LY75 Suppression in Mesenchymal Epithelial Ovarian Cancer Cells Generates a Stable Hybrid EOC Cellular Phenotype, Associated with Enhanced Tumor Initiation, Spreading and Resistance to Treatment in Orthotopic Xenograft Mouse Model

 , , , and
, , , and
Abstract
1. Introduction
2. Results
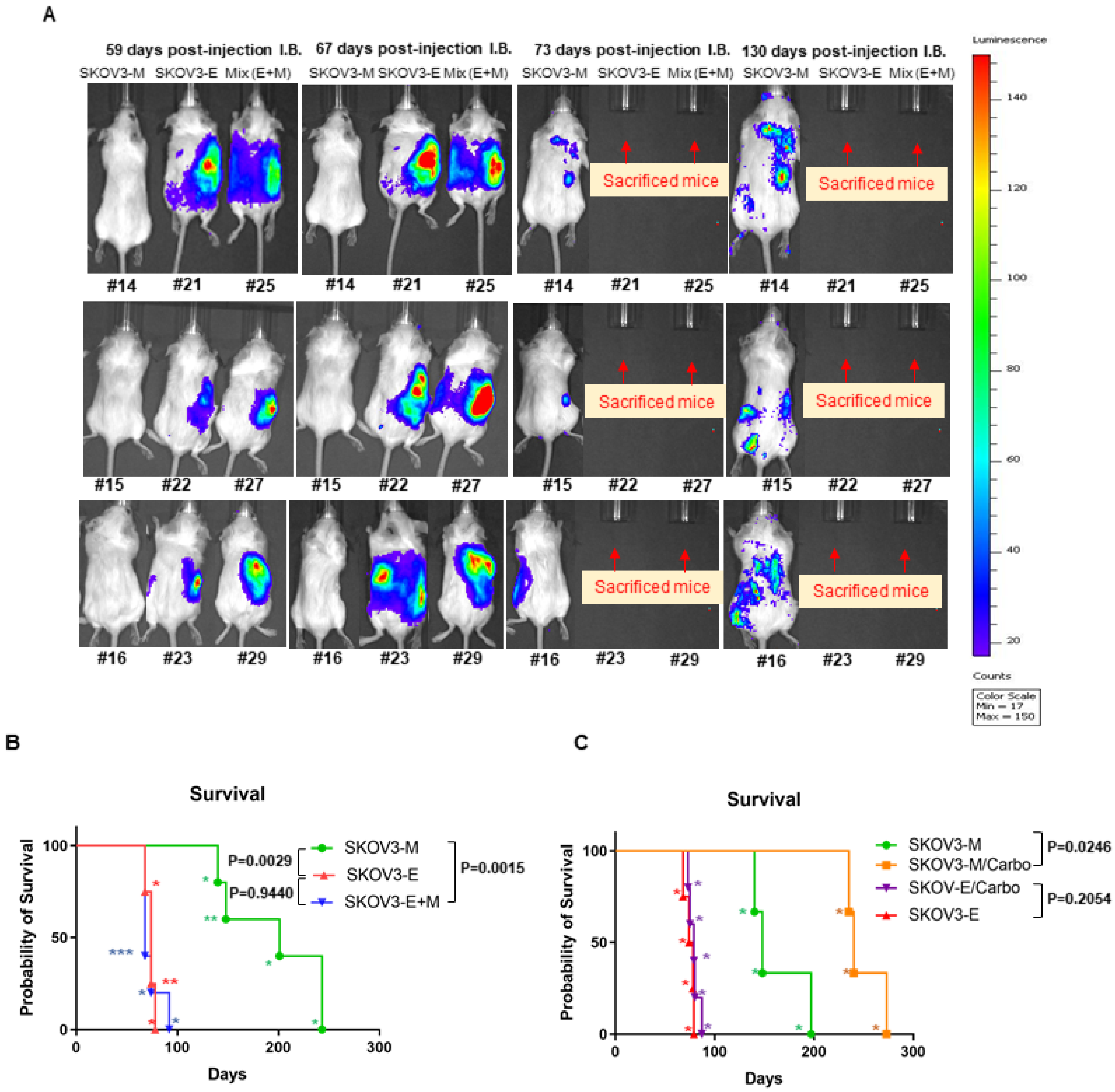
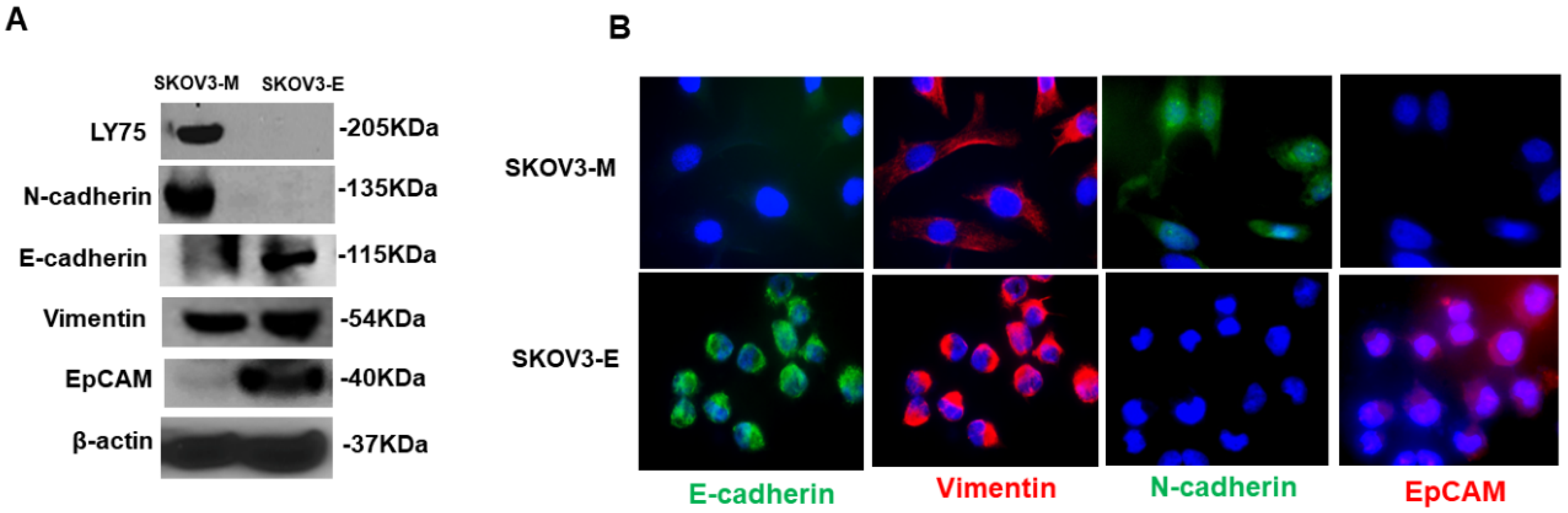
2.1. Ly75KD SKOV3 Cells with Epithelial Phenotype (SKOV3-E) Display Enhanced EOC Initiation, Spread, and Resistance to Treatment in Severe Combined Immunodeficiency (SCID) Mice
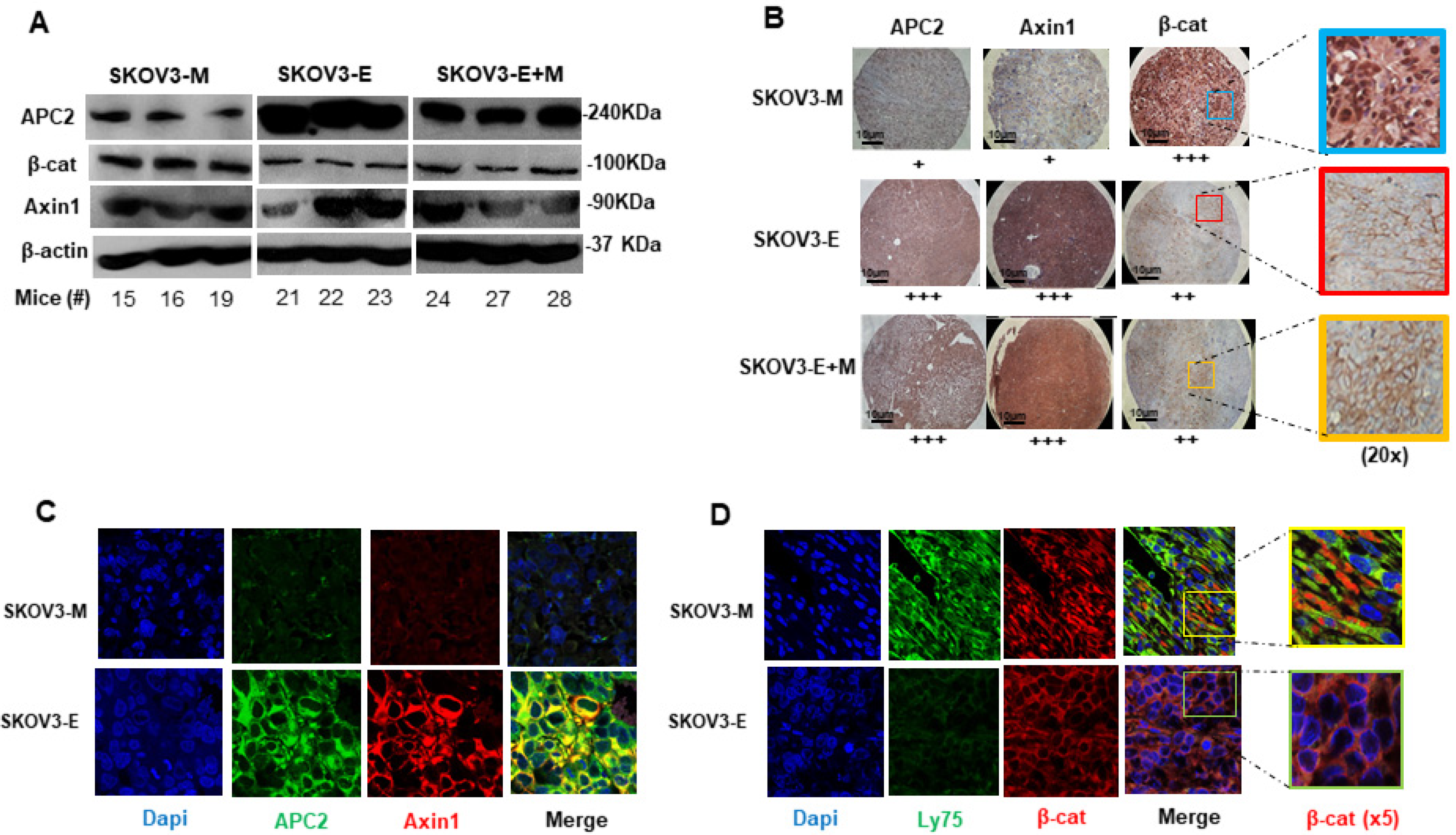
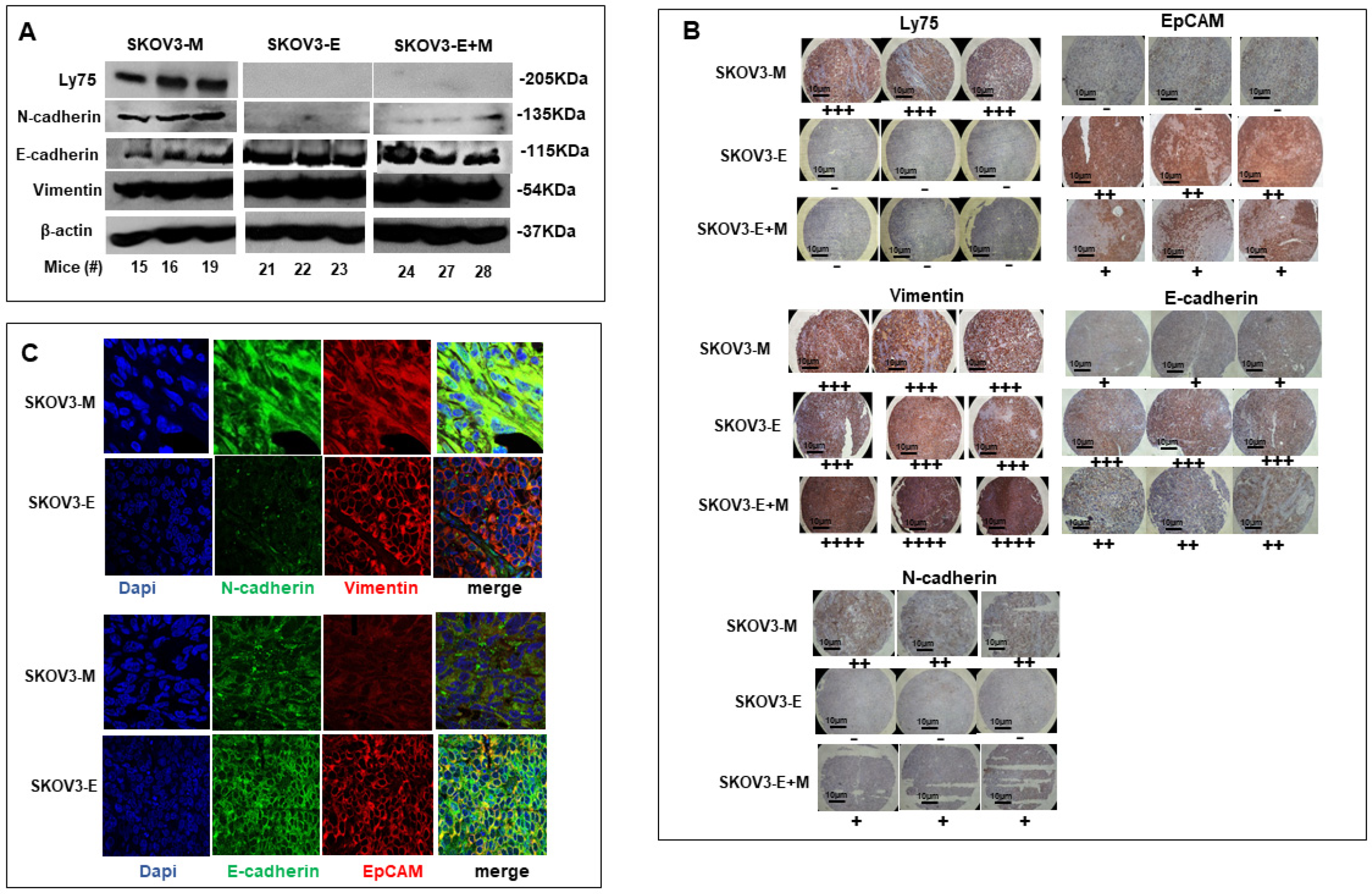
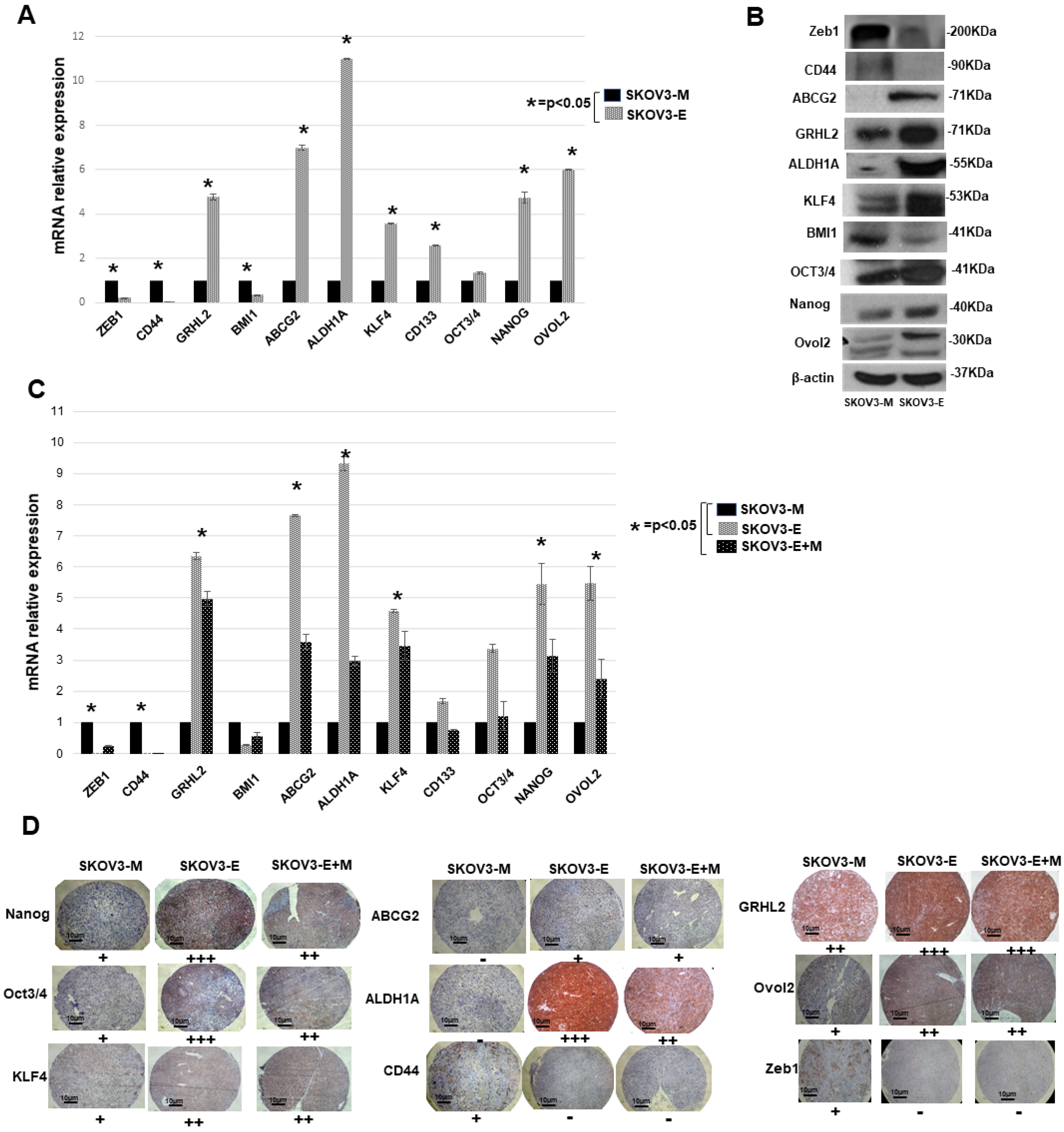
2.2. SKOV3-E Cells Display Hybrid (E/M) Phenotype and Cancer Stem Cells (CSCs) Features
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. Firefly Luciferase Stable Transfection
4.3. Animal Experiments
4.3.1. Study Design
4.3.2. Intra-Bursal Injections
4.3.3. In Vivo Bioluminescence Imaging
4.3.4. Carboplatin Treatment
4.3.5. Data Analysis
4.4. Statistical Analysis
4.5. Immunohistochemistry (IHC)
4.6. Immunofluorescence (IF)
4.7. Western Blot
4.8. Quantitative PCR (qPCR)
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| EOC | Epithelial ovarian cancer |
| EMT | Epithelial mesenchymal transition |
| MET | Mesenchymal epithelial transition |
| EMP | Epithelial mesenchymal plasticity |
| SCID mice | Severe combined immunodeficient mice |
| IB injection | Intra-bursal injection |
| CSCs | Cancer stem cells |
| IHC | Immunohistochemistry |
| IF | Immunofluorescence |
| qPCR | Quantitative PCR |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | q-PCR Primers | Sequence (5′-3′) |
|---|---|---|
| ZEB1 (NM_001128128.3) | Forward | CATTTCAGGGAAGCCTGGGT |
| Reverse | CACCCTGTTAGGCAGTGAGG | |
| Product lengths (pb) | 158 | |
| NANOG (NM_024865.4) | Forward | ATATTGCATGCCTCCTGGGG |
| Reverse | GTGCCGGAAGCTTTTGTCTG | |
| Product lengths (pb) | 104 | |
| POU5F1(Oct4) (NM_001285986.1) | Forward | AGATGCTTTGAGCTCCCTCTG |
| Reverse | TTGGCTGAATACCTTCCCTGG | |
| Product lengths (pb) | 124 | |
| CD44 (NM_001001391.2) | Forward | TCCCTGCTACCAGAGACCAA |
| Reverse | GCTCCACCTTCTTGACTCCC | |
| Product lengths (pb) | 106 | |
| CD133 (Prom1) (NM_001371406.1) | Forward | CCCCGCAGGAGTGAATCTTT |
| Reverse | GAAGGACTCGTTGCTGGTGA | |
| Product lengths (pb) | 137 | |
| ABCG2 (NM_004827.3) | Forward | GAGTTAACTGAGCTGGGGCA |
| Reverse | TGACAGAATGTCAGGGCACC | |
| Product lengths (pb) | 98 | |
| BMI1 (NM_005180.9) | Forward | TGCTTTGGTCGAACTTGGTG |
| Reverse | TGCAGACTGGGGACAATGAA | |
