Mice Lacking Connective Tissue Growth Factor in the Forebrain Exhibit Delayed Seizure Response, Reduced C-Fos Expression and Different Microglial Phenotype Following Acute PTZ Injection

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Expression of Ctgf in the Mouse Brain
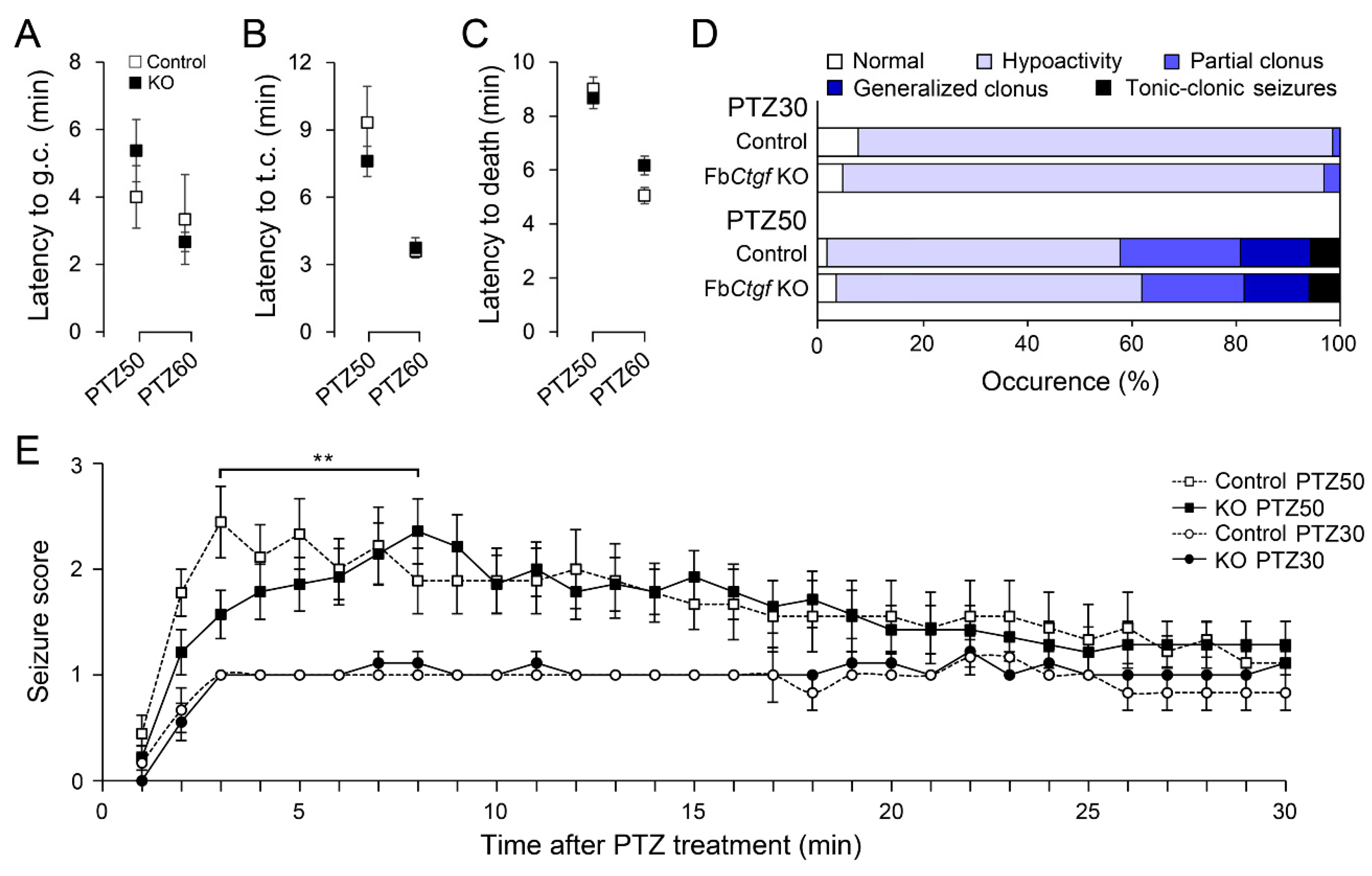
2.2. PTZ-Induced Seizures in Mice
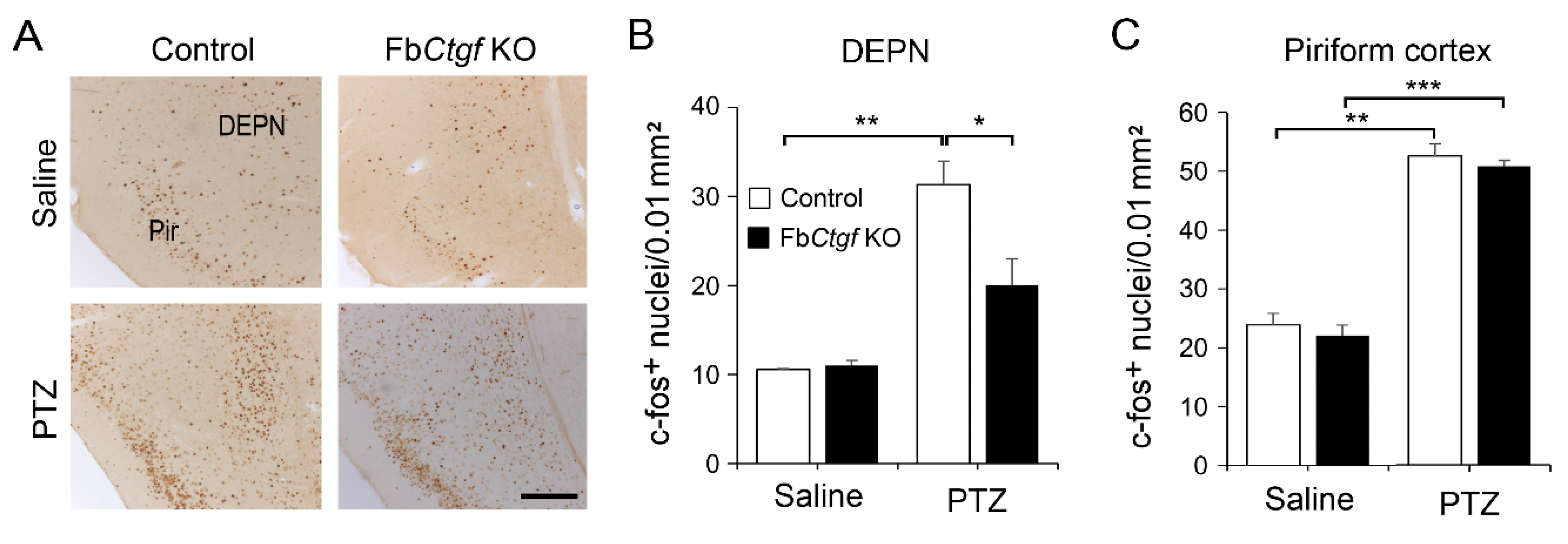
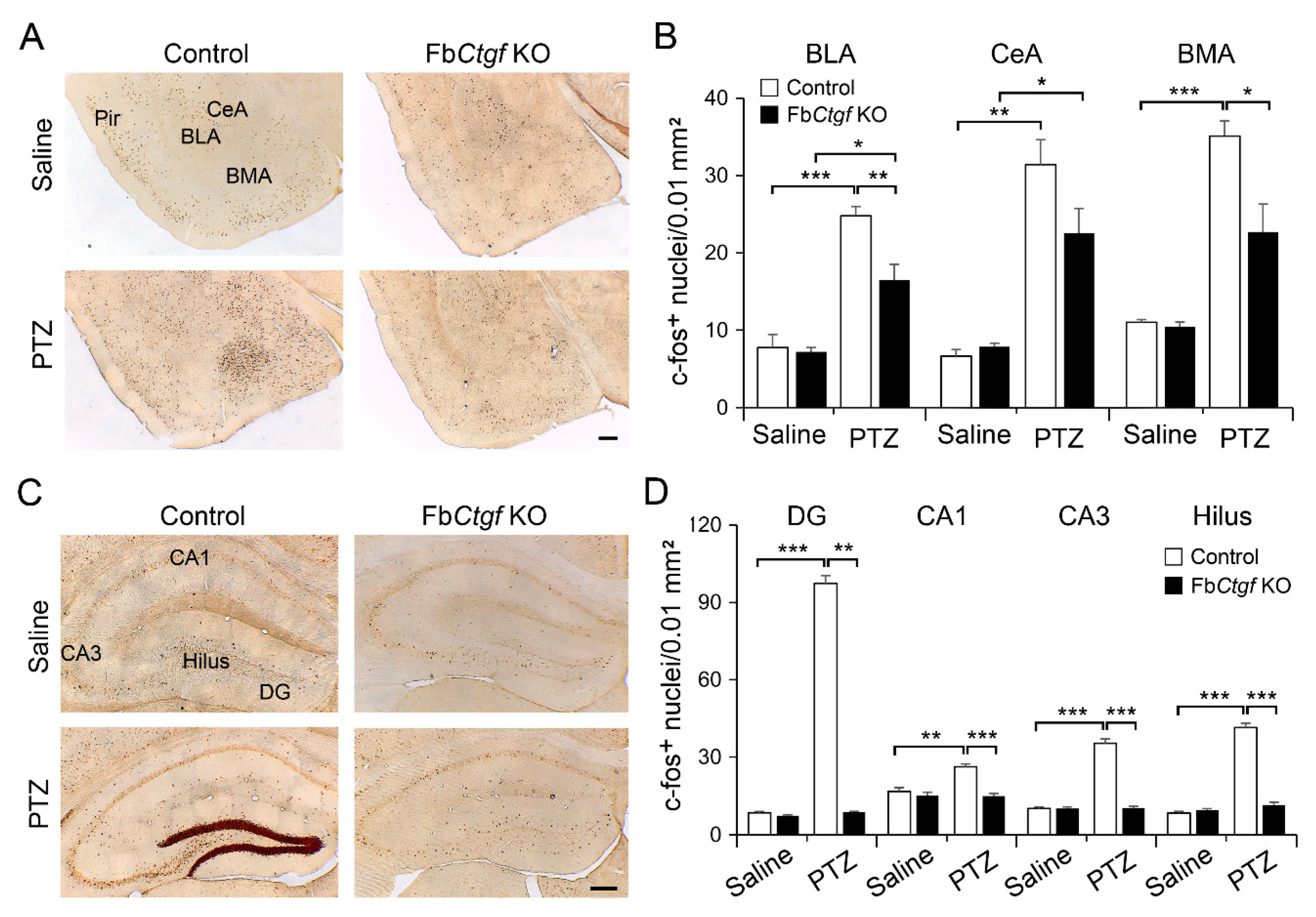
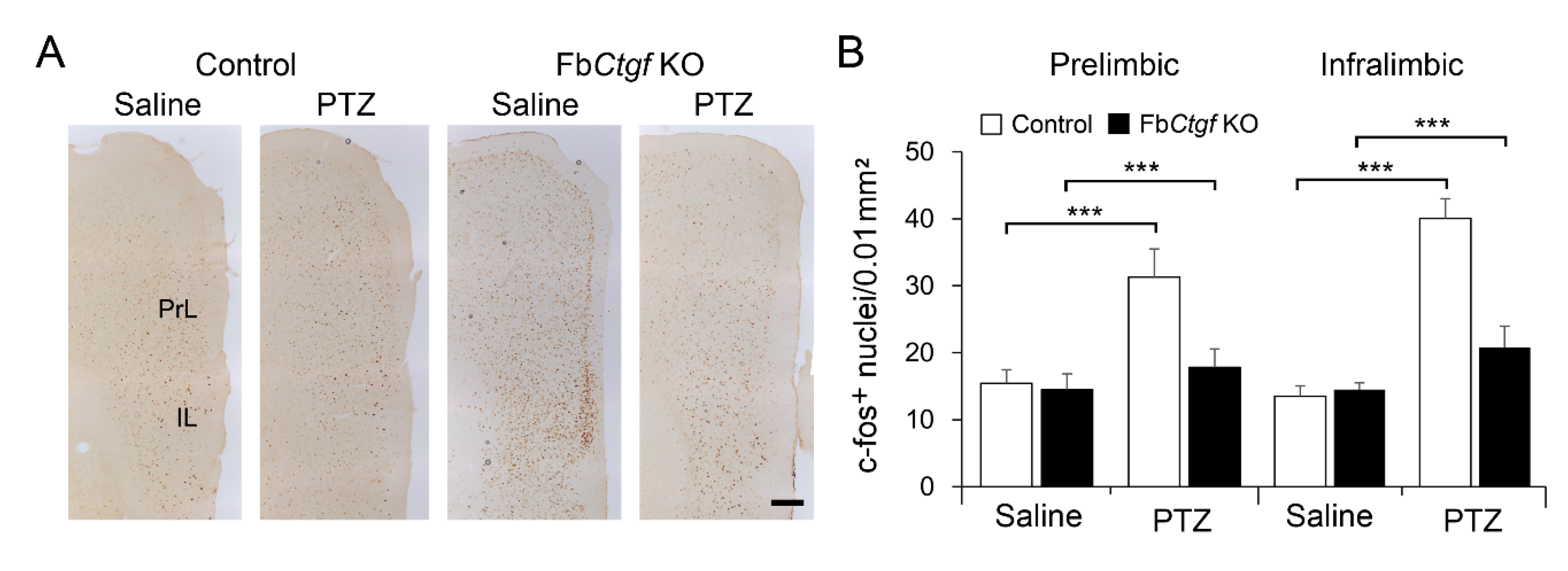
2.3. Expression of c-Fos in the Brain after Acute PTZ Treatment
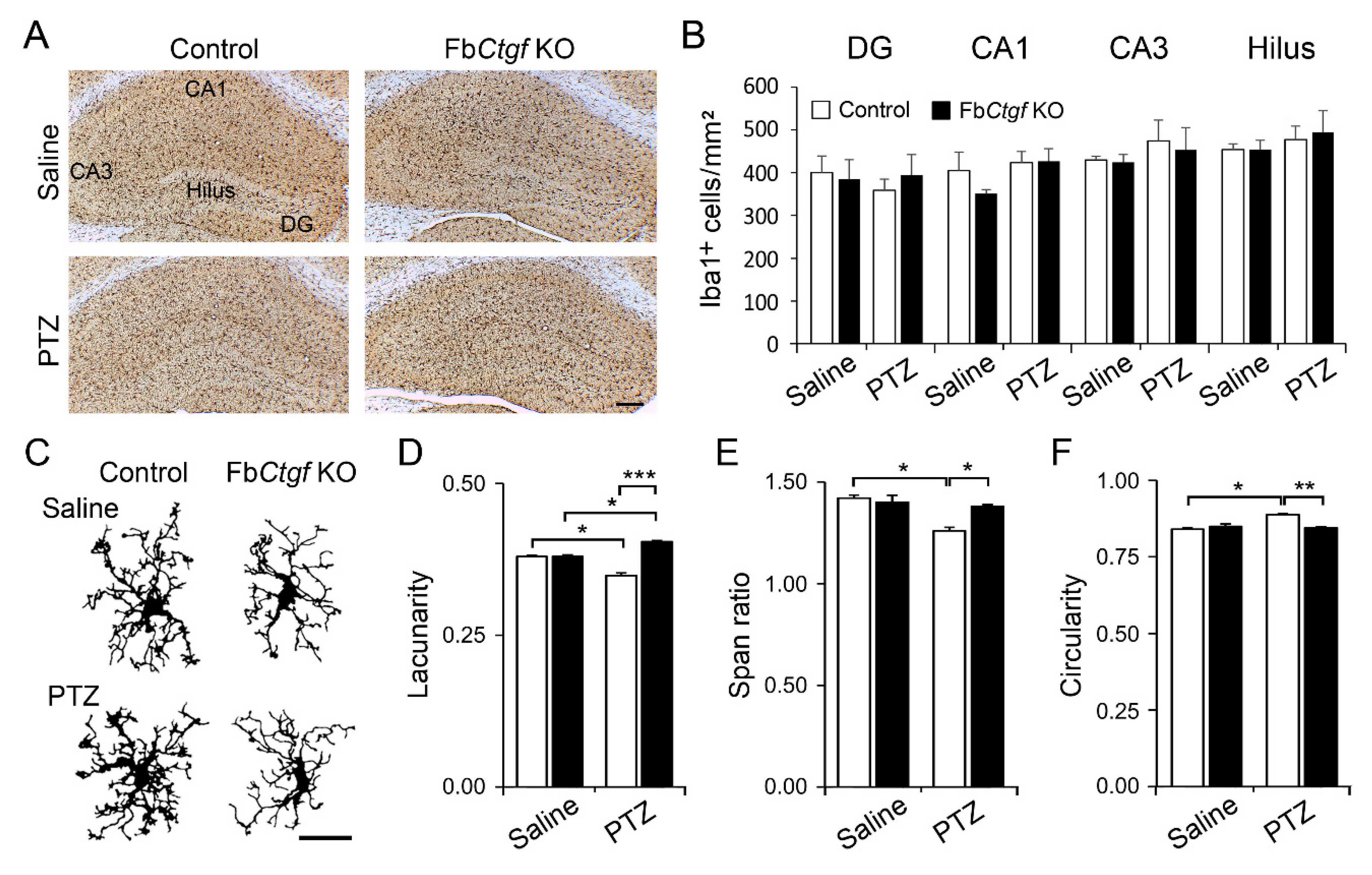
2.4. Glial Reactions in the Hippocampus after Acute PTZ Treatment
3. Discussion
3.1. CTGF Involves in Various Fundamental Biological Process in the Living Organisms
3.2. Beyond Gliosis: The Emerging Role of CTGF in Regulating Brain Function
3.3. The Role of DEPN-Derived CTGF in the Regulation of PTZ-Induced Seizure Reactions
3.4. Glial Reaction and Epilepsy
3.5. Inhibition of CTGF May Have an Antiepileptic Potential
4. Materials and Methods
4.1. Animals
4.2. Pentylenetetrazole (PTZ)-Induced Seizure Model
4.3. Immunohistochemistry
4.4. Cell Density Quantification
4.5. Reconstruction and Morphometric Analysis of Microglia
4.6. Statistical Analyses
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ALS | Amyotrophic lateral sclerosis |
| CCN2 | Cyr61/CTGF/NOV family protein 2 |
| CTGF | Connective tissue growth factor |
| DG | dentate gyrus |
| DMD | Duchene muscular dystrophy |
| ECM | extracellular matrix |
| DEPN | dorsal endopiriform nucleus |
| FbCtgf KO | forebrain-specific Ctgf knockout |
| GFAP | glial fibrillary acidic protein |
| Iba1 | ionized calcium-binding adapter molecule 1 |
| PTZ | pentylenetetrazole |
References
- Stafstrom, C.E.; Carmant, L. Seizures and epilepsy: An overview for neuroscientists. Cold Spring Harb Perspect. Med. 2015, 5, a022426. [Google Scholar] [CrossRef]
- Moshé, S.L.; Perucca, E.; Ryvlin, P.; Tomson, T. Epilepsy: New advances. Lancet 2015, 385, 884–898. [Google Scholar] [CrossRef]
