Exploring Drug Treatment Patterns Based on the Action of Drug and Multilayer Network Model



Abstract
1. Introduction
- For the first time, we proposed a network-based model to analyze the drug treatment patterns.
- The new framework to study the treatment pattern of drugs was based on the action of drug and multilayer network model.
- Taking drug TSA as a case, we found two modules from a tissue-specific multilayer protein-protein network as TSA’s treatment patterns.
- By analyzing the significance, composition, and functions, the two modules were proven to be the potential treatment patterns of TSA.
- Analysis of the treatment patterns of the drug through the network method provides novel solutions for disease treatment.
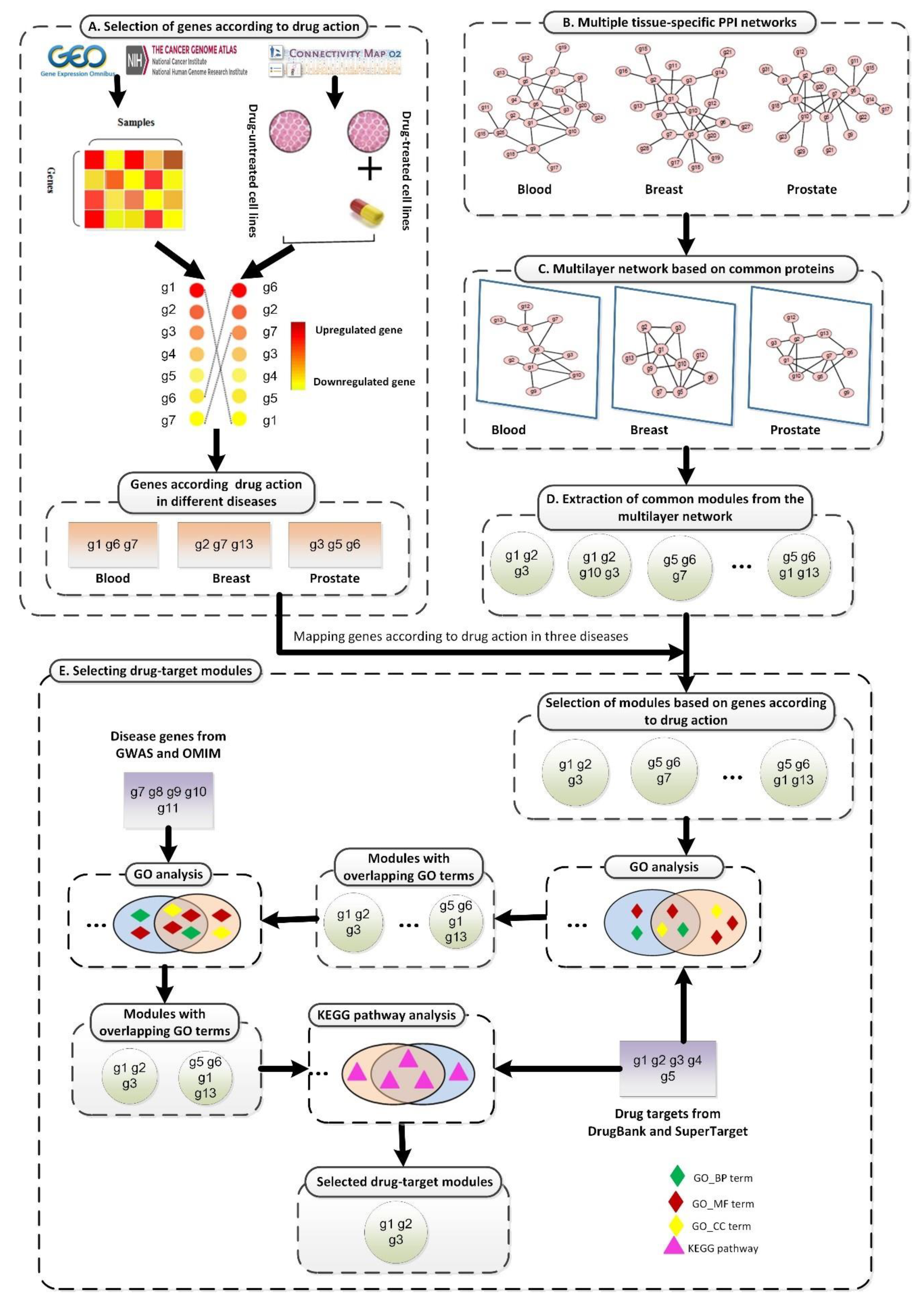
2. Materials and Methods
2.1. Datasets