| Product lengths (pb) | 129 | |
| KLF4 (NM_001314052.2) | Forward | CCAAAAATGCGACCGAGCAT |
| Reverse | TGTAGTGCTTTCTGGCTGGG | |
| Product lengths (pb) | 174 | |
| ALDH1A1 (NM_000689.5) | Forward | TTGGACCAGTGCAGCAAATC |
| Reverse | CGCCATAGCAATTCACCCAC | |
| Product lengths (pb) | 171 |
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. Ca A Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.L.; Monk, B.J.; Sood, A.K.; Herzog, T.J. Latest research and treatment of advanced-stage epithelial ovarian cancer. Nat. Rev. Clin. Oncol. 2013, 10, 211–224. [Google Scholar] [CrossRef]
- Yao, D.; Dai, C.; Peng, S. Mechanism of the mesenchymal-epithelial transition and its relationship with metastatic tumor formation. Mol. Cancer. Res. 2011, 9, 1608–1620. [Google Scholar] [CrossRef] [PubMed]
- Suhail, Y.; Cain, M.P.; Vanaja, K.; Kurywchak, P.A.; Levchenko, A.; Kalluri, R. Systems Biology of Cancer Metastasis. Cell Syst. 2019, 9, 109–127. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer. 2002, 2, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Nagafuchi, A.; Shirayoshi, Y.; Okazaki, K.; Yasuda, K.; Takeichi, M. Transformation of cell adhesion properties by exogenously introduced E-cadherin cDNA. Nature 1987, 329, 341–343. [Google Scholar] [CrossRef]
- Perl, A.K.; Wilgenbus, P.; Dahl, U.; Semb, H.; Christofori, G. A causal role for E-cadherin in the transition from adenoma to carcinoma. Nature 1998, 392, 190–193. [Google Scholar] [CrossRef]
- Hojo, N.; Huisken, A.; Wang, H.; Chirshev, E.; Kim, N.; Nguyen, S.; Campos, H.; Glackin, C.; Ioffe, Y.; Unternaehrer, J. Snail knockdown reverses stemness and inhibits tumour growth in ovarian cancer. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef]
- Brabletz, T.; Jung, A.; Spaderna, S.; Hlubek, F.; Kirchner, T. Migrating cancer stem cells—an integrated concept of malignant tumour progression. Nat. Rev. Cancer 2005, 5, 744–749. [Google Scholar] [CrossRef]
- Wu, Z.-Q.; Li, X.-Y.; Hu, C.Y.; Ford, M.; Kleer, C.G.; Weiss, S.J. Canonical Wnt signaling regulates Slug activity and links epithelial–mesenchymal transition with epigenetic Breast Cancer 1, Early Onset (BRCA1) repression. Proc. Natl. Acad. Sci. USA 2012, 109, 16654–16659. [Google Scholar] [CrossRef]
- Yook, J.I.; Li, X.-Y.; Ota, I.; Fearon, E.R.; Weiss, S.J. Wnt-dependent regulation of the E-cadherin repressor snail. J. Biol. Chem. 2005, 280, 11740–11748. [Google Scholar] [CrossRef] [PubMed]