- Kandratavicius, L.; Balista, P.A.; Lopes-Aguiar, C.; Ruggiero, R.N.; Umeoka, E.H.; Garcia-Cairasco, N.; Bueno-Junior, L.S.; Leite, J.P. Animal models of epilepsy: Use and limitations. Neuropsychiatr. Dis. Treat. 2014, 10, 1693–1705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Löscher, W. The Search for New Screening models of pharmacoresistant epilepsy: Is induction of acute seizures in epileptic rodents a suitable approach? Neurochem. Res. 2017, 42, 1926–1938. [Google Scholar] [CrossRef] [PubMed]
- Van Erum, J.; Van Dam, D.; De Deyn, P.P. PTZ-induced seizures in mice require a revised Racine scale. Epilepsy Behav. 2019, 95, 51–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perbal, B. The concept of the CCN protein family revisited: A centralized coordination network. J. Cell Commun. Signal. 2018, 12, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Canalis, E.; Zanotti, S.; Beamer, W.G.; Economides, A.N.; Smerdel-Ramoya, A. Connective tissue growth factor is required for skeletal development and postnatal skeletal homeostasis in male mice. Endocrinology 2010, 151, 3490–3501. [Google Scholar] [CrossRef] [Green Version]
- Hall-Glenn, F.; De Young, R.A.; Huang, B.L.; van Handel, B.; Hofmann, J.J.; Chen, T.T.; Choi, A.; Ong, J.R.; Benya, P.D.; Mikkola, H.; et al. CCN2/connective tissue growth factor is essential for pericyte adhesion and endothelial basement membrane formation during angiogenesis. PLoS ONE 2012, 7, e30562. [Google Scholar] [CrossRef] [Green Version]
- Heuer, H.; Christ, S.; Friedrichsen, S.; Brauer, D.; Winckler, M.; Bauer, K.; Raivich, G. Connective tissue growth factor: A novel marker of layer VII neurons in the rat cerebral cortex. Neuroscience 2003, 119, 43–52. [Google Scholar] [CrossRef]
- Piredda, S.; Gale, K. A crucial epileptogenic site in the deep prepiriform cortex. Nature 1985, 317, 623–625. [Google Scholar] [CrossRef]
