Role of Inflammation in Pathophysiology of Colonic Disease: An Update



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
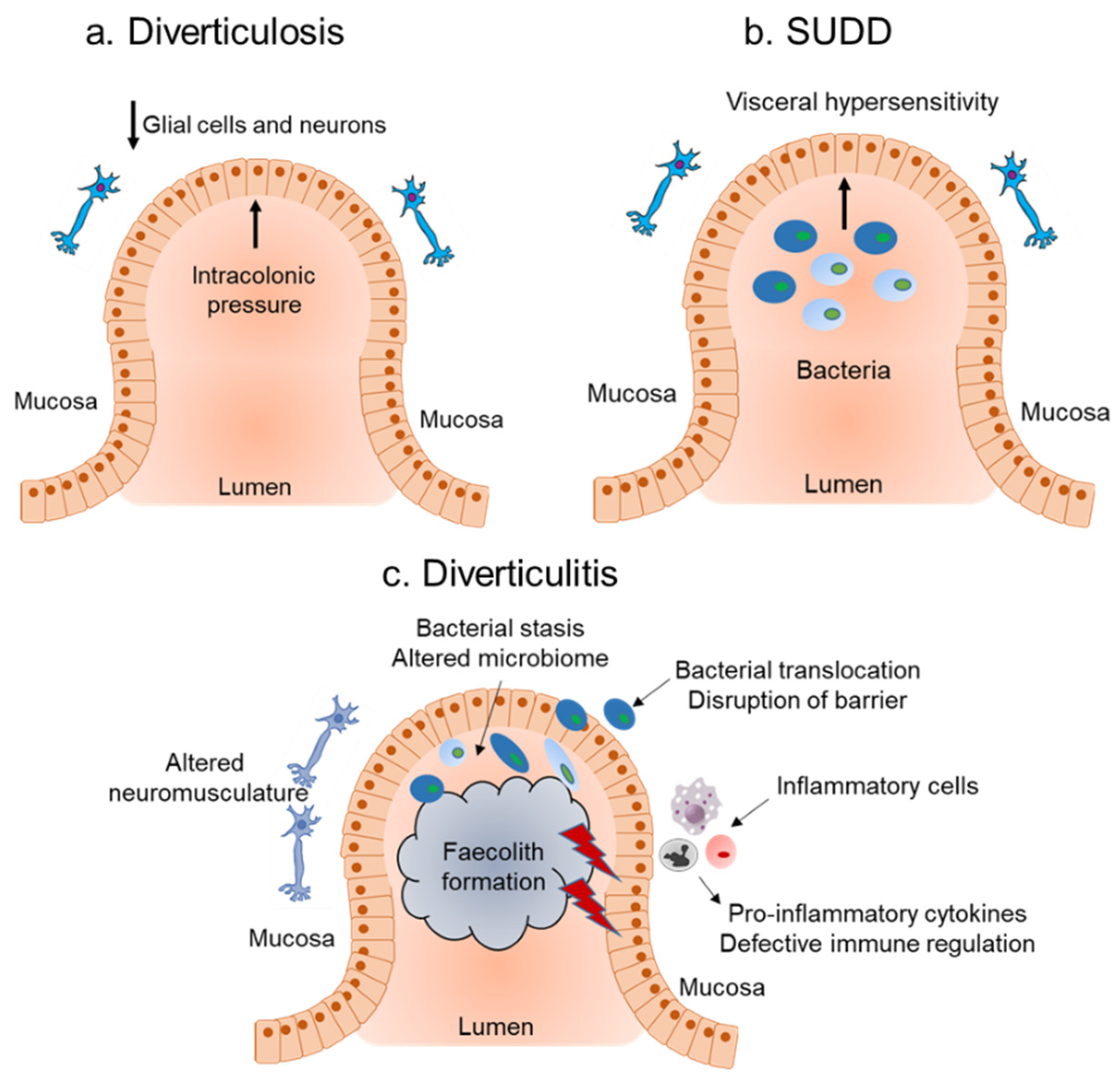
2. Diverticular Disease
2.1. Host Immune Response and Role of Inflammation in Diverticular Disease
2.2. Influence of the Microbiota on Inflammation Associated with Diverticular Disease
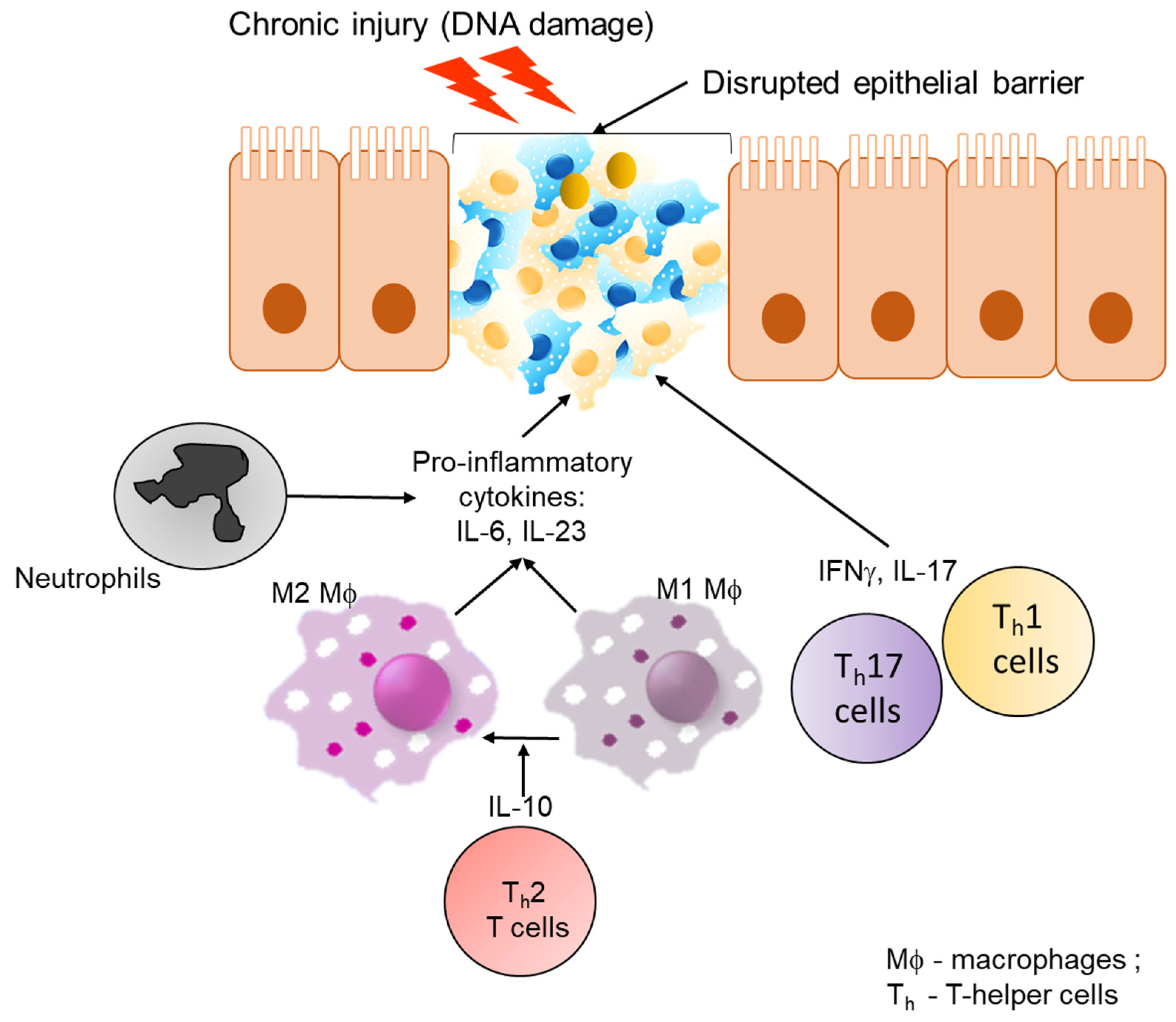
3. Inflammatory Bowel Disease
3.1. Host Immune Response and Role of Inflammation in IBD
3.2. Influence of the Microbiota in Inflammation Associated with IBD
3.3. Irriteable Bowel Syndrome Link to IBD
4. Colorectal Cancer
4.1. Host Immune Response and Inflammation in Colon Cancer
4.2. Influence of Microbiota in Inflammation Associated with Colon Cancer
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AOM | Azoxymethane |
| ARGHGA15 ATG16L1 | Rho-GTPase-activating protein 15 Autophagy related 16 like 1 |
| ATM b-FGF | Ataxia Telangiectasia Mutated basic-Fibroblast growth factor |
| CAC | Colitis associated cancer |
| CD | Crohn’s Disease |
| COL3A1 | Collagen type III alpha I chain |
| COLQ | Acetylcholinesterase |
| CRC | Colorectal cancer |
| DAMP | Danger associated molecular pattern |
| DD | Diverticular disease |
| DSS | Dextran sulfate sodium |
| FAP | Familial adenomatous polyposis |
| FGID | Functional Gastrointestinal disorders |
| FODMAP FMT | Fermentable oligo-, di-, monosaccharides and polyols Fecal microbiota transplantation |
| GBP-1 | Guanylate binding protein-1 |
| GI | Gastrointestinal |
| GWAS | Genome wide association studies |
| HNPCC | Hereditary non-polyposis colorectal cancer |
| IBD | Inflammatory bowel disease |
| IBS | Inflammatory bowel syndrome |
| IFNg | Interferon-gamma |
| IL-10 | Interleukin-10 |
| IL-6 | Interleukin-6 |
| iNOS | Nitric oxide synthase |
| INPP5D IRGM | Inositol polyphosphate-5-phophatase D Immunity Related GTPase M |
| LAMB4 | Laminin beta 4 |
| MYD88 NK | Myeloid differentiation factor 88 Natural Killer |
| NK1 | Neurokinin 1 |
| PAMP | Pathogen associated molecular pattern |
| PAR2 | Protease activated receptor-2 |
| PDGF | Platelet-derived growth factor |
| PRR | Pattern recognition receptor |
| PTPRC | Protein tyrosine phosphatase, receptor type C |
| RASAL3 | RAS protein activator-like 3 |
| ROS | Reactive oxygen species |
| SASH3 | SAM and SH3 domain-containing 3 |
| SCFA | Short chain fatty acid |
| SD1 | Syndecan-1 |
| SNP | Single nucleotide polymorphism |
| STING SUDD | Stimulator of interferon genes Symptomatic uncomplicated diverticular disease |
| Th1 | T-helper cells 1 |
| Th17 | T-helper cells 17 |
| TL1A | TNF-like ligand 1A |
| TLR | Toll-like receptor |
| TNF1A | Tumour necrosis factor-like cytokine 1A |
| TNFSF15 | Tumour necrosis factor superfamily 15 |
| TNF-α | Tumour necrosis factor-alpha |
| Tregs | Regulatory T cells |
| TRPM | Melastatin-like transient receptor potential |
| UC | Ulcerative colitis |
| VEGFA | Vascular endothelial growth factor A |
References
- Ahmed Nasef, N.; Ferguson, L.R. Dietary interactions with the bacterial sensing machinery in the intestine: The plant polyphenol case. Front. Genet. 2014, 5, 64. [Google Scholar] [CrossRef] [PubMed]
- Nasef, N.A.; Mehta, S.; Ferguson, L.R. Susceptibility to chronic inflammation: An update. Arch. Toxicol. 2017, 91, 1131–1141. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Takeuchi, M.; Hashimoto, S.; Mizuno, K.I.; Furukawa, K.; Sato, A.; Yokoyama, J.; Terai, S. Esophageal diverticulum: New perspectives in the era of minimally invasive endoscopic treatment. World J. Gastroenterol. 2019, 25, 1457–1464. [Google Scholar] [CrossRef] [PubMed]
- Shah, J.; Patel, K.; Sunkara, T.; Papafragkakis, C.; Shahidullah, A. Gastric Diverticulum: A Comprehensive Review. Inflamm. Intestig. Dis. 2019, 3, 161–166. [Google Scholar] [CrossRef]
- Rangan, V.; Lamont, J.T. Small Bowel Diverticulosis: Pathogenesis, Clinical Management, and New Concepts. Curr. Gastroenterol. Rep. 2020, 22, 4. [Google Scholar] [CrossRef]
- Schieffer, K.M.; Kline, B.P.; Yochum, G.S.; Koltun, W.A. Pathophysiology of diverticular disease. Expert Rev. Gastroenterol. Hepatol. 2018, 12, 683–692. [Google Scholar] [CrossRef]
- Tursi, A.; Scarpignato, C.; Strate, L.L.; Lanas, A.; Kruis, W.; Lahat, A.; Danese, S. Colonic diverticular disease. Nat. Rev. Dis. Primers 2020, 6, 20. [Google Scholar] [CrossRef]
- Strate, L.L.; Modi, R.; Cohen, E.; Spiegel, B.M. Diverticular disease as a chronic illness: Evolving epidemiologic and clinical insights. Am. J. Gastroenterol. 2012, 107, 1486–1493. [Google Scholar] [CrossRef]
- Warner, E.; Crighton, E.J.; Moineddin, R.; Mamdani, M.; Upshur, R. Fourteen-year study of hospital admissions for diverticular disease in Ontario. Can. J. Gastroenterol. 2007, 21, 97–99. [Google Scholar] [CrossRef]
- Violi, A.; Cambiè, G.; Miraglia, C.; Barchi, A.; Nouvenne, A.; Capasso, M.; Leandro, G.; Meschi, T.; De’ Angelis, G.L.; Di Mario, F. Epidemiology and risk factors for diverticular disease. Acta Biomed. 2018, 89, 107–112. [Google Scholar]
- Reichert, M.C.; Kupcinskas, J.; Krawczyk, M.; Jüngst, C.; Casper, M.; Grünhage, F.; Appenrodt, B.; Zimmer, V.; Weber, S.N.; Tamelis, A.; et al. A Variant of COL3A1 (rs3134646) Is Associated With Risk of Developing Diverticulosis in White Men. Dis. Colon Rectum 2018, 61, 604–611. [Google Scholar] [CrossRef]
- Peery, A.F.; Keku, T.O.; Addamo, C.; McCoy, A.N.; Martin, C.F.; Galanko, J.A.; Sandler, R.S. Colonic Diverticula Are Not Associated With Mucosal Inflammation or Chronic Gastrointestinal Symptoms. Clin. Gastroenterol. Hepatol. 2018, 16, 884–891.e1. [Google Scholar] [CrossRef]