2.1.1. Gene Expression Data for TSA Activity
2.1.2. Gene Expression Data According to Disease State
2.1.3. TSPPI Networks
2.2. Standardizing Networks
2.3. Selecting Differentially Expressed Genes
2.4. Mining Modules from the Multilayer Network
2.5. Quantifying the Overlap between Modules
3. Results
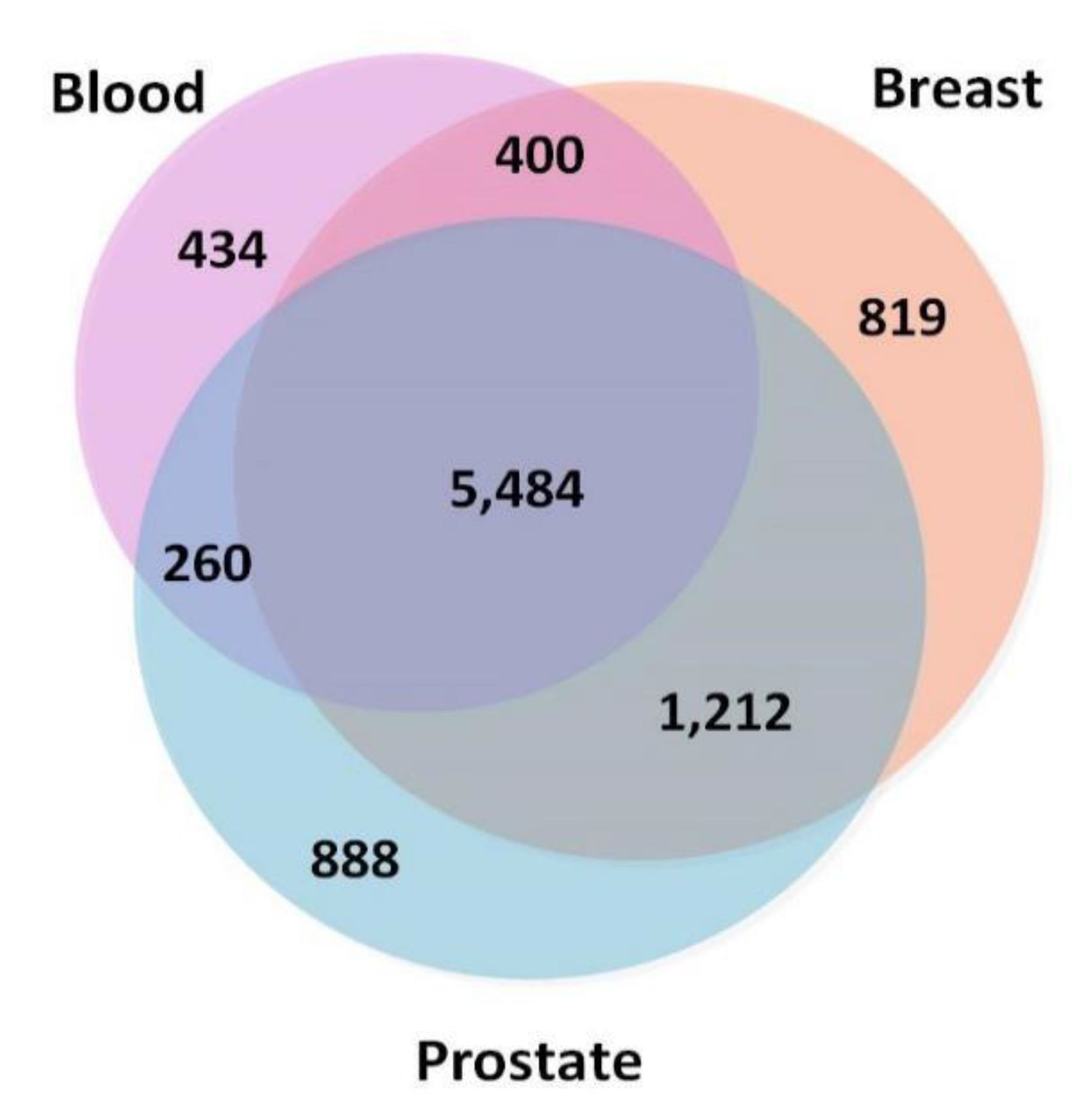
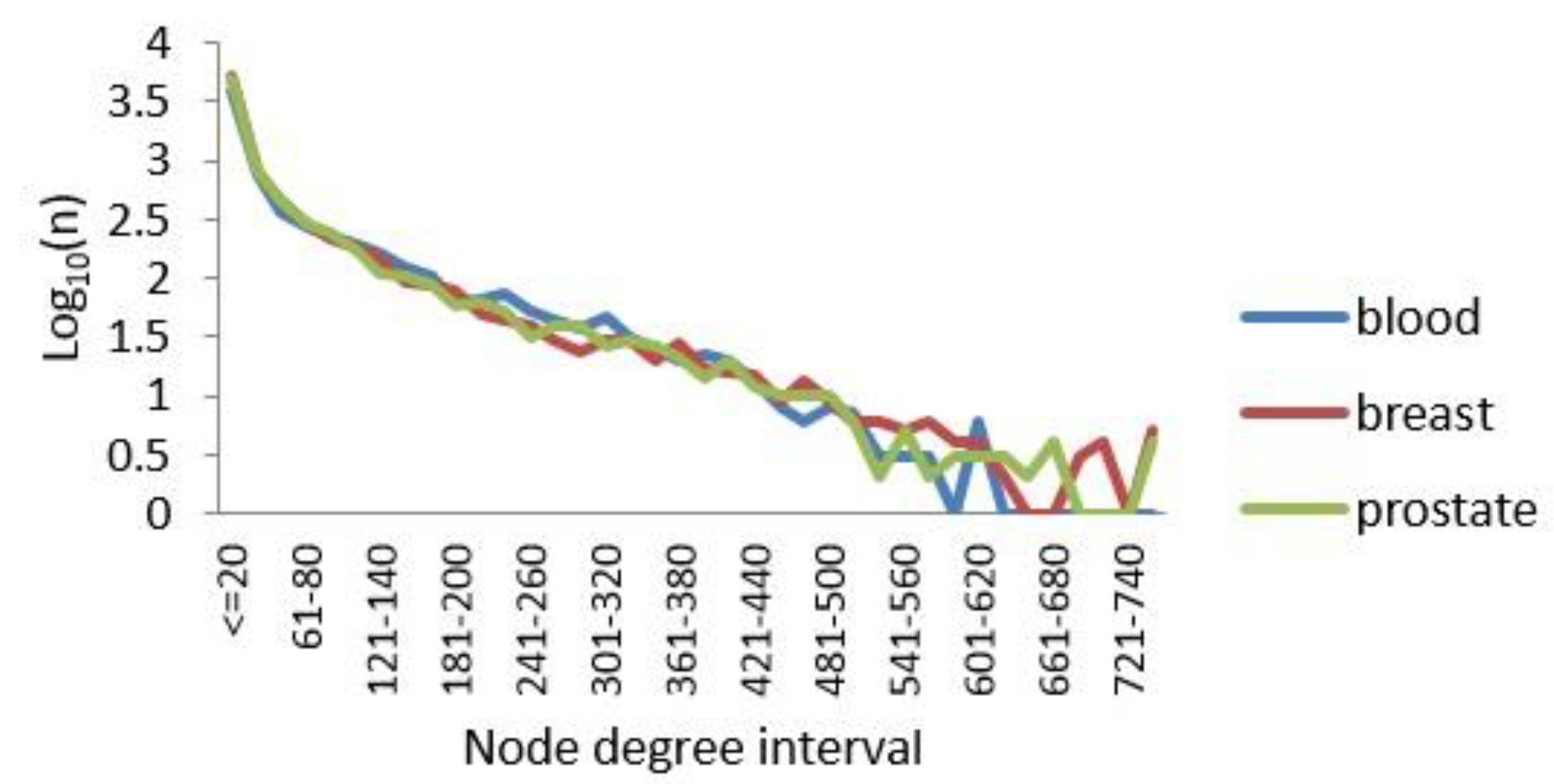
