Absolute Binding Free Energy Calculations for Highly Flexible Protein MDM2 and Its Inhibitors



Abstract
:1. Introduction
2. Results and Discussion
2.1. Absolute Binding Free Energy Calculations for MDMX-Ligand Systems
2.2. Absolute Binding Free Energy Calculations for MDM2-Ligand Systems
2.3. Free Energy Landscape and Improved Prediction of Binding Free Energy
2.4. Absolute Binding Free Energy and Drug Design
3. Materials and Methods
3.1. Dataset
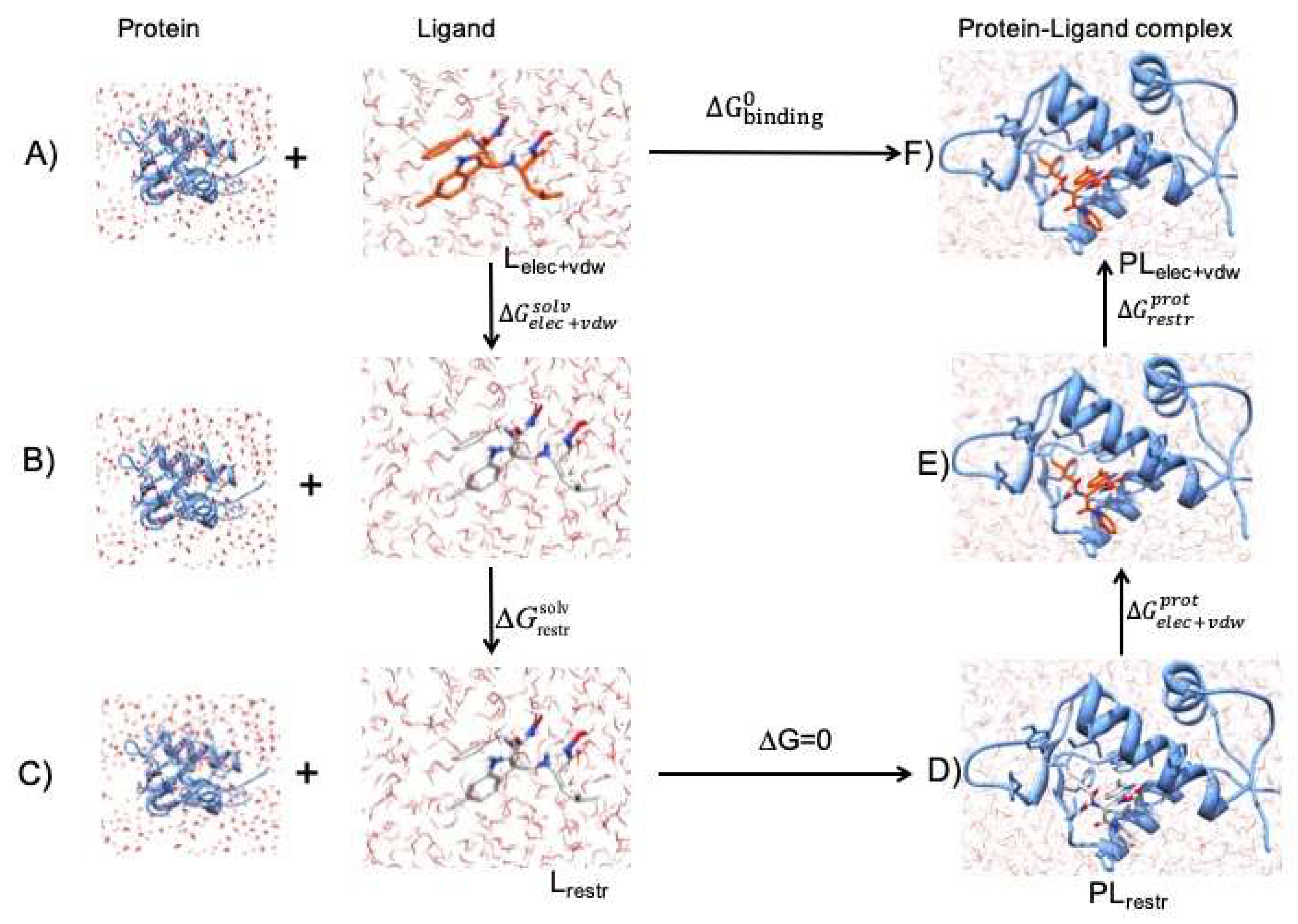
3.2. Free Energy Simulations
3.3. Restraints
3.4. Data Analysis
3.5. Free Energy Landscape Generation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Chodera, J.D.; Mobley, D.L.; Shirts, M.R.; Dixon, R.W.; Branson, K.; Pande, V.S. Alchemical free energy methods for drug discovery: Progress and challenges. Curr. Opin. Struct. Biol. 2011, 21, 150–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bissantz, C.; Kuhn, B.; Stahl, M. A medicinal chemist’s guide to molecular interactions. J. Med. Chem. 2010, 53, 5061–5084. [Google Scholar] [CrossRef] [PubMed]
- Chodera, J.D.; Mobley, D.L. Entropy-enthalpy compensation: Role and ramifications in biomolecular ligand recognition and design. Annu. Rev. Biophys. 2013, 42, 121–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wereszczynski, J.; McCammon, J.A. Statistical mechanics and molecular dynamics in evaluating thermodynamic properties of biomolecular recognition. Q. Rev. Biophys. 2011, 45, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Homeyer, N.; Stoll, F.; Hillisch, A.; Gohlke, H. Binding Free Energy Calculations for Lead Optimization: Assessment of Their Accuracy in an Industrial Drug Design Context. J. Chem. Theory Comput. 2014, 10, 3331–3344. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wu, Y.; Deng, Y.; Kim, B.; Pierce, L.; Krilov, G.; Lupyan, D.; Robinson, S.; Dahlgren, M.K.; Greenwood, J.; et al. Accurate and Reliable Prediction of Relative Ligand Binding Potency in Prospective Drug Discovery by Way of a Modern Free-Energy Calculation Protocol and Force Field. J. Am. Chem. Soc. 2015, 137, 2695–2703. [Google Scholar] [CrossRef] [Green Version]
- Mobley, D.L.; Gilson, M.K. Predicting Binding Free Energies: Frontiers and Benchmarks. Annu. Rev. Biophys. 2017, 46, 531–558. [Google Scholar] [CrossRef] [Green Version]
- Michel, J.; Essex, J.W. Prediction of protein–ligand binding affinity by free energy simulations: Assumptions, pitfalls, and expectations. J. Comput. Mol. Des. 2010, 24, 639–658. [Google Scholar] [CrossRef]
