Nonsense Suppression Therapy: New Hypothesis for the Treatment of Inherited Bone Marrow Failure Syndromes

 , ,
, ,
Abstract
:1. Introduction
2. Inherited Bone Marrow Failure Syndromes
2.1. Shwachman–Diamond Syndrome
2.2. Diamond–Blackfan Anemia
2.3. Fanconi Anemia
2.4. Dyskeratosis Congenita
2.5. Severe Congenital Neutropenia
3. The Nonsense Suppression Therapy
3.1. Aminoglycoside Compounds
3.2. Aminoglycoside Derivatives
3.3. Readthrough Enhancer Molecules
3.4. Ataluren and Analogues
3.5. NMD Inhibitors
3.6. Modified t-RNA
4. Discussion and Perspectives
5. Patents
Funding
Conflicts of Interest
Abbreviations
| IBMFS | Inherited Bone Marrow Failure syndromes |
| SDS | Shwachman–Diamond syndrome |
| DBA | Diamond–Blackfan anemia |
| FA | Fanconi Anemia |
| DC | Dyskeratosis congenita |
| SCN | Severe congenital neutropenia |
| MDS | Myelodysplastic syndrome |
| AML | Acute Myeloid Leukemia |
| CF | Cystic Fibrosis |
| AA | Aplastic anemia |
| PHN | paroxysmal nocturnal hemoglobinuria |
| SBDS | Shwachman–Bodian–Diamond syndrome (gene) |
| CFTR | Cystic Fibrosis Transmembrane conductance Regulator (gene) |
| NMD | Nonsense mediated decay |
| Nc-tRNA | Near-cognate transfer RNA |
References
- Giri, N.; Stratton, P.; Savage, S.A.; Alter, B.P. Pregnancies in patients with inherited bone marrow failure syndromes in the NCI cohort. Blood 2017, 130, 1674–1676. [Google Scholar] [CrossRef] [Green Version]
- Hashmi, S.K.; Allen, C.; Klaassen, R.; Fernandez, C.V.; Yanofsky, R.; Shereck, E.; Champagne, J.; Silva, M.; Lipton, J.H.; Brossard, J.; et al. Comparative analysis of Shwachman-Diamond syndrome to other inherited bone marrow failure syndromes and genotype-phenotype correlation. Clin. Genet. 2011, 79, 448–458. [Google Scholar] [CrossRef] [PubMed]
- Calado, R.T.; Clé, D.V. Treatment of inherited bone marrow failure syndromes beyond transplantation. Hematol. Am. Soc. Hematol. Educ. Program. 2017, 2017, 96–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lejeune, F. Nonsense-mediated mRNA decay at the crossroads of many cellular pathways. BMB Rep. 2017, 50, 175–185. [Google Scholar] [CrossRef] [Green Version]
- Borgatti, M.; Altamura, E.; Salvatori, F.; D’Aversa, E.; Altamura, N. Screening Readthrough Compounds to Suppress Nonsense Mutations: Possible Application to β-Thalassemia. J. Clin. Med. 2020, 9, 289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peltz, S.W.; Welch, E.M.; Jacobson, A.; Trotta, C.R.; Naryshkin, N.; Sweeney, H.L.; Bedwell, D.M. Nonsense suppression activity of PTC124 (ataluren). Proc. Natl. Acad. Sci. USA 2009, 106, E64–E65. [Google Scholar] [CrossRef] [Green Version]
- Savage, S.A.; Dufour, C. Classical inherited bone marrow failure syndromes with high risk for myelodysplastic syndrome and acute myelogenous leukemia. Semin. Hematol. 2017, 54, 105–114. [Google Scholar] [CrossRef]
- Ajore, R.; Raiser, D.; McConkey, M.; Jöud, M.; Boidol, B.; Mar, B.; Saksena, G.; Weinstock, D.M.; Armstrong, S.; Ellis, S.R.; et al. Deletion of ribosomal protein genes is a common vulnerability in human cancer, especially in concert with. EMBO Mol. Med. 2017, 9, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Sulima, S.O.; Kampen, K.R.; De Keersmaecker, K. Cancer Biogenesis in Ribosomopathies. Cells 2019, 8, 229. [Google Scholar] [CrossRef] [Green Version]
- Armistead, J.; Triggs-Raine, B. Diverse diseases from a ubiquitous process: The ribosomopathy paradox. FEBS Lett. 2014, 588, 1491–1500. [Google Scholar] [CrossRef] [Green Version]
- Dror, Y. P53 protein overexpression in Shwachman-Diamond syndrome. Arch. Pathol. Lab. Med. 2002, 126, 1157–1158. [Google Scholar] [CrossRef] [PubMed]
- Elghetany, M.T.; Alter, B.P. p53 protein overexpression in bone marrow biopsies of patients with Shwachman-Diamond syndrome has a prevalence similar to that of patients with refractory anemia. Arch. Pathol. Lab. Med. 2002, 126, 452–455. [Google Scholar] [CrossRef] [PubMed]
- Engidaye, G.; Melku, M.; Enawgaw, B. Diamond Blackfan Anemia: Genetics, Pathogenesis, Diagnosis and Treatment. EJIFCC 2019, 30, 67–81. [Google Scholar] [PubMed]
- Link, D.C. Mechanisms of leukemic transformation in congenital neutropenia. Curr. Opin. Hematol. 2019, 26, 34–40. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef] [Green Version]
- Shwachman, H.; Diamond, L.K.; Oski, F.A.; Khaw, K.T. The syndrome of pancreatic insufficiency and bone marrow dysfunction. J. Pediatr. 1964, 65, 645–663. [Google Scholar] [CrossRef]
- Goobie, S.; Popovic, M.; Morrison, J.; Ellis, L.; Ginzberg, H.; Boocock, G.R.; Ehtesham, N.; Bétard, C.; Brewer, C.G.; Roslin, N.M.; et al. Shwachman-Diamond syndrome with exocrine pancreatic dysfunction and bone marrow failure maps to the centromeric region of chromosome 7. Am. J. Hum. Genet. 2001, 68, 1048–1054. [Google Scholar] [CrossRef] [Green Version]
- Mercuri, A.; Cannata, E.; Perbellini, O.; Cugno, C.; Balter, R.; Zaccaron, A.; Tridello, G.; Pizzolo, G.; De Bortoli, M.; Krampera, M.; et al. Immunophenotypic analysis of hematopoiesis in patients suffering from Shwachman-Bodian-Diamond Syndrome. Eur. J. Haematol. 2015, 95, 308–315. [Google Scholar] [CrossRef]
- Dror, Y.; Durie, P.; Ginzberg, H.; Herman, R.; Banerjee, A.; Champagne, M.; Shannon, K.; Malkin, D.; Freedman, M.H. Clonal evolution in marrows of patients with Shwachman-Diamond syndrome: A prospective 5-year follow-up study. Exp. Hematol. 2002, 30, 659–669. [Google Scholar] [CrossRef]
- Maserati, E.; Minelli, A.; Pressato, B.; Valli, R.; Crescenzi, B.; Stefanelli, M.; Menna, G.; Sainati, L.; Poli, F.; Panarello, C.; et al. Shwachman syndrome as mutator phenotype responsible for myeloid dysplasia/neoplasia through karyotype instability and chromosomes 7 and 20 anomalies. Genes Chromosomes Cancer 2006, 45, 375–382. [Google Scholar] [CrossRef]
- Valli, R.; De Paoli, E.; Nacci, L.; Frattini, A.; Pasquali, F.; Maserati, E. Novel recurrent chromosome anomalies in Shwachman-Diamond syndrome. Pediatr. Blood Cancer 2017, 64. [Google Scholar] [CrossRef] [PubMed]
- Maserati, E.; Minelli, A.; Olivieri, C.; Bonvini, L.; Marchi, A.; Bozzola, M.; Danesino, C.; Scappaticci, S.; Pasquali, F. Isochromosome (7)(q10) in Shwachman syndrome without MDS/AML and role of chromosome 7 anomalies in myeloproliferative disorders. Cancer Genet. Cytogenet 2000, 121, 167–171. [Google Scholar] [CrossRef]
- Bezzerri, V.; Cipolli, M. Shwachman-Diamond Syndrome: Molecular Mechanisms and Current Perspectives. Mol. Diagn. Ther. 2018, 23, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Warren, A.J. Molecular basis of the human ribosomopathy Shwachman-Diamond syndrome. Adv. Biol. Regul. 2018, 67, 109–127. [Google Scholar] [CrossRef] [PubMed]
- Boocock, G.R.; Morrison, J.A.; Popovic, M.; Richards, N.; Ellis, L.; Durie, P.R.; Rommens, J.M. Mutations in SBDS are associated with Shwachman-Diamond syndrome. Nat. Genet. 2003, 33, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Dhanraj, S.; Matveev, A.; Li, H.; Lauhasurayotin, S.; Jardine, L.; Cada, M.; Zlateska, B.; Tailor, C.S.; Zhou, J.; Mendoza-Londono, R.; et al. Biallelic mutations in DNAJC21 cause Shwachman-Diamond syndrome. Blood 2017. [Google Scholar] [CrossRef]
- Carapito, R.; Konantz, M.; Paillard, C.; Miao, Z.; Pichot, A.; Leduc, M.S.; Yang, Y.; Bergstrom, K.L.; Mahoney, D.H.; Shardy, D.L.; et al. Mutations in signal recognition particle SRP54 cause syndromic neutropenia with Shwachman-Diamond-like features. J. Clin. Investig. 2017, 127, 4090–4103. [Google Scholar] [CrossRef]
- Stepensky, P.; Chacón-Flores, M.; Kim, K.H.; Abuzaitoun, O.; Bautista-Santos, A.; Simanovsky, N.; Siliqi, D.; Altamura, D.; Méndez-Godoy, A.; Gijsbers, A.; et al. Mutations in EFL1, an SBDS partner, are associated with infantile pancytopenia, exocrine pancreatic insufficiency and skeletal anomalies in aShwachman-Diamond like syndrome. J. Med. Genet. 2017, 54, 558–566. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.; Kermasson, L.; Hoslin, A.; Jaako, P.; Faille, A.; Acevedo-Arozena, A.; Lengline, E.; Ranta, D.; Poirée, M.; Fenneteau, O.; et al. EFL1 mutations impair eIF6 release to cause Shwachman-Diamond syndrome. Blood 2019, 134, 277–290. [Google Scholar] [CrossRef] [Green Version]
- Donadieu, J.; Fenneteau, O.; Beaupain, B.; Beaufils, S.; Bellanger, F.; Mahlaoui, N.; Lambilliotte, A.; Aladjidi, N.; Bertrand, Y.; Mialou, V.; et al. Classification of and risk factors for hematologic complications in a French national cohort of 102 patients with Shwachman-Diamond syndrome. Haematologica 2012, 97, 1312–1319. [Google Scholar] [CrossRef] [Green Version]
- Dror, Y.; Donadieu, J.; Koglmeier, J.; Dodge, J.; Toiviainen-Salo, S.; Makitie, O.; Kerr, E.; Zeidler, C.; Shimamura, A.; Shah, N.; et al. Draft consensus guidelines for diagnosis and treatment of Shwachman-Diamond syndrome. Ann. N. Y. Acad. Sci. 2011, 1242, 40–55. [Google Scholar] [CrossRef] [PubMed]