- Abbott, S.E.; Camacho, F.; Peres, L.C.; Alberg, A.J.; Bandera, E.V.; Bondy, M.; Cote, M.L.; Funkhouser, E.; Moorman, P.G.; Peters, E.S.; et al. Recreational physical activity and survival in African-American women with ovarian cancer. Cancer Causes Control 2018, 29, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Wang, L.; Hou, H.; Zhou, J.; Li, X. Epigenetic regulation of IQGAP2 promotes ovarian cancer progression via activating Wnt/β-catenin signaling. Int. J. Oncol. 2016, 48, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Bernaudo, S.; Salem, M.; Qi, X.; Zhou, W.; Zhang, C.; Yang, W.; Rosman, D.; Deng, Z.; Ye, G.; Yang, B. Cyclin G2 inhibits epithelial-to-mesenchymal transition by disrupting Wnt/β-catenin signaling. Oncogene 2016, 35, 4816–4827. [Google Scholar] [CrossRef] [PubMed]
- Elloul, S.; Vaksman, O.; Stavnes, H.T.; Trope, C.G.; Davidson, B.; Reich, R. Mesenchymal-to-epithelial transition determinants as characteristics of ovarian carcinoma effusions. Clin. Exp. Metastasis 2010, 27, 161–172. [Google Scholar] [CrossRef]
- Wells, A.; Yates, C.; Shepard, C.R. E-cadherin as an indicator of mesenchymal to epithelial reverting transitions during the metastatic seeding of disseminated carcinomas. Clin. Exp. Metastasis 2008, 25, 621–628. [Google Scholar] [CrossRef]
- Jolly, M.K.; Boareto, M.; Huang, B.; Jia, D.; Lu, M.; Ben-Jacob, E.; Onuchic, J.N.; Levine, H. Implications of the Hybrid Epithelial/Mesenchymal Phenotype in Metastasis. Front. Oncol. 2015, 5, 155. [Google Scholar] [CrossRef]
- Grosse-Wilde, A.; Fouquier d’Herouel, A.; McIntosh, E.; Ertaylan, G.; Skupin, A.; Kuestner, R.E.; del Sol, A.; Walters, K.A.; Huang, S. Stemness of the hybrid Epithelial/Mesenchymal State in Breast Cancer and Its Association with Poor Survival. PLoS ONE 2015, 10, e0126522. [Google Scholar] [CrossRef]
- Andriani, F.; Bertolini, G.; Facchinetti, F.; Baldoli, E.; Moro, M.; Casalini, P.; Caserini, R.; Milione, M.; Leone, G.; Pelosi, G.; et al. Conversion to stem-cell state in response to microenvironmental cues is regulated by balance between epithelial and mesenchymal features in lung cancer cells. Mol. Oncol. 2016, 10, 253–271. [Google Scholar] [CrossRef] [PubMed]
- Strauss, R.; Li, Z.Y.; Liu, Y.; Beyer, I.; Persson, J.; Sova, P.; Moller, T.; Pesonen, S.; Hemminki, A.; Hamerlik, P.; et al. Analysis of epithelial and mesenchymal markers in ovarian cancer reveals phenotypic heterogeneity and plasticity. PLoS ONE 2011, 6, e16186. [Google Scholar] [CrossRef]
- Klymenko, Y.; Johnson, J.; Bos, B.; Lombard, R.; Campbell, L.; Loughran, E.; Stack, M.S. Heterogeneous Cadherin Expression and Multicellular Aggregate Dynamics in Ovarian Cancer Dissemination. Neoplasia 2017, 19, 549–563. [Google Scholar] [CrossRef] [PubMed]