- Liu, S.; Wu, Y.; Lin, T.; Abel, R.; Redmann, J.P.; Summa, C.M.; Jaber, V.R.; Lim, N.; Mobley, D.L. Lead optimization mapper: Automating free energy calculations for lead optimization. J. Comput. Mol. Des. 2013, 27, 755–770. [Google Scholar] [CrossRef]
- Mikulskis, P.; Genheden, S.; Ryde, U. A Large-Scale Test of Free-Energy Simulation Estimates of Protein–Ligand Binding Affinities. J. Chem. Inf. Model. 2014, 54, 2794–2806. [Google Scholar] [CrossRef]
- Boresch, S.; Tettinger, F.; Leitgeb, M.; Karplus, M. Absolute Binding Free Energies: A Quantitative Approach for Their Calculation. J. Phys. Chem. B 2003, 107, 9535–9551. [Google Scholar] [CrossRef]
- Aldeghi, M.; Heifetz, A.; Bodkin, M.J.; Knapp, S.; Biggin, P.C. Accurate calculation of the absolute free energy of binding for drug molecules. Chem. Sci. 2016, 7, 207–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mobley, D.L.; Graves, A.P.; Chodera, J.D.; McReynolds, A.C.; Shoichet, B.K.; Dill, K.A. Predicting Absolute Ligand Binding Free Energies to a Simple Model Site. J. Mol. Biol. 2007, 371, 1118–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyce, S.E.; Mobley, D.L.; Rocklin, G.J.; Graves, A.P.; Dill, K.A.; Shoichet, B.K. Predicting Ligand Binding Affinity with Alchemical Free Energy Methods in a Polar Model Binding Site. J. Mol. Biol. 2009, 394, 747–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, B.; Nguyen, T.H.; Minh, D.D.L. Absolute Binding Free Energies between T4 Lysozyme and 141 Small Molecules: Calculations Based on Multiple Rigid Receptor Configurations. J. Chem. Theory Comput. 2017, 13, 2930–2944. [Google Scholar] [CrossRef]
- Fujitani, H.; Tanida, Y.; Ito, M.; Jayachandran, G.; Snow, C.D.; Shirts, M.R.; Sorin, E.J.; Pande, V.S. Direct calculation of the binding free energies of FKBP ligands. J. Chem. Phys. 2005, 123, 84108. [Google Scholar] [CrossRef]
- Wang, J.; Deng, Y.; Roux, B. Absolute Binding Free Energy Calculations Using Molecular Dynamics Simulations with Restraining Potentials. Biophys. J. 2006, 91, 2798–2814. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wang, X.; Wang, G.; Yang, Y.; Yuan, Y.; Ouyang, L. The past, present and future of potential small-molecule drugs targeting p53-MDM2/MDMX for cancer therapy. Eur. J. Med. Chem. 2019, 176, 92–104. [Google Scholar] [CrossRef]
- Kussie, P.H.; Gorina, S.; Marechal, V.; Elenbaas, B.; Moreau, J.; Levine, A.J.; Pavletich, N.P. Structure of the MDM2 Oncoprotein Bound to the p53 Tumor Suppressor Transactivation Domain. Science 1996, 274, 948–953. [Google Scholar] [CrossRef]
