2D Coordination Network of Trioxotriangulene with Multiple Redox Abilities and Its Rechargeable Battery Performance


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
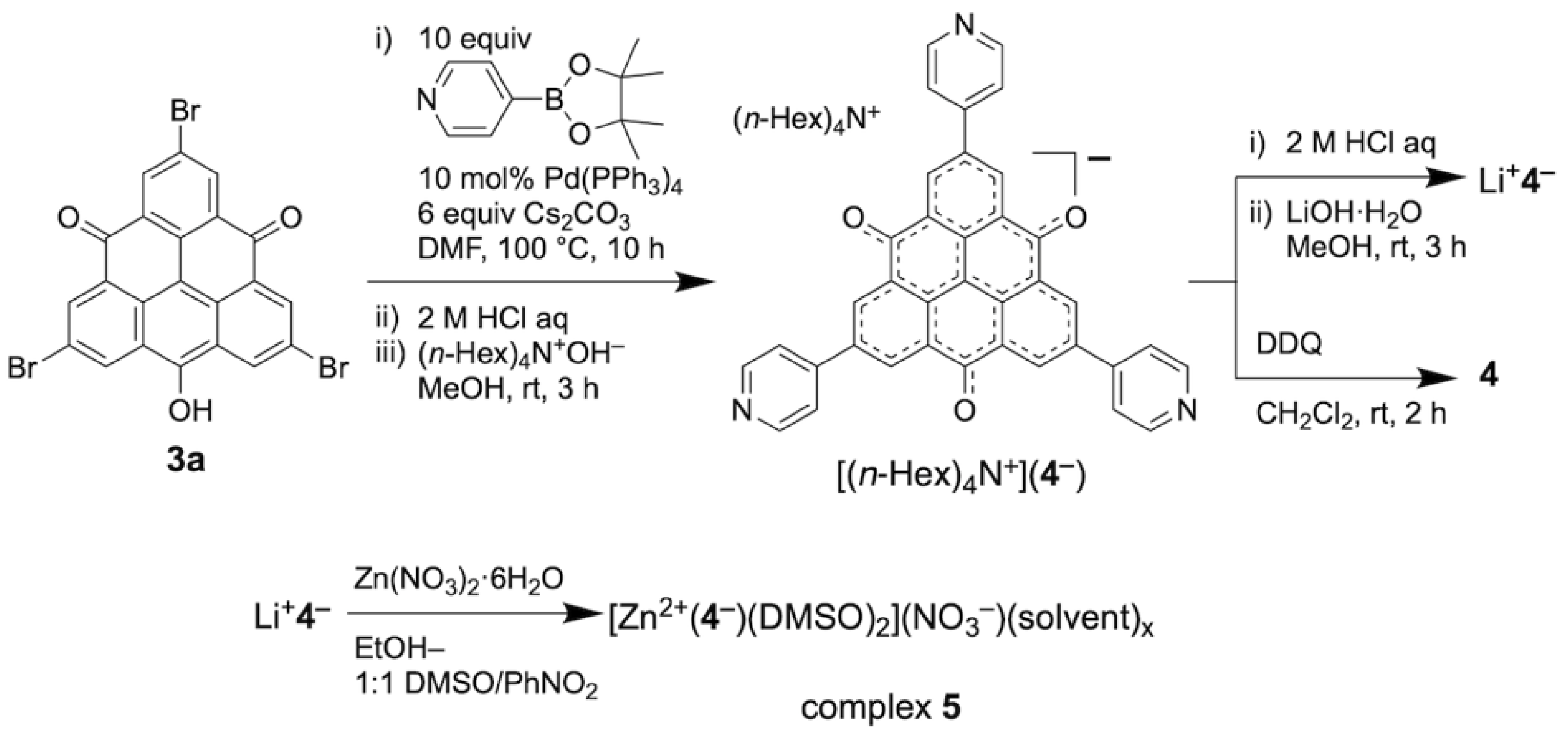
2.1. Synthesis and Structures of TOT with Three Pyridyl Moieties
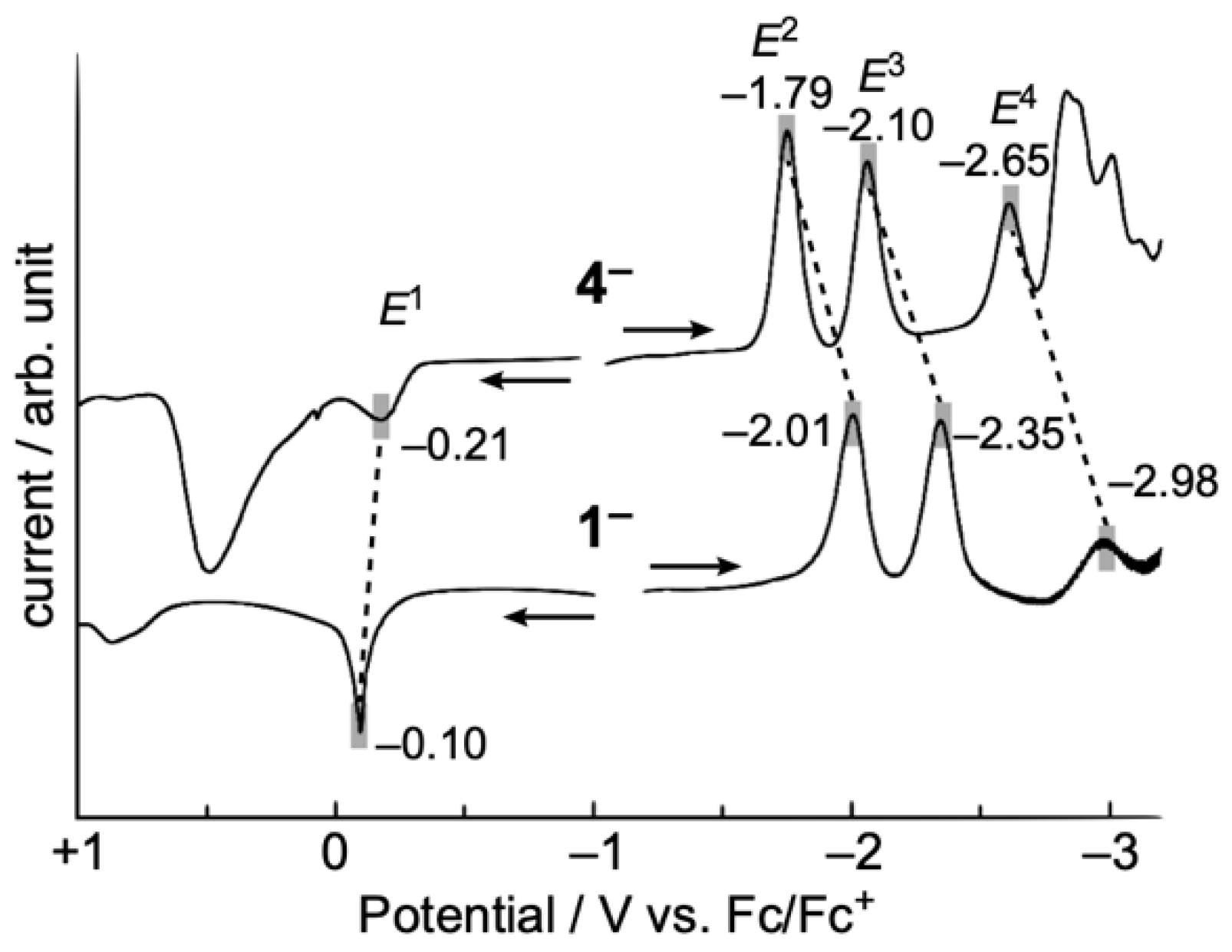
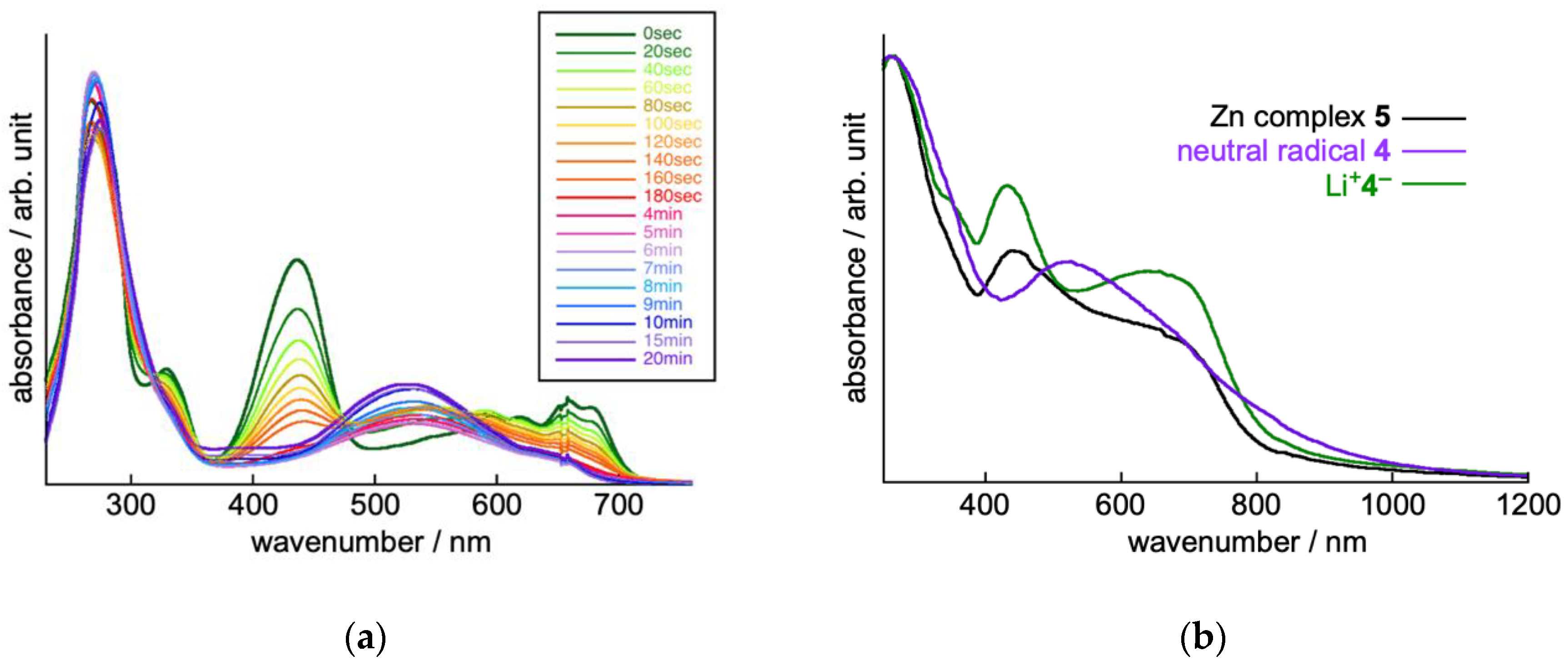
2.2. Electrochemical Properties of TOT with Three Pyridyl Moieties
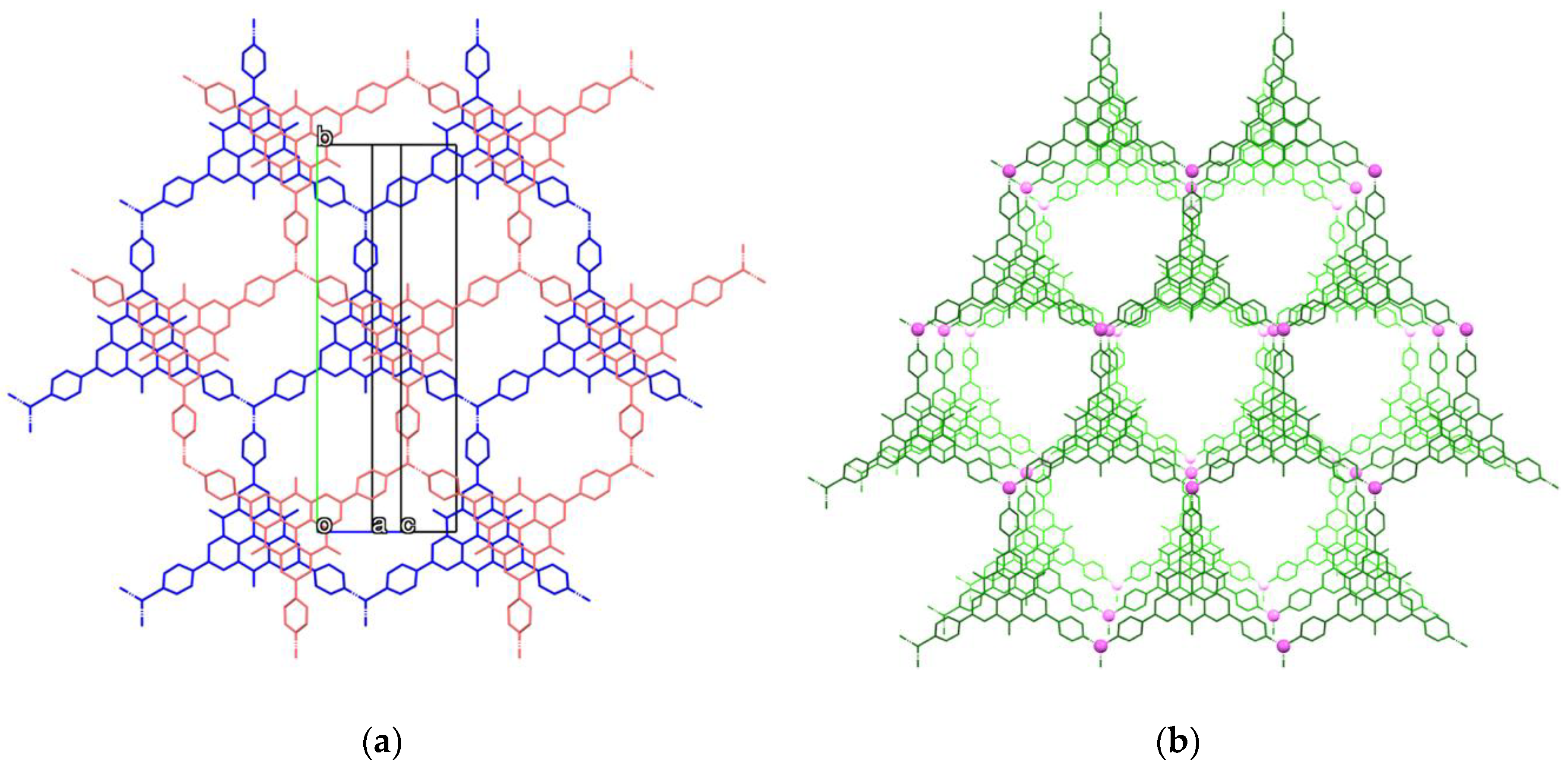
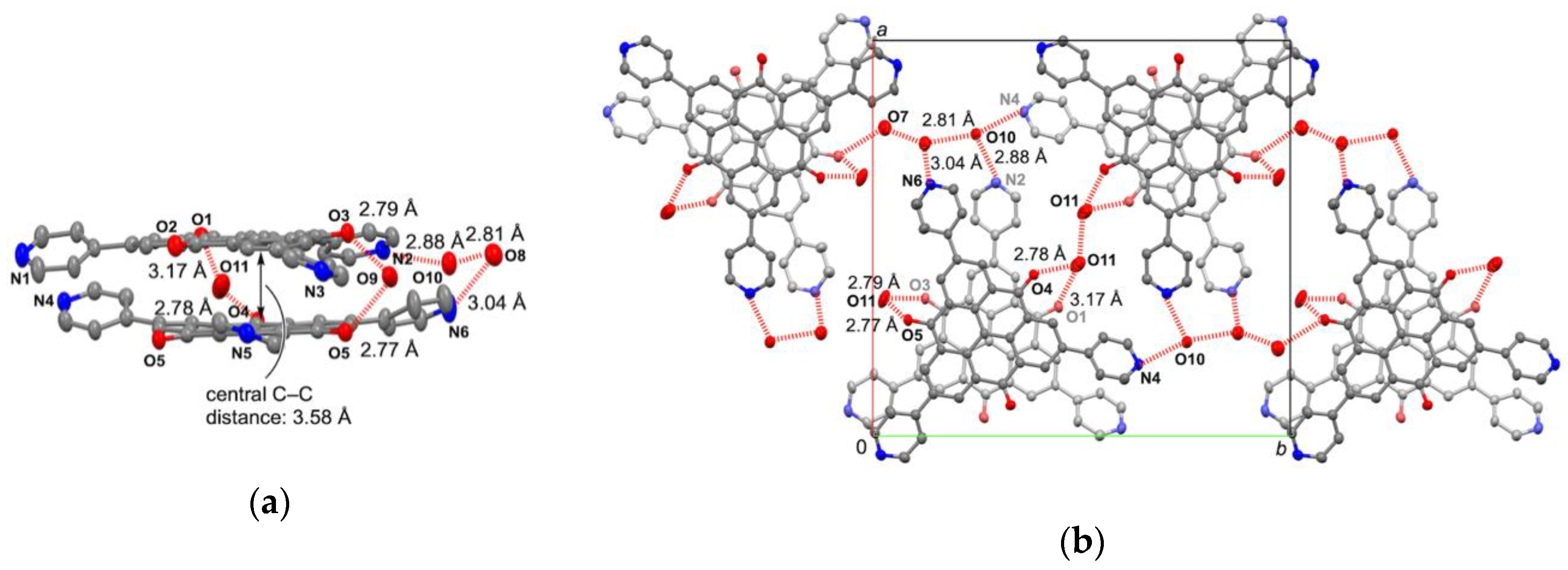
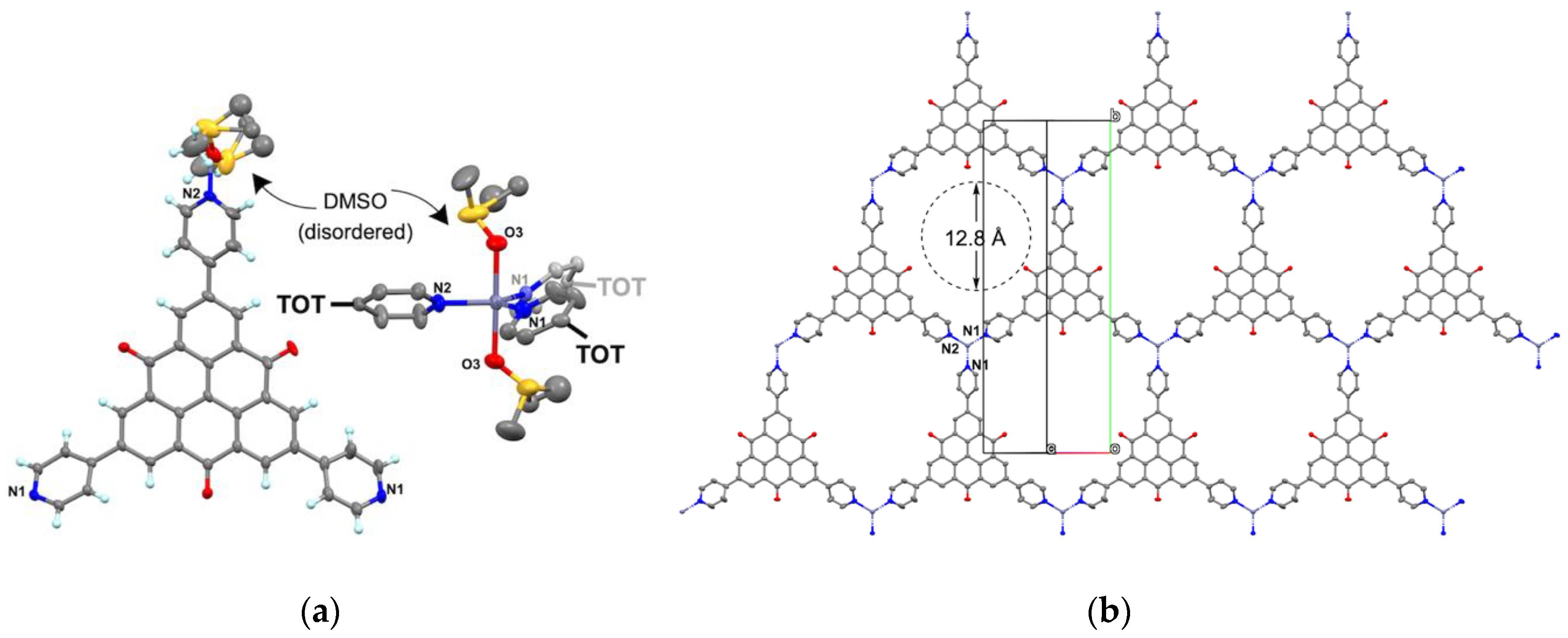
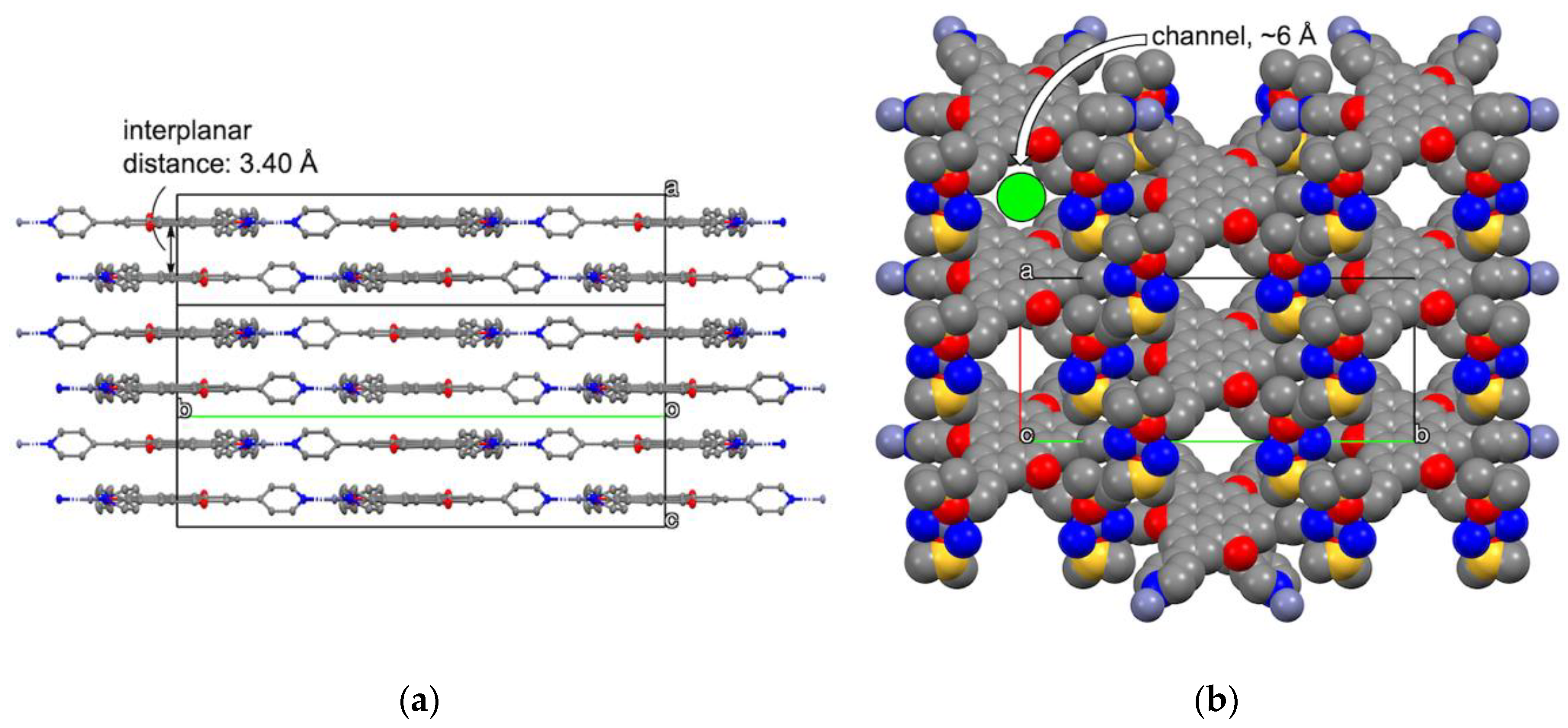
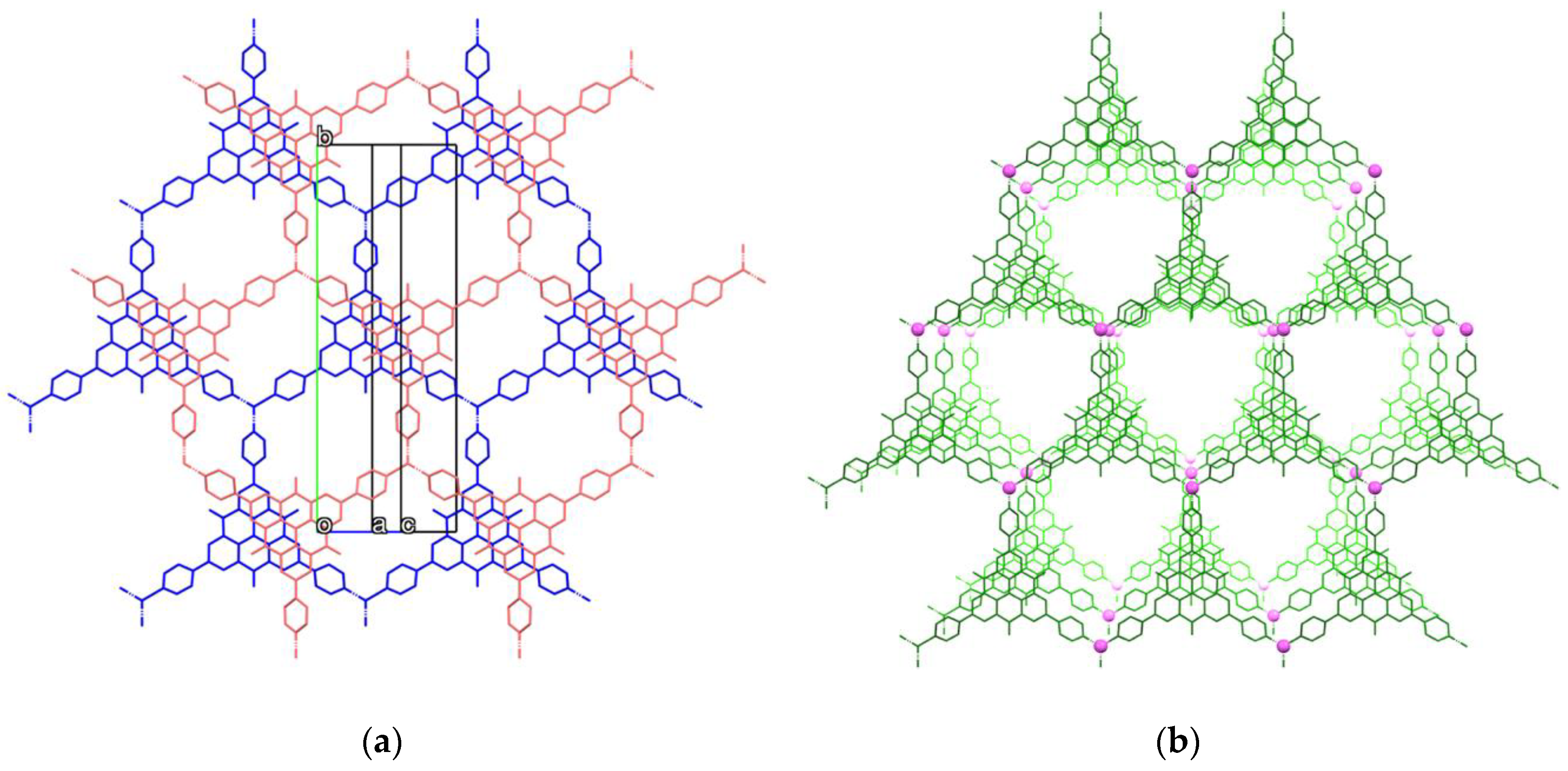
2.3. Crystal Structure of Zinc(II) Complex 5
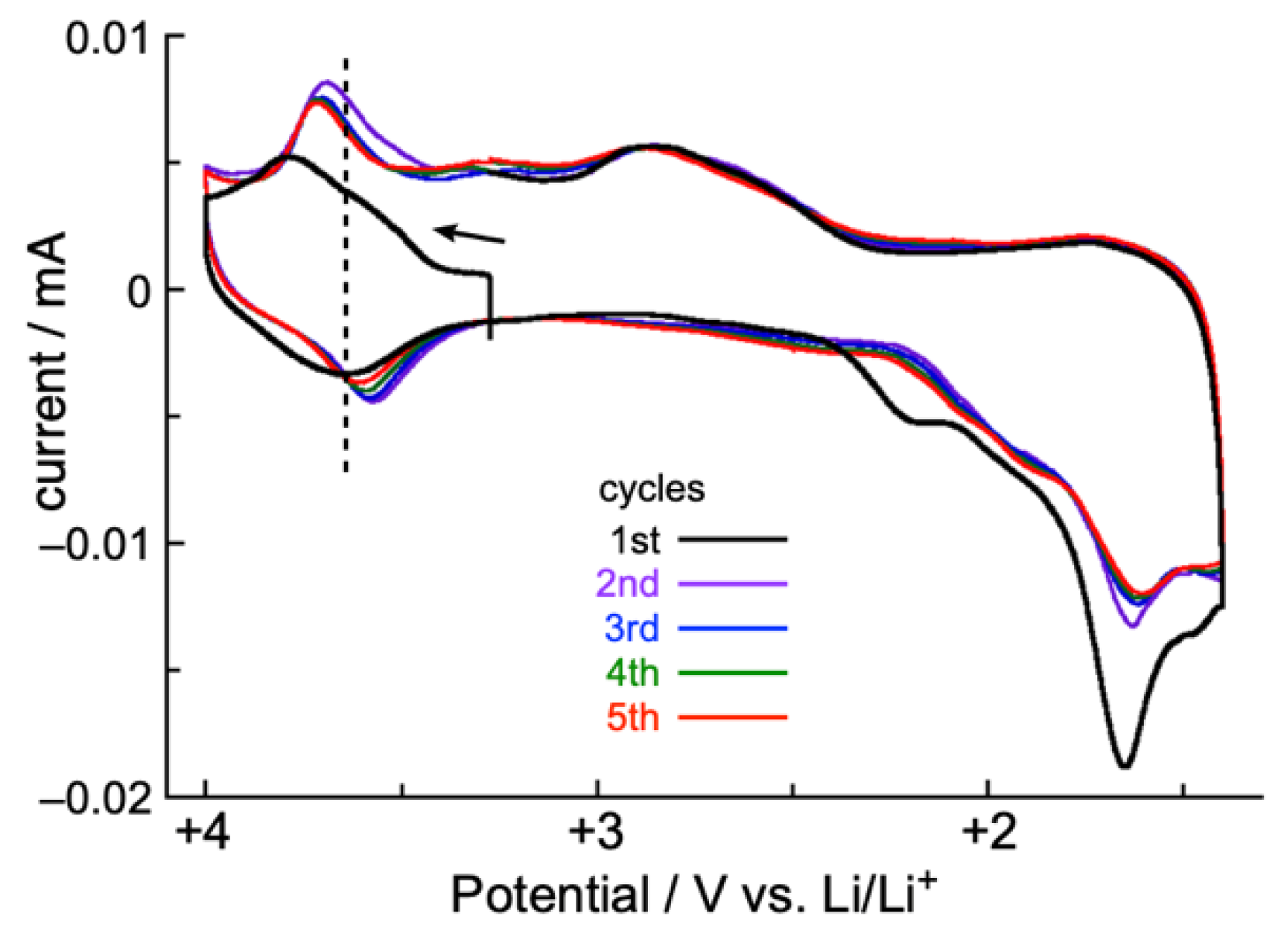
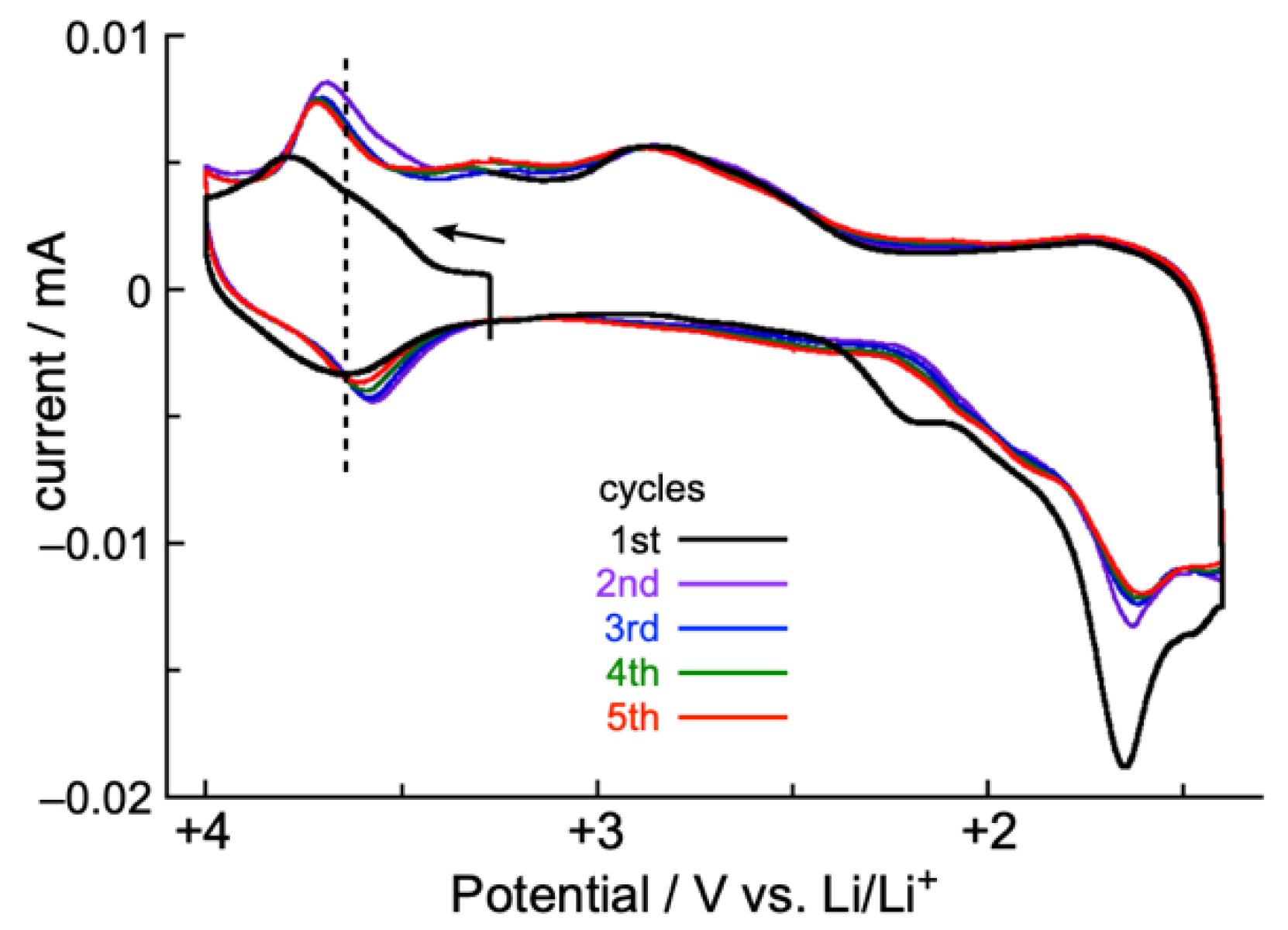
2.4. Solid-State CV and Battery Performance of Zn(II) Complex 5
3. Discussion
4. Materials and Methods
4.1. Synthesis and Characterization
4.2. Synthesis
4.2.1. Synthesis of Tetra-n-hexylammonium 2,6,10-tri-4′-pyridyl-4,8-dioxo-4H,8H-dibenzo[cd,mn] pyren-12-olate [(n-Hex)4N+)(4−)]
4.2.2. Synthesis of Lithium 2,6,10-tri-4′-pyridyl-4,8-dioxo-4H,8H-dibenzo[cd,mn]pyren-12-olate [Li+4−]
4.2.3. Synthesis of 2,6,10-tri-4′-pyridyl-4,8-dioxo-4H,8H-dibenzo[cd,mn]pyren-12-oxyl [4]
4.2.4. Synthesis of [Zn2+(4−)(DMSO)2](NO3−)(solvent)x [zinc(II) complex 5]
4.3. X-Ray Crystallography
4.3.1. Crystal Data for [(n-Hex)4N+)](4−)(H2O)2.5(DME)0.5
4.3.2. Crystal Data for [Zn2+(4−)(DMSO)2](NO3−)(solvent)x 5
4.4. Electrochemical Measurements
4.5. Electrochemical Absorption Spectroscopy
4.6. Battery Fabrication, Solid-State CV Measurement, and Charge–Discharge Experiments
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tarascon, J.-M.; Armand, M. Issues and challenges facing rechargeable lithium batteries. Nature 2001, 414, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Armand, M.; Tarascon, J.-M. Building better batteries. Nature 2008, 451, 652–657. [Google Scholar] [CrossRef]
- Liang, Y.; Tao, Z.; Chen, J. Organic Electrode Materials for Rechargeable Lithium Batteries. Adv. Energy Mater. 2012, 2, 742–769. [Google Scholar] [CrossRef]