- Tseng, G.F.; Haberly, L.B. Deep neurons in piriform cortex. I. Morphology and synaptically evoked responses including a unique high-amplitude paired shock facilitation. J. Neurophysiol. 1989, 62, 369–385. [Google Scholar] [CrossRef] [PubMed]
- Demir, R.; Haberly, L.B.; Jackson, M.B. Sustained and accelerating activity at two discrete sites generate epileptiform discharges in slices of piriform cortex. J. Neurosci. 1999, 19, 1294–1306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brevi, S.; de Curtis, M.; Magistretti, J. Pharmacological and biophysical characterization of voltage-gated calcium currents in the endopiriform nucleus of the guinea pig. J. Neurophysiol. 2001, 85, 2076–2087. [Google Scholar] [CrossRef]
- Majak, K.; Moryś, J. Endopiriform nucleus connectivities: The implications for epileptogenesis and epilepsy. Folia Morphol. (Warsz). 2007, 66, 267–271. [Google Scholar] [PubMed]
- Vismer, M.S.; Forcelli, P.A.; Skopin, M.D.; Gale, K.; Koubeissi, M.Z. The piriform, perirhinal, and entorhinal cortex in seizure generation. Front. Neural Circuits 2015, 9, 27. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, W.H.; Haberly, L.B. Role of synaptic excitation in the generation of bursting-induced epileptiform potentials in the endopiriform nucleus and piriform cortex. J. Neurophysiol. 1993, 70, 2550–2561. [Google Scholar] [CrossRef] [PubMed]
- Yu, I.S.; Chang, H.C.; Chen, K.C.; Lu, Y.L.; Shy, H.T.; Chen, C.Y.; Lee, K.Y.; Lee, L.J. Genetic elimination of connective tissue growth factor in the forebrain affects subplate neurons in the cortex and oligodendrocytes in the underlying white matter. Front. Neuroanat. 2019, 13, 16. [Google Scholar] [CrossRef]
- Siracusa, R.; Fusco, R.; Cuzzocrea, S. Astrocytes: Role and functions in brain pathologies. Front. Pharmacol. 2019, 10, 1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szepesi, Z.; Manouchehrian, O.; Bachiller, S.; Deierborg, T. Bidirectional microglia-neuron communication in health and disease. Front. Cell Neurosci. 2018, 12, 323. [Google Scholar] [CrossRef]