- Showalter, S.A.; Bruschweiler-Li, L.; Johnson, E.; Zhang, F.; Brüschweiler, R. Quantitative Lid Dynamics of MDM2 Reveals Differential Ligand Binding Modes of the p53-Binding Cleft. J. Am. Chem. Soc. 2008, 130, 6472–6478. [Google Scholar] [CrossRef]
- Popowicz, G.M.; Czarna, A.; Wolf, S.; Wang, K.; Wang, W.; Dömling, A.; Holak, T.A. Structures of low molecular weight inhibitors bound to MDMX and MDM2 reveal new approaches for p53-MDMX/MDM2 antagonist drug discovery. Cell Cycle 2010, 9, 1104–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.-L.; Meng, Y.; Jiang, W.; Roux, B. Explaining why Gleevec is a specific and potent inhibitor of Abl kinase. Proc. Natl. Acad. Sci. USA 2013, 110, 1664–1669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mobley, D.L.; Chodera, J.D.; Dill, K.A. Confine-and-Release Method: Obtaining Correct Binding Free Energies in the Presence of Protein Conformational Change. J. Chem. Theory Comput. 2007, 3, 1231–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapelosa, M.; Gallicchio, E.; Levy, R.M. Conformational Transitions and Convergence of Absolute Binding Free Energy Calculations. J. Chem. Theory Comput. 2011, 8, 47–60. [Google Scholar] [CrossRef]
- Jiang, W.; Roux, B. Free Energy Perturbation Hamiltonian Replica-Exchange Molecular Dynamics (FEP/H-REMD) for Absolute Ligand Binding Free Energy Calculations. J. Chem. Theory Comput. 2010, 6, 2559–2565. [Google Scholar] [CrossRef] [Green Version]
- Joseph, T.L.; Madhumalar, A.; Brown, C.J.; Lane, D.P.; Verma, C.S. Differential binding of p53 and nutlin to MDM2 and MDMX: Computational studies. Cell Cycle 2010, 9, 1167–1181. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Roux, B. Calculation of Standard Binding Free Energies: Aromatic Molecules in the T4 Lysozyme L99A Mutant. J. Chem. Theory Comput. 2006, 2, 1255–1273. [Google Scholar] [CrossRef]
- Lee, H.S.; Jo, S.; Lim, H.-S.; Im, W. Application of Binding Free Energy Calculations to Prediction of Binding Modes and Affinities of MDM2 and MDMX Inhibitors. J. Chem. Inf. Model. 2012, 52, 1821–1832. [Google Scholar] [CrossRef] [Green Version]
- Marine, J.-C.; Dyer, M.A.; Jochemsen, A.G. MDMX: From bench to bedside. J. Cell Sci. 2007, 120, 371–378. [Google Scholar] [CrossRef] [Green Version]
- Shadfan, M.; Lopez-Pajares, V.; Yuan, Z.-M. MDM2 and MDMX: Alone and together in regulation of p53. Transl. Cancer Res. 2012, 1, 88–89. [Google Scholar]