- Lindsley, R.C.; Saber, W.; Mar, B.G.; Redd, R.; Wang, T.; Haagenson, M.D.; Grauman, P.V.; Hu, Z.H.; Spellman, S.R.; Lee, S.J.; et al. Prognostic Mutations in Myelodysplastic Syndrome after Stem-Cell Transplantation. N. Engl. J. Med. 2017, 376, 536–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauhasurayotin, S.; Cuvelier, G.D.; Klaassen, R.J.; Fernandez, C.V.; Pastore, Y.D.; Abish, S.; Rayar, M.; Steele, M.; Jardine, L.; Breakey, V.R.; et al. Reanalysing genomic data by normalized coverage values uncovers CNVs in bone marrow failure gene panels. NPJ Genom. Med. 2019, 4, 30. [Google Scholar] [CrossRef] [PubMed]
- Willig, T.N.; Draptchinskaia, N.; Dianzani, I.; Ball, S.; Niemeyer, C.; Ramenghi, U.; Orfali, K.; Gustavsson, P.; Garelli, E.; Brusco, A.; et al. Mutations in ribosomal protein S19 gene and diamond blackfan anemia: Wide variations in phenotypic expression. Blood 1999, 94, 4294–4306. [Google Scholar]
- Choesmel, V.; Bacqueville, D.; Rouquette, J.; Noaillac-Depeyre, J.; Fribourg, S.; Crétien, A.; Leblanc, T.; Tchernia, G.; Da Costa, L.; Gleizes, P.E. Impaired ribosome biogenesis in Diamond-Blackfan anemia. Blood 2007, 109, 1275–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gazda, H.T.; Zhong, R.; Long, L.; Niewiadomska, E.; Lipton, J.M.; Ploszynska, A.; Zaucha, J.M.; Vlachos, A.; Atsidaftos, E.; Viskochil, D.H.; et al. RNA and protein evidence for haplo-insufficiency in Diamond-Blackfan anaemia patients with RPS19 mutations. Br. J. Haematol. 2004, 127, 105–113. [Google Scholar] [CrossRef]
- Tsangaris, E.; Klaassen, R.; Fernandez, C.V.; Yanofsky, R.; Shereck, E.; Champagne, J.; Silva, M.; Lipton, J.H.; Brossard, J.; Michon, B.; et al. Genetic analysis of inherited bone marrow failure syndromes from one prospective, comprehensive and population-based cohort and identification of novel mutations. J. Med. Genet. 2011, 48, 618–628. [Google Scholar] [CrossRef] [Green Version]
- Boria, I.; Garelli, E.; Gazda, H.T.; Aspesi, A.; Quarello, P.; Pavesi, E.; Ferrante, D.; Meerpohl, J.J.; Kartal, M.; Da Costa, L.; et al. The ribosomal basis of Diamond-Blackfan Anemia: Mutation and database update. Hum. Mutat. 2010, 31, 1269–1279. [Google Scholar] [CrossRef] [Green Version]
- Proust, A.; Da Costa, L.; Rince, P.; Landois, A.; Tamary, H.; Zaizov, R.; Tchernia, G.; Delaunay, J.; DBA, S.W.G.o. Ten novel Diamond-Blackfan anemia mutations and three polymorphisms within the rps19 gene. Hematol. J. 2003, 4, 132–136. [Google Scholar] [CrossRef]
- Pospisilova, D.; Cmejlova, J.; Ludikova, B.; Stary, J.; Cerna, Z.; Hak, J.; Timr, P.; Petrtylova, K.; Blatny, J.; Vokurka, S.; et al. The Czech National Diamond-Blackfan Anemia Registry: Clinical data and ribosomal protein mutations update. Blood Cells Mol. Dis. 2012, 48, 209–218. [Google Scholar] [CrossRef]
- Gazda, H.T.; Sheen, M.R.; Vlachos, A.; Choesmel, V.; O’Donohue, M.F.; Schneider, H.; Darras, N.; Hasman, C.; Sieff, C.A.; Newburger, P.E.; et al. Ribosomal protein L5 and L11 mutations are associated with cleft palate and abnormal thumbs in Diamond-Blackfan anemia patients. Am. J. Hum. Genet. 2008, 83, 769–780. [Google Scholar] [CrossRef] [Green Version]
- Draptchinskaia, N.; Gustavsson, P.; Andersson, B.; Pettersson, M.; Willig, T.N.; Dianzani, I.; Ball, S.; Tchernia, G.; Klar, J.; Matsson, H.; et al. The gene encoding ribosomal protein S19 is mutated in Diamond-Blackfan anaemia. Nat. Genet. 1999, 21, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Idol, R.A.; Robledo, S.; Du, H.Y.; Crimmins, D.L.; Wilson, D.B.; Ladenson, J.H.; Bessler, M.; Mason, P.J. Cells depleted for RPS19, a protein associated with Diamond Blackfan Anemia, show defects in 18S ribosomal RNA synthesis and small ribosomal subunit production. Blood Cells Mol. Dis. 2007, 39, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Delaporta, P.; Sofocleous, C.; Economou, M.; Makis, A.; Kostaridou, S.; Kattamis, A. The Greek Registry of Shwachman Diamond-Syndrome: Molecular and clinical data. Pediatr. Blood Cancer 2017, 64. [Google Scholar] [CrossRef]
- Delaporta, P.; Sofocleous, C.; Stiakaki, E.; Polychronopoulou, S.; Economou, M.; Kossiva, L.; Kostaridou, S.; Kattamis, A. Clinical phenotype and genetic analysis of RPS19, RPL5, and RPL11 genes in Greek patients with Diamond Blackfan Anemia. Pediatr. Blood Cancer 2014, 61, 2249–2255. [Google Scholar] [CrossRef] [PubMed]
- Ramenghi, U.; Campagnoli, M.F.; Garelli, E.; Carando, A.; Brusco, A.; Bagnara, G.P.; Strippoli, P.; Izzi, G.C.; Brandalise, S.; Riccardi, R.; et al. Diamond-Blackfan anemia: Report of seven further mutations in the RPS19 gene and evidence of mutation heterogeneity in the Italian population. Blood Cells Mol. Dis. 2000, 26, 417–422. [Google Scholar] [CrossRef]
- Gerrard, G.; Valgañón, M.; Foong, H.E.; Kasperaviciute, D.; Iskander, D.; Game, L.; Müller, M.; Aitman, T.J.; Roberts, I.; de la Fuente, J.; et al. Target enrichment and high-throughput sequencing of 80 ribosomal protein genes to identify mutations associated with Diamond-Blackfan anaemia. Br. J. Haematol. 2013, 162, 530–536. [Google Scholar] [CrossRef]
- Quarello, P.; Garelli, E.; Carando, A.; Brusco, A.; Calabrese, R.; Dufour, C.; Longoni, D.; Misuraca, A.; Vinti, L.; Aspesi, A.; et al. Diamond-Blackfan anemia: Genotype-phenotype correlations in Italian patients with RPL5 and RPL11 mutations. Haematologica 2010, 95, 206–213. [Google Scholar] [CrossRef] [Green Version]
- Gripp, K.W.; Curry, C.; Olney, A.H.; Sandoval, C.; Fisher, J.; Chong, J.X.; Pilchman, L.; Sahraoui, R.; Stabley, D.L.; Sol-Church, K.; et al. Diamond-Blackfan anemia with mandibulofacial dystostosis is heterogeneous, including the novel DBA genes TSR2 and RPS28. Am. J. Med. Genet. A 2014, 164A, 2240–2249. [Google Scholar] [CrossRef] [Green Version]
- Cmejla, R.; Cmejlova, J.; Handrkova, H.; Petrak, J.; Petrtylova, K.; Mihal, V.; Stary, J.; Cerna, Z.; Jabali, Y.; Pospisilova, D. Identification of mutations in the ribosomal protein L5 (RPL5) and ribosomal protein L11 (RPL11) genes in Czech patients with Diamond-Blackfan anemia. Hum. Mutat. 2009, 30, 321–327. [Google Scholar] [CrossRef]
- Volejnikova, J.; Vojta, P.; Urbankova, H.; Mojzíkova, R.; Horvathova, M.; Hochova, I.; Cermak, J.; Blatny, J.; Sukova, M.; Bubanska, E.; et al. Czech and Slovak Diamond-Blackfan Anemia (DBA) Registry update: Clinical data and novel causative genetic lesions. Blood Cells Mol. Dis. 2020, 81, 102380. [Google Scholar] [CrossRef] [PubMed]
- Farrar, J.E.; Nater, M.; Caywood, E.; McDevitt, M.A.; Kowalski, J.; Takemoto, C.M.; Talbot, C.C.; Meltzer, P.; Esposito, D.; Beggs, A.H.; et al. Abnormalities of the large ribosomal subunit protein, Rpl35a, in Diamond-Blackfan anemia. Blood 2008, 112, 1582–1592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulirsch, J.C.; Verboon, J.M.; Kazerounian, S.; Guo, M.H.; Yuan, D.; Ludwig, L.S.; Handsaker, R.E.; Abdulhay, N.J.; Fiorini, C.; Genovese, G.; et al. The Genetic Landscape of Diamond-Blackfan Anemia. Am. J. Hum. Genet. 2019, 104, 356. [Google Scholar] [CrossRef] [Green Version]
- Nelson, A.S.; Myers, K.C. Diagnosis, Treatment, and Molecular Pathology of Shwachman-Diamond Syndrome. Hematol. Oncol. Clin. N. Am. 2018, 32, 687–700. [Google Scholar] [CrossRef]
- Sasa, G.S.; Ribes-Zamora, A.; Nelson, N.D.; Bertuch, A.A. Three novel truncating TINF2 mutations causing severe dyskeratosis congenita in early childhood. Clin. Genet. 2012, 81, 470–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savage, S.A.; Walsh, M.F. Myelodysplastic Syndrome, Acute Myeloid Leukemia, and Cancer Surveillance in Fanconi Anemia. Hematol. Oncol. Clin. N. Am. 2018, 32, 657–668. [Google Scholar] [CrossRef] [PubMed]
- Speckmann, C.; Sahoo, S.S.; Rizzi, M.; Hirabayashi, S.; Karow, A.; Serwas, N.K.; Hoemberg, M.; Damatova, N.; Schindler, D.; Vannier, J.B.; et al. Clinical and Molecular Heterogeneity of RTEL1 Deficiency. Front. Immunol. 2017, 8, 449. [Google Scholar] [CrossRef] [Green Version]
- Norberg, A.; Rosén, A.; Raaschou-Jensen, K.; Kjeldsen, L.; Moilanen, J.S.; Paulsson-Karlsson, Y.; Baliakas, P.; Lohi, O.; Ahmed, A.; Kittang, A.O.; et al. Novel variants in Nordic patients referred for genetic testing of telomere-related disorders. Eur. J. Hum. Genet. 2018, 26, 858–867. [Google Scholar] [CrossRef] [Green Version]