- Tursi, A.; Brandimarte, G.; Elisei, W.; Giorgetti, G.M.; Inchingolo, C.D.; Danese, S.; Aiello, F. Assessment and grading of mucosal inflammation in colonic diverticular disease. J. Clin. Gastroenterol. 2008, 42, 699–703. [Google Scholar] [CrossRef]
- von Rahden, B.H.; Kircher, S.; Thiery, S.; Landmann, D.; Jurowich, C.F.; Germer, C.T.; Grimm, M. Association of steroid use with complicated sigmoid diverticulitis: Potential role of activated CD68+/CD163+ macrophages. Langenbecks Arch. Surg. 2011, 396, 759–768. [Google Scholar] [CrossRef] [PubMed]
- Barbara, G.; Scaioli, E.; Barbaro, M.R.; Biagi, E.; Laghi, L.; Cremon, C.; Marasco, G.; Colecchia, A.; Picone, G.; Salfi, N.; et al. Gut microbiota, metabolome and immune signatures in patients with uncomplicated diverticular disease. Gut 2017, 66, 1252–1261. [Google Scholar] [CrossRef] [PubMed]
- Tursi, A.; Elisei, W.; Brandimarte, G.; Giorgetti, G.M.; Inchingolo, C.D.; Nenna, R.; Picchio, M.; Giorgio, F.; Ierardi, E. Musosal tumour necrosis factor α in diverticular disease of the colon is overexpressed with disease severity. Colorectal Dis. 2012, 14, e258–e263. [Google Scholar] [CrossRef] [PubMed]
- Humes, D.J.; Simpson, J.; Smith, J.; Sutton, P.; Zaitoun, A.; Bush, D.; Bennett, A.; Scholefield, J.H.; Spiller, R.C. Visceral hypersensitivity in symptomatic diverticular disease and the role of neuropeptides and low grade inflammation. Neurogastroenterol. Motil. 2012, 24, 318-e163. [Google Scholar] [CrossRef] [PubMed]
- Tursi, A.; Mastromarino, P.; Capobianco, D.; Elisei, W.; Picchio, M.; Brandimarte, G. No changes in Interleukin-10 expression in symptomatic uncomplicated diverticular disease of the colon. J. Gastrointest. Liver Dis. 2018, 27, 476–477. [Google Scholar] [CrossRef] [PubMed]
- Tursi, A.; Elisei, W.; Giorgetti, G.M.; Inchingolo, C.D.; Nenna, R.; Picchio, M.; Giorgio, F.; Ierardi, E.; Brandimarte, G. Expression of basic fibroblastic growth factor, syndecan 1 and tumour necrosis factor α in resected acute colonic diverticulitis. Colorectal Dis. 2014, 16, O98–O103. [Google Scholar] [CrossRef]
- Connelly, T.M.; Berg, A.S.; Hegarty, J.P.; Deiling, S.; Brinton, D.; Poritz, L.S.; Koltun, W.A. The TNFSF15 gene single nucleotide polymorphism rs7848647 is associated with surgical diverticulitis. Ann. Surg. 2014, 259, 1132–1137. [Google Scholar] [CrossRef]
- Sigurdsson, S.; Alexandersson, K.F.; Sulem, P.; Feenstra, B.; Gudmundsdottir, S.; Halldorsson, G.H.; Olafsson, S.; Sigurdsson, A.; Rafnar, T.; Thorgeirsson, T.; et al. Sequence variants in ARHGAP15, COLQ and FAM155A associate with diverticular disease and diverticulitis. Nat. Commun. 2017, 8, 15789. [Google Scholar] [CrossRef] [PubMed]
- Coble, J.L.; Sheldon, K.E.; Yue, F.; Salameh, T.J.; Harris, L.R., III; Deiling, S.; Ruggiero, F.M.; Eshelman, M.A.; Yochum, G.S.; Koltun, W.A.; et al. Identification of a rare LAMB4 variant associated with familial diverticulitis through exome sequencing. Hum. Mol. Genet. 2017, 26, 3212–3220. [Google Scholar] [CrossRef]
- Maguire, L.H.; Handelman, S.K.; Du, X.; Chen, Y.; Pers, T.H.; Speliotes, E.K. Genome-wide association analyses identify 39 new susceptibility loci for diverticular disease. Nat. Genet. 2018, 50, 1359–1365. [Google Scholar] [CrossRef]
- Schafmayer, C.; Harrison, J.W.; Buch, S.; Lange, C.; Reichert, M.C.; Hofer, P.; Cossais, F.; Kupcinskas, J.; von Schönfels, W.; Schniewind, B.; et al. Genome-wide association analysis of diverticular disease points towards neuromuscular, connective tissue and epithelial pathomechanisms. Gut 2019, 68, 854–865. [Google Scholar] [CrossRef] [PubMed]
- Schieffer, K.M.; Choi, C.S.; Emrich, S.; Harris, L.; Deiling, S.; Karamchandani, D.M.; Salzberg, A.; Kawasawa, Y.I.; Yochum, G.S.; Koltun, W.A. RNA-seq implicates deregulation of the immune system in the pathogenesis of diverticulitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 313, G277–G284. [Google Scholar] [CrossRef] [PubMed]
- Tursi, A.; Mastromarino, P.; Capobianco, D.; Elisei, W.; Miccheli, A.; Capuani, G.; Tomassini, A.; Campagna, G.; Picchio, M.; Giorgetti, G.; et al. Assessment of Fecal Microbiota and Fecal Metabolome in Symptomatic Uncomplicated Diverticular Disease of the Colon. J. Clin. Gastroenterol. 2016, 50 (Suppl. 1), S9–S12. [Google Scholar] [CrossRef]
- Jones, R.B.; Fodor, A.A.; Peery, A.F.; Tsilimigras, M.C.B.; Winglee, K.; McCoy, A.; Sioda, M.; Sandler, R.S.; Keku, T.O. An Aberrant Microbiota is not Strongly Associated with Incidental Colonic Diverticulosis. Sci. Rep. 2018, 8, 4951. [Google Scholar] [CrossRef]
- Lopetuso, L.R.; Petito, V.; Graziani, C.; Schiavoni, E.; Paroni Sterbini, F.; Poscia, A.; Gaetani, E.; Franceschi, F.; Cammarota, G.; Sanguinetti, M.; et al. Gut Microbiota in Health, Diverticular Disease, Irritable Bowel Syndrome, and Inflammatory Bowel Diseases: Time for Microbial Marker of Gastrointestinal Disorders. Dig. Dis. 2018, 36, 56–65. [Google Scholar] [CrossRef]
- Jiang, F.; Meng, D.; Weng, M.; Zhu, W.; Wu, W.; Kasper, D.; Walker, W.A. The symbiotic bacterial surface factor polysaccharide A on Bacteroides fragilis inhibits IL-1β-induced inflammation in human fetal enterocytes via toll receptors 2 and 4. PLoS ONE 2017, 12, e0172738. [Google Scholar] [CrossRef]
- Linninge, C.; Roth, B.; Erlanson-Albertsson, C.; Molin, G.; Toth, E.; Ohlsson, B. Abundance of Enterobacteriaceae in the colon mucosa in diverticular disease. World J. Gastrointest. Pathophysiol. 2018, 9, 18–27. [Google Scholar] [CrossRef]