- Wade, M.; Wahl, G.M. Targeting Mdm2 and Mdmx in cancer therapy: Better living through medicinal chemistry? Mol. Cancer Res. 2009, 7, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macchiarulo, A.; Pellicciari, R. MDM2/MDMX inhibitor peptide: WO2008106507. Expert Opin. Ther. Patents 2009, 19, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Williams-Noonan, B.J.; Yuriev, E.; Chalmers, D.K. Free Energy Methods in Drug Design: Prospects of “Alchemical Perturbation” in Medicinal Chemistry. J. Med. Chem. 2018, 61, 638–649. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Liu, S.; Lee, T.-S.; Ji, B.; Man, V.H.; York, D.M.; Wang, J. Fast, Accurate, and Reliable Protocols for Routine Calculations of Protein–Ligand Binding Affinities in Drug Design Projects Using AMBER GPU-TI with ff14SB/GAFF. ACS Omega 2020, 5, 4611–4619. [Google Scholar] [CrossRef]
- Grace, C.R.; Bán, D.; Min, J.; Mayasundari, A.; Min, L.; Finch, K.E.; Griffiths, L.; Bharatham, N.; Bashford, D.; Guy, R.K.; et al. Monitoring Ligand-Induced Protein Ordering in Drug Discovery. J. Mol. Biol. 2016, 428, 1290–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaupel, A.; Holzer, P.; Ferretti, S.; Guagnano, V.; Kallen, J.; Mah, R.; Masuya, K.; Ruetz, S.; Rynn, C.; Schlapbach, A.; et al. In vitro and in vivo characterization of a novel, highly potent p53-MDM2 inhibitor. Bioorg. Med. Chem. Lett. 2018, 28, 3404–3408. [Google Scholar] [CrossRef]
- Jeay, S.; Ferretti, S.; Holzer, P.; Fuchs, J.; Chapeau, E.A.; Wartmann, M.; Sterker, D.; Romanet, V.; Murakami, M.; Kerr, G.; et al. Dose and Schedule Determine Distinct Molecular Mechanisms Underlying the Efficacy of the p53–MDM2 Inhibitor HDM201. Cancer Res. 2018, 78, 6257–6267. [Google Scholar] [CrossRef] [Green Version]
- Holzer, P.; Masuya, K.; Furet, P.; Kallen, J.; Valat-Stachyra, T.; Ferretti, S.; Berghausen, J.; Bouisset-Leonard, M.; Buschmann, N.; Pissot-Soldermann, C.; et al. Discovery of a Dihydroisoquinolinone Derivative (NVP-CGM097): A Highly Potent and Selective MDM2 Inhibitor Undergoing Phase 1 Clinical Trials in p53wt Tumors. J. Med. Chem. 2015, 58, 6348–6358. [Google Scholar] [CrossRef]
- Gollner, A.; Rudolph, D.; Arnhof, H.; Bauer, M.; Blake, S.M.; Boehmelt, G.; Cockroft, X.L.; Dahmann, G.; Ettmayer, P.; Gerstberger, T.; et al. Discovery of Novel Spiro[3 H-indole-3,2’-pyrrolidin]-2(1H)-one Compounds as Chemically Stable and Orally Active Inhibitors of the MDM2-p53 Interaction. J. Med. Chem. 2016, 59, 10147–10162. [Google Scholar] [CrossRef]
- Huang, Y.; Wolf, S.; Beck, B.; Köhler, L.-M.; Khoury, K.; Popowicz, G.M.; Goda, S.K.; Subklewe, M.; Twarda, A.; Holak, T.A.; et al. Discovery of Highly Potent p53-MDM2 Antagonists and Structural Basis for Anti-Acute Myeloid Leukemia Activities. ACS Chem. Biol. 2014, 9, 802–811. [Google Scholar] [CrossRef]