- Song, Z.; Zhou, H. Towards sustainable and versatile energy storage devices: An overview of organic electrode materials. Energy Environ. Sci. 2013, 6, 2280. [Google Scholar] [CrossRef]
- Häupler, B.; Wild, A.; Schubert, U.S. Carbonyls: Powerful Organic Materials for Secondary Batteries. Adv. Energy Mater. 2015, 5, 1402034. [Google Scholar] [CrossRef]
- Muench, S.; Wild, A.; Friebe, C.; Häupler, B.; Janoschka, T.; Schubert, U.S. Polymer-Based Organic Batteries. Chem. Rev. 2016, 116, 9438–9484. [Google Scholar] [CrossRef]
- Schon, T.B.; McAllister, B.T.; Li, P.; Seferos, D.S. The rise of organic electrode materials for energy storage. Chem. Soc. Rev. 2016, 45, 6345–6404. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Zhu, Z.; Chen, J. Molecular Engineering with Organic Carbonyl Electrode Materials for Advanced Stationary and Redox Flow Rechargeable Batteries. Adv. Mater. 2017, 29, 1607007. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zeng, R.; Nan, J.; Shu, D.; Qiu, Y.; Chou, S. Quinone Electrode Materials for Rechargeable Lithium/Sodium Ion Batteries. Adv. Energy Mater. 2017, 7, 1700278. [Google Scholar] [CrossRef]
- Kim, K.C. Design Strategies for Promising Organic Positive Electrodes in Lithium-Ion Batteries: Quinones and Carbon Materials. Ind. Eng. Chem. Res. 2017, 56, 12009–12023. [Google Scholar] [CrossRef]
- Friebe, C.; Schubert, U.S. High-Power-Density Organic Radical Batteries. Top. Curr. Chem. 2017, 375, 19. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Yao, Y. Positioning Organic Electrode Materials in the Battery Landscape. Joule 2018, 2, 1690–1706. [Google Scholar] [CrossRef]
- Lu, Y.; Zhang, Q.; Li, L.; Niu, Z.; Chen, J. Design Strategies toward Enhancing the Performance of Organic Electrode Materials in Metal-Ion Batteries. Chem 2018, 4, 2786–2813. [Google Scholar] [CrossRef] [Green Version]
- Mauger, A.; Julien, C.M.; Paolella, A.; Armand, M.; Zaghib, K. Recent Progress on Organic Electrodes Materials for Rechargeable Batteries and Supercapacitors. Materials 2019, 12, 1770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Ding, G.; Xie, L.; Cao, X.; Liu, J.; Lei, X.; Ma, J. Conjugated Carbonyl Compounds as High-Performance Cathode Materials for Rechargeable Batteries. Chem. Mater. 2019, 31, 8582–8612. [Google Scholar] [CrossRef]