- Andoh, M.; Ikegaya, Y.; Koyama, R. Synaptic pruning by microglia in epilepsy. J. Clin. Med. 2019, 8, 2170. [Google Scholar] [CrossRef] [Green Version]
- Nikolic, L.; Nobili, P.; Shen, W.; Audinat, E. Role of astrocyte purinergic signaling in epilepsy. Glia 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Hadera, M.G.; Eloqayli, H.; Jaradat, S.; Nehlig, A.; Sonnewald, U. Astrocyte-neuronal interactions in epileptogenesis. J. Neurosci. Res. 2015, 93, 1157–1164. [Google Scholar] [CrossRef]
- Zhang, B.; Zou, J.; Han, L.; Beeler, B.; Friedman, J.L.; Griffin, E.; Piao, Y.S.; Rensing, N.R.; Wong, M. The specificity and role of microglia in epileptogenesis in mouse models of tuberous sclerosis complex. Epilepsia 2018, 59, 1796–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Liao, Y.; Morgan, S.; Mathur, R.; Feustel, P.; Mazurkiewicz, J.; Qian, J.; Chang, J.; Mathern, G.W.; Adamo, M.A.; et al. Noninflammatory changes of microglia are sufficient to cause epilepsy. Cell Rep. 2018, 22, 2080–2093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, D.C.; Tewari, B.P.; Chaunsali, L.; Sontheimer, H. Neuron-glia interactions in the pathophysiology of epilepsy. Nat. Rev. Neurosci. 2019, 20, 282–297. [Google Scholar] [CrossRef] [PubMed]
- Binder, D.K.; Oshio, K.; Ma, T.; Verkman, A.S.; Manley, G.T. Increased seizure threshold in mice lacking aquaporin-4 water channels. Neuroreport 2004, 15, 259–262. [Google Scholar] [CrossRef] [PubMed]
- Lüttjohann, A.; Fabene, P.F.; van Luijtelaar, G. A revised Racine’s scale for PTZ-induced seizures in rats. Physiol. Behav. 2009, 98, 579–586. [Google Scholar] [CrossRef]
- Schallier, A.; Massie, A.; Loyens, E.; Moechars, D.; Drinkenburg, W.; Michotte, Y.; Smolders, I. vGLUT2 heterozygous mice show more susceptibility to clonic seizures induced by pentylenetetrazol. Neurochem. Int. 2009, 55, 41–44. [Google Scholar] [CrossRef]