- Richards, L.A.; Kumari, A.; Knezevic, K.; Thoms, J.A.; von Jonquieres, G.; Napier, C.E.; Ali, Z.; O’Brien, R.; Marks-Bluth, J.; Maritz, M.F.; et al. DKC1 is a transcriptional target of GATA1 and drives upregulation of telomerase activity in normal human erythroblasts. Haematologica 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walne, A.J.; Bhagat, T.; Kirwan, M.; Gitiaux, C.; Desguerre, I.; Leonard, N.; Nogales, E.; Vulliamy, T.; Dokal, I.S. Mutations in the telomere capping complex in bone marrow failure and related syndromes. Haematologica 2013, 98, 334–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walne, A.J.; Vulliamy, T.; Kirwan, M.; Plagnol, V.; Dokal, I. Constitutional mutations in RTEL1 cause severe dyskeratosis congenita. Am. J. Hum. Genet. 2013, 92, 448–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballew, B.J.; Yeager, M.; Jacobs, K.; Giri, N.; Boland, J.; Burdett, L.; Alter, B.P.; Savage, S.A. Germline mutations of regulator of telomere elongation helicase 1, RTEL1, in Dyskeratosis congenita. Hum. Genet. 2013, 132, 473–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Rocco, D.; Bottega, R.; Cappelli, E.; Cavani, S.; Criscuolo, M.; Nicchia, E.; Corsolini, F.; Greco, C.; Borriello, A.; Svahn, J.; et al. Molecular analysis of Fanconi anemia: The experience of the Bone Marrow Failure Study Group of the Italian Association of Pediatric Onco-Hematology. Haematologica 2014, 99, 1022–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, M.; Ramanagoudr-Bhojappa, R.; van Twest, S.; Rosti, R.O.; Murphy, V.; Tan, W.; Donovan, F.X.; Lach, F.P.; Kimble, D.C.; Jiang, C.S.; et al. Association of clinical severity with FANCB variant type in Fanconi anemia. Blood 2020, 135, 1588–1602. [Google Scholar] [CrossRef]
- Kimble, D.C.; Lach, F.P.; Gregg, S.Q.; Donovan, F.X.; Flynn, E.K.; Kamat, A.; Young, A.; Vemulapalli, M.; Thomas, J.W.; Mullikin, J.C.; et al. A comprehensive approach to identification of pathogenic FANCA variants in Fanconi anemia patients and their families. Hum. Mutat. 2018, 39, 237–254. [Google Scholar] [CrossRef]
- Donovan, F.X.; Kimble, D.C.; Kim, Y.; Lach, F.P.; Harper, U.; Kamat, A.; Jones, M.; Sanborn, E.M.; Tryon, R.; Wagner, J.E.; et al. Paternal or Maternal Uniparental Disomy of Chromosome 16 Resulting in Homozygosity of a Mutant Allele Causes Fanconi Anemia. Hum. Mutat. 2016, 37, 465–468. [Google Scholar] [CrossRef] [Green Version]
- Bottega, R.; Nicchia, E.; Cappelli, E.; Ravera, S.; De Rocco, D.; Faleschini, M.; Corsolini, F.; Pierri, F.; Calvillo, M.; Russo, G.; et al. Hypomorphic FANCA mutations correlate with mild mitochondrial and clinical phenotype in Fanconi anemia. Haematologica 2018, 103, 417–426. [Google Scholar] [CrossRef] [Green Version]
- Wijker, M.; Morgan, N.V.; Herterich, S.; van Berkel, C.G.; Tipping, A.J.; Gross, H.J.; Gille, J.J.; Pals, G.; Savino, M.; Altay, C.; et al. Heterogeneous spectrum of mutations in the Fanconi anaemia group A gene. Eur. J. Hum. Genet. 1999, 7, 52–59. [Google Scholar] [CrossRef]
- Auerbach, A.D. Fanconi anemia and its diagnosis. Mutat. Res. 2009, 668, 4–10. [Google Scholar] [CrossRef] [Green Version]
- Callén, E.; Casado, J.A.; Tischkowitz, M.D.; Bueren, J.A.; Creus, A.; Marcos, R.; Dasí, A.; Estella, J.M.; Muñoz, A.; Ortega, J.J.; et al. A common founder mutation in FANCA underlies the world’s highest prevalence of Fanconi anemia in Gypsy families from Spain. Blood 2005, 105, 1946–1949. [Google Scholar] [CrossRef] [Green Version]
- Castella, M.; Pujol, R.; Callén, E.; Trujillo, J.P.; Casado, J.A.; Gille, H.; Lach, F.P.; Auerbach, A.D.; Schindler, D.; Benítez, J.; et al. Origin, functional role, and clinical impact of Fanconi anemia FANCA mutations. Blood 2011, 117, 3759–3769. [Google Scholar] [CrossRef] [PubMed]
- Aftab, I.; Iram, S.; Khaliq, S.; Israr, M.; Ali, N.; Jahan, S.; Hussain, S.; Mohsin, S. Analysis of FANCC gene mutations (IVS4+4A>T, del322G, and R548X)in patients with Fanconi anemia in Pakistan. Turk. J. Med. Sci. 2017, 47, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekharappa, S.C.; Chinn, S.B.; Donovan, F.X.; Chowdhury, N.I.; Kamat, A.; Adeyemo, A.A.; Thomas, J.W.; Vemulapalli, M.; Hussey, C.S.; Reid, H.H.; et al. Assessing the spectrum of germline variation in Fanconi anemia genes among patients with head and neck carcinoma before age 50. Cancer 2017, 123, 3943–3954. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekharappa, S.C.; Lach, F.P.; Kimble, D.C.; Kamat, A.; Teer, J.K.; Donovan, F.X.; Flynn, E.; Sen, S.K.; Thongthip, S.; Sanborn, E.; et al. Massively parallel sequencing, aCGH, and RNA-Seq technologies provide a comprehensive molecular diagnosis of Fanconi anemia. Blood 2013, 121, e138–e148. [Google Scholar] [CrossRef] [PubMed]
- Ramus, S.J.; Song, H.; Dicks, E.; Tyrer, J.P.; Rosenthal, A.N.; Intermaggio, M.P.; Fraser, L.; Gentry-Maharaj, A.; Hayward, J.; Philpott, S.; et al. Germline Mutations in the BRIP1, BARD1, PALB2, and NBN Genes in Women With Ovarian Cancer. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef]
- McCauley, J.; Masand, N.; McGowan, R.; Rajagopalan, S.; Hunter, A.; Michaud, J.L.; Gibson, K.; Robertson, J.; Vaz, F.; Abbs, S.; et al. X-linked VACTERL with hydrocephalus syndrome: Further delineation of the phenotype caused by FANCB mutations. Am. J. Med. Genet. A 2011, 155A, 2370–2380. [Google Scholar] [CrossRef]
- Frey, M.K.; Kopparam, R.V.; Ni Zhou, Z.; Fields, J.C.; Buskwofie, A.; Carlson, A.D.; Caputo, T.; Holcomb, K.; Chapman-Davis, E. Prevalence of nonfounder BRCA1/2 mutations in Ashkenazi Jewish patients presenting for genetic testing at a hereditary breast and ovarian cancer center. Cancer 2019, 125, 690–697. [Google Scholar] [CrossRef]
- Weber-Lassalle, N.; Hauke, J.; Ramser, J.; Richters, L.; Groß, E.; Blümcke, B.; Gehrig, A.; Kahlert, A.K.; Müller, C.R.; Hackmann, K.; et al. BRIP1 loss-of-function mutations confer high risk for familial ovarian cancer, but not familial breast cancer. Breast Cancer Res. 2018, 20, 7. [Google Scholar] [CrossRef]
- Yin, H.; Ma, H.; Hussain, S.; Zhang, H.; Xie, X.; Jiang, L.; Jiang, X.; Iqbal, F.; Bukhari, I.; Jiang, H.; et al. A homozygous FANCM frameshift pathogenic variant causes male infertility. Genet. Med. 2019, 21, 62–70. [Google Scholar] [CrossRef] [Green Version]
- Ghazwani, Y.; AlBalwi, M.; Al-Abdulkareem, I.; Al-Dress, M.; Alharbi, T.; Alsudairy, R.; Alomari, A.; Aljamaan, K.; Essa, M.; Al-Zahrani, M.; et al. Clinical characteristics and genetic subtypes of Fanconi anemia in Saudi patients. Cancer Genet. 2016, 209, 171–176. [Google Scholar] [CrossRef]
- Leung, C.C.; Gong, Z.; Chen, J.; Glover, J.N. Molecular basis of BACH1/FANCJ recognition by TopBP1 in DNA replication checkpoint control. J. Biol. Chem. 2011, 286, 4292–4301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, B.; Dorsman, J.C.; Ameziane, N.; de Vries, Y.; Rooimans, M.A.; Sheng, Q.; Pals, G.; Errami, A.; Gluckman, E.; Llera, J.; et al. Fanconi anemia is associated with a defect in the BRCA2 partner PALB2. Nat. Genet. 2007, 39, 159–161. [Google Scholar] [CrossRef] [PubMed]
- Lhota, F.; Zemankova, P.; Kleiblova, P.; Soukupova, J.; Vocka, M.; Stranecky, V.; Janatova, M.; Hartmannova, H.; Hodanova, K.; Kmoch, S.; et al. Hereditary truncating mutations of DNA repair and other genes in BRCA1/BRCA2/PALB2-negatively tested breast cancer patients. Clin. Genet. 2016, 90, 324–333. [Google Scholar] [CrossRef]
- Horwitz, M.S.; Duan, Z.; Korkmaz, B.; Lee, H.H.; Mealiffe, M.E.; Salipante, S.J. Neutrophil elastase in cyclic and severe congenital neutropenia. Blood 2007, 109, 1817–1824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, J.; Bolyard, A.A.; Rodger, E.; Stein, S.; Aprikyan, A.A.; Dale, D.C.; Link, D.C. Prevalence of mutations in ELANE, GFI1, HAX1, SBDS, WAS and G6PC3 in patients with severe congenital neutropenia. Br. J. Haematol. 2009, 147, 535–542. [Google Scholar] [CrossRef] [Green Version]
- Gong, R.L.; Wu, J.; Chen, T.X. Clinical, Laboratory, and Molecular Characteristics and Remission Status in Children With Severe Congenital and Non-congenital Neutropenia. Front. Pediatr. 2018, 6, 305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dale, D.C.; Cottle, T.E.; Fier, C.J.; Bolyard, A.A.; Bonilla, M.A.; Boxer, L.A.; Cham, B.; Freedman, M.H.; Kannourakis, G.; Kinsey, S.E.; et al. Severe chronic neutropenia: Treatment and follow-up of patients in the Severe Chronic Neutropenia International Registry. Am. J. Hematol. 2003, 72, 82–93. [Google Scholar] [CrossRef]
- Dale, D.C.; Person, R.E.; Bolyard, A.A.; Aprikyan, A.G.; Bos, C.; Bonilla, M.A.; Boxer, L.A.; Kannourakis, G.; Zeidler, C.; Welte, K.; et al. Mutations in the gene encoding neutrophil elastase in congenital and cyclic neutropenia. Blood 2000, 96, 2317–2322. [Google Scholar] [CrossRef]
- Shiohara, M.; Shigemura, T.; Saito, S.; Tanaka, M.; Yanagisawa, R.; Sakashita, K.; Asada, H.; Ishii, E.; Koike, K.; Chin, M.; et al. Ela2 mutations and clinical manifestations in familial congenital neutropenia. J. Pediatr. Hematol. Oncol. 2009, 31, 319–324. [Google Scholar] [CrossRef]
- Ishikawa, N.; Okada, S.; Miki, M.; Shirao, K.; Kihara, H.; Tsumura, M.; Nakamura, K.; Kawaguchi, H.; Ohtsubo, M.; Yasunaga, S.; et al. Neurodevelopmental abnormalities associated with severe congenital neutropenia due to the R86X mutation in the HAX1 gene. J. Med. Genet. 2008, 45, 802–807. [Google Scholar] [CrossRef]