- Kvasnovsky, C.L.; Leong, L.E.X.; Choo, J.M.; Abell, G.C.J.; Papagrigoriadis, S.; Bruce, K.D.; Rogers, G.B. Clinical and symptom scores are significantly correlated with fecal microbiota features in patients with symptomatic uncomplicated diverticular disease: A pilot study. Eur. J. Gastroenterol. Hepatol. 2018, 30, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Nogales, A.; Algieri, F.; Garrido-Mesa, J.; Vezza, T.; Utrilla, M.P.; Chueca, N.; Garcia, F.; Olivares, M.; Rodríguez-Cabezas, M.E.; Gálvez, J. Differential intestinal anti-inflammatory effects of Lactobacillus fermentum and Lactobacillus salivarius in DSS mouse colitis: Impact on microRNAs expression and microbiota composition. Mol. Nutr. Food Res. 2017, 61, 1700144. [Google Scholar] [CrossRef] [PubMed]
- Ponziani, F.R.; Scaldaferri, F.; Petito, V.; Sterbini, F.P.; Pecere, S.; Lopetuso, L.R.; Palladini, A.; Gerardi, V.; Masucci, L.; Pompili, M. The role of antibiotics in gut microbiota modulation: The eubiotic effects of rifaximin. Dig. Dis. 2016, 34, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Derrien, M.; Belzer, C.; de Vos, W.M. Akkermansia muciniphila and its role in regulating host functions. Microb. Pathog. 2017, 106, 171–181. [Google Scholar] [CrossRef]
- Gueimonde, M.; Ouwehand, A.; Huhtinen, H.; Salminen, E.; Salminen, S. Qualitative and quantitative analyses of the bifidobacterial microbiota in the colonic mucosa of patients with colorectal cancer, diverticulitis and inflammatory bowel disease. World J. Gastroenterol. WJG 2007, 13, 3985. [Google Scholar] [CrossRef]
- Daniels, L.; Budding, A.E.; de Korte, N.; Eck, A.; Bogaards, J.A.; Stockmann, H.B.; Consten, E.C.; Savelkoul, P.H.; Boermeester, M.A. Fecal microbiome analysis as a diagnostic test for diverticulitis. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 1927–1936. [Google Scholar] [CrossRef]
- Graham, D.B.; Xavier, R.J. Pathway paradigms revealed from the genetics of inflammatory bowel disease. Nature 2020, 578, 527–539. [Google Scholar] [CrossRef]
- Ng, S.C.; Shi, H.Y.; Hamidi, N.; Underwood, F.E.; Tang, W.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Wu, J.C.; Chan, F.K. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: A systematic review of population-based studies. Lancet 2017, 390, 2769–2778. [Google Scholar] [CrossRef]
- Alatab, S.; Sepanlou, S.G.; Ikuta, K.; Vahedi, H.; Bisignano, C.; Safiri, S.; Sadeghi, A.; Nixon, M.R.; Abdoli, A.; Abolhassani, H. The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2020, 5, 17–30. [Google Scholar] [CrossRef]
- Ananthakrishnan, A.N.; Khalili, H.; Konijeti, G.G.; Higuchi, L.M.; De Silva, P.; Korzenik, J.R.; Fuchs, C.S.; Willett, W.C.; Richter, J.M.; Chan, A.T. A prospective study of long-term intake of dietary fiber and risk of Crohn’s disease and ulcerative colitis. Gastroenterology 2013, 145, 970–977. [Google Scholar] [CrossRef] [PubMed]
- Racine, A.; Carbonnel, F.; Chan, S.S.; Hart, A.R.; Bueno-de-Mesquita, H.B.; Oldenburg, B.; Van Schaik, F.D.; Tjønneland, A.; Olsen, A.; Dahm, C.C. Dietary patterns and risk of inflammatory bowel disease in Europe: Results from the EPIC study. Inflamm. Bowel Dis. 2016, 22, 345–354. [Google Scholar] [CrossRef]
- Ananthakrishnan, A.N.; Khalili, H.; Konijeti, G.G.; Higuchi, L.M.; de Silva, P.; Fuchs, C.S.; Willett, W.C.; Richter, J.M.; Chan, A.T. Long-term intake of dietary fat and risk of ulcerative colitis and Crohn’s disease. Gut 2014, 63, 776–784. [Google Scholar] [CrossRef] [PubMed]
- Ananthakrishnan, A.N. Impact of Diet on Risk of IBD. Crohn’s Colitis 2020, 2, otz054. [Google Scholar] [CrossRef]
- de Silva, P.S.; Olsen, A.; Christensen, J.; Schmidt, E.B.; Overvaad, K.; Tjonneland, A.; Hart, A.R. An association between dietary arachidonic acid, measured in adipose tissue, and ulcerative colitis. Gastroenterology 2010, 139, 1912–1917. [Google Scholar] [CrossRef] [PubMed]
- Jonkers, D.; Penders, J.; Masclee, A.; Pierik, M. Probiotics in the management of inflammatory bowel disease. Drugs 2012, 72, 803–823. [Google Scholar] [CrossRef] [PubMed]
- Lima, S.; Gogokhia, L.; Viladomiu, M.; Crawford, C.; Jacob, V.; Scherl, E.; Rosenthal, M.; Brown, S.-E.; Hambor, J.; Longman, R. P076 transferable immune reactive microbiota determine clinical and immunologic outcome of fecal microbiota transplantation for ulcerative colitis. Gastroenterology 2020, 158, S63–S64. [Google Scholar] [CrossRef]
- Caër, C.; Wick, M.J. Human Intestinal Mononuclear Phagocytes in Health and Inflammatory Bowel Disease. Front. Immunol. 2020, 11, 410. [Google Scholar] [CrossRef]
- Chapuy, L.; Bsat, M.; Sarkizova, S.; Rubio, M.; Therrien, A.; Wassef, E.; Bouin, M.; Orlicka, K.; Weber, A.; Hacohen, N. Two distinct colonic CD14+ subsets characterized by single-cell RNA profiling in Crohn’s disease. Mucosal Immunol. 2019, 12, 703–719. [Google Scholar] [CrossRef]
- Thiesen, S.; Janciauskiene, S.; Uronen-Hansson, H.; Agace, W.; Högerkorp, C.M.; Spee, P.; Håkansson, K.; Grip, O. CD14hiHLA-DRdim macrophages, with a resemblance to classical blood monocytes, dominate inflamed mucosa in Crohn’s disease. J. Leukoc. Biol. 2014, 95, 531–541. [Google Scholar] [CrossRef]
- Baillie, J.K.; Arner, E.; Daub, C.; De Hoon, M.; Itoh, M.; Kawaji, H.; Lassmann, T.; Carninci, P.; Forrest, A.R.; Hayashizaki, Y. Analysis of the human monocyte-derived macrophage transcriptome and response to lipopolysaccharide provides new insights into genetic aetiology of inflammatory bowel disease. PLoS Genet. 2017, 13, e1006641. [Google Scholar] [CrossRef]
- Papoutsopoulou, S.; Burkitt, M.D.; Bergey, F.; England, H.; Hough, R.; Schmidt, L.; Spiller, D.G.; White, M.R.H.; Paszek, P.; Jackson, D.A. Macrophage-specific NF-kB activation dynamics can segregate inflammatory bowel disease patients. Front. Immunol. 2019, 10, 2168. [Google Scholar] [CrossRef]
- Andreou, N.-P.; Legaki, E.; Gazouli, M. Inflammatory bowel disease pathobiology: The role of the interferon signature. Ann. Gastroenterol. 2020, 33, 125–133. [Google Scholar] [CrossRef]