- Francart, M.E.; Lambert, J.; Vanwynsberghe, A.M.; Thompson, E.W.; Bourcy, M.; Polette, M.; Gilles, C. Epithelial-mesenchymal plasticity and circulating tumor cells: Travel companions to metastases. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2018, 247, 432–450. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.; Monkman, J.; Toh, A.K.L.; Nagaraj, S.H.; Thompson, E.W. Targeting epithelial-mesenchymal plasticity in cancer: clinical and preclinical advances in therapy and monitoring. Biochem. J. 2017, 474, 3269–3306. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef]
- Faddaoui, A.; Bachvarova, M.; Plante, M.; Gregoire, J.; Renaud, M.C.; Sebastianelli, A.; Gobeil, S.; Morin, C.; Macdonald, E.; Vanderhyden, B.; et al. The mannose receptor LY75 (DEC205/CD205) modulates cellular phenotype and metastatic potential of ovarian cancer cells. Oncotarget 2016, 7, 14125–14142. [Google Scholar] [CrossRef]
- Mehdi, S.; Bachvarova, M.; Scott-Boyer, M.-P.; Droit, A.; Bachvarov, D. LY75 Ablation Mediates Mesenchymal-Epithelial Transition (MET) in Epithelial Ovarian Cancer (EOC) Cells Associated with DNA Methylation Alterations and Suppression of the Wnt/β-Catenin Pathway. Int. J. Mol. Sci. 2020, 21, 1848. [Google Scholar] [CrossRef]
- Killion, J.J.; Radinsky, R.; Fidler, I.J. Orthotopic models are necessary to predict therapy of transplantable tumors in mice. Cancer Metastasis Rev. 1998, 17, 279–284. [Google Scholar] [CrossRef]
- Kuo, T.H.; Kubota, T.; Watanabe, M.; Furukawa, T.; Kase, S.; Tanino, H.; Saikawa, Y.; Ishibiki, K.; Kitajima, M.; Hoffman, R.M. Site-specific chemosensitivity of human small-cell lung carcinoma growing orthotopically compared to subcutaneously in SCID mice: the importance of orthotopic models to obtain relevant drug evaluation data. Anticancer Res. 1993, 13, 627–630. [Google Scholar]
- Lu, W.; Kang, Y. Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Dev. Cell 2019, 49, 361–374. [Google Scholar] [CrossRef]
- Van Staalduinen, J.; Baker, D.; Ten Dijke, P.; van Dam, H. Epithelial-mesenchymal-transition-inducing transcription factors: new targets for tackling chemoresistance in cancer? Oncogene 2018, 37, 6195–6211. [Google Scholar] [CrossRef] [PubMed]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef] [PubMed]
- Garg, M. Epithelial, mesenchymal and hybrid epithelial/mesenchymal phenotypes and their clinical relevance in cancer metastasis. Expert Rev. Mol. Med. 2017, 19, e3. [Google Scholar] [CrossRef] [PubMed]