- Klein, C.; Grudzien, M.; Appaswamy, G.; Germeshausen, M.; Sandrock, I.; Schäffer, A.A.; Rathinam, C.; Boztug, K.; Schwinzer, B.; Rezaei, N.; et al. HAX1 deficiency causes autosomal recessive severe congenital neutropenia (Kostmann disease). Nat. Genet. 2007, 39, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Boztug, K.; Appaswamy, G.; Ashikov, A.; Schäffer, A.A.; Salzer, U.; Diestelhorst, J.; Germeshausen, M.; Brandes, G.; Lee-Gossler, J.; Noyan, F.; et al. A syndrome with congenital neutropenia and mutations in G6PC3. N. Engl. J. Med. 2009, 360, 32–43. [Google Scholar] [CrossRef] [Green Version]
- Klimiankou, M.; Mellor-Heineke, S.; Zeidler, C.; Welte, K.; Skokowa, J. Role of CSF3R mutations in the pathomechanism of congenital neutropenia and secondary acute myeloid leukemia. Ann. N. Y. Acad. Sci. 2016, 1370, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Banka, S.; Newman, W.G. A clinical and molecular review of ubiquitous glucose-6-phosphatase deficiency caused by G6PC3 mutations. Orphanet J. Rare Dis. 2013, 8, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulácsy, V.; Soltész, B.; Petrescu, C.; Bataneant, M.; Gyimesi, E.; Serban, M.; Maródi, L.; Tóth, B. A novel large deletion and single nucleotide insertion in the Wiskott-Aldrich syndrome protein gene. Eur. J. Haematol. 2015, 95, 93–98. [Google Scholar] [CrossRef] [Green Version]
- Gulácsy, V.; Freiberger, T.; Shcherbina, A.; Pac, M.; Chernyshova, L.; Avcin, T.; Kondratenko, I.; Kostyuchenko, L.; Prokofjeva, T.; Pasic, S.; et al. Genetic characteristics of eighty-seven patients with the Wiskott-Aldrich syndrome. Mol. Immunol. 2011, 48, 788–792. [Google Scholar] [CrossRef]
- Campagnoli, M.F.; Garelli, E.; Quarello, P.; Carando, A.; Varotto, S.; Nobili, B.; Longoni, D.; Pecile, V.; Zecca, M.; Dufour, C.; et al. Molecular basis of Diamond-Blackfan anemia: New findings from the Italian registry and a review of the literature. Haematologica 2004, 89, 480–489. [Google Scholar]
- Mirabello, L.; Khincha, P.P.; Ellis, S.R.; Giri, N.; Brodie, S.; Chandrasekharappa, S.C.; Donovan, F.X.; Zhou, W.; Hicks, B.D.; Boland, J.F.; et al. Novel and known ribosomal causes of Diamond-Blackfan anaemia identified through comprehensive genomic characterisation. J. Med. Genet. 2017, 54, 417–425. [Google Scholar] [CrossRef]
- Ludwig, L.S.; Gazda, H.T.; Eng, J.C.; Eichhorn, S.W.; Thiru, P.; Ghazvinian, R.; George, T.I.; Gotlib, J.R.; Beggs, A.H.; Sieff, C.A.; et al. Altered translation of GATA1 in Diamond-Blackfan anemia. Nat. Med. 2014, 20, 748–753. [Google Scholar] [CrossRef] [Green Version]
- Boria, I.; Quarello, P.; Avondo, F.; Garelli, E.; Aspesi, A.; Carando, A.; Campagnoli, M.F.; Dianzani, I.; Ramenghi, U. A new database for ribosomal protein genes which are mutated in Diamond-Blackfan Anemia. Hum. Mutat. 2008, 29, E263–E270. [Google Scholar] [CrossRef]
- Campagnoli, M.F.; Ramenghi, U.; Armiraglio, M.; Quarello, P.; Garelli, E.; Carando, A.; Avondo, F.; Pavesi, E.; Fribourg, S.; Gleizes, P.E.; et al. RPS19 mutations in patients with Diamond-Blackfan anemia. Hum. Mutat. 2008, 29, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Chatr-Aryamontri, A.; Angelini, M.; Garelli, E.; Tchernia, G.; Ramenghi, U.; Dianzani, I.; Loreni, F. Nonsense-mediated and nonstop decay of ribosomal protein S19 mRNA in Diamond-Blackfan anemia. Hum. Mutat. 2004, 24, 526–533. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, L.; Narla, A.; Mohandas, N. An update on the pathogenesis and diagnosis of Diamond-Blackfan anemia. F1000Res 2018, 7. [Google Scholar] [CrossRef]
- Shimamura, A.; Alter, B.P. Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev. 2010, 24, 101–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santucci, M.A.; Bagnara, G.P.; Strippoli, P.; Bonsi, L.; Vitale, L.; Tonelli, R.; Locatelli, F.; Gabutti, V.; Ramenghi, U.; D’Avanzo, M.; et al. Long-term bone marrow cultures in Diamond-Blackfan anemia reveal a defect of both granulomacrophage and erythroid progenitors. Exp. Hematol. 1999, 27, 9–18. [Google Scholar] [CrossRef]
- Zhang, Y.; Ear, J.; Yang, Z.; Morimoto, K.; Zhang, B.; Lin, S. Defects of protein production in erythroid cells revealed in a zebrafish Diamond-Blackfan anemia model for mutation in RPS19. Cell Death Dis. 2014, 5, e1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Signer, R.A.; Magee, J.A.; Salic, A.; Morrison, S.J. Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature 2014, 509, 49–54. [Google Scholar] [CrossRef]
- Vlachos, A.; Rosenberg, P.S.; Atsidaftos, E.; Alter, B.P.; Lipton, J.M. Incidence of neoplasia in Diamond Blackfan anemia: A report from the Diamond Blackfan Anemia Registry. Blood 2012, 119, 3815–3819. [Google Scholar] [CrossRef] [Green Version]
- Vlachos, A.; Rosenberg, P.S.; Atsidaftos, E.; Kang, J.; Onel, K.; Sharaf, R.N.; Alter, B.P.; Lipton, J.M. Increased risk of colon cancer and osteogenic sarcoma in Diamond-Blackfan anemia. Blood 2018, 132, 2205–2208. [Google Scholar] [CrossRef] [Green Version]
- Maung, K.Z.Y.; Leo, P.J.; Bassal, M.; Casolari, D.A.; Gray, J.X.; Bray, S.C.; Pederson, S.; Singhal, D.; Samaraweera, S.E.; Nguyen, T.; et al. Rare variants in Fanconi anemia genes are enriched in acute myeloid leukemia. Blood Cancer J. 2018, 8, 50. [Google Scholar] [CrossRef]
- Kee, Y.; D’Andrea, A.D. Molecular pathogenesis and clinical management of Fanconi anemia. J. Clin. Investig. 2012, 122, 3799–3806. [Google Scholar] [CrossRef] [Green Version]
- Risitano, A.M.; Marotta, S.; Calzone, R.; Grimaldi, F.; Zatterale, A.; Contributors, R. Twenty years of the Italian Fanconi Anemia Registry: Where we stand and what remains to be learned. Haematologica 2016, 101, 319–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alter, B.P.; Greene, M.H.; Velazquez, I.; Rosenberg, P.S. Cancer in Fanconi anemia. Blood 2003, 101, 2072. [Google Scholar] [CrossRef]
- Butturini, A.; Gale, R.P.; Verlander, P.C.; Adler-Brecher, B.; Gillio, A.P.; Auerbach, A.D. Hematologic abnormalities in Fanconi anemia: An International Fanconi Anemia Registry study. Blood 1994, 84, 1650–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, P.S.; Alter, B.P.; Ebell, W. Cancer risks in Fanconi anemia: Findings from the German Fanconi Anemia Registry. Haematologica 2008, 93, 511–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alter, B.P.; Giri, N.; Savage, S.A.; Rosenberg, P.S. Cancer in the National Cancer Institute inherited bone marrow failure syndrome cohort after fifteen years of follow-up. Haematologica 2018, 103, 30–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat. Rev. Genet. 2007, 8, 735–748. [Google Scholar] [CrossRef] [PubMed]
- Naim, V.; Rosselli, F. The FANC pathway and mitosis: A replication legacy. Cell Cycle 2009, 8, 2907–2911. [Google Scholar] [CrossRef]
- Kottemann, M.C.; Smogorzewska, A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature 2013, 493, 356–363. [Google Scholar] [CrossRef] [Green Version]
- Alter, B.P. Fanconi anemia and the development of leukemia. Best Pract. Res. Clin. Haematol. 2014, 27, 214–221. [Google Scholar] [CrossRef] [Green Version]
- Bertuch, A.A. The molecular genetics of the telomere biology disorders. RNA Biol. 2016, 13, 696–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, S. Evaluation and Management of Hematopoietic Failure in Dyskeratosis Congenita. Hematol. Oncol. Clin. N. Am. 2018, 32, 669–685. [Google Scholar] [CrossRef]
- Alter, B.P.; Rosenberg, P.S.; Giri, N.; Baerlocher, G.M.; Lansdorp, P.M.; Savage, S.A. Telomere length is associated with disease severity and declines with age in dyskeratosis congenita. Haematologica 2012, 97, 353–359. [Google Scholar] [CrossRef] [Green Version]
- Mason, P.J.; Bessler, M. The genetics of dyskeratosis congenita. Cancer Genet. 2011, 204, 635–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.; Jing, S.; Dai, C.; Tu, C.; Tan, Z.; Du, J.; Lu, G.X.; Lin, G.; Zeng, S. Telomerase insufficiency induced telomere erosion accumulation in successive generations in dyskeratosis congenita family. Mol. Genet. Genom. Med. 2019, 7, e00709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scahill, C.M.; Digby, Z.; Sealy, I.M.; White, R.J.; Wali, N.; Collins, J.E.; Stemple, D.L.; Busch-Nentwich, E.M. The age of heterozygous. Wellcome Open Res. 2017, 2, 77. [Google Scholar] [CrossRef] [Green Version]
- Donadieu, J.; Beaupain, B.; Fenneteau, O.; Bellanné-Chantelot, C. Congenital neutropenia in the era of genomics: Classification, diagnosis, and natural history. Br. J. Haematol. 2017, 179, 557–574. [Google Scholar] [CrossRef] [Green Version]
- Kostmann, R. Infantile genetic agranulocytosis; agranulocytosis infantilis hereditaria. Acta Paediatr. 1956, 45, 309–310. [Google Scholar] [CrossRef]
- Lanciotti, M.; Caridi, G.; Rosano, C.; Pigullo, S.; Lanza, T.; Dufour, C. Severe congenital neutropenia: A negative synergistic effect of multiple mutations of ELANE (ELA2) gene. Br. J. Haematol. 2009, 146, 578–580. [Google Scholar] [CrossRef]
- Person, R.E.; Li, F.Q.; Duan, Z.; Benson, K.F.; Wechsler, J.; Papadaki, H.A.; Eliopoulos, G.; Kaufman, C.; Bertolone, S.J.; Nakamoto, B.; et al. Mutations in proto-oncogene GFI1 cause human neutropenia and target ELA2. Nat. Genet. 2003, 34, 308–312. [Google Scholar] [CrossRef] [Green Version]