- Haep, L.; Britzen-Laurent, N.; Weber, T.G.; Naschberger, E.; Schaefer, A.; Kremmer, E.; Foersch, S.; Vieth, M.; Scheuer, W.; Wirtz, S. Interferon gamma counteracts the angiogenic switch and induces vascular permeability in dextran sulfate sodium colitis in mice. Inflamm. Bowel Dis. 2015, 21, 2360–2371. [Google Scholar] [CrossRef] [PubMed]
- Langer, V.; Vivi, E.; Regensburger, D.; Winkler, T.H.; Waldner, M.J.; Rath, T.; Schmid, B.; Skottke, L.; Lee, S.; Jeon, N.L. IFN-γ drives inflammatory bowel disease pathogenesis through VE-cadherin–directed vascular barrier disruption. J. Clin. Investig. 2019, 129, 4691–4707. [Google Scholar] [CrossRef] [PubMed]
- Guan, Q. A Comprehensive Review and Update on the Pathogenesis of Inflammatory Bowel Disease. J. Immunol. Res. 2019, 2019, 7247238. [Google Scholar] [CrossRef] [PubMed]
- Castellanos, J.G.; Woo, V.; Viladomiu, M.; Putzel, G.; Lima, S.; Diehl, G.E.; Marderstein, A.R.; Gandara, J.; Perez, A.R.; Withers, D.R. Microbiota-induced TNF-like ligand 1A drives group 3 innate lymphoid cell-mediated barrier protection and intestinal T cell activation during colitis. Immunity 2018, 49, 1077–1089.e5. [Google Scholar] [CrossRef]
- Kamada, N.; Hisamatsu, T.; Honda, H.; Kobayashi, T.; Chinen, H.; Takayama, T.; Kitazume, M.T.; Okamoto, S.; Koganei, K.; Sugita, A. TL1A produced by lamina propria macrophages induces Th1 and Th17 immune responses in cooperation with IL-23 in patients with Crohn’s disease. Inflamm. Bowel Dis. 2010, 16, 568–575. [Google Scholar] [CrossRef]
- Richard, A.C.; Peters, J.E.; Savinykh, N.; Lee, J.C.; Hawley, E.T.; Meylan, F.; Siegel, R.M.; Lyons, P.A.; Smith, K.G. Reduced monocyte and macrophage TNFSF15/TL1A expression is associated with susceptibility to inflammatory bowel disease. PLoS Genet. 2018, 14, e1007458. [Google Scholar] [CrossRef]
- Gordon, H.; Trier Moller, F.; Andersen, V.; Harbord, M. Heritability in inflammatory bowel disease: From the first twin study to genome-wide association studies. Inflamm. Bowel Dis. 2015, 21, 1428–1434. [Google Scholar] [CrossRef]
- Chen, G.-B.; Lee, S.H.; Brion, M.-J.A.; Montgomery, G.W.; Wray, N.R.; Radford-Smith, G.L.; Visscher, P.M.; Consortium, I.I.G. Estimation and partitioning of (co) heritability of inflammatory bowel disease from GWAS and immunochip data. Hum. Mol. Genet. 2014, 23, 4710–4720. [Google Scholar] [CrossRef]
- de Lange, K.M.; Moutsianas, L.; Lee, J.C.; Lamb, C.A.; Luo, Y.; Kennedy, N.A.; Jostins, L.; Rice, D.L.; Gutierrez-Achury, J.; Ji, S.-G. Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat. Genet. 2017, 49, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Khosravi, A.; Kusumawardhani, I.P.; Kwon, A.H.; Vasconcelos, A.C.; Cunha, L.D.; Mayer, A.E.; Shen, Y.; Wu, W.-L.; Kambal, A. Gene-microbiota interactions contribute to the pathogenesis of inflammatory bowel disease. Science 2016, 352, 1116–1120. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Z.; Van Sommeren, S.; Huang, H.; Ng, S.C.; Alberts, R.; Takahashi, A.; Ripke, S.; Lee, J.C.; Jostins, L.; Shah, T. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 2015, 47, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Aschard, H.; Laville, V.; Tchetgen, E.T.; Knights, D.; Imhann, F.; Seksik, P.; Zaitlen, N.; Silverberg, M.S.; Cosnes, J.; Weersma, R.K. Genetic effects on the commensal microbiota in inflammatory bowel disease patients. PLoS Genet. 2019, 15, e1008018. [Google Scholar] [CrossRef]
- Knights, D.; Silverberg, M.S.; Weersma, R.K.; Gevers, D.; Dijkstra, G.; Huang, H.; Tyler, A.D.; Van Sommeren, S.; Imhann, F.; Stempak, J.M. Complex host genetics influence the microbiome in inflammatory bowel disease. Genome Med. 2014, 6, 107. [Google Scholar] [CrossRef]
- Sadabad, M.S.; Regeling, A.; de Goffau, M.C.; Blokzijl, T.; Weersma, R.K.; Penders, J.; Faber, K.N.; Harmsen, H.J.; Dijkstra, G. The ATG16L1–T300A allele impairs clearance of pathosymbionts in the inflamed ileal mucosa of Crohn’s disease patients. Gut 2015, 64, 1546–1552. [Google Scholar] [CrossRef]
- Grainger, J.R.; Wohlfert, E.A.; Fuss, I.J.; Bouladoux, N.; Askenase, M.H.; Legrand, F.; Koo, L.Y.; Brenchley, J.M.; Fraser, I.D.; Belkaid, Y. Inflammatory monocytes regulate pathologic responses to commensals during acute gastrointestinal infection. Nat. Med. 2013, 19, 713–721. [Google Scholar] [CrossRef]
- Atarashi, K.; Tanoue, T.; Shima, T.; Imaoka, A.; Kuwahara, T.; Momose, Y.; Cheng, G.; Yamasaki, S.; Saito, T.; Ohba, Y. Induction of colonic regulatory T cells by indigenous Clostridium species. Science 2011, 331, 337–341. [Google Scholar] [CrossRef]
- Longman, R.S.; Diehl, G.E.; Victorio, D.A.; Huh, J.R.; Galan, C.; Miraldi, E.R.; Swaminath, A.; Bonneau, R.; Scherl, E.J.; Littman, D.R. CX3CR1+ mononuclear phagocytes support colitis-associated innate lymphoid cell production of IL-22. J. Exp. Med. 2014, 211, 1571–1583. [Google Scholar] [CrossRef]
- Kim, M.; Galan, C.; Hill, A.A.; Wu, W.-J.; Fehlner-Peach, H.; Song, H.W.; Schady, D.; Bettini, M.L.; Simpson, K.W.; Longman, R.S. Critical role for the microbiota in CX3CR1+ intestinal mononuclear phagocyte regulation of intestinal T cell responses. Immunity 2018, 49, 151–163.e5. [Google Scholar] [CrossRef]
- Alameddine, J.; Godefroy, E.; Papargyris, L.; Sarrabayrouse, G.; Tabiasco, J.; Bridonneau, C.; Yazdanbakhsh, K.; Sokol, H.; Altare, F.; Jotereau, F. Faecalibacterium prausnitzii skews human DC to prime IL10-producing T cells through TLR2/6/JNK signaling and IL-10, IL-27, CD39, and IDO-1 induction. Front. Immunol. 2019, 10, 143. [Google Scholar] [CrossRef] [PubMed]