- Klymenko, Y.; Kim, O.; Loughran, E.; Yang, J.; Lombard, R.; Alber, M.; Stack, M.S. Cadherin composition and multicellular aggregate invasion in organotypic models of epithelial ovarian cancer intraperitoneal metastasis. Oncogene 2017, 36, 5840–5851. [Google Scholar] [CrossRef] [PubMed]
- Loret, N.; Denys, H.; Tummers, P.; Berx, G. The Role of Epithelial-to-Mesenchymal Plasticity in Ovarian Cancer Progression and Therapy Resistance. Cancers 2019, 11, 838. [Google Scholar] [CrossRef]
- Garson, K.; Shaw, T.J.; Clark, K.V.; Yao, D.S.; Vanderhyden, B.C. Models of ovarian cancer--are we there yet? Mol. Cell. Endocrinol. 2005, 239, 15–26. [Google Scholar] [CrossRef]
- Ribatti, D.; Tamma, R.; Annese, T. Epithelial-Mesenchymal Transition in Cancer: A Historical Overview. Transl. Oncol. 2020, 13, 100773. [Google Scholar] [CrossRef]
- Jolly, M.K.; Tripathi, S.C.; Jia, D.; Mooney, S.M.; Celiktas, M.; Hanash, S.M.; Mani, S.A.; Pienta, K.J.; Ben-Jacob, E.; Levine, H. Stability of the hybrid epithelial/mesenchymal phenotype. Oncotarget 2016, 7, 27067–27084. [Google Scholar] [CrossRef]
- Jordan, N.V.; Johnson, G.L.; Abell, A.N. Tracking the intermediate stages of epithelial-mesenchymal transition in epithelial stem cells and cancer. Cell Cycle 2011, 10, 2865–2873. [Google Scholar] [CrossRef]
- Pastushenko, I.; Brisebarre, A.; Sifrim, A.; Fioramonti, M.; Revenco, T.; Boumahdi, S.; Van Keymeulen, A.; Brown, D.; Moers, V.; Lemaire, S. Identification of the tumour transition states occurring during EMT. Nature 2018, 556, 463–468. [Google Scholar] [CrossRef]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef]
- Satelli, A.; Li, S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell. Mol. Life Sci. Cmls 2011, 68, 3033–3046. [Google Scholar] [CrossRef]
- Hudson, L.G.; Zeineldin, R.; Stack, M.S. Phenotypic plasticity of neoplastic ovarian epithelium: unique cadherin profiles in tumor progression. Clin. Exp. Metastasis 2008, 25, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Kenda Suster, N.; Smrkolj, S.; Virant-Klun, I. Putative stem cells and epithelial-mesenchymal transition revealed in sections of ovarian tumor in patients with serous ovarian carcinoma using immunohistochemistry for vimentin and pluripotency-related markers. J. Ovarian. Res. 2017, 10, 11. [Google Scholar] [CrossRef] [PubMed]