- Makaryan, V.; Rosenthal, E.A.; Bolyard, A.A.; Kelley, M.L.; Below, J.E.; Bamshad, M.J.; Bofferding, K.M.; Smith, J.D.; Buckingham, K.; Boxer, L.A.; et al. TCIRG1-associated congenital neutropenia. Hum. Mutat. 2014, 35, 824–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boztug, K.; Järvinen, P.M.; Salzer, E.; Racek, T.; Mönch, S.; Garncarz, W.; Gertz, E.M.; Schäffer, A.A.; Antonopoulos, A.; Haslam, S.M.; et al. JAGN1 deficiency causes aberrant myeloid cell homeostasis and congenital neutropenia. Nat. Genet. 2014, 46, 1021–1027. [Google Scholar] [CrossRef]
- McDermott, D.H.; De Ravin, S.S.; Jun, H.S.; Liu, Q.; Priel, D.A.; Noel, P.; Takemoto, C.M.; Ojode, T.; Paul, S.M.; Dunsmore, K.P.; et al. Severe congenital neutropenia resulting from G6PC3 deficiency with increased neutrophil CXCR4 expression and myelokathexis. Blood 2010, 116, 2793–2802. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Z.Q.; Lu, C.X.; Wang, W.; Qiu, J.J.; Wei, M. Mutation in the SLC37A4 gene of glycogen storage disease type Ib in 15 families of the mainland of China. Zhonghua Er Ke Za Zhi 2011, 49, 203–208. [Google Scholar] [PubMed]
- Stepensky, P.; Saada, A.; Cowan, M.; Tabib, A.; Fischer, U.; Berkun, Y.; Saleh, H.; Simanovsky, N.; Kogot-Levin, A.; Weintraub, M.; et al. The Thr224Asn mutation in the VPS45 gene is associated with the congenital neutropenia and primary myelofibrosis of infancy. Blood 2013, 121, 5078–5087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilboux, T.; Lev, A.; Malicdan, M.C.; Simon, A.J.; Järvinen, P.; Racek, T.; Puchalka, J.; Sood, R.; Carrington, B.; Bishop, K.; et al. A congenital neutrophil defect syndrome associated with mutations in VPS45. N. Engl. J. Med. 2013, 369, 54–65. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, P.A.; Gorlin, R.J.; Lukens, J.N.; Taniuchi, S.; Bohinjec, J.; Francois, F.; Klotman, M.E.; Diaz, G.A. Mutations in the chemokine receptor gene CXCR4 are associated with WHIM syndrome, a combined immunodeficiency disease. Nat. Genet. 2003, 34, 70–74. [Google Scholar] [CrossRef]
- Auer, P.L.; Teumer, A.; Schick, U.; O’Shaughnessy, A.; Lo, K.S.; Chami, N.; Carlson, C.; de Denus, S.; Dubé, M.P.; Haessler, J.; et al. Rare and low-frequency coding variants in CXCR2 and other genes are associated with hematological traits. Nat. Genet. 2014, 46, 629–634. [Google Scholar] [CrossRef]
- Halacli, S.O.; Ayvaz, D.C.; Sun-Tan, C.; Erman, B.; Uz, E.; Yilmaz, D.Y.; Ozgul, K.; Tezcan, İ.; Sanal, O. STK4 (MST1) deficiency in two siblings with autoimmune cytopenias: A novel mutation. Clin. Immunol. 2015, 161, 316–323. [Google Scholar] [CrossRef]
- Pasquet, M.; Bellanné-Chantelot, C.; Tavitian, S.; Prade, N.; Beaupain, B.; Larochelle, O.; Petit, A.; Rohrlich, P.; Ferrand, C.; Van Den Neste, E.; et al. High frequency of GATA2 mutations in patients with mild chronic neutropenia evolving to MonoMac syndrome, myelodysplasia, and acute myeloid leukemia. Blood 2013, 121, 822–829. [Google Scholar] [CrossRef]
- Devriendt, K.; Kim, A.S.; Mathijs, G.; Frints, S.G.; Schwartz, M.; Van Den Oord, J.J.; Verhoef, G.E.; Boogaerts, M.A.; Fryns, J.P.; You, D.; et al. Constitutively activating mutation in WASP causes X-linked severe congenital neutropenia. Nat. Genet. 2001, 27, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Bellanné-Chantelot, C.; Clauin, S.; Leblanc, T.; Cassinat, B.; Rodrigues-Lima, F.; Beaufils, S.; Vaury, C.; Barkaoui, M.; Fenneteau, O.; Maier-Redelsperger, M.; et al. Mutations in the ELA2 gene correlate with more severe expression of neutropenia: A study of 81 patients from the French Neutropenia Register. Blood 2004, 103, 4119–4125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makaryan, V.; Zeidler, C.; Bolyard, A.A.; Skokowa, J.; Rodger, E.; Kelley, M.L.; Boxer, L.A.; Bonilla, M.A.; Newburger, P.E.; Shimamura, A.; et al. The diversity of mutations and clinical outcomes for ELANE-associated neutropenia. Curr. Opin. Hematol. 2015, 22, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Germeshausen, M.; Zeidler, C.; Stuhrmann, M.; Lanciotti, M.; Ballmaier, M.; Welte, K. Digenic mutations in severe congenital neutropenia. Haematologica 2010, 95, 1207–1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Germeshausen, M.; Grudzien, M.; Zeidler, C.; Abdollahpour, H.; Yetgin, S.; Rezaei, N.; Ballmaier, M.; Grimbacher, B.; Welte, K.; Klein, C. Novel HAX1 mutations in patients with severe congenital neutropenia reveal isoform-dependent genotype-phenotype associations. Blood 2008, 111, 4954–4957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yılmaz Karapınar, D.; Patıroğlu, T.; Metin, A.; Çalışkan, Ü.; Celkan, T.; Yılmaz, B.; Karakaş, Z.; Karapınar, T.H.; Akıncı, B.; Özkınay, F.; et al. Homozygous c.130-131 ins A (pW44X) mutation in the HAX1 gene as the most common cause of congenital neutropenia in Turkey: Report from the Turkish Severe Congenital Neutropenia Registry. Pediatr. Blood Cancer 2019, 66, e27923. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, P.S.; Zeidler, C.; Bolyard, A.A.; Alter, B.P.; Bonilla, M.A.; Boxer, L.A.; Dror, Y.; Kinsey, S.; Link, D.C.; Newburger, P.E.; et al. Stable long-term risk of leukaemia in patients with severe congenital neutropenia maintained on G-CSF therapy. Br. J. Haematol. 2010, 150, 196–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skokowa, J.; Dale, D.C.; Touw, I.P.; Zeidler, C.; Welte, K. Severe congenital neutropenias. Nat. Rev. Dis. Prim. 2017, 3, 17032. [Google Scholar] [CrossRef]
- Keeling, K.M.; Wang, D.; Conard, S.E.; Bedwell, D.M. Suppression of premature termination codons as a therapeutic approach. Crit. Rev. Biochem. Mol. Biol. 2012, 47, 444–463. [Google Scholar] [CrossRef] [Green Version]
- Bonetti, B.; Fu, L.; Moon, J.; Bedwell, D.M. The efficiency of translation termination is determined by a synergistic interplay between upstream and downstream sequences in Saccharomyces cerevisiae. J. Mol. Biol. 1995, 251, 334–345. [Google Scholar] [CrossRef] [PubMed]
- Cassan, M.; Rousset, J.P. UAG readthrough in mammalian cells: Effect of upstream and downstream stop codon contexts reveal different signals. BMC Mol. Biol. 2001, 2, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manuvakhova, M.; Keeling, K.; Bedwell, D.M. Aminoglycoside antibiotics mediate context-dependent suppression of termination codons in a mammalian translation system. RNA 2000, 6, 1044–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dabrowski, M.; Bukowy-Bieryllo, Z.; Zietkiewicz, E. Translational readthrough potential of natural termination codons in eucaryotes--The impact of RNA sequence. RNA Biol. 2015, 12, 950–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fearon, K.; McClendon, V.; Bonetti, B.; Bedwell, D.M. Premature translation termination mutations are efficiently suppressed in a highly conserved region of yeast Ste6p, a member of the ATP-binding cassette (ABC) transporter family. J. Biol. Chem. 1994, 269, 17802–17808. [Google Scholar]
- McCaughan, K.K.; Brown, C.M.; Dalphin, M.E.; Berry, M.J.; Tate, W.P. Translational termination efficiency in mammals is influenced by the base following the stop codon. Proc. Natl. Acad. Sci. USA 1995, 92, 5431–5435. [Google Scholar] [CrossRef] [Green Version]
- Jungreis, I.; Lin, M.F.; Spokony, R.; Chan, C.S.; Negre, N.; Victorsen, A.; White, K.P.; Kellis, M. Evidence of abundant stop codon readthrough in Drosophila and other metazoa. Genome Res. 2011, 21, 2096–2113. [Google Scholar] [CrossRef] [Green Version]
- Loughran, G.; Chou, M.Y.; Ivanov, I.P.; Jungreis, I.; Kellis, M.; Kiran, A.M.; Baranov, P.V.; Atkins, J.F. Evidence of efficient stop codon readthrough in four mammalian genes. Nucleic Acids Res. 2014, 42, 8928–8938. [Google Scholar] [CrossRef] [Green Version]
- Beznosková, P.; Gunišová, S.; Valášek, L.S. Rules of UGA-N decoding by near-cognate tRNAs and analysis of readthrough on short uORFs in yeast. RNA 2016, 22, 456–466. [Google Scholar] [CrossRef] [Green Version]
- Dabrowski, M.; Bukowy-Bieryllo, Z.; Zietkiewicz, E. Advances in therapeutic use of a drug-stimulated translational readthrough of premature termination codons. Mol. Med. 2018, 24, 25. [Google Scholar] [CrossRef] [Green Version]
- Krause, K.M.; Serio, A.W.; Kane, T.R.; Connolly, L.E. Aminoglycosides: An Overview. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Mingeot-Leclercq, M.P.; Glupczynski, Y.; Tulkens, P.M. Aminoglycosides: Activity and resistance. Antimicrob. Agents Chemother. 1999, 43, 727–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magnet, S.; Blanchard, J.S. Molecular insights into aminoglycoside action and resistance. Chem. Rev. 2005, 105, 477–498. [Google Scholar] [CrossRef] [PubMed]