- Mannon, P.J.; Hornung, R.L.; Yang, Z.; Yi, C.; Groden, C.; Friend, J.; Yao, M.; Strober, W.; Fuss, I.J. Suppression of inflammation in ulcerative colitis by interferon-β-1a is accompanied by inhibition of IL-13 production. Gut 2011, 60, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Butera, A.; Di Paola, M.; Vitali, F.; De Nitto, D.; Covotta, F.; Borrini, F.; Pica, R.; De Filippo, C.; Cavalieri, D.; Giuliani, A. IL-13 mRNA tissue content identifies two subsets of adult ulcerative colitis patients with different clinical and mucosa-associated microbiota profiles. J. Crohn’s Colitis 2020, 14, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Paramsothy, S.; Kamm, M.A.; Kaakoush, N.O.; Walsh, A.J.; van den Bogaerde, J.; Samuel, D.; Leong, R.W.; Connor, S.; Ng, W.; Paramsothy, R. Multidonor intensive faecal microbiota transplantation for active ulcerative colitis: A randomised placebo-controlled trial. Lancet 2017, 389, 1218–1228. [Google Scholar] [CrossRef]
- Paramsothy, S.; Nielsen, S.; Kamm, M.A.; Deshpande, N.P.; Faith, J.J.; Clemente, J.C.; Paramsothy, R.; Walsh, A.J.; van den Bogaerde, J.; Samuel, D. Specific bacteria and metabolites associated with response to fecal microbiota transplantation in patients with ulcerative colitis. Gastroenterology 2019, 156, 1440–1454.e2. [Google Scholar] [CrossRef]
- Morgan, X.C.; Kabakchiev, B.; Waldron, L.; Tyler, A.D.; Tickle, T.L.; Milgrom, R.; Stempak, J.M.; Gevers, D.; Xavier, R.J.; Silverberg, M.S. Associations between host gene expression, the mucosal microbiome, and clinical outcome in the pelvic pouch of patients with inflammatory bowel disease. Genome Biol. 2015, 16, 67. [Google Scholar] [CrossRef]
- Kaakoush, N.O. Sutterella Species, IgA-degrading Bacteria in Ulcerative Colitis. Trends Microbiol. 2020, 28, 519–522. [Google Scholar] [CrossRef]
- Canavan, C.; West, J.; Card, T. The epidemiology of irritable bowel syndrome. Clin. Epidemiol. 2014, 6, 71. [Google Scholar]
- Burns, G.; Carroll, G.; Mathe, A.; Horvat, J.; Foster, P.; Walker, M.M.; Talley, N.J.; Keely, S. Evidence for local and systemic immune activation in functional dyspepsia and the irritable bowel syndrome: A systematic review. Am. J. Gastroenterol. 2018, 11, 429–436. [Google Scholar] [CrossRef]
- Said, H.M. Physiology of the Gastrointestinal Tract, 6th ed.; Academic Press: London, UK, 2018; Chapter 67; pp. 1643–1668. [Google Scholar]
- Keohane, J.; O’mahony, C.; O’mahony, L.; O’mahony, S.; Quigley, E.M.; Shanahan, F. Irritable bowel syndrome–type symptoms in patients with inflammatory bowel disease: A real association or reflection of occult inflammation? Am. J. Gastroenterol. 2010, 105, 1789–1794. [Google Scholar] [CrossRef]
- Ng, Q.X.; Soh, A.Y.S.; Loke, W.; Lim, D.Y.; Yeo, W.-S. The role of inflammation in irritable bowel syndrome (IBS). J. Inflamm. Res. 2018, 11, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Wang, B.; Jia, Q.; Duan, L. Candidate single nucleotide polymorphisms of irritable bowel syndrome: A systemic review and meta-analysis. BMC Gastroenterol. 2019, 19, 165. [Google Scholar] [CrossRef]
- Sinagra, E.; Pompei, G.; Tomasello, G.; Cappello, F.; Morreale, G.C.; Amvrosiadis, G.; Rossi, F.; Monte, A.I.L.; Rizzo, A.G.; Raimondo, D. Inflammation in irritable bowel syndrome: Myth or new treatment target? World J. Gastroenterol. 2016, 22, 2242–2255. [Google Scholar] [CrossRef] [PubMed]
- Seyedmirzaee, S.; Hayatbakhsh, M.M.; Ahmadi, B.; Baniasadi, N.; Rafsanjani, A.M.B.; Nikpoor, A.R.; Mohammadi, M. Serum immune biomarkers in irritable bowel syndrome. Clin. Res. Hepatol. Gastroenterol. 2016, 40, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Bashashati, M.; Moradi, M.; Sarosiek, I. Interleukin-6 in irritable bowel syndrome: A systematic review and meta-analysis of IL-6 (-G174C) and circulating IL-6 levels. Cytokine 2017, 99, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Klem, F.; Wadhwa, A.; Prokop, L.J.; Sundt, W.J.; Farrugia, G.; Camilleri, M.; Singh, S.; Grover, M. Prevalence, risk factors, and outcomes of irritable bowel syndrome after infectious enteritis: A systematic review and meta-analysis. Gastroenterology 2017, 152, 1042–1054.e1. [Google Scholar] [CrossRef]
- Sadeghi, A.; Biglari, M.; Moghaddam, S.N. Post-infectious Irritable Bowel Syndrome: A Narrative Review. Middle East J. Dig. Dis. 2019, 11, 69–75. [Google Scholar] [CrossRef]
- Walker, M.M.; Talley, N.J.; Inganäs, L.; Engstrand, L.; Jones, M.P.; Nyhlin, H.; Agréus, L.; Kjellstrom, L.; Öst, Å.; Andreasson, A. Colonic spirochetosis is associated with colonic eosinophilia and irritable bowel syndrome in a general population in Sweden. Hum. Pathol. 2015, 46, 277–283. [Google Scholar] [CrossRef]
- Jalanka, J.; Salonen, A.; Fuentes, S.; de Vos, W.M. Microbial signatures in post-infectious irritable bowel syndrome–toward patient stratification for improved diagnostics and treatment. Gut Microbes 2015, 6, 364–369. [Google Scholar] [CrossRef]
- Maxwell, P.; Rink, E.; Kumar, D.; Mendall, M. Antibiotics increase functional abdominal symptoms. Am. J. Gastroenterol. 2002, 97, 104–108. [Google Scholar] [CrossRef]
- Paula, H.; Grover, M.; Halder, S.L.; Locke III, G.R.; Schleck, C.D.; Zinsmeister, A.R.; Talley, N.J. Non-enteric infections, antibiotic use, and risk of development of functional gastrointestinal disorders. Neurogastroenterol. Motil. 2015, 27, 1580–1586. [Google Scholar] [CrossRef]
- Pimentel, M.; Lembo, A. Microbiome and Its Role in Irritable Bowel Syndrome. Dig. Dis. Sci. 2020, 65, 829–839. [Google Scholar] [CrossRef]
- Pimentel, M.; Chang, C.; Chua, K.S.; Mirocha, J.; DiBaise, J.; Rao, S.; Amichai, M. Antibiotic treatment of constipation-predominant irritable bowel syndrome. Dig. Dis. Sci. 2014, 59, 1278–1285. [Google Scholar] [CrossRef] [PubMed]