- Tsikouras, P.; Tsagias, N.; Pinidis, P.; Csorba, R.; Vrachnis, N.; Dafopoulos, A.; Bouchlariotou, S.; Liberis, A.; Teichmann, A.T.; von Tempelhoff, G.F. The contribution of catumaxomab in the treatment of malignant ascites in patients with ovarian cancer: a review of the literature. Arch. Gynecol. Obs. 2013, 288, 581–585. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, T.; Zhu, J.; Zhang, H.; Jiang, H.; Sun, H. High ALDH activity defines ovarian cancer stem-like cells with enhanced invasiveness and EMT progress which are responsible for tumor invasion. Biochem. Biophys. Res. Commun. 2018, 495, 1081–1088. [Google Scholar] [CrossRef]
- Li, X.; Zou, Z.; Tang, J.; Zheng, Y.; Liu, Y.; Luo, Y.; Liu, Q.; Wang, Y. NOS1 upregulates ABCG2 expression contributing to DDP chemoresistance in ovarian cancer cells. Oncol. Lett. 2019, 17, 1595–1602. [Google Scholar] [CrossRef]
- Roy, L.; Bobbs, A.; Sattler, R.; Kurkewich, J.L.; Dausinas, P.B.; Nallathamby, P.; Cowden Dahl, K.D. CD133 Promotes Adhesion to the Ovarian Cancer Metastatic Niche. Cancer Growth Metastasis 2018, 11, 1179064418767882. [Google Scholar] [CrossRef]
- Meng, E.; Mitra, A.; Tripathi, K.; Finan, M.A.; Scalici, J.; McClellan, S.; Madeira da Silva, L.; Reed, E.; Shevde, L.A.; Palle, K.; et al. ALDH1A1 maintains ovarian cancer stem cell-like properties by altered regulation of cell cycle checkpoint and DNA repair network signaling. PLoS ONE 2014, 9, e107142. [Google Scholar] [CrossRef]
- Hu, L.; McArthur, C.; Jaffe, R. Ovarian cancer stem-like side-population cells are tumourigenic and chemoresistant. Br. J. Cancer 2010, 102, 1276–1283. [Google Scholar] [CrossRef]
- Pan, Y.; Jiao, J.; Zhou, C.; Cheng, Q.; Hu, Y.; Chen, H. Nanog is highly expressed in ovarian serous cystadenocarcinoma and correlated with clinical stage and pathological grade. Pathobiology 2010, 77, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Silva, I.A.; Bai, S.; McLean, K.; Yang, K.; Griffith, K.; Thomas, D.; Ginestier, C.; Johnston, C.; Kueck, A.; Reynolds, R.K.; et al. Aldehyde dehydrogenase in combination with CD133 defines angiogenic ovarian cancer stem cells that portend poor patient survival. Cancer Res. 2011, 71, 3991–4001. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, M.; D’Alterio, C.; Camerlingo, R.; Tirino, V.; Consales, C.; Riccio, A.; Ierano, C.; Cecere, S.C.; Losito, N.S.; Greggi, S.; et al. Identification of a distinct population of CD133(+)CXCR4(+) cancer stem cells in ovarian cancer. Sci. Rep. 2015, 5, 10357. [Google Scholar] [CrossRef]
- Faddaoui, A.; Sheta, R.; Bachvarova, M.; Plante, M.; Gregoire, J.; Renaud, M.C.; Sebastianelli, A.; Gobeil, S.; Morin, C.; Ghani, K.; et al. Suppression of the grainyhead transcription factor 2 gene (GRHL2) inhibits the proliferation, migration, invasion and mediates cell cycle arrest of ovarian cancer cells. Cell Cycle 2017, 16, 693–706. [Google Scholar] [CrossRef] [PubMed]