- Bista, M.; Wolf, S.; Khoury, K.; Kowalska, K.; Huang, Y.; Wrona, E.; Arciniega, M.; Popowicz, G.M.; Holak, T.A.; Dömling, A. Transient Protein States in Designing Inhibitors of the MDM2-p53 Interaction. Structure 2013, 21, 2143–2151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Turiso, F.G.-L.; Sun, D.; Rew, Y.; Bartberger, M.D.; Beck, H.P.; Canon, J.; Chen, A.; Chow, D.; Correll, T.L.; Huang, X.; et al. Rational Design and Binding Mode Duality of MDM2–p53 Inhibitors. J. Med. Chem. 2013, 56, 4053–4070. [Google Scholar] [CrossRef] [PubMed]
- Mobley, D.L.; Klimovich, P.V. Perspective: Alchemical free energy calculations for drug discovery. J. Chem. Phys. 2012, 137, 230901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falconer, R. Applications of isothermal titration calorimetry—The research and technical developments from 2011 to 2015. J. Mol. Recognit. 2016, 29, 504–515. [Google Scholar] [CrossRef] [Green Version]
- Moraca, F.; Negri, A.; de Oliveira, C.; Abel, R. Application of Free Energy Perturbation (FEP+) to Understanding Ligand Selectivity: A Case Study to Assess Selectivity Between Pairs of Phosphodiesterases (PDE’s). J. Chem. Inf. Model. 2019, 59, 2729–2740. [Google Scholar] [CrossRef]
- Aldeghi, M.; Heifetz, A.; Bodkin, M.J.; Knapp, S.; Biggin, P.C. Predictions of Ligand Selectivity from Absolute Binding Free Energy Calculations. J. Am. Chem. Soc. 2017, 139, 946–957. [Google Scholar] [CrossRef] [Green Version]
- Vu, B.; Wovkulich, P.; Pizzolato, G.; Lovey, A.; Ding, Q.; Jiang, N.; Liu, J.-J.; Zhao, C.; Glenn, K.; Wen, Y.; et al. Discovery of RG7112: A Small-Molecule MDM2 Inhibitor in Clinical Development. ACS Med. Chem. Lett. 2013, 4, 466–469. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Sun, W.; Zhao, Y.; McEachern, N.; Meaux, I.; Barrière, C.; Stuckey, J.A.; Meagher, J.L.; Bai, L.; Liu, L.; et al. SAR405838: An optimized inhibitor of MDM2-p53 interaction that induces complete and durable tumor regression. Cancer Res. 2014, 74, 5855–5865. [Google Scholar] [CrossRef] [Green Version]
- Gessier, F.; Kallen, J.; Jacoby, E.; Chène, P.; Stachyra-Valat, T.; Ruetz, S.; Jeay, S.; Holzer, P.; Masuya, K.; Furet, P. Discovery of dihydroisoquinolinone derivatives as novel inhibitors of the p53–MDM2 interaction with a distinct binding mode. Bioorg. Med. Chem. Lett. 2015, 25, 3621–3625. [Google Scholar] [CrossRef]