- Hanyu, Y.; Honma, I. Rechargeable quasi-solid state lithium battery with organic crystalline cathode. Sci. Rep. 2012, 2, 453. [Google Scholar] [CrossRef] [Green Version]
- Janoschka, T.; Hager, M.D.; Schubert, U.S. Powering up the Future: Radical Polymers for Battery Applications. Adv. Mater. 2012, 24, 6397–6409. [Google Scholar] [CrossRef]
- Shimizu, A.; Kuramoto, H.; Tsujii, Y.; Nokami, T.; Inatomi, Y.; Hojo, N.; Suzuki, H.; Yoshida, J.-I. Introduction of two lithiooxycarbonyl groups enhances cyclability of lithium batteries with organic cathode materials. J. Power Sources 2014, 260, 211–217. [Google Scholar] [CrossRef] [Green Version]
- Kitagawa, S.; Kitaura, R.; Noro, S.-I. Functional Porous Coordination Polymers. Angew. Chem. Int. Ed. 2004, 43, 2334–2375. [Google Scholar] [CrossRef]
- Lee, J.; Farha, O.K.; Roberts, J.M.; Scheidt, K.A.; Nguyen, S.T.; Hupp, J.T. Metal–organic framework materials as catalysts. Chem. Soc. Rev. 2009, 38, 1450–1459. [Google Scholar] [CrossRef]
- Furukawa, H.; Cordova, K.E.; O’Keeffe, M.; Yaghi, O.M. The Chemistry and Applications of Metal-Organic Frameworks. Science 2013, 341, 1230444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slater, A.G.; Cooper, A.I. Function-led design of new porous materials. Science 2015, 348, aaa8075. [Google Scholar] [CrossRef]
- D’Alessandro, D.M. Exploiting redox activity in metal–organic frameworks: Concepts, trends and perspectives. Chem. Commun. 2016, 52, 8957–8971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakamoto, R.; Takada, K.; Pal, T.; Maeda, H.; Kambe, T.; Nishihara, H. Coordination nanosheets (CONASHs): Strategies, structures and functions. Chem. Commun. 2017, 53, 5781–5801. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Nai, J.; Yu, L.; Lou, X.W. (David) Metal-Organic-Framework-Based Materials as Platforms for Renewable Energy and Environmental Applications. Joule 2017, 1, 77–107. [Google Scholar] [CrossRef] [Green Version]