- Rezaie, A.; Buresi, M.; Lembo, A.; Lin, H.; McCallum, R.; Rao, S.; Schmulson, M.; Valdovinos, M.; Zakko, S.; Pimentel, M. Hydrogen and methane-based breath testing in gastrointestinal disorders: The North American Consensus. Am. J. Gastroenterol. 2017, 112, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Spiller, R.; Major, G. IBS and IBD—Separate entities or on a spectrum? Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Moraes, L.; Magnusson, M.K.; Mavroudis, G.; Polster, A.; Jonefjäll, B.; Törnblom, H.; Sundin, J.; Simrén, M.; Strid, H.; Öhman, L. Systemic Inflammatory Protein Profiles Distinguish Irritable Bowel Syndrome (IBS) and Ulcerative Colitis, Irrespective of Inflammation or IBS-Like Symptoms. Inflamm. Bowel Dis. 2020, 26, 874–884. [Google Scholar] [CrossRef] [PubMed]
- Howlader, N.; Noone, A.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D. SEER Cancer Statistics Review, 1975–2016; National Cancer Institute: Bethesda, MD, USA, 2019. [Google Scholar]
- Dulai, P.S.; Sandborn, W.J.; Gupta, S. Colorectal cancer and dysplasia in inflammatory bowel disease: A review of disease epidemiology, pathophysiology, and management. Cancer Prev. Res. 2016, 9, 887–894. [Google Scholar] [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; De Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Friis, S.; Riis, A.H.; Erichsen, R.; Baron, J.A.; Sørensen, H.T. Low-dose aspirin or nonsteroidal anti-inflammatory drug use and colorectal cancer risk: A population-based, case–control study. Ann. Intern. Med. 2015, 163, 347–355. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Chung, A.Y.; Li, Q.; Blair, S.J.; De Jesus, M.; Dennis, K.L.; LeVea, C.; Yao, J.; Sun, Y.; Conway, T.F.; Virtuoso, L.P. Oral interleukin-10 alleviates polyposis via neutralization of pathogenic T-regulatory cells. Cancer Res. 2014, 74, 5377–5385. [Google Scholar] [CrossRef] [PubMed]
- Tsiaoussis, G.I.; Assimakopoulos, S.F.; Tsamandas, A.C.; Triantos, C.K.; Thomopoulos, K.C. Intestinal barrier dysfunction in cirrhosis: Current concepts in pathophysiology and clinical implications. World J. Hepatol. 2015, 7, 2058–2068. [Google Scholar] [CrossRef] [PubMed]
- Salim, S.Y.; Söderholm, J.D. Importance of disrupted intestinal barrier in inflammatory bowel diseases. Inflamm. Bowel Dis. 2011, 17, 362–381. [Google Scholar] [CrossRef] [PubMed]
- Velcich, A.; Yang, W.; Heyer, J.; Fragale, A.; Nicholas, C.; Viani, S.; Kucherlapati, R.; Lipkin, M.; Yang, K.; Augenlicht, L. Colorectal cancer in mice genetically deficient in the mucin Muc2. Science 2002, 295, 1726–1729. [Google Scholar] [CrossRef] [PubMed]
- Lasry, A.; Zinger, A.; Ben-Neriah, Y. Inflammatory networks underlying colorectal cancer. Nat. Immunol. 2016, 17, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Okayasu, I.; Hatakeyama, S.; Yamada, M.; Ohkusa, T.; Inagaki, Y.; Nakaya, R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology 1990, 98, 694–702. [Google Scholar] [CrossRef]
- Cooper, H.S.; Murthy, S.; Shah, R.; Sedergran, D. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab. Investig. J. Tech. Methods Pathol. 1993, 69, 238–249. [Google Scholar]
- Spadoni, I.; Zagato, E.; Bertocchi, A.; Paolinelli, R.; Hot, E.; Di Sabatino, A.; Caprioli, F.; Bottiglieri, L.; Oldani, A.; Viale, G. A gut-vascular barrier controls the systemic dissemination of bacteria. Science 2015, 350, 830–834. [Google Scholar] [CrossRef]
- Cromer, W.E.; Mathis, J.M.; Granger, D.N.; Chaitanya, G.V.; Alexander, J.S. Role of the endothelium in inflammatory bowel diseases. World J. Gastroenterol. WJG 2011, 17, 578–593. [Google Scholar] [CrossRef]
- Alkim, C.; Alkim, H.; Koksal, A.R.; Boga, S.; Sen, I. Angiogenesis in inflammatory bowel disease. Int. J. Inflamm. 2015, 2015, 970890. [Google Scholar] [CrossRef]
- Naschberger, E.; Croner, R.S.; Merkel, S.; Dimmler, A.; Tripal, P.; Amann, K.U.; Kremmer, E.; Brueckl, W.M.; Papadopoulos, T.; Hohenadl, C. Angiostatic immune reaction in colorectal carcinoma: Impact on survival and perspectives for antiangiogenic therapy. Int. J. Cancer 2008, 123, 2120–2129. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Wang, K.; Mucida, D.; Stewart, C.A.; Schnabl, B.; Jauch, D.; Taniguchi, K.; Yu, G.-Y.; Österreicher, C.H.; Hung, K.E. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 2012, 491, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [PubMed]
- De Simone, V.; Pallone, F.; Monteleone, G.; Stolfi, C. Role of TH17 cytokines in the control of colorectal cancer. Oncoimmunology 2013, 2, e26617. [Google Scholar] [CrossRef]
- Song, X.; Gao, H.; Lin, Y.; Yao, Y.; Zhu, S.; Wang, J.; Liu, Y.; Yao, X.; Meng, G.; Shen, N. Alterations in the microbiota drive interleukin-17C production from intestinal epithelial cells to promote tumorigenesis. Immunity 2014, 40, 140–152. [Google Scholar] [CrossRef]
- Kirchberger, S.; Royston, D.J.; Boulard, O.; Thornton, E.; Franchini, F.; Szabady, R.L.; Harrison, O.; Powrie, F. Innate lymphoid cells sustain colon cancer through production of interleukin-22 in a mouse model. J. Exp. Med. 2013, 210, 917–931. [Google Scholar] [CrossRef]
- Bollrath, J.; Phesse, T.J.; von Burstin, V.A.; Putoczki, T.; Bennecke, M.; Bateman, T.; Nebelsiek, T.; Lundgren-May, T.; Canli, Ö.; Schwitalla, S. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell 2009, 15, 91–102. [Google Scholar] [CrossRef]
- Sanchez-Lopez, E.; Flashner-Abramson, E.; Shalapour, S.; Zhong, Z.; Taniguchi, K.; Levitzki, A.; Karin, M. Targeting colorectal cancer via its microenvironment by inhibiting IGF-1 receptor-insulin receptor substrate and STAT3 signaling. Oncogene 2016, 35, 2634–2644. [Google Scholar] [CrossRef]