- Furet, P.; Masuya, K.; Kallen, J.; Stachyra-Valat, T.; Ruetz, S.; Guagnano, V.; Holzer, P.; Mah, R.; Stutz, S.; Vaupel, A.; et al. Discovery of a novel class of highly potent inhibitors of the p53–MDM2 interaction by structure-based design starting from a conformational argument. Bioorganic Med. Chem. Lett. 2016, 26, 4837–4841. [Google Scholar] [CrossRef]
- Vassilev, L.; Vu, B.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In Vivo Activation of the p53 Pathway by Small-Molecule Antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grasberger, B.L.; Lu, T.; Schubert, C.; Parks, D.J.; Carver, T.E.; Koblish, H.K.; Cummings, M.D.; Lafrance, L.V.; Milkiewicz, K.L.; Calvo, R.R.; et al. Discovery and Cocrystal Structure of Benzodiazepinedione HDM2 Antagonists That Activate p53 in Cells. J. Med. Chem. 2005, 48, 909–912. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.E.M.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2009, 31, 671–690. [Google Scholar] [CrossRef] [Green Version]
- Vanommeslaeghe, K.; Raman, E.P.; MacKerell, A.D. Automation of the CHARMM General Force Field (CGenFF) II: Assignment of Bonded Parameters and Partial Atomic Charges. J. Chem. Inf. Model. 2012, 52, 3155–3168. [Google Scholar] [CrossRef] [Green Version]
- Beutler, T.C.; Mark, A.E.; van Schaik, R.C.; Gerber, P.R.; van Gunsteren, W.F. Avoiding singularities and numerical instabilities in free energy calculations based on molecular simulations. Chem. Phys. Lett. 1994, 222, 529–539. [Google Scholar] [CrossRef]
- Goga, N.; Rzepiela, A.J.; de Vries, A.H.; Marrink, S.-J.; Berendsen, H.J. Efficient Algorithms for Langevin and DPD Dynamics. J. Chem. Theory Comput. 2012, 8, 3637–3649. [Google Scholar] [CrossRef] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.; Fraaije, J.G. LINCS: A linear constraint solver for molecular simulations. J. Comput.l Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Bennett, C.H. Efficient estimation of free energy differences from Monte Carlo data. J. Comput. Phys. 1976, 22, 245–268. [Google Scholar] [CrossRef]
- De Ruiter, A.; Boresch, S.; Oostenbrink, C. Comparison of thermodynamic integration and Bennett acceptance ratio for calculating relative protein-ligand binding free energies. J. Comput. Chem. 2013, 34, 1024–1034. [Google Scholar] [CrossRef] [PubMed]
- Klimovich, P.V.; Shirts, M.R.; Mobley, D.L. Guidelines for the analysis of free energy calculations. J. Comput. Mol. Des. 2015, 29, 397–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhrínová, S.; Uhrin, D.; Powers, H.; Watt, K.; Zheleva, D.; Fischer, P.M.; McInnes, C.; Barlow, P.N. Structure of Free MDM2 N-terminal Domain Reveals Conformational Adjustments that Accompany p53-binding. J. Mol. Biol. 2005, 350, 587–598. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S.no. | PDBID/Ligand | ΔGcalculated (kcal/mol) | ΔGexperimental (kcal/mol) | IC50 (µM) | Ref. |