- Dhakshinamoorthy, A.; Asiri, A.M.; Garcia, H. 2D Metal–Organic Frameworks as Multifunctional Materials in Heterogeneous Catalysis and Electro/Photocatalysis. Adv. Mater. 2019, 31, e1900617. [Google Scholar] [CrossRef] [PubMed]
- Morozan, A.; Jaouen, F. Metal organic frameworks for electrochemical applications. Energy Environ. Sci. 2012, 5, 9269. [Google Scholar] [CrossRef]
- Xia, W.; Mahmood, A.; Zou, R.; Xu, Q. Metal–organic frameworks and their derived nanostructures for electrochemical energy storage and conversion. Energy Environ. Sci. 2015, 8, 1837–1866. [Google Scholar] [CrossRef]
- Wang, L.; Han, Y.; Feng, X.; Zhou, J.; Qi, P.; Wang, B. Metal-Organic Frameworks for Energy Storage: Batteries and Super-capacitors. Coord. Chem. Rev. 2016, 307, 361–381. [Google Scholar] [CrossRef]
- Baumann, A.E.; Burns, D.A.; Liu, B.; Thoi, V.S. Metal-organic framework functionalization and design strategies for advanced electrochemical energy storage devices. Commun. Chem. 2019, 2, 86. [Google Scholar] [CrossRef] [Green Version]
- Kong, L.; Zhong, M.; Shuang, W.; Xu, Y.; Bu, X.-H. Electrochemically active sites inside crystalline porous materials for energy storage and conversion. Chem. Soc. Rev. 2020, 49, 2378–2407. [Google Scholar] [CrossRef] [PubMed]
- Feŕey, G.; Millange, F.; Morcrette, M.; Serre, C.; Doublet, M.-L.; Greneche, J.-M.; Tarascon, J.-M. Mixed-Valence Li/Fe-Based Metal-Organic Frameworks with Both Reversible Redox and Sorption Properties. Angew. Chem. Int. Ed. 2007, 46, 3259–3263. [Google Scholar]
- Nagarathinam, M.; Saravanan, K.; Phua, E.J.; Reddy, M.V.; Chowdari, B.V.; Vittal, J.J. Redox-Active Metal-Centered Oxalato Phosphate Open Framework Cathode Materials for Lithium Ion Batteries. Angew. Chem. Int. Ed. 2012, 51, 5866–5870. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.M.; Jeong, H.M.; Park, J.H.; Zhang, Y.-B.; Kang, J.K.; Yaghi, O.M. Supercapacitors of Nanocrystalline Metal–Organic Frameworks. ACS Nano 2014, 8, 7451–7457. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yoshikawa, H.; Awaga, K. Monitoring the Solid-State Electrochemistry of Cu(2,7-AQDC) (AQDC = Anthraquinone Dicarboxylate) in a Lithium Battery: Coexistence of Metal and Ligand Redox Activities in a Metal–Organic Framework. J. Am. Chem. Soc. 2014, 136, 16112–16115. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Awaga, K. Redox-active metal–organic frameworks as electrode materials for batteries. MRS Bull. 2016, 41, 883–889. [Google Scholar] [CrossRef]