- Putoczki, T.L.; Thiem, S.; Loving, A.; Busuttil, R.A.; Wilson, N.J.; Ziegler, P.K.; Nguyen, P.M.; Preaudet, A.; Farid, R.; Edwards, K.M. Interleukin-11 is the dominant IL-6 family cytokine during gastrointestinal tumorigenesis and can be targeted therapeutically. Cancer Cell 2013, 24, 257–271. [Google Scholar] [CrossRef]
- Dennis, K.L.; Saadalla, A.; Blatner, N.R.; Wang, S.; Venkateswaran, V.; Gounari, F.; Cheroutre, H.; Weaver, C.T.; Roers, A.; Egilmez, N.K. T-cell expression of IL10 is essential for tumor immune surveillance in the small intestine. Cancer Immunol. Res. 2015, 3, 806–814. [Google Scholar] [CrossRef][Green Version]
- Dennis, K.L.; Wang, Y.; Blatner, N.R.; Wang, S.; Saadalla, A.; Trudeau, E.; Roers, A.; Weaver, C.T.; Lee, J.J.; Gilbert, J.A. Adenomatous polyps are driven by microbe-instigated focal inflammation and are controlled by IL-10–producing T cells. Cancer Res. 2013, 73, 5905–5913. [Google Scholar] [CrossRef] [PubMed]
- Parker, B.S.; Rautela, J.; Hertzog, P.J. Antitumour actions of interferons: Implications for cancer therapy. Nat. Rev. Cancer 2016, 16, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, Y.; Song, Z.; Chu, J.; Qu, X. Deficiency of interferon-gamma or its receptor promotes colorectal cancer development. J. Interferon Cytokine Res. 2015, 35, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Soleimani, A.; Rahmani, F.; Ferns, G.A.; Ryzhikov, M.; Avan, A.; Hassanian, S.M. Role of the NF-κB signaling pathway in the pathogenesis of colorectal cancer. Gene 2019, 144132. [Google Scholar] [CrossRef]
- Shaked, H.; Hofseth, L.J.; Chumanevich, A.; Chumanevich, A.A.; Wang, J.; Wang, Y.; Taniguchi, K.; Guma, M.; Shenouda, S.; Clevers, H. Chronic epithelial NF-κB activation accelerates APC loss and intestinal tumor initiation through iNOS up-regulation. Proc. Natl. Acad. Sci. USA 2012, 109, 14007–14012. [Google Scholar] [CrossRef]
- Koliaraki, V.; Pasparakis, M.; Kollias, G. IKKβ in intestinal mesenchymal cells promotes initiation of colitis-associated cancer. J. Exp. Med. 2015, 212, 2235–2251. [Google Scholar] [CrossRef]
- Bergstrom, K.; Liu, X.; Zhao, Y.; Gao, N.; Wu, Q.; Song, K.; Cui, Y.; Li, Y.; McDaniel, J.M.; McGee, S. Defective intestinal mucin-type O-glycosylation causes spontaneous colitis-associated cancer in mice. Gastroenterology 2016, 151, 152–164.e11. [Google Scholar] [CrossRef]
- Chen, K.; Liu, M.; Liu, Y.; Yoshimura, T.; Shen, W.; Le, Y.; Durum, S.; Gong, W.; Wang, C.; Gao, J.-L. Formylpeptide receptor-2 contributes to colonic epithelial homeostasis, inflammation, and tumorigenesis. J. Clin. Investig. 2013, 123, 1694–1704. [Google Scholar] [CrossRef]
- Salcedo, R.; Worschech, A.; Cardone, M.; Jones, Y.; Gyulai, Z.; Dai, R.-M.; Wang, E.; Ma, W.; Haines, D.; O’hUigin, C. MyD88-mediated signaling prevents development of adenocarcinomas of the colon: Role of interleukin 18. J. Exp. Med. 2010, 207, 1625–1636. [Google Scholar] [CrossRef]
- Rakoff-Nahoum, S.; Medzhitov, R. Regulation of spontaneous intestinal tumorigenesis through the adaptor protein MyD88. Science 2007, 317, 124–127. [Google Scholar] [CrossRef]
- Zeng, M.; Inohara, N.; Nuñez, G. Mechanisms of inflammation-driven bacterial dysbiosis in the gut. Mucosal Immunol. 2017, 10, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Son, J.S.; Khair, S.; Pettet III, D.W.; Ouyang, N.; Tian, X.; Zhang, Y.; Zhu, W.; Mackenzie, G.G.; Robertson, C.E.; Ir, D. Altered interactions between the gut microbiome and colonic mucosa precede polyposis in APCMin/+ mice. PLoS ONE 2015, 10, e0127985. [Google Scholar] [CrossRef] [PubMed]
- Man, S.M.; Zhu, Q.; Zhu, L.; Liu, Z.; Karki, R.; Malik, A.; Sharma, D.; Li, L.; Malireddi, R.S.; Gurung, P. Critical role for the DNA sensor AIM2 in stem cell proliferation and cancer. Cell 2015, 162, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Couturier-Maillard, A.; Secher, T.; Rehman, A.; Normand, S.; De Arcangelis, A.; Haesler, R.; Huot, L.; Grandjean, T.; Bressenot, A.; Delanoye-Crespin, A. NOD2-mediated dysbiosis predisposes mice to transmissible colitis and colorectal cancer. J. Clin. Investig. 2013, 123, 700–711. [Google Scholar] [CrossRef]
- Harrison, C. Cancer: IL-22: Linking inflammation and cancer. Nat. Rev. Drug Discov. 2013, 12, 505. [Google Scholar] [CrossRef]
- Malik, A.; Sharma, D.; Zhu, Q.; Karki, R.; Guy, C.S.; Vogel, P.; Kanneganti, T.-D. IL-33 regulates the IgA-microbiota axis to restrain IL-1α–dependent colitis and tumorigenesis. J. Clin. Investig. 2016, 126, 4469–4481. [Google Scholar] [CrossRef]
- Wu, M.; Wu, Y.; Deng, B.; Li, J.; Cao, H.; Qu, Y.; Qian, X.; Zhong, G. Isoliquiritigenin decreases the incidence of colitis-associated colorectal cancer by modulating the intestinal microbiota. Oncotarget 2016, 7, 85318–85331. [Google Scholar] [CrossRef]
- Lenoir, M.; Del Carmen, S.; Cortes-Perez, N.G.; Lozano-Ojalvo, D.; Muñoz-Provencio, D.; Chain, F.; Langella, P.; de LeBlanc, A.d.M.; LeBlanc, J.G.; Bermúdez-Humarán, L.G. Lactobacillus casei BL23 regulates T reg and Th17 T-cell populations and reduces DMH-associated colorectal cancer. J. Gastroenterol. 2016, 51, 862–873. [Google Scholar] [CrossRef]
- Kuugbee, E.D.; Shang, X.; Gamallat, Y.; Bamba, D.; Awadasseid, A.; Suliman, M.A.; Zang, S.; Ma, Y.; Chiwala, G.; Xin, Y. Structural change in microbiota by a probiotic cocktail enhances the gut barrier and reduces cancer via TLR2 signaling in a rat model of colon cancer. Dig. Dis. Sci. 2016, 61, 2908–2920. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Liang, S.; Jia, H.; Stadlmayr, A.; Tang, L.; Lan, Z.; Zhang, D.; Xia, H.; Xu, X.; Jie, Z. Gut microbiome development along the colorectal adenoma–carcinoma sequence. Nat. Commun. 2015, 6, 6528. [Google Scholar] [CrossRef]