- Samokhina, E.; Samokhin, A. Neuropathological profile of the pentylenetetrazol (PTZ) kindling model. Int. J. Neurosci. 2018, 128, 1086–1096. [Google Scholar] [CrossRef]
- Shapiro, L.; Wong, J.C.; Escayg, A. Reduced cannabinoid 2 receptor activity increases susceptibility to induced seizures in mice. Epilepsia 2019, 60, 2359–2369. [Google Scholar] [CrossRef]
- Tilelli, C.Q.; Flôres, L.R.; Cota, V.R.; Castro, O.W.; Garcia-Cairasco, N. Amygdaloid complex anatomopathological findings in animal models of status epilepticus. Epilepsy Behav. 2019, 106831. [Google Scholar] [CrossRef] [PubMed]
- Watson, G.D.R.; Smith, J.B.; Alloway, K.D. Interhemispheric connections between the infralimbic and entorhinal cortices: The endopiriform nucleus has limbic connections that parallel the sensory and motor connections of the claustrum. J. Comp. Neurol. 2017, 525, 1363–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Arjona, M.D.M.; Grondona, J.M.; Granados-Durán, P.; Fernández-Llebrez, P.; López-Ávalos, M.D. Microglia morphological categorization in a rat model of neuroinflammation by hierarchical cluster and principal components analysis. Front. Cell Neurosci. 2017, 11, 235. [Google Scholar] [CrossRef]
- Karperien, A.; Jelinek, H.; Milosevic, N.T. Reviewing lacunarity analysis and classification of microglia in neuroscience. In Proceedings of the 8th European Conference on Mathematical and Theoretical Biology; 2011. [Google Scholar]
- Perbal, B. CCN proteins: Multifunctional signalling regulators. Lancet 2004, 363, 62–64. [Google Scholar] [CrossRef]
- Ungvari, Z.; Valcarcel-Ares, M.N.; Tarantini, S.; Yabluchanskiy, A.; Fülöp, G.A.; Kiss, T.; Csiszar, A. Connective tissue growth factor (CTGF) in age-related vascular pathologies. Geroscience 2017, 39, 491–498. [Google Scholar] [CrossRef]
- Lipson, K.E.; Wong, C.; Teng, Y.; Spong, S. CTGF is a central mediator of tissue remodeling and fibrosis and its inhibition can reverse the process of fibrosis. Fibrogenesis Tissue Repair. 2012, 5 (Suppl. 1), S24. [Google Scholar] [CrossRef] [Green Version]