- Wada, K.; Sakaushi, K.; Sasaki, S.; Nishihara, H. Multielectron-Transfer-Based Rechargeable Energy Storage of Two-Dimensional Coordination Frameworks with Non-Innocent Ligands. Angew. Chem. Int. Ed. 2018, 57, 8886−8890. [Google Scholar] [CrossRef]
- Kapaev, R.R.; Zhidkov, I.S.; Kurmaev, E.Z.; Stevenson, K.J.; Troshin, P.A. A nickel coordination polymer derived from 1,2,4,5-tetraaminobenzene for fast and stable potassium battery anodes. Chem. Commun. 2020, 56, 1541–1544. [Google Scholar] [CrossRef]
- Morita, Y.; Nishida, S. Stable Radicals: Fundamental and Applied Aspects of Odd-Electron Compounds; Hicks, R.G., Ed.; Wiley: Chichester, UK, 2010; Chapter 3; pp. 81–145. [Google Scholar] [CrossRef]
- Morita, Y.; Suzuki, S.; Sato, K.; Takui, T. Synthetic organic spin chemistry for structurally well-defined open-shell graphene fragments. Nat. Chem. 2011, 3, 197–204. [Google Scholar] [CrossRef]
- Morita, Y.; Murata, T.; Ueda, A.; Yamada, C.; Kanzaki, Y.; Shiomi, D.; Sato, K.; Takui, T. Trioxotriangulene: Air- and Thermally Stable Organic Carbon-Centered Neutral π-Radical without Steric Protection. Bull. Chem. Soc. Jpn. 2018, 91, 922–931. [Google Scholar] [CrossRef] [Green Version]
- Ikabata, Y.; Wang, Q.; Yoshikawa, T.; Ueda, A.; Murata, T.; Kariyazono, K.; Moriguchi, M.; Okamoto, H.; Morita, Y.; Nakai, H. Near-infrared absorption of π-stacking columns composed of trioxotriangulene neutral radicals. NPJ Quantum Mater. 2017, 2, 27. [Google Scholar] [CrossRef]
- Murata, T.; Yamada, C.; Furukawa, K.; Morita, Y. Mixed valence salts based on carbon-centered neutral radical crystals. Commun. Chem. 2018, 1, 47. [Google Scholar] [CrossRef] [Green Version]