- Hong, T.; Watanabe, K.; Ta, C.H.; Villarreal-Ponce, A.; Nie, Q.; Dai, X. An Ovol2-Zeb1 Mutual Inhibitory Circuit Governs Bidirectional and Multi-step Transition between Epithelial and Mesenchymal States. PLoS Comput. Biol. 2015, 11, e1004569. [Google Scholar] [CrossRef]
- Basu, S.; Cheriyamundath, S.; Ben-Ze’ev, A. Cell-cell adhesion: linking Wnt/beta-catenin signaling with partial EMT and stemness traits in tumorigenesis. F1000Research 2018, 7. [Google Scholar] [CrossRef]
- Uka, R.; Britschgi, C.; Krattli, A.; Matter, C.; Mihic, D.; Okoniewski, M.J.; Gualandi, M.; Stupp, R.; Cinelli, P.; Dummer, R.; et al. Temporal activation of WNT/beta-catenin signaling is sufficient to inhibit SOX10 expression and block melanoma growth. Oncogene 2020, 39, 4132–4154. [Google Scholar] [CrossRef]
- Arend, R.C.; Londoño-Joshi, A.I.; Straughn, J.M.; Buchsbaum, D.J. The Wnt/β-catenin pathway in ovarian cancer: A review. Gynecol. Oncol. 2013, 131, 772–779. [Google Scholar] [CrossRef]
- Nagaraj, A.B.; Joseph, P.; Kovalenko, O.; Singh, S.; Armstrong, A.; Redline, R.; Resnick, K.; Zanotti, K.; Waggoner, S.; DiFeo, A. Critical role of Wnt/β-catenin signaling in driving epithelial ovarian cancer platinum resistance. Oncotarget 2015, 6, 23720. [Google Scholar] [CrossRef]
- Nguyen, V.H.L.; Hough, R.; Bernaudo, S.; Peng, C. Wnt/beta-catenin signalling in ovarian cancer: Insights into its hyperactivation and function in tumorigenesis. J. Ovarian Res. 2019, 12, 122. [Google Scholar] [CrossRef]
- Kuchenbaecker, K.B.; Ramus, S.J.; Tyrer, J.; Lee, A.; Shen, H.C.; Beesley, J.; Lawrenson, K.; McGuffog, L.; Healey, S.; Lee, J.M.; et al. Identification of six new susceptibility loci for invasive epithelial ovarian cancer. Nat. Genet. 2015, 47, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, K.; Berger, H.; Kulbe, H.; Thillainadarasan, S.; Mollenkopf, H.-J.; Zemojtel, T.; Taube, E.; Darb-Esfahani, S.; Mangler, M.; Sehouli, J.; et al. Stable expansion of high-grade serous ovarian cancer organoids requires a low-Wnt environment. Embo J. 2020, 39, e104013. [Google Scholar] [CrossRef] [PubMed]
- Sheta, R.; Bachvarova, M.; Macdonald, E.; Gobeil, S.; Vanderhyden, B.; Bachvarov, D. The polypeptide GALNT6 Displays Redundant Functions upon Suppression of its Closest Homolog GALNT3 in Mediating Aberrant O-Glycosylation, Associated with Ovarian Cancer Progression. Int. J. Mol. Sci. 2019, 20, 2264. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]