- Richeldi, L.; Fernández Pérez, E.R.; Costabel, U.; Albera, C.; Lederer, D.J.; Flaherty, K.R.; Ettinger, N.; Perez, R.; Scholand, M.B.; Goldin, J.; et al. Pamrevlumab, an anti-connective tissue growth factor therapy, for idiopathic pulmonary fibrosis (PRAISE): A phase 2, randomised, double-blind, placebo-controlled trial. Lancet. Respir. Med. 2020, 8, 25–33. [Google Scholar] [CrossRef]
- Kuiper, E.J.; Van Nieuwenhoven, F.A.; de Smet, M.D.; van Meurs, J.C.; Tanck, M.W.; Oliver, N.; Klaassen, I.; Van Noorden, C.J.; Goldschmeding, R.; Schlingemann, R.O. The angio-fibrotic switch of VEGF and CTGF in proliferative diabetic retinopathy. PLoS ONE 2008, 3, e2675. [Google Scholar] [CrossRef]
- Ivkovic, S.; Yoon, B.S.; Popoff, S.N.; Safadi, F.F.; Libuda, D.E.; Stephenson, R.C.; Daluiski, A.; Lyons, K.M. Connective tissue growth factor coordinates chondrogenesis and angiogenesis during skeletal development. Development 2003, 130, 2779–2791. [Google Scholar] [CrossRef] [Green Version]
- Stritt, C.; Stern, S.; Harting, K.; Manke, T.; Sinske, D.; Schwarz, H.; Vingron, M.; Nordheim, A.; Knöll, B. Paracrine control of oligodendrocyte differentiation by SRF-directed neuronal gene expression. Nat. Neurosci. 2009, 12, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Khodosevich, K.; Lazarini, F.; von Engelhardt, J.; Kaneko, H.; Lledo, P.M.; Monyer, H. Connective tissue growth factor regulates interneuron survival and information processing in the olfactory bulb. Neuron 2013, 79, 1136–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, A.R.; Liszewska, E.; Jaworski, J. Matricellular proteins of the Cyr61/CTGF/NOV (CCN) family and the nervous system. Front. Cell Neurosci. 2015, 9, 237. [Google Scholar] [CrossRef] [Green Version]