- Murata, T.; Asakura, N.; Ukai, S.; Ueda, A.; Kanzaki, Y.; Sato, K.; Takui, T.; Morita, Y. Intramolecular Magnetic Interaction of Spin-Delocalized Neutral Radicals through m -Phenylene Spacers. ChemPlusChem 2019, 84, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Enozawa, H.; Ukai, S.; Ito, H.; Murata, T.; Morita, Y. Colored Ionic Liquid Based on Stable Polycyclic Anion Salt Showing Halochromism with HCl Vapor. Org. Lett. 2019, 21, 2161–2165. [Google Scholar] [CrossRef] [PubMed]
- Murata, T.; Kotsuki, K.; Murayama, H.; Tsuji, R.; Morita, Y. Metal-free electrocatalysts for oxygen reduction reaction based on trioxotriangulene. Commun. Chem. 2019, 2, 46. [Google Scholar] [CrossRef]
- Murata, T.; Kariyazono, K.; Ukai, S.; Ueda, A.; Kanzaki, Y.; Shiomi, D.; Sato, K.; Takui, T.; Morita, Y. Trioxotriangulene with carbazole: A donor–acceptor molecule showing strong near-infrared absorption exceeding 1000 nm. Org. Chem. Front. 2019, 6, 3107–3115. [Google Scholar] [CrossRef] [Green Version]
- Ito, H.; Murata, T.; Miyata, T.; Morita, M.; Tsuji, R.; Morita, Y. Air-Stable Thin Films with High and Anisotropic Electrical Conductivities Composed of a Carbon-Centered Neutral π-Radical. ACS Omega 2019, 4, 17569–17575. [Google Scholar] [CrossRef]
- Morita, Y.; Nishida, S.; Murata, T.; Moriguchi, M.; Ueda, A.; Satoh, M.; Arifuku, K.; Sato, K.; Takui, T. Organic tailored batteries materials using stable open-shell molecules with degenerate frontier orbitals. Nat. Mater. 2011, 10, 947–951. [Google Scholar] [CrossRef]
- Nishida, S.; Morita, Y. Organic Redox Systems; Nishinaga, T., Ed.; Wiley: Hoboken, NJ, USA, 2015; Chapter 6; pp. 177–243. [Google Scholar]
- Luo, Z.; Liu, L.; Zhao, Q.; Li, F.; Chen, J. An Insoluble Benzoquinone-Based Organic Cathode for Use in Rechargeable Lithium-Ion Batteries. Angew. Chem. Int. Ed. 2017, 56, 12561–12565. [Google Scholar] [CrossRef]
- Sieuw, L.; Jouhara, A.; Quarez, E.; Auger, C.; Gohy, J.; Poizot, P.; Vlad, A. A H-bond stabilized quinone electrode material for Li–organic batteries: The strength of weak bonds. Chem. Sci. 2018, 10, 418–426. [Google Scholar] [CrossRef] [Green Version]
- Tuttle, M.R.; Zhang, S. Bisthiazolyl Quinones: Stabilizing Organic Electrode Materials with Sulfur-Rich Thiazyl Motifs. Chem. Mater. 2019, 32, 255–261. [Google Scholar] [CrossRef]