| Cells Injected IB | Animal ID (#) | Survival Days | Metastasis Sites |
|---|---|---|---|
| SKOV3-M (shControl) | 14 | 243 | Tumor nodules in gut mesentery Tumor nodules in pancreas/omentum Tumor nodules attached to dorsal wall |
| 15 | 148 | ||
| 16 | 201 | ||
| 17 | 140 | ||
| 18 | 243 | ||
| Median survival | 201 | ||
| SKOV3-E (shSKOV3-LY75-KD) | 20 | 78 | Tumor in gut mesentery Tumor nodules in pancreas/omentun Tumor nodules attached to dorsal wall |
| 21 | 68 | ||
| 22 | 74 | ||
| 23 | 74 | ||
| Median survival | 74 | ||
| SKOV3-M/SKOV3-E (mixed; SKOV3-E+M) | 24 | 92 | Small nodules in mesentery Tumor nodules in pancreas/omentum Invasion of liver Nodules attached to dorsal wall |
| 25 | 68 | ||
| 27 | 68 | ||
| 28 | 68 | ||
| 29 | 74 | ||
| Median survival | 68 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mehdi, S.; Macdonald, E.; Galpin, K.; Landry, D.A.; Rodriguez, G.; Vanderhyden, B.; Bachvarov, D. LY75 Suppression in Mesenchymal Epithelial Ovarian Cancer Cells Generates a Stable Hybrid EOC Cellular Phenotype, Associated with Enhanced Tumor Initiation, Spreading and Resistance to Treatment in Orthotopic Xenograft Mouse Model. Int. J. Mol. Sci. 2020, 21, 4992. https://doi.org/10.3390/ijms21144992
Mehdi S, Macdonald E, Galpin K, Landry DA, Rodriguez G, Vanderhyden B, Bachvarov D. LY75 Suppression in Mesenchymal Epithelial Ovarian Cancer Cells Generates a Stable Hybrid EOC Cellular Phenotype, Associated with Enhanced Tumor Initiation, Spreading and Resistance to Treatment in Orthotopic Xenograft Mouse Model. International Journal of Molecular Sciences. 2020; 21(14):4992. https://doi.org/10.3390/ijms21144992
Chicago/Turabian StyleMehdi, Sadia, Elizabeth Macdonald, Kristianne Galpin, David A. Landry, Galaxia Rodriguez, Barbara Vanderhyden, and Dimcho Bachvarov. 2020. "LY75 Suppression in Mesenchymal Epithelial Ovarian Cancer Cells Generates a Stable Hybrid EOC Cellular Phenotype, Associated with Enhanced Tumor Initiation, Spreading and Resistance to Treatment in Orthotopic Xenograft Mouse Model" International Journal of Molecular Sciences 21, no. 14: 4992. https://doi.org/10.3390/ijms21144992
APA StyleMehdi, S., Macdonald, E., Galpin, K., Landry, D. A., Rodriguez, G., Vanderhyden, B., & Bachvarov, D. (2020). LY75 Suppression in Mesenchymal Epithelial Ovarian Cancer Cells Generates a Stable Hybrid EOC Cellular Phenotype, Associated with Enhanced Tumor Initiation, Spreading and Resistance to Treatment in Orthotopic Xenograft Mouse Model. International Journal of Molecular Sciences, 21(14), 4992. https://doi.org/10.3390/ijms21144992