- Ercan, E.; Han, J.M.; Di Nardo, A.; Winden, K.; Han, M.J.; Hoyo, L.; Saffari, A.; Leask, A.; Geschwind, D.H.; Sahin, M. Neuronal CTGF/CCN2 negatively regulates myelination in a mouse model of tuberous sclerosis complex. J. Exp. Med. 2017, 214, 681–697. [Google Scholar] [CrossRef] [Green Version]
- Turner, C.A.; Sharma, V.; Hagenauer, M.H.; Chaudhury, S.; O’Connor, A.M.; Hebda-Bauer, E.K.; Thompson, R.C.; Myers, R.M.; Bunney, W.E.; Barchas, J.D.; et al. Connective tissue growth factor is a novel prodepressant. Biol. Psychiatry 2018, 84, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Conrad, S.; Schluesener, H.J.; Adibzahdeh, M.; Schwab, J.M. Spinal cord injury induction of lesional expression of profibrotic and angiogenic connective tissue growth factor confined to reactive astrocytes, invading fibroblasts and endothelial cells. J. Neurosurg. Spine 2005, 2, 319–326. [Google Scholar] [CrossRef]
- Spliet, W.G.; Aronica, E.; Ramkema, M.; Aten, J.; Troost, D. Increased expression of connective tissue growth factor in amyotrophic lateral sclerosis human spinal cord. Acta Neuropathol. 2003, 106, 449–457. [Google Scholar] [CrossRef]
- Gonzalez, D.; Brandan, E. CTGF/CCN2 from skeletal muscle to nervous system: Impact on neurodegenerative diseases. Mol. Neurobiol. 2019, 56, 5911–5916. [Google Scholar] [CrossRef]
- Pan, L.H.; Beppu, T.; Kurose, A.; Yamauchi, K.; Sugawara, A.; Suzuki, M.; Ogawa, A.; Sawai, T. Neoplastic cells and proliferating endothelial cells express connective tissue growth factor (CTGF) in glioblastoma. Neurol. Res. 2002, 24, 677–683. [Google Scholar] [CrossRef]
- Ueberham, U.; Ueberham, E.; Gruschka, H.; Arendt, T. Connective tissue growth factor in Alzheimer’s disease. Neuroscience 2003, 116, 1–6. [Google Scholar] [CrossRef]
- Goldberg, E.M.; Coulter, D.A. Mechanisms of epileptogenesis: A convergence on neural circuit dysfunction. Nat. Rev. Neurosci. 