- Nakashima, K.; Shimizu, T.; Kamakura, Y.; Hinokimoto, A.; Kitagawa, Y.; Yoshikawa, H.; Tanaka, D. A new design strategy for redox-active molecular assemblies with crystalline porous structures for lithium-ion batteries. Chem. Sci. 2019, 11, 37–43. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Xiong, P.; Shi, Y.; Sun, P.; Wang, Z.; Chen, Z.; Xu, Y. Rational Molecular Design of Benzoquinone-Derived Cathode Materials for High-Performance Lithium-Ion Batteries. Adv. Funct. Mater. 2020, 30. [Google Scholar] [CrossRef]
- Murata, T.; Yokoyama, M.; Ueda, A.; Kanzaki, Y.; Shiomi, D.; Sato, K.; Takui, T.; Morita, Y. Synthesis of Trioxotriangulene Stable Neutral π-Radicals Having Alkyl Substituent Groups, and Their Effects on Electronic-spin and π-Stacking Structures. Chem. Lett. 2020, 49, 95–98. [Google Scholar] [CrossRef]
- Yoshikawa, H.; Kazama, C.; Awaga, K.; Satoh, M.; Wada, J. Rechargeable molecular cluster batteries. Chem. Commun. 2007, 3169–3170. [Google Scholar] [CrossRef]
- Matsuda, Y. Behavior of Some Ions in Mixed Organic Electrolytes of High Energy Density Batteries. J. Electrochem. Soc. 1981, 128, 2552. [Google Scholar] [CrossRef]
- Kitaura, R.; Kitagawa, S.; Kubota, Y.; Kobayashi, T.C.; Kindo, K.; Mita, Y.; Matsuo, A.; Kobayashi, M.; Chang, H.-C.; Ozawa, T.C.; et al. Formation of a One-Dimensional Array of Oxygen in a Microporous Metal-Organic Solid. Science 2002, 298, 2358–2361. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, R.; Kitaura, R.; Kitagawa, S.; Kubota, Y.; Belosludov, R.V.; Kobayashi, T.C.; Sakamoto, H.; Chiba, T.; Takata, M.; Kawazoe, Y.; et al. Highly controlled acetylene accommodation in a metal–organic microporous material. Nature 2005, 436, 238–241. [Google Scholar] [CrossRef]
- Uemura, T.; Kitaura, R.; Ohta, Y.; Nagaoka, M.; Kitagawa, S. Nanochannel-Promoted Polymerization of Substituted Acetylenes in Porous Coordination Polymers. Angew. Chem. Int. Ed. 2006, 45, 4112–4116. [Google Scholar] [CrossRef]
- Shimomura, S.; Matsuda, R.; Tsujino, T.; Kawamura, T.; Kitagawa, S. TCNQ Dianion-Based Coordination Polymer Whose Open Framework Shows Charge-Transfer Type Guest Inclusion. J. Am. Chem. Soc. 2006, 128, 16416–16417. [Google Scholar] [CrossRef]