|---|---|---|---|---|---|
| 1 | 3LBJ | −6.56 ± 0.387 | −6.76 | 11.0 | [21] |
| 2 | WK23 | −5.90 ± 0.341 | −6.05 | 36.0 | [21] |
| 3 | 2N14 | −5.95 ± 0.38 | −7.3 ± 0.04 | 7.0 | [35] |
| 4 | 2N0U | −10.48 ± 0.45 | −8.6 ± 0.2 | 8.6 | [35] |
| 5 | 2N06 | −7.20 ± 0.38 | −6.7 ± 0.2 | 24.9 | [35] |
| S. No. | Complex/PDBID | IC50/Ki (nM) | ΔGcalculated a (Kcal/mol) | ΔGcorrection (Kcal/mol) | ΔGcorrected b (Kcal/mol) | ∆Gexperimental (Kcal/mol) | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | 6GGN | 0.08 | −17.05 ± 0.33 | 1.04 ± 0.07 | −16.01 ± 0.4 | −13.76 c | [36] |
| 2 | 5OC8 | 0.21 | −14.91 ± 0.78 | 1.15 ± 0.08 | −13.76 ± 0.86 | −13.18 d | [37] |
| 3 | 4ZYF | 1.3 | −12.89 ± 0.39 | 1.34 ± 0.29 | −11.55 ± 0.68 | −12.11 d | [38] |
| 4 | 5LAZ | 4 | −12.57 ± 0.35 | 1.0 ± 0.03 | −11.57 ± 0.38 | −11.44 c | [39] |
| 5 | 5LAY | 34 | −12.04 ± 0.29 | 0.98 ± 0.1 | −11.06 ± 0.39 | −10.19 c | [39] |
| 6 | 5LAW | 80 | −11.06 ± 0.36 | 1.00 ± 0.03 | −10.06 ± 0.39 | −9.68 c | [39] |
| 7 | WK298 | 109 | −10.885 ± 0.33 | 1.15 ± 0.08 | −9.73 ± 0.41 | −9.49 d | [21] |
| 8 | 3TJ2 | 300 | −11.64 ± 0.39 | 1.028 ± 0.09 | −10.64 ± 0.48 | −8.89 d | [40] |
| 9 | 4MDN | 600 | −9.79 ±0.40 | 1.19 ± 0.18 | −8.6 ± 0.58 | −8.47 d | [41] |
| 10 | 5LAV | 819 | −15.80 ± 0.25 | 1.15 ± 0.08 | −14.65 ± 0.33 | −8.29 c | [39] |
| 11 | 4MDQ | 900 | −12.52 ± 0.42 | 1.66 ± 0.18 | −10.86 ± 0.6 | −8.24 d | [41] |
| 12 | 3LBK | 916 | −10.49 ± 0.28 | 1.12 ± 0.03 | −9.37 ± 0.31 | −8.22 d | [21] |
| 13 | 4JV7 | 1000 | −14.56 ± 0.29 | 1.42 ± 0.18 | −13.14 ± 0.47 | −8.63 c | [42] |
| 14 | 4JV9 | 1800 | −15.73 ± 0.29 | 1.62 ± 0.27 | −14.11 ± 0.56 | −8.27 c | [42] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, N.; Li, W. Absolute Binding Free Energy Calculations for Highly Flexible Protein MDM2 and Its Inhibitors. Int. J. Mol. Sci. 2020, 21, 4765. https://doi.org/10.3390/ijms21134765
Singh N, Li W. Absolute Binding Free Energy Calculations for Highly Flexible Protein MDM2 and Its Inhibitors. International Journal of Molecular Sciences. 2020; 21(13):4765. https://doi.org/10.3390/ijms21134765
Chicago/Turabian StyleSingh, Nidhi, and Wenjin Li. 2020. "Absolute Binding Free Energy Calculations for Highly Flexible Protein MDM2 and Its Inhibitors" International Journal of Molecular Sciences 21, no. 13: 4765. https://doi.org/10.3390/ijms21134765