3.1. Constructing Three-Layer Tissue-Specific Networks
3.1.1. Nodes and Edges
3.1.2. Degree Distribution
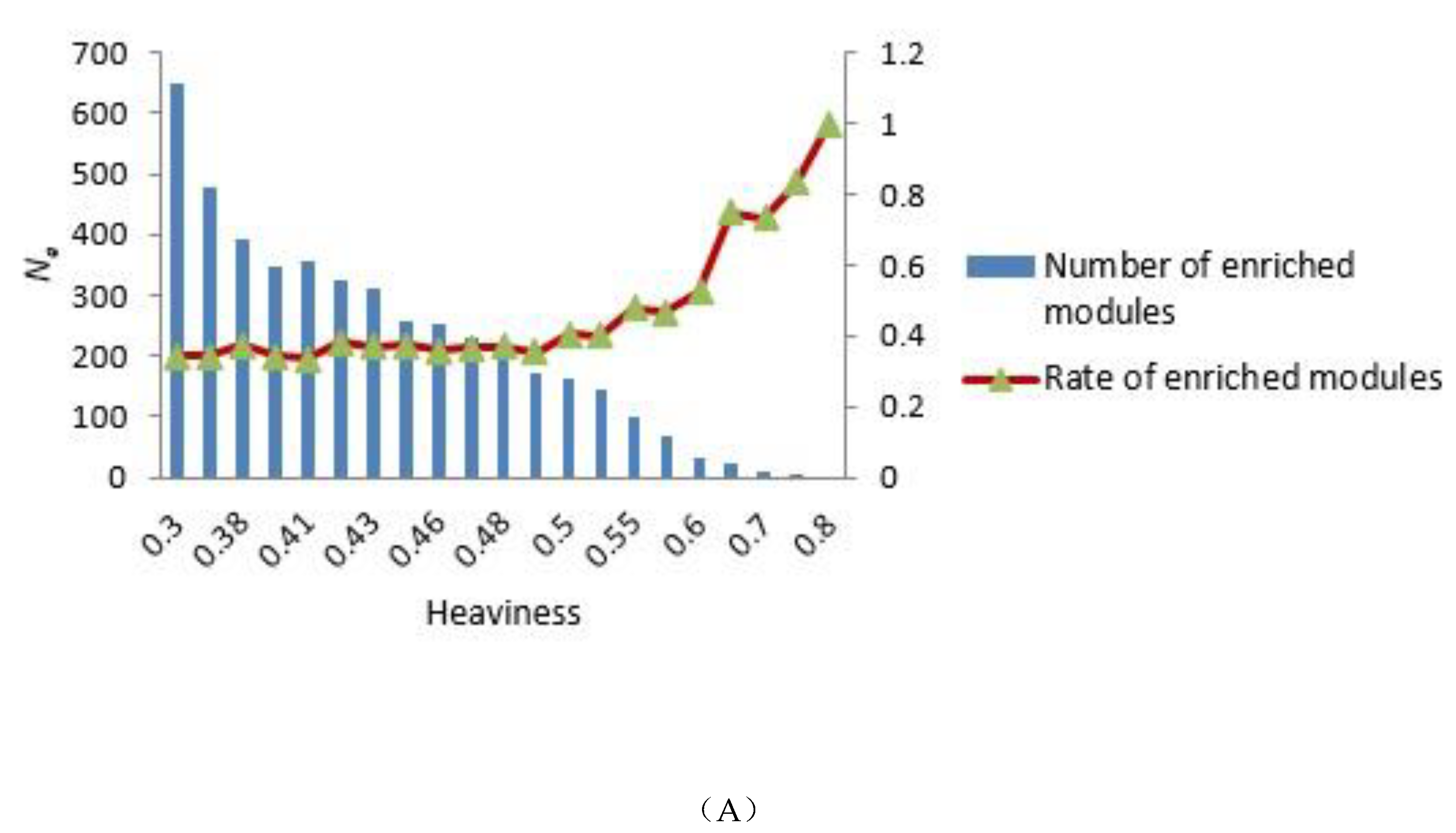
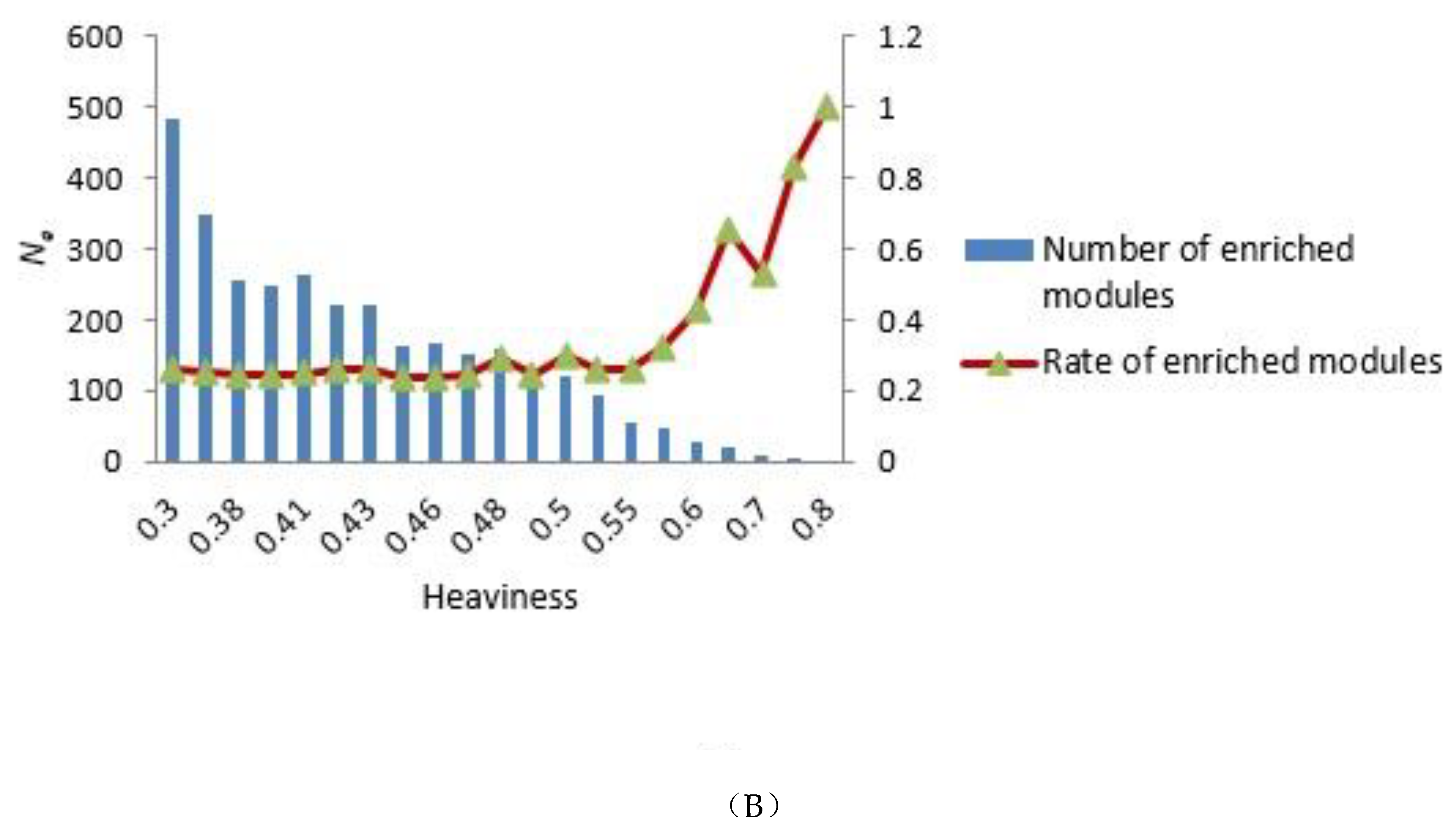
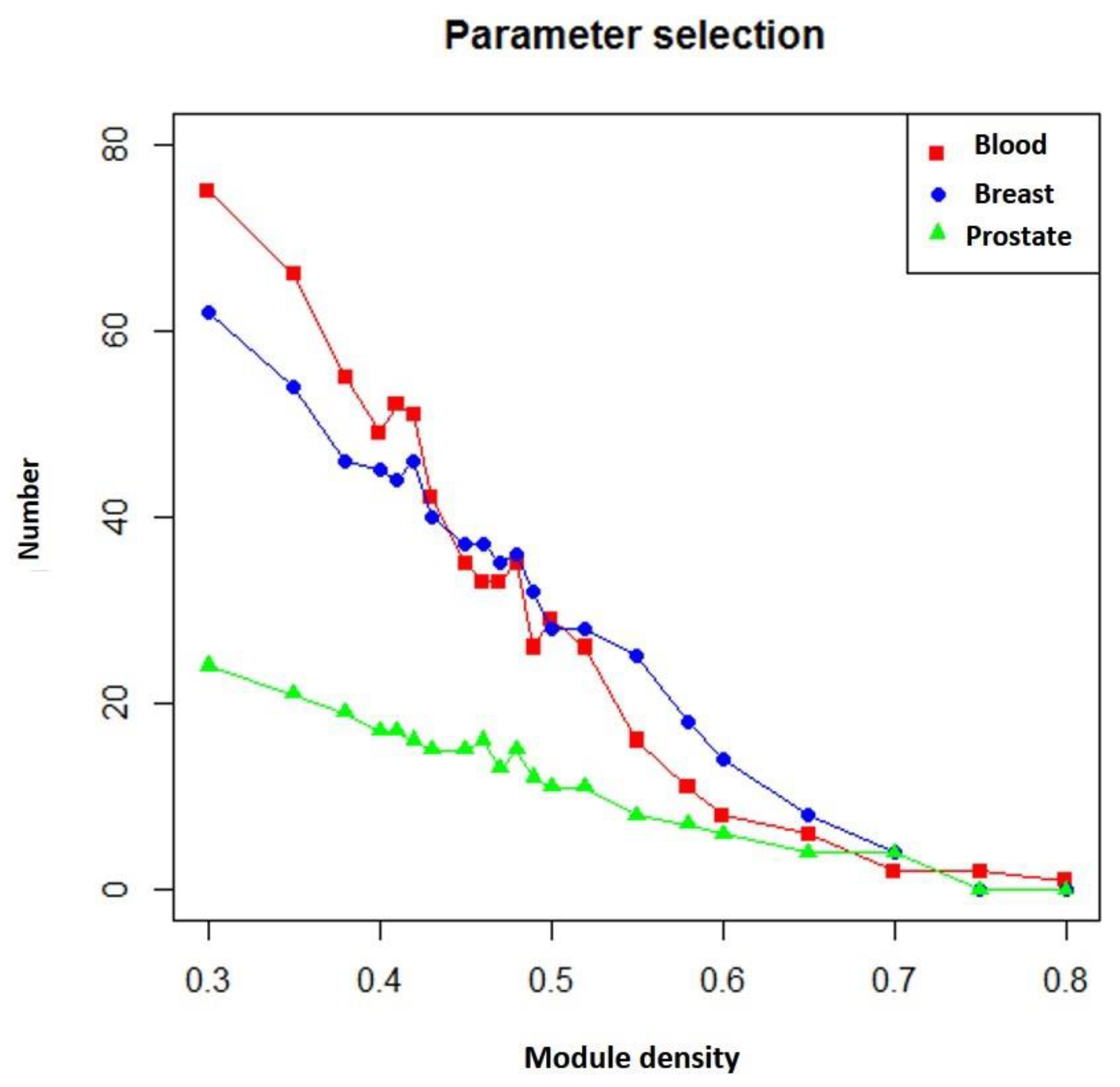
