, P.; Román-Roldán, R.; Oliver, J.L. Compositional segmentation and long-range fractal correlations in DNA sequences. Phys. Rev. E 1996, 53, 5181–5189. [Google Scholar] [CrossRef] [PubMed]
- Arneodo, A.; d’Aubenton Carafa, Y.; Bacry, E.; Graves, P.V.; Muzy, J.F.; Thermes, C. Wavelet based fractal analysis of DNA sequences. Phys. D Nonlinear Phenom. 1996, 96, 291–320. [Google Scholar] [CrossRef]
- Jeffrey, H.J. Chaos game representation of gene structure. Nucleic Acids Res. 1990, 18, 2163–2170. [Google Scholar] [CrossRef] [Green Version]
- Hoang, T.; Yin, C.; Yau, S.S.T. Numerical encoding of DNA sequences by chaos game representation with application in similarity comparison. Genomics 2016, 108, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, J.M.; Rodriguez, M.A.; Abramson, G. Multifractal analysis of DNA sequences using a novel chaos-game representation. Phys. A Stat. Mech. Appl. 2001, 300, 271–284. [Google Scholar] [CrossRef]
- Han, J.-J.; Fu, W.-J. Wavelet-based multifractal analysis of DNA sequences by using chaos-game representation. Chin. Phys. B 2010, 19, 010205. [Google Scholar] [CrossRef]
- Yu, Z.G.; Anh, V.; Lau, K.S. Chaos game representation of protein sequences based on the detailed HP model and their multifractal and correlation analyses. J. Theor. Biol. 2004, 226, 341–348. [Google Scholar] [CrossRef] [Green Version]
- Zu-Guo, Y.; Qian-Jun, X.; Long, S.; Jun-Wu, Y.; Anh, V. Chaos game representation of functional protein sequences, and simulation and multifractal analysis of induced measures. Chin. Phys. B 2010, 19, 068701. [Google Scholar] [CrossRef]
- Pal, M.; Kiran, V.S.; Rao, P.M.; Manimaran, P. Multifractal detrended cross-correlation analysis of genome sequences using chaos-game representation. Phys. A Stat. Mech. Appl. 2016, 456, 288–293. [Google Scholar] [CrossRef]
- Zaia, A.; Maponi, P.; Zannotti, M.; Casoli, T. Biocomplexity and Fractality in the Search of Biomarkers of Aging and Pathology: Mitochondrial DNA Profiling of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feigin, L.A.; Svergun, D.I. Structure Analysis by Small-Angle X-Ray and Neutron Scattering; Springer: Boston, MA, USA, 1987; p. 335. [Google Scholar] [CrossRef]
- Martin, J.E.; Hurd, A.J. Scattering from fractals. J. Appl. Cryst. 1987, 20, 61–78. [Google Scholar] [CrossRef]
- Schmidt, P.W. Small-angle scattering studies of disordered, porous and fractal systems. J. Appl. Cryst. 1991, 24, 414–435. [Google Scholar] [CrossRef]
- Cherny, A.Y.; Anitas, E.M.; Osipov, V.A.; Kuklin, A.I. Deterministic fractals: Extracting additional information from small-angle scattering data. Phys. Rev. E 2011, 84, 036203. [Google Scholar] [CrossRef] [Green Version]
- Anitas, E.M.; Slyamov, A. Structural characterization of chaos game fractals using small-angle scattering analysis. PLoS ONE 2017, 12, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Debye, P. Zerstreuung von Röntgenstrahlen. Ann. Phys. 1915, 351, 809–823. [Google Scholar] [CrossRef] [Green Version]
- Provata, A.; Almirantis, Y. Fractal Cantor Patterns in the Sequence Structure of DNA. Fractals 2000, 8, 15–27. [Google Scholar] [CrossRef]
- Barnsley, M.F. Fractals Everywhere, 2nd ed.; Morgan Kaufmann: Burlington, MA, USA, 2000. [Google Scholar]