- Ko, M.; Mendecki, L.; Mirica, K.A. Conductive two-dimensional metal–organic frameworks as multifunctional materials. Chem. Commun. 2018, 54, 7873–7891. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.S.; Skorupskii, G.; Dincă, M. Electrically Conductive Metal-Organic Frameworks. Chem. Rev. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. A short history ofSHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2007, 64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.F.; Xia, B.Y.; Zang, S.-O.; Lou, X.W. Metal-organic frameworks based electrocatalysts for the oxygen reduction reaction. Angew. Chem. Int. Ed. 2020, 59, 4634–4650. [Google Scholar] [CrossRef]
- Thomas, S.; Li, H.; Bredas, J.-L. Emergence of an Antiferromagnetic Mott Insulating Phase in Hexagonal π-Conjugated Covalent Organic Frameworks. Adv. Mater. 2019, 31. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murata, T.; Koide, T.; Nobukuni, H.; Tsuji, R.; Morita, Y. 2D Coordination Network of Trioxotriangulene with Multiple Redox Abilities and Its Rechargeable Battery Performance. Int. J. Mol. Sci. 2020, 21, 4723. https://doi.org/10.3390/ijms21134723
Murata T, Koide T, Nobukuni H, Tsuji R, Morita Y. 2D Coordination Network of Trioxotriangulene with Multiple Redox Abilities and Its Rechargeable Battery Performance. International Journal of Molecular Sciences. 2020; 21(13):4723. https://doi.org/10.3390/ijms21134723
Chicago/Turabian StyleMurata, Tsuyoshi, Taro Koide, Hirofumi Nobukuni, Ryotaro Tsuji, and Yasushi Morita. 2020. "2D Coordination Network of Trioxotriangulene with Multiple Redox Abilities and Its Rechargeable Battery Performance" International Journal of Molecular Sciences 21, no. 13: 4723. https://doi.org/10.3390/ijms21134723
APA StyleMurata, T., Koide, T., Nobukuni, H., Tsuji, R., & Morita, Y. (2020). 2D Coordination Network of Trioxotriangulene with Multiple Redox Abilities and Its Rechargeable Battery Performance. International Journal of Molecular Sciences, 21(13), 4723. https://doi.org/10.3390/ijms21134723