- Wachino, J.; Arakawa, Y. Exogenously acquired 16S rRNA methyltransferases found in aminoglycoside-resistant pathogenic Gram-negative bacteria: An update. Drug Resist. Updates 2012, 15, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Kotra, L.P.; Haddad, J.; Mobashery, S. Aminoglycosides: Perspectives on mechanisms of action and resistance and strategies to counter resistance. Antimicrob. Agents Chemother. 2000, 44, 3249–3256. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, M.S.; Tolmasky, M.E. Aminoglycoside modifying enzymes. Drug Resist. Updates 2010, 13, 151–171. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, H.M.; Siddiqui, M.A.; Kanneganti, S.; Sharmin, N.; Chowdhury, M.W.; Nasim, M.T. Aminoglycoside-mediated promotion of translation readthrough occurs through a non-stochastic mechanism that competes with translation termination. Hum. Mol. Genet. 2018, 27, 373–384. [Google Scholar] [CrossRef] [Green Version]
- Prokhorova, I.; Altman, R.B.; Djumagulov, M.; Shrestha, J.P.; Urzhumtsev, A.; Ferguson, A.; Chang, C.T.; Yusupov, M.; Blanchard, S.C.; Yusupova, G. Aminoglycoside interactions and impacts on the eukaryotic ribosome. Proc. Natl. Acad. Sci. USA 2017, 114, E10899–E10908. [Google Scholar] [CrossRef] [Green Version]
- Howard, M.; Frizzell, R.A.; Bedwell, D.M. Aminoglycoside antibiotics restore CFTR function by overcoming premature stop mutations. Nat. Med. 1996, 2, 467–469. [Google Scholar] [CrossRef]
- Prayle, A.; Smyth, A.R. Aminoglycoside use in cystic fibrosis: Therapeutic strategies and toxicity. Curr. Opin. Pulm. Med. 2010, 16, 604–610. [Google Scholar] [CrossRef]
- Burke, J.F.; Mogg, A.E. Suppression of a nonsense mutation in mammalian cells in vivo by the aminoglycoside antibiotics G-418 and paromomycin. Nucleic Acids Res. 1985, 13, 6265–6272. [Google Scholar] [CrossRef]
- Kamei, M.; Kasperski, K.; Fuller, M.; Parkinson-Lawrence, E.J.; Karageorgos, L.; Belakhov, V.; Baasov, T.; Hopwood, J.J.; Brooks, D.A. Aminoglycoside-Induced Premature Stop Codon Read-Through of Mucopolysaccharidosis Type I Patient Q70X and W402X Mutations in Cultured Cells. JIMD Rep. 2014, 13, 139–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fazzari, M.; Frasca, A.; Bifari, F.; Landsberger, N. Aminoglycoside drugs induce efficient read-through of CDKL5 nonsense mutations, slightly restoring its kinase activity. RNA Biol. 2019, 16, 1414–1423. [Google Scholar] [CrossRef] [PubMed]
- Sleat, D.E.; Sohar, I.; Gin, R.M.; Lobel, P. Aminoglycoside-mediated suppression of nonsense mutations in late infantile neuronal ceroid lipofuscinosis. Eur. J. Paediatr. Neurol. 2001, 5 (Suppl. A), 57–62. [Google Scholar] [CrossRef] [PubMed]
- Keeling, K.M.; Brooks, D.A.; Hopwood, J.J.; Li, P.; Thompson, J.N.; Bedwell, D.M. Gentamicin-mediated suppression of Hurler syndrome stop mutations restores a low level of alpha-L-iduronidase activity and reduces lysosomal glycosaminoglycan accumulation. Hum. Mol. Genet. 2001, 10, 291–299. [Google Scholar] [CrossRef] [Green Version]
- Keeling, K.M.; Bedwell, D.M. Clinically relevant aminoglycosides can suppress disease-associated premature stop mutations in the IDUA and P53 cDNAs in a mammalian translation system. J. Mol. Med. (Berl) 2002, 80, 367–376. [Google Scholar] [CrossRef]
- Brendel, C.; Belakhov, V.; Werner, H.; Wegener, E.; Gärtner, J.; Nudelman, I.; Baasov, T.; Huppke, P. Readthrough of nonsense mutations in Rett syndrome: Evaluation of novel aminoglycosides and generation of a new mouse model. J. Mol. Med. (Berl) 2011, 89, 389–398. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Liu, X.; Huang, J.; Zhang, Y.; Hu, R.; Pu, J. Comparison of read-through effects of aminoglycosides and PTC124 on rescuing nonsense mutations of HERG gene associated with long QT syndrome. Int. J. Mol. Med. 2014, 33, 729–735. [Google Scholar] [CrossRef]
- Lincoln, V.; Cogan, J.; Hou, Y.; Hirsch, M.; Hao, M.; Alexeev, V.; De Luca, M.; De Rosa, L.; Bauer, J.W.; Woodley, D.T.; et al. Gentamicin induces. Proc. Natl. Acad. Sci. USA 2018, 115, E6536–E6545. [Google Scholar] [CrossRef] [Green Version]
- Harada, N.; Hatakeyama, A.; Okuyama, M.; Miyatake, Y.; Nakagawa, T.; Kuroda, M.; Masumoto, S.; Tsutsumi, R.; Nakaya, Y.; Sakaue, H. Readthrough of ACTN3 577X nonsense mutation produces full-length α-actinin-3 protein. Biochem. Biophys. Res. Commun. 2018, 502, 422–428. [Google Scholar] [CrossRef]
- Goldmann, T.; Overlack, N.; Möller, F.; Belakhov, V.; van Wyk, M.; Baasov, T.; Wolfrum, U.; Nagel-Wolfrum, K. A comparative evaluation of NB30, NB54 and PTC124 in translational read-through efficacy for treatment of an USH1C nonsense mutation. EMBO Mol. Med. 2012, 4, 1186–1199. [Google Scholar] [CrossRef]
- Nudelman, I.; Rebibo-Sabbah, A.; Cherniavsky, M.; Belakhov, V.; Hainrichson, M.; Chen, F.; Schacht, J.; Pilch, D.S.; Ben-Yosef, T.; Baasov, T. Development of novel aminoglycoside (NB54) with reduced toxicity and enhanced suppression of disease-causing premature stop mutations. J. Med. Chem. 2009, 52, 2836–2845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowe, S.M.; Sloane, P.; Tang, L.P.; Backer, K.; Mazur, M.; Buckley-Lanier, J.; Nudelman, I.; Belakhov, V.; Bebok, Z.; Schwiebert, E.; et al. Suppression of CFTR premature termination codons and rescue of CFTR protein and function by the synthetic aminoglycoside NB54. J. Mol. Med. (Berl) 2011, 89, 1149–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vecsler, M.; Ben Zeev, B.; Nudelman, I.; Anikster, Y.; Simon, A.J.; Amariglio, N.; Rechavi, G.; Baasov, T.; Gak, E. Ex vivo treatment with a novel synthetic aminoglycoside NB54 in primary fibroblasts from Rett syndrome patients suppresses MECP2 nonsense mutations. PLoS ONE 2011, 6, e20733. [Google Scholar] [CrossRef] [PubMed]
- Nudelman, I.; Glikin, D.; Smolkin, B.; Hainrichson, M.; Belakhov, V.; Baasov, T. Repairing faulty genes by aminoglycosides: Development of new derivatives of geneticin (G418) with enhanced suppression of diseases-causing nonsense mutations. Bioorg. Med. Chem. 2010, 18, 3735–3746. [Google Scholar] [CrossRef]
- Leubitz, A.; Frydman-Marom, A.; Sharpe, N.; van Duzer, J.; Campbell, K.C.M.; Vanhoutte, F. Safety, Tolerability, and Pharmacokinetics of Single Ascending Doses of ELX-02, a Potential Treatment for Genetic Disorders Caused by Nonsense Mutations, in Healthy Volunteers. Clin. Pharmacol. Drug Dev. 2019, 8, 984–994. [Google Scholar] [CrossRef]
- Crawford, D.; Alroy, I.; Sharpe, N.; Goddeeris, M.; Williams, G. ELX-02 generates protein via premature stop codon read-through without inducing native stop codon read-through proteins. J. Pharmacol. Exp. Ther. 2020. [Google Scholar] [CrossRef]
- Mattis, V.B.; Fosso, M.Y.; Chang, C.W.; Lorson, C.L. Subcutaneous administration of TC007 reduces disease severity in an animal model of SMA. BMC Neurosci. 2009, 10, 142. [Google Scholar] [CrossRef] [Green Version]
- Mattis, V.B.; Ebert, A.D.; Fosso, M.Y.; Chang, C.W.; Lorson, C.L. Delivery of a read-through inducing compound, TC007, lessens the severity of a spinal muscular atrophy animal model. Hum. Mol. Genet. 2009, 18, 3906–3913. [Google Scholar] [CrossRef] [Green Version]
- McDonald, C.M.; Campbell, C.; Torricelli, R.E.; Finkel, R.S.; Flanigan, K.M.; Goemans, N.; Heydemann, P.; Kaminska, A.; Kirschner, J.; Muntoni, F.; et al. Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD): A multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1489–1498. [Google Scholar] [CrossRef]
- Konstan, M.W.; VanDevanter, D.R.; Rowe, S.M.; Wilschanski, M.; Kerem, E.; Sermet-Gaudelus, I.; DiMango, E.; Melotti, P.; McIntosh, J.; De Boeck, K.; et al. Efficacy and safety of ataluren in patients with nonsense-mutation cystic fibrosis not receiving chronic inhaled aminoglycosides: The international, randomized, double-blind, placebo-controlled Ataluren Confirmatory Trial in Cystic Fibrosis (ACT CF). J. Cyst. Fibros. 2020. [Google Scholar] [CrossRef]
- Peabody Lever, J.E.; Mutyam, V.; Hathorne, H.Y.; Peng, N.; Sharma, J.; Edwards, L.J.; Rowe, S.M. Ataluren/ivacaftor combination therapy: Two N-of-1 trials in cystic fibrosis patients with nonsense mutations. Pediatr. Pulmonol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Finkel, R.S.; Flanigan, K.M.; Wong, B.; Bönnemann, C.; Sampson, J.; Sweeney, H.L.; Reha, A.; Northcutt, V.J.; Elfring, G.; Barth, J.; et al. Phase 2a study of ataluren-mediated dystrophin production in patients with nonsense mutation Duchenne muscular dystrophy. PLoS ONE 2013, 8, e81302. [Google Scholar] [CrossRef] [PubMed]
- Kerem, E.; Konstan, M.W.; De Boeck, K.; Accurso, F.J.; Sermet-Gaudelus, I.; Wilschanski, M.; Elborn, J.S.; Melotti, P.; Bronsveld, I.; Fajac, I.; et al. Ataluren for the treatment of nonsense-mutation cystic fibrosis: A randomised, double-blind, placebo-controlled phase 3 trial. Lancet Respir. Med. 2014, 2, 539–547. [Google Scholar] [CrossRef] [Green Version]
- Kong, R.; Laskin, O.L.; Kaushik, D.; Jin, F.; Ma, J.; McIntosh, J.; Souza, M.; Almstead, N. Ataluren Pharmacokinetics in Healthy Japanese and Caucasian Subjects. Clin. Pharmacol. Drug Dev. 2019, 8, 172–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lentini, L.; Melfi, R.; Di Leonardo, A.; Spinello, A.; Barone, G.; Pace, A.; Palumbo Piccionello, A.; Pibiri, I. Toward a rationale for the PTC124 (Ataluren) promoted readthrough of premature stop codons: A computational approach and GFP-reporter cell-based assay. Mol. Pharm. 2014, 11, 653–664. [Google Scholar] [CrossRef]