- Pfalzer, A.C.; Nesbeth, P.-D.C.; Parnell, L.D.; Iyer, L.K.; Liu, Z.; Kane, A.V.; Chen, C.O.; Tai, A.K.; Bowman, T.A.; Obin, M.S. Diet-and genetically-induced obesity differentially affect the fecal microbiome and metabolome in Apc1638N mice. PLoS ONE 2015, 10, e0135758. [Google Scholar] [CrossRef] [PubMed]
- Burns, M.B.; Lynch, J.; Starr, T.K.; Knights, D.; Blekhman, R. Virulence genes are a signature of the microbiome in the colorectal tumor microenvironment. Genome Med. 2015, 7, 55. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.G.; Ge, Z.; Whary, M.T.; Erdman, S.E.; Horwitz, B.H. Helicobacter hepaticus infection in mice: Models for understanding lower bowel inflammation and cancer. Mucosal Immunol. 2011, 4, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Mangerich, A.; Knutson, C.G.; Parry, N.M.; Muthupalani, S.; Ye, W.; Prestwich, E.; Cui, L.; McFaline, J.L.; Mobley, M.; Ge, Z. Infection-induced colitis in mice causes dynamic and tissue-specific changes in stress response and DNA damage leading to colon cancer. Proc. Natl. Acad. Sci. USA 2012, 109, E1820–E1829. [Google Scholar] [CrossRef]
- Erdman, S.; Rao, V.; Poutahidis, T.; Rogers, A.; Taylor, C.; Jackson, E.; Ge, Z.; Lee, C.; Schauer, D.; Wogan, G. Nitric oxide and TNF-α trigger colonic inflammation and carcinogenesis in Helicobacter hepaticus-infected, Rag2-deficient mice. Proc. Natl. Acad. Sci. USA 2009, 106, 1027–1032. [Google Scholar] [CrossRef]
- Maggio-Price, L.; Treuting, P.; Zeng, W.; Tsang, M.; Bielefeldt-Ohmann, H.; Iritani, B.M. Helicobacter infection is required for inflammation and colon cancer in SMAD3-deficient mice. Cancer Res. 2006, 66, 828–838. [Google Scholar] [CrossRef]
- Housseau, F.; Wu, S.; Wick, E.C.; Fan, H.; Wu, X.; Llosa, N.J.; Smith, K.N.; Tam, A.; Ganguly, S.; Wanyiri, J.W. Redundant innate and adaptive sources of IL17 production drive colon tumorigenesis. Cancer Res. 2016, 76, 2115–2124. [Google Scholar] [CrossRef]
- Wu, S.; Rhee, K.-J.; Albesiano, E.; Rabizadeh, S.; Wu, X.; Yen, H.-R.; Huso, D.L.; Brancati, F.L.; Wick, E.; McAllister, F. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat. Med. 2009, 15, 1016–1022. [Google Scholar] [CrossRef]
- Purcell, R.V.; Pearson, J.; Aitchison, A.; Dixon, L.; Frizelle, F.A.; Keenan, J.I. Colonization with enterotoxigenic Bacteroides fragilis is associated with early-stage colorectal neoplasia. PLoS ONE 2017, 12, e0171602. [Google Scholar] [CrossRef]
- Viljoen, K.S.; Dakshinamurthy, A.; Goldberg, P.; Blackburn, J.M. Quantitative profiling of colorectal cancer-associated bacteria reveals associations between fusobacterium spp. enterotoxigenic Bacteroides fragilis (ETBF) and clinicopathological features of colorectal cancer. PLoS ONE 2015, 10, e0119462. [Google Scholar] [CrossRef]
- Boleij, A.; Hechenbleikner, E.M.; Goodwin, A.C.; Badani, R.; Stein, E.M.; Lazarev, M.G.; Ellis, B.; Carroll, K.C.; Albesiano, E.; Wick, E.C. The Bacteroides fragilis toxin gene is prevalent in the colon mucosa of colorectal cancer patients. Clin. Infect. Dis. 2015, 60, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Mima, K.; Nishihara, R.; Qian, Z.R.; Cao, Y.; Sukawa, Y.; Nowak, J.A.; Yang, J.; Dou, R.; Masugi, Y.; Song, M. Fusobacterium nucleatum in colorectal carcinoma tissue and patient prognosis. Gut 2016, 65, 1973–1980. [Google Scholar] [CrossRef] [PubMed]
- Borroni, E.M.; Qehajaj, D.; Farina, F.M.; Yiu, D.; Bresalier, R.S.; Chiriva-Internati, M.; Mirandola, L.; Štifter, S.; Laghi, L.; Grizzi, F. Fusobacterium nucleatum and the Immune System in Colorectal Cancer. Curr. Colorectal Cancer Rep. 2019, 15, 149–156. [Google Scholar] [CrossRef]
- Lu, R.; Wu, S.; Zhang, Y.; Xia, Y.; Liu, X.; Zheng, Y.; Chen, H.; Schaefer, K.; Zhou, Z.; Bissonnette, M. Enteric bacterial protein AvrA promotes colonic tumorigenesis and activates colonic beta-catenin signaling pathway. Oncogenesis 2014, 3, e105. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Huycke, M.M. Extracellular superoxide production by Enterococcus faecalis promotes chromosomal instability in mammalian cells. Gastroenterology 2007, 132, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Cuevas-Ramos, G.; Petit, C.R.; Marcq, I.; Boury, M.; Oswald, E.; Nougayrède, J.-P. Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc. Natl. Acad. Sci. USA 2010, 107, 11537–11542. [Google Scholar] [CrossRef] [PubMed]
- Pleguezuelos-Manzano, C.; Puschhof, J.; Huber, A.R.; van Hoeck, A.; Wood, H.M.; Nomburg, J.; Gurjao, C.; Manders, F.; Dalmasso, G.; Stege, P.B. Mutational signature in colorectal cancer caused by genotoxic pks+ E. coli. Nature 2020, 580, 269–273. [Google Scholar] [CrossRef]
- Bonnet, M.; Buc, E.; Sauvanet, P.; Darcha, C.; Dubois, D.; Pereira, B.; Déchelotte, P.; Bonnet, R.; Pezet, D.; Darfeuille-Michaud, A. Colonization of the human gut by E. coli and colorectal cancer risk. Clin. Cancer Res. 2014, 20, 859–867. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nasef, N.A.; Mehta, S. Role of Inflammation in Pathophysiology of Colonic Disease: An Update. Int. J. Mol. Sci. 2020, 21, 4748. https://doi.org/10.3390/ijms21134748
Nasef NA, Mehta S. Role of Inflammation in Pathophysiology of Colonic Disease: An Update. International Journal of Molecular Sciences. 2020; 21(13):4748. https://doi.org/10.3390/ijms21134748
Chicago/Turabian StyleNasef, Noha Ahmed, and Sunali Mehta. 2020. "Role of Inflammation in Pathophysiology of Colonic Disease: An Update" International Journal of Molecular Sciences 21, no. 13: 4748. https://doi.org/10.3390/ijms21134748
APA StyleNasef, N. A., & Mehta, S. (2020). Role of Inflammation in Pathophysiology of Colonic Disease: An Update. International Journal of Molecular Sciences, 21(13), 4748. https://doi.org/10.3390/ijms21134748