- Rogers, C.A. Hausdorff Measures; Cambridge University Press: Cambridge, UK, 1970; p. 179. [Google Scholar]
- Dimension und äußeres Maß. Mathematische Annalen 1918, 79, 157–179. [CrossRef]
- Gouyet, J.F. Physics and Fractal Structures; Masson: Paris, France, 1996; p. 234. [Google Scholar]
- Arneodo, A.; Decoster, N.; Roux, S. A wavelet-based method for multifractal image analysis. I. Methodology and test applications on isotropic and anisotropic random rough surfaces. Eur. Phys. J. B 2000, 15, 567–600. [Google Scholar] [CrossRef]
- Decoster, N.; Roux, S.; Arnéodo, A. A wavelet-based method for multifractal image analysis. II. Applications to synthetic multifractal rough surfaces. Eur. Phys. J. B 2000, 15, 739–764. [Google Scholar] [CrossRef]
- Muzy, J.F.; Bacry, E.; Arneodo, A. Multifractal formalism for fractal signals: The structure-function approach versus the wavelet-transform modulus-maxima method. Phys. Rev. E 1993, 47, 875–884. [Google Scholar] [CrossRef] [Green Version]
- Chhabra, A.; Jensen, R.V. Direct Determination of the f (alpha) Singularity Spectrum. Phys. Rev. Lett. 1989, 62, 1327. [Google Scholar] [CrossRef] [PubMed]
- Pantos, E.; van Garderen, H.F.; Hilbers, P.A.J.; Beelen, T.P.M.; van Santen, R.A. Simulation of small-angle scattering from large assemblies of multi-type scatterer particle. J. Mol. Struct. 1996, 383, 303. [Google Scholar] [CrossRef] [Green Version]
- Meakin, P. Diffusion-limited aggregation on multifractal lattices: A model for fluid-fluid displacement in porous media. Phys. Rev. A 1987, 36, 2833–2837. [Google Scholar] [CrossRef] [PubMed]
- Martinez, V.J.; Jones, B.J.T.; Dominguez-Tenreiro, R.; van de Weygaert, R. Clustering Paradigms and Multifractal Measures. Astrophys. J. 1990, 357. [Google Scholar] [CrossRef] [Green Version]
- Tarquis, A.M.; Losada, J.C.; Benito, R.M.; Borondo, F. Multifractal analysis of tori destruction in a molecular Hamiltonian system. Phys. Rev. E 2001, 65, 016213. [Google Scholar] [CrossRef]
- Hao, B.; Xie, H.; Yu, Z.; Chen, G. Avoided Strings in Bacterial Complete Genomes and a Related Combinatorial Problem. Annals of Combinatorics. Ann. Comb. 2000, 4, 247–255. [Google Scholar] [CrossRef]
- Hao, B.L.; Lee, H.; Zhang, S.Y. Fractals related to long DNA sequences and complete genomes. Chaos Solitons Fract. 2000, 11, 825–836. [Google Scholar] [CrossRef]
- Hao, B.; Xie, H.; Yu, Z.; Chen, G.Y. Factorizable language: from dynamics to bacterial complete genomes. Physica A 2000, 288, 10–20. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, P. DNA Sequences with Forbidden Words and the Generalized Cantor Set. J. Appl. Math. Phys. 2019, 7, 1687–1696. [Google Scholar] [CrossRef] [Green Version]
- Cherny, A.Y.; Anitas, E.M.; Kuklin, A.I.; Balasoiu, M.; Osipov, V.A. Scattering from generalized Cantor fractals. J. Appl. Cryst. 2010, 43, 790–797. [Google Scholar] [CrossRef] [Green Version]
- Anitas, E.M. Small-Angle Scattering from Fractals: Differentiating between Various Types of Structures. Symmetry 2020, 12, 65. [Google Scholar] [CrossRef] [Green Version]
- NCBI. PLN Phospholamban [Homo Sapiens (Human)]. Available online: https://www.ncbi.nlm.nih.gov/gene/5350 (accessed on 29 June 2020).