- Roy, B.; Friesen, W.J.; Tomizawa, Y.; Leszyk, J.D.; Zhuo, J.; Johnson, B.; Dakka, J.; Trotta, C.R.; Xue, X.; Mutyam, V.; et al. Ataluren stimulates ribosomal selection of near-cognate tRNAs to promote nonsense suppression. Proc. Natl. Acad. Sci. USA 2016, 113, 12508–12513. [Google Scholar] [CrossRef] [Green Version]
- Bezzerri, V.; Bardelli, D.; Morini, J.; Vella, A.; Cesaro, S.; Sorio, C.; Biondi, A.; Danesino, C.; Farruggia, P.; Assael, B.M.; et al. Ataluren-driven Restoration of Shwachman-Bodian-Diamond Syndrome Protein Function in Shwachman-Diamond Syndrome Bone Marrow Cells. Am. J. Hematol. 2017, 93, 527–536. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Gregory-Evans, K.; Wasan, K.M.; Sivak, O.; Shan, X.; Gregory-Evans, C.Y. Efficacy of Postnatal In Vivo Nonsense Suppression Therapy in a Pax6 Mouse Model of Aniridia. Mol. Ther. Nucleic Acids 2017, 7, 417–428. [Google Scholar] [CrossRef] [Green Version]
- Mercuri, E.; Muntoni, F.; Osorio, A.N.; Tulinius, M.; Buccella, F.; Morgenroth, L.P.; Gordish-Dressman, H.; Jiang, J.; Trifillis, P.; Zhu, J.; et al. Safety and effectiveness of ataluren: Comparison of results from the STRIDE Registry and CINRG DMD Natural History Study. J. Comp. Eff. Res. 2020, 9, 341–360. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhang, Y.; Zhang, B.; Gao, H.; Qiu, C. Nonsense suppression induced readthrough of a novel PAX6 mutation in patient-derived cells of congenital aniridia. Mol. Genet. Genomic. Med. 2020, 8, e1198. [Google Scholar] [CrossRef] [Green Version]
- Pibiri, I.; Lentini, L.; Melfi, R.; Tutone, M.; Baldassano, S.; Ricco Galluzzo, P.; Di Leonardo, A.; Pace, A. Rescuing the CFTR protein function: Introducing 1,3,4-oxadiazoles as translational readthrough inducing drugs. Eur. J. Med. Chem. 2018, 159, 126–142. [Google Scholar] [CrossRef]
- Baradaran-Heravi, A.; Balgi, A.D.; Zimmerman, C.; Choi, K.; Shidmoossavee, F.S.; Tan, J.S.; Bergeaud, C.; Krause, A.; Flibotte, S.; Shimizu, Y.; et al. Novel small molecules potentiate premature termination codon readthrough by aminoglycosides. Nucleic Acids Res. 2016, 44, 6583–6598. [Google Scholar] [CrossRef]
- Miller, J.N.; Chan, C.H.; Pearce, D.A. The role of nonsense-mediated decay in neuronal ceroid lipofuscinosis. Hum. Mol. Genet. 2013, 22, 2723–2734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, M.; Keeling, K.M.; Fan, L.; Liu, X.; Bedwell, D.M. Poly-L-aspartic acid enhances and prolongs gentamicin-mediated suppression of the CFTR-G542X mutation in a cystic fibrosis mouse model. J. Biol. Chem. 2009, 284, 6885–6892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Hilarion, S.; Beghyn, T.; Jia, J.; Debreuck, N.; Berte, G.; Mamchaoui, K.; Mouly, V.; Gruenert, D.C.; Déprez, B.; Lejeune, F. Rescue of nonsense mutations by amlexanox in human cells. Orphanet J. Rare Dis. 2012, 7, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atanasova, V.S.; Jiang, Q.; Prisco, M.; Gruber, C.; Piñón Hofbauer, J.; Chen, M.; Has, C.; Bruckner-Tuderman, L.; McGrath, J.A.; Uitto, J.; et al. Amlexanox Enhances Premature Termination Codon Read-Through in COL7A1 and Expression of Full Length Type VII Collagen: Potential Therapy for Recessive Dystrophic Epidermolysis Bullosa. J. Investig. Dermatol. 2017, 137, 1842–1849. [Google Scholar] [CrossRef] [Green Version]
- Lentini, L.; Melfi, R.; Cancemi, P.; Pibiri, I.; Di Leonardo, A. Caffeine boosts Ataluren’s readthrough activity. Heliyon 2019, 5, e01963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, H.; Mitsios, A.; Smart, M.; Skinner, J.; Welch, A.A.; Kalatzis, V.; Coffey, P.J.; Dubis, A.M.; Webster, A.R.; Moosajee, M. Nonsense-mediated mRNA decay efficiency varies in choroideremia providing a target to boost small molecule therapeutics. Hum. Mol. Genet. 2019, 28, 1865–1871. [Google Scholar] [CrossRef] [PubMed]
- Usuki, F.; Yamashita, A.; Higuchi, I.; Ohnishi, T.; Shiraishi, T.; Osame, M.; Ohno, S. Inhibition of nonsense-mediated mRNA decay rescues the phenotype in Ullrich’s disease. Ann. Neurol. 2004, 55, 740–744. [Google Scholar] [CrossRef]
- Lee, H.L.; Dougherty, J.P. Pharmaceutical therapies to recode nonsense mutations in inherited diseases. Pharmacol. Ther. 2012, 136, 227–266. [Google Scholar] [CrossRef]
- Chang, C.W.; Hui, Y.; Elchert, B.; Wang, J.; Li, J.; Rai, R. Pyranmycins, a novel class of aminoglycosides with improved acid stability: The SAR of D-pyranoses on ring III of pyranmycin. Org. Lett. 2002, 4, 4603–4606. [Google Scholar] [CrossRef] [PubMed]
- Campofelice, A.; Lentini, L.; Di Leonardo, A.; Melfi, R.; Tutone, M.; Pace, A.; Pibiri, I. Strategies against Nonsense: Oxadiazoles as Translational Readthrough-Inducing Drugs (TRIDs). Int. J. Mol. Sci. 2019, 20, 3329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, X.; Mutyam, V.; Tang, L.; Biswas, S.; Du, M.; Jackson, L.A.; Dai, Y.; Belakhov, V.; Shalev, M.; Chen, F.; et al. Synthetic aminoglycosides efficiently suppress cystic fibrosis transmembrane conductance regulator nonsense mutations and are enhanced by ivacaftor. Am. J. Respir. Cell Mol. Biol. 2014, 50, 805–816. [Google Scholar] [CrossRef] [Green Version]
- Peltz, S.W.; Morsy, M.; Welch, E.M.; Jacobson, A. Ataluren as an agent for therapeutic nonsense suppression. Annu. Rev. Med. 2013, 64, 407–425. [Google Scholar] [CrossRef] [Green Version]
- Pranke, I.; Bidou, L.; Martin, N.; Blanchet, S.; Hatton, A.; Karri, S.; Cornu, D.; Costes, B.; Chevalier, B.; Tondelier, D.; et al. Factors influencing readthrough therapy for frequent cystic fibrosis premature termination codons. ERJ Open Res. 2018, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McElroy, S.P.; Nomura, T.; Torrie, L.S.; Warbrick, E.; Gartner, U.; Wood, G.; McLean, W.H. A lack of premature termination codon read-through efficacy of PTC124 (Ataluren) in a diverse array of reporter assays. PLoS Biol. 2013, 11, e1001593. [Google Scholar] [CrossRef] [Green Version]
- Thada, V.; Miller, J.N.; Kovács, A.D.; Pearce, D.A. Tissue-specific variation in nonsense mutant transcript level and drug-induced read-through efficiency in the Cln1(R151X) mouse model of INCL. J. Cell Mol. Med. 2016, 20, 381–385. [Google Scholar] [CrossRef]
- Tutone, M.; Pibiri, I.; Lentini, L.; Pace, A.; Almerico, A.M. Deciphering the Nonsense Readthrough Mechanism of Action of Ataluren: An. ACS Med. Chem. Lett. 2019, 10, 522–527. [Google Scholar] [CrossRef]
- Zainal Abidin, N.; Haq, I.J.; Gardner, A.I.; Brodlie, M. Ataluren in cystic fibrosis: Development, clinical studies and where are we now? Expert Opin. Pharmacother. 2017, 18, 1363–1371. [Google Scholar] [CrossRef]
- Kerem, E.; Hirawat, S.; Armoni, S.; Yaakov, Y.; Shoseyov, D.; Cohen, M.; Nissim-Rafinia, M.; Blau, H.; Rivlin, J.; Aviram, M.; et al. Effectiveness of PTC124 treatment of cystic fibrosis caused by nonsense mutations: A prospective phase II trial. Lancet 2008, 372, 719–727. [Google Scholar] [CrossRef]
- Ryan, N.J.; Lo, J.H. Delamanid: First global approval. Drugs 2014, 74, 1041–1045. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi-Fakhari, D.; Dillmann, U.; Flotats-Bastardas, M.; Poryo, M.; Abdul-Khaliq, H.; Shamdeen, M.G.; Mischo, B.; Zemlin, M.; Meyer, S. Off-Label Use of Ataluren in Four Non-ambulatory Patients With Nonsense Mutation Duchenne Muscular Dystrophy: Effects on Cardiac and Pulmonary Function and Muscle Strength. Front. Pediatr. 2018, 6, 316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratner, D.; Lennerz, J.K. Implementing Keytruda/Pembrolizumab Testing in Clinical Practice. Oncologist 2018, 23, 647–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bezzerri, V.; Vella, A.; Calcaterra, E.; Finotti, A.; Gasparello, J.; Gambari, R.; Assael, B.M.; Cipolli, M.; Sorio, C. New insights into the Shwachman-Diamond Syndrome-related haematological disorder: Hyper-activation of mTOR and STAT3 in leukocytes. Sci. Rep. 2016, 6, 33165. [Google Scholar] [CrossRef] [Green Version]
- Dror, Y.; Freedman, M.H. Shwachman-Diamond syndrome marrow cells show abnormally increased apoptosis mediated through the Fas pathway. Blood 2001, 97, 3011–3016. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, K.; Ambekar, C.; Wang, H.; Ciccolini, A.; Schimmer, A.D.; Dror, Y. SBDS-deficiency results in specific hypersensitivity to Fas stimulation and accumulation of Fas at the plasma membrane. Apoptosis 2009, 14, 77–89. [Google Scholar] [CrossRef]
- Pibiri, I.; Lentini, L.; Melfi, R.; Gallucci, G.; Pace, A.; Spinello, A.; Barone, G.; Di Leonardo, A. Enhancement of premature stop codon readthrough in the CFTR gene by Ataluren (PTC124) derivatives. Eur. J. Med. Chem. 2015, 101, 236–244. [Google Scholar] [CrossRef]