2013, 14, 337–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eyo, U.B.; Murugan, M.; Wu, L.J. Microglia-neuron communication in epilepsy. Glia 2017, 65, 5–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorski, J.A.; Talley, T.; Qiu, M.; Puelles, L.; Rubenstein, J.L.; Jones, K.R. Cortical excitatory neurons and glia, but not GABAergic neurons, are produced in the Emx1-expressing lineage. J. Neurosci. 2002, 22, 6309–6314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferraro, T.N.; Golden, G.T.; Smith, G.G.; Jean, P.S.; Schork, N.J.; Mulholland, N.; Ballas, C.; Schill, J.; Buono, R.J.; Berrettini, W.H. Mapping loci for pentylenetetrazol-induced seizure susceptibility in mice. J. Neurosci. 1999, 19, 6733–6739. [Google Scholar] [CrossRef] [Green Version]
- Young, K.; Morrison, H. Quantifying Microglia morphology from photomicrographs of immunohistochemistry prepared tissue using ImageJ. J. Vis. Exp. 2018, 57648. [Google Scholar] [CrossRef] [PubMed] [Green Version]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siow, P.-F.; Tsao, C.-Y.; Chang, H.-C.; Chen, C.-Y.; Yu, I.-S.; Lee, K.-Y.; Lee, L.-J. Mice Lacking Connective Tissue Growth Factor in the Forebrain Exhibit Delayed Seizure Response, Reduced C-Fos Expression and Different Microglial Phenotype Following Acute PTZ Injection. Int. J. Mol. Sci. 2020, 21, 4921. https://doi.org/10.3390/ijms21144921
Siow P-F, Tsao C-Y, Chang H-C, Chen C-Y, Yu I-S, Lee K-Y, Lee L-J. Mice Lacking Connective Tissue Growth Factor in the Forebrain Exhibit Delayed Seizure Response, Reduced C-Fos Expression and Different Microglial Phenotype Following Acute PTZ Injection. International Journal of Molecular Sciences. 2020; 21(14):4921. https://doi.org/10.3390/ijms21144921
Chicago/Turabian StyleSiow, Pei-Fen, Chih-Yu Tsao, Ho-Ching Chang, Chwen-Yu Chen, I-Shing Yu, Kuang-Yung Lee, and Li-Jen Lee. 2020. "Mice Lacking Connective Tissue Growth Factor in the Forebrain Exhibit Delayed Seizure Response, Reduced C-Fos Expression and Different Microglial Phenotype Following Acute PTZ Injection" International Journal of Molecular Sciences 21, no. 14: 4921. https://doi.org/10.3390/ijms21144921