- Bibb, M.J.; Van Etten, R.A.; Wright, C.T.; Walberg, M.W.; Clayton, D.A. Sequence and gene organization of mouse mitochondrial DNA. Cell 1981, 26, 167–180. [Google Scholar] [CrossRef]
- Brooks, J.T.; Sowers, E.G.; Wells, J.G.; Green, K.D.; Griffin, P.M.; Hoekstra, P.M.; Strockbine, N.A. Non-O157 Shiga toxin-producing Escherichia coli infections in the United States. J. Infect. Dis. 2005, 192, 1422–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, K.; Murase, K.; Sato, M.P.; Toyoda, A.; Itoh, T.; Mainil, J.G.; Piérard, D.; Yoshino, S.; Kimata, K.; Isobe, J.; et al. Differential dynamics and impacts of prophages and plasmids on the pangenome and virulence factor repertoires of Shiga toxin-producing Escherichia coli O145:H28. Microb. Genom. 2020, 6, 1–13. [Google Scholar] [CrossRef]
- Feng, J.; Wang, T.-M. A 3D graphical representation of RNA secondary structures based on chaos game representation. Chem. Phys. Lett. 2008, 454, 355–361. [Google Scholar] [CrossRef]
- Anitas, E.M.; Marcelli, G.; Szakacs, Z.; Todoran, R.; Todoran, D. Structural Properties of Vicsek-like Deterministic Multifractals. Symmetry 2019, 11, 806. [Google Scholar] [CrossRef] [Green Version]
- Berthelsen, C.L.; Glazier, J.A.; Skolnick, M.H. Global fractal dimension of human DNA sequences treated as pseudorandom walks. Phys. Rev. A 1992, 45, 8902–8913. [Google Scholar] [CrossRef]
- Yu, Z.G.; Anh, V.; Lau, K.S. Measure representation and multifractal analysis of complete genomes. Phys. Rev. E 2001, 64, 031903. [Google Scholar] [CrossRef] [Green Version]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| w | a | b | c | d | e | f | p |
|---|---|---|---|---|---|---|---|
| 1 | 1/2 | 0 | 0 | 1/2 | 0 | 0 | 1/4 |
| 2 | 1/2 | 0 | 0 | 1/2 | 0 | 1/2 | 1/4 |
| 3 | 1/2 | 0 | 0 | 1/2 | 1/2 | 0 | 1/4 |
| 4 | 1/2 | 0 | 0 | 1/2 | 1/2 | 1/2 | 1/4 |
| Model | ||||
|---|---|---|---|---|
| M1 | 1 | 1 | 1 | 0.5 |
| M2 | 1 | 1 | 0.5 | 0.5 |
| M3 | 1 | 0.75 | 0.75 | 0.75 |
| M4 | 1 | 1 | 1 | 0 |
| M5 | 1 | 1 | 0.5 | 0.25 |
| M6 | 0.5 | 1 | 1 | 0.25 |
| M7 | 1 | 1 | 1 | 1 |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anitas, E.M. Small-Angle Scattering and Multifractal Analysis of DNA Sequences. Int. J. Mol. Sci. 2020, 21, 4651. https://doi.org/10.3390/ijms21134651
Anitas EM. Small-Angle Scattering and Multifractal Analysis of DNA Sequences. International Journal of Molecular Sciences. 2020; 21(13):4651. https://doi.org/10.3390/ijms21134651
Chicago/Turabian StyleAnitas, Eugen Mircea. 2020. "Small-Angle Scattering and Multifractal Analysis of DNA Sequences" International Journal of Molecular Sciences 21, no. 13: 4651. https://doi.org/10.3390/ijms21134651
APA StyleAnitas, E. M. (2020). Small-Angle Scattering and Multifractal Analysis of DNA Sequences. International Journal of Molecular Sciences, 21(13), 4651. https://doi.org/10.3390/ijms21134651