- Pibiri, I.; Lentini, L.; Tutone, M.; Melfi, R.; Pace, A.; Di Leonardo, A. Exploring the readthrough of nonsense mutations by non-acidic Ataluren analogues selected by ligand-based virtual screening. Eur. J. Med. Chem. 2016, 122, 429–435. [Google Scholar] [CrossRef]
- Kurosaki, T.; Popp, M.W.; Maquat, L.E. Quality and quantity control of gene expression by nonsense-mediated mRNA decay. Nat. Rev. Mol. Cell Biol. 2019, 20, 406–420. [Google Scholar] [CrossRef]
- Causier, B.; Li, Z.; De Smet, R.; Lloyd, J.P.B.; Van de Peer, Y.; Davies, B. Conservation of Nonsense-Mediated mRNA Decay Complex Components Throughout Eukaryotic Evolution. Sci. Rep. 2017, 7, 16692. [Google Scholar] [CrossRef]
- Keenan, M.M.; Huang, L.; Jordan, N.J.; Wong, E.; Cheng, Y.; Valley, H.C.; Mahiou, J.; Liang, F.; Bihler, H.; Mense, M.; et al. Nonsense-mediated RNA Decay Pathway Inhibition Restores Expression and Function of W1282X CFTR. Am. J. Respir. Cell Mol. Biol. 2019, 61, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Usuki, F.; Yamashita, A.; Shiraishi, T.; Shiga, A.; Onodera, O.; Higuchi, I.; Ohno, S. Inhibition of SMG-8, a subunit of SMG-1 kinase, ameliorates nonsense-mediated mRNA decay-exacerbated mutant phenotypes without cytotoxicity. Proc. Natl. Acad. Sci. USA 2013, 110, 15037–15042. [Google Scholar] [CrossRef] [Green Version]
- Pal, M.; Ishigaki, Y.; Nagy, E.; Maquat, L.E. Evidence that phosphorylation of human Upfl protein varies with intracellular location and is mediated by a wortmannin-sensitive and rapamycin-sensitive PI 3-kinase-related kinase signaling pathway. RNA 2001, 7, 5–15. [Google Scholar] [CrossRef]
- Keeling, K.M.; Wang, D.; Dai, Y.; Murugesan, S.; Chenna, B.; Clark, J.; Belakhov, V.; Kandasamy, J.; Velu, S.E.; Baasov, T.; et al. Attenuation of nonsense-mediated mRNA decay enhances in vivo nonsense suppression. PLoS ONE 2013, 8, e60478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Temple, G.F.; Dozy, A.M.; Roy, K.L.; Kan, Y.W. Construction of a functional human suppressor tRNA gene: An approach to gene therapy for beta-thalassaemia. Nature 1982, 296, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Panchal, R.G.; Wang, S.; McDermott, J.; Link, C.J. Partial functional correction of xeroderma pigmentosum group A cells by suppressor tRNA. Hum. Gene Ther. 1999, 10, 2209–2219. [Google Scholar] [CrossRef]
- Sako, Y.; Usuki, F.; Suga, H. A novel therapeutic approach for genetic diseases by introduction of suppressor tRNA. Nucleic Acids Symp. Ser. (Oxf) 2006, 239–240. [Google Scholar] [CrossRef] [Green Version]
- Buvoli, M.; Buvoli, A.; Leinwand, L.A. Suppression of nonsense mutations in cell culture and mice by multimerized suppressor tRNA genes. Mol. Cell Biol. 2000, 20, 3116–3124. [Google Scholar] [CrossRef] [Green Version]
- Lueck, J.D.; Yoon, J.S.; Perales-Puchalt, A.; Mackey, A.L.; Infield, D.T.; Behlke, M.A.; Pope, M.R.; Weiner, D.B.; Skach, W.R.; McCray, P.B.; et al. Engineered transfer RNAs for suppression of premature termination codons. Nat. Commun. 2019, 10, 822. [Google Scholar] [CrossRef] [Green Version]
- Osborn, M.J.; Gabriel, R.; Webber, B.R.; DeFeo, A.P.; McElroy, A.N.; Jarjour, J.; Starker, C.G.; Wagner, J.E.; Joung, J.K.; Voytas, D.F.; et al. Fanconi anemia gene editing by the CRISPR/Cas9 system. Hum. Gene Ther. 2015, 26, 114–126. [Google Scholar] [CrossRef] [Green Version]
- Osborn, M.; Lonetree, C.L.; Webber, B.R.; Patel, D.; Dunmire, S.; McElroy, A.N.; DeFeo, A.P.; MacMillan, M.L.; Wagner, J.; Balzar, B.R.; et al. CRISPR/Cas9 Targeted Gene Editing and Cellular Engineering in Fanconi Anemia. Stem Cells Dev. 2016, 25, 1591–1603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skvarova Kramarzova, K.; Osborn, M.J.; Webber, B.R.; DeFeo, A.P.; McElroy, A.N.; Kim, C.J.; Tolar, J. CRISPR/Cas9-Mediated Correction of the FANCD1 Gene in Primary Patient Cells. Int. J. Mol. Sci. 2017, 18, 1269. [Google Scholar] [CrossRef] [PubMed]
- Rio, P.; Baños, R.; Lombardo, A.; Quintana-Bustamante, O.; Alvarez, L.; Garate, Z.; Genovese, P.; Almarza, E.; Valeri, A.; Díez, B.; et al. Targeted gene therapy and cell reprogramming in Fanconi anemia. EMBO Mol. Med. 2014, 6, 835–848. [Google Scholar] [CrossRef] [PubMed]
- Nasri, M.; Ritter, M.; Mir, P.; Dannenmann, B.; Aghaallaei, N.; Amend, D.; Makaryan, V.; Xu, Y.; Fletcher, B.; Bernhard, R.; et al. CRISPR/Cas9-mediated. Haematologica 2020, 105, 598–609. [Google Scholar] [CrossRef] [Green Version]
- Sürün, D.; von Melchner, H.; Schnütgen, F. CRISPR/Cas9 genome engineering in hematopoietic cells. Drug Discov. Today Technol. 2018, 28, 33–39. [Google Scholar] [CrossRef]
- Payne, E.M.; Virgilio, M.; Narla, A.; Sun, H.; Levine, M.; Paw, B.H.; Berliner, N.; Look, A.T.; Ebert, B.L.; Khanna-Gupta, A. L-Leucine improves the anemia and developmental defects associated with Diamond-Blackfan anemia and del(5q) MDS by activating the mTOR pathway. Blood 2012, 120, 2214–2224. [Google Scholar] [CrossRef]
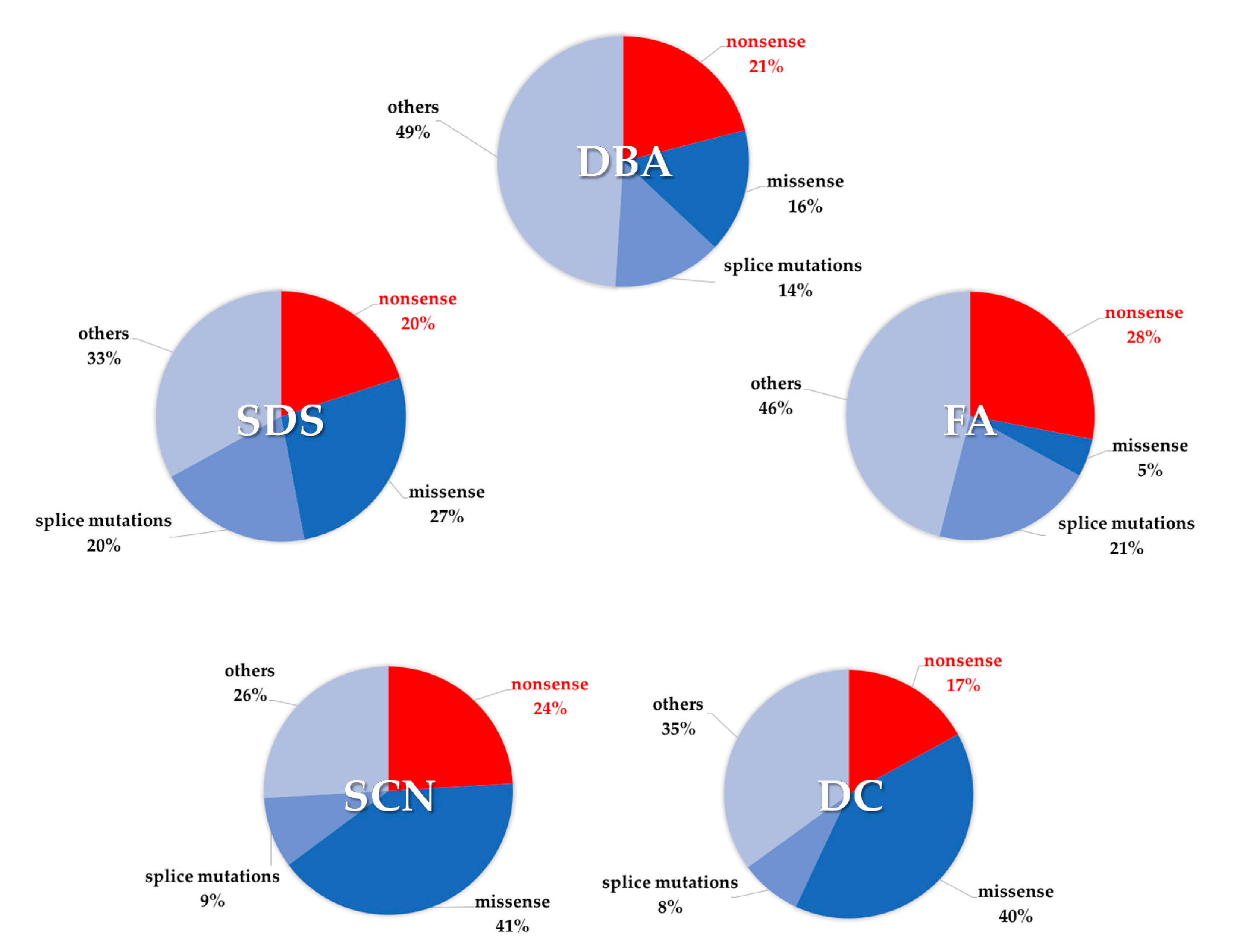

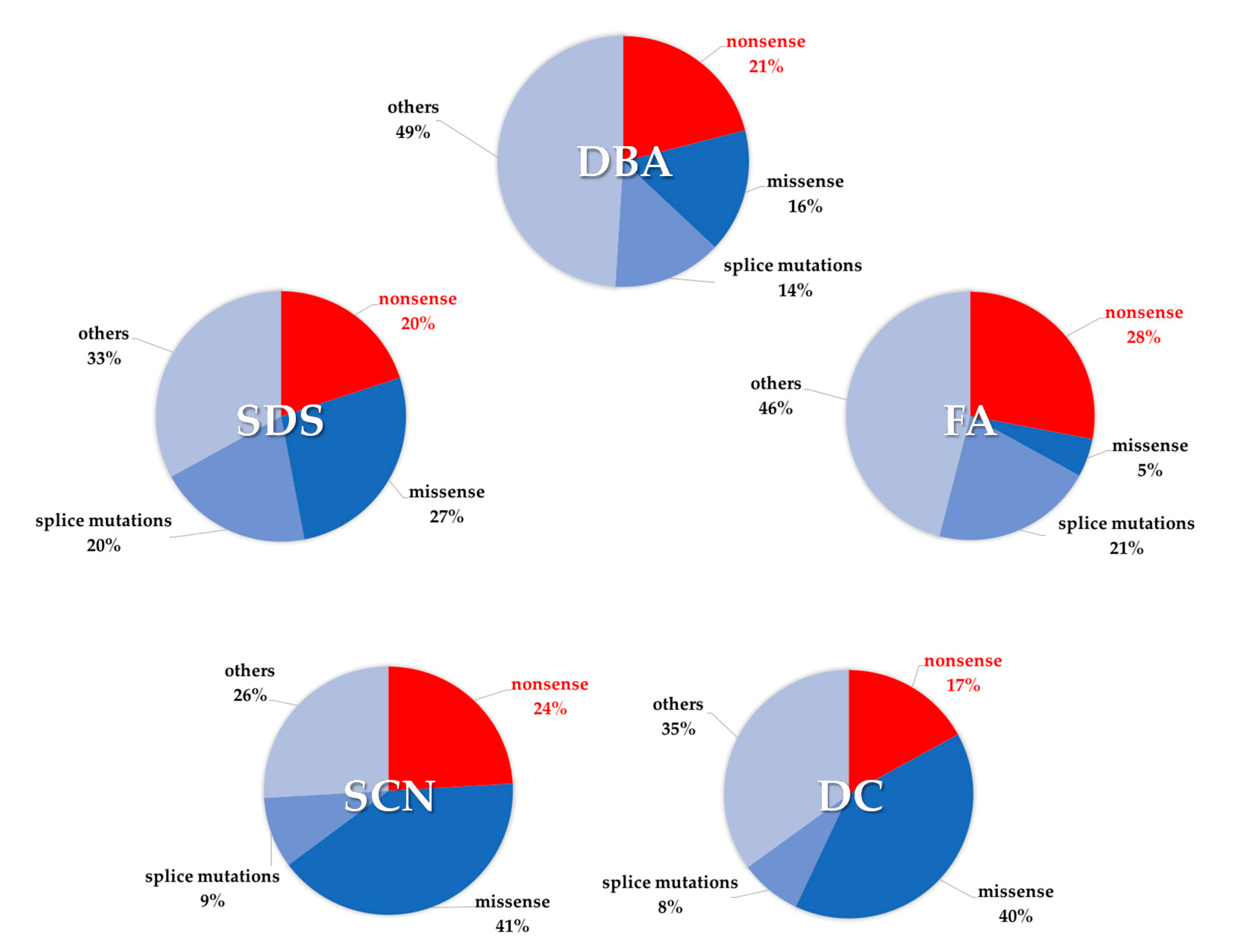{kind=link}
| Disease | Gene | Incidence of Nonsense Mutations | Gene Function | Ref |
|---|---|---|---|---|
| Diamond–Blackfan anemia | RPL5 RPL11 RPL35A RPS10 RPS17 RPS19 RPS24 RPS26 | 33% 7% 22% 17% 25% 20% 25% 20% | Pre-rRNA processing of the 18 rRNA, formation of the 40S or 60S ribosome subunit | [33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53] |
| Shwachman–Diamond syndrome | SBDS | 20% | 60S ribosome assembly | [23,24,25,54] |
| Dyskeratosis Congenita | CTC1 PARN RTEL1 TERT TINF2 | 28% 21% 26% 27% 18% | Telomere protection Poly(A)-specific ribonuclease Stability and elongation of telomeres Telomerase reverse transcriptase Stability and elongation of telomeres | [55,56,57,58,59,60,61,62] |
| Fanconi anemia | FANCA FANCB FANCC FANCD1 FANCE FANCF FANCG FANCI FANCJ FANCL FANCN | 14% 16% 33% 59% 43% 25% 24% 29% 30% 14% 37% | DNA cross-link repair, chromosome stability | [33,37,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83] |
| Severe congenital neutropenia | CSF3R ELANE G6PC3 HAX1 JAGN1 | 40% 8% 33% 29% 14% | G-CSF receptor Neutrophil elastase Hydrolysis of glucose 6-phosphate Apoptosis control, cytoskeletal development, myeloid regulator Neutrophil differentiation and survival | [33,84,85,86,87,88,89,90,91,92,93,94,95,96] |
| Molecule | Function | Clinical Trials | Ref |
|---|---|---|---|
| G418 (geneticin) | Readthrough inducer | None | [152,168,170,171,172] |
| Gentamicin | Readthrough inducer | Phase II in CF (NCT00376428) | [168,171,173,174,175,176,177,178,179] |
| NB30 | Readthrough inducer | None | [176,180] |
| NB54 | Readthrough inducer | None | [171,176,180,181,182,183] |
| NB84 | Readthrough inducer | None | [176,184] |
| ELX-02 | Readthrough inducer | Phase II in NephrophaticCystinosis (NCT04069260) Phase II in CF (NCT04126473) | [185,186] |
| Pyramycin (TC007) | Readthrough inducer | None | [187,188] |
| Ataluren (PTC124) | Readthrough inducer | Phase IIa in HA and HB (NCT00947193) Phase IIa in methylmalonic academia (NCT01141075) Phase II in Dravet syndrome (NCT02758626) Phase II in aniridia (NCT02647359) Phase II in CF (NCT00351078; NCT00237380; NCT00458341; NCT00234663) Phase II in DMD (NCT00264888) Phase III in CF (NCT02139306; NCT02107859; NCT02456103) Phase III in DMD(NCT02456103; NCT02139306; NCT00803205; NCT01140451; NCT02107859) Phase IV in CF (NCT03256968; NCT03256799) | [189,190,191,192,193,194,195,196,197,198,199,200] |
| NV2445 | Readthrough inducer | None | [201] |
| CDX3, CDX4, CDX5, CDX10, CDX11 | Readthrough enhancer | None | [202,203] |
| Poly-L-aspartic acid | Readthrough enhancer | None | [204] |
| Amlexanox | NMD inhibitor/ Readthrough inducer | None | [205,206] |
| Caffeine | NMD inhibitor | None | [179,207,208] |
| Wortmannin | NMD inhibitor | None | [209] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bezzerri, V.; Api, M.; Allegri, M.; Fabrizzi, B.; Corey, S.J.; Cipolli, M. Nonsense Suppression Therapy: New Hypothesis for the Treatment of Inherited Bone Marrow Failure Syndromes. Int. J. Mol. Sci. 2020, 21, 4672. https://doi.org/10.3390/ijms21134672
Bezzerri V, Api M, Allegri M, Fabrizzi B, Corey SJ, Cipolli M. Nonsense Suppression Therapy: New Hypothesis for the Treatment of Inherited Bone Marrow Failure Syndromes. International Journal of Molecular Sciences. 2020; 21(13):4672. https://doi.org/10.3390/ijms21134672
Chicago/Turabian StyleBezzerri, Valentino, Martina Api, Marisole Allegri, Benedetta Fabrizzi, Seth J. Corey, and Marco Cipolli. 2020. "Nonsense Suppression Therapy: New Hypothesis for the Treatment of Inherited Bone Marrow Failure Syndromes" International Journal of Molecular Sciences 21, no. 13: 4672. https://doi.org/10.3390/ijms21134672
APA StyleBezzerri, V., Api, M., Allegri, M., Fabrizzi, B., Corey, S. J., & Cipolli, M. (2020). Nonsense Suppression Therapy: New Hypothesis for the Treatment of Inherited Bone Marrow Failure Syndromes. International Journal of Molecular Sciences, 21(13), 4672. https://doi.org/10.3390/ijms21134672