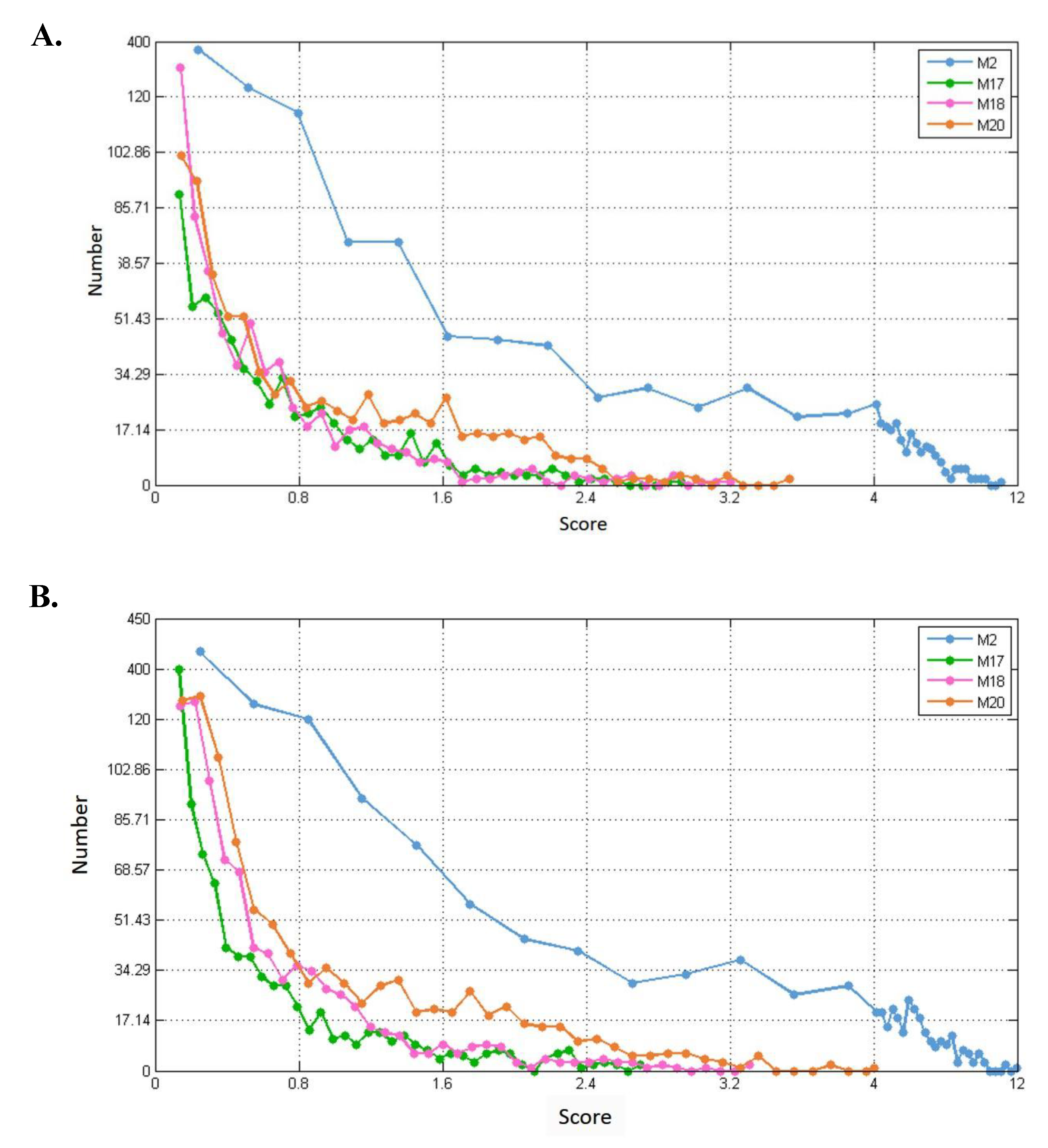
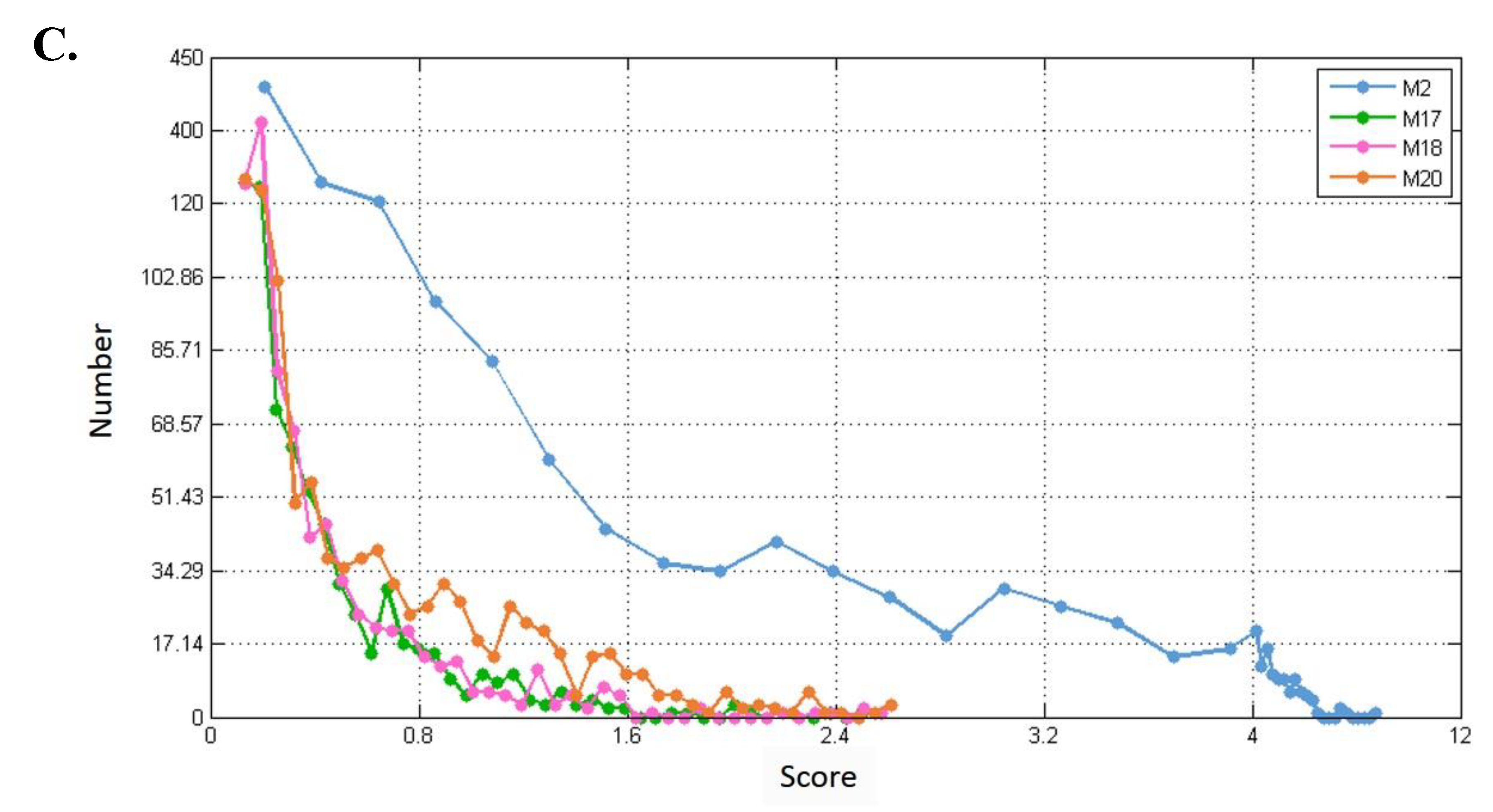
3.2. Selecting Parameter Heaviness
3.3. Comparison of Predicted Modules between Three-Layer and Single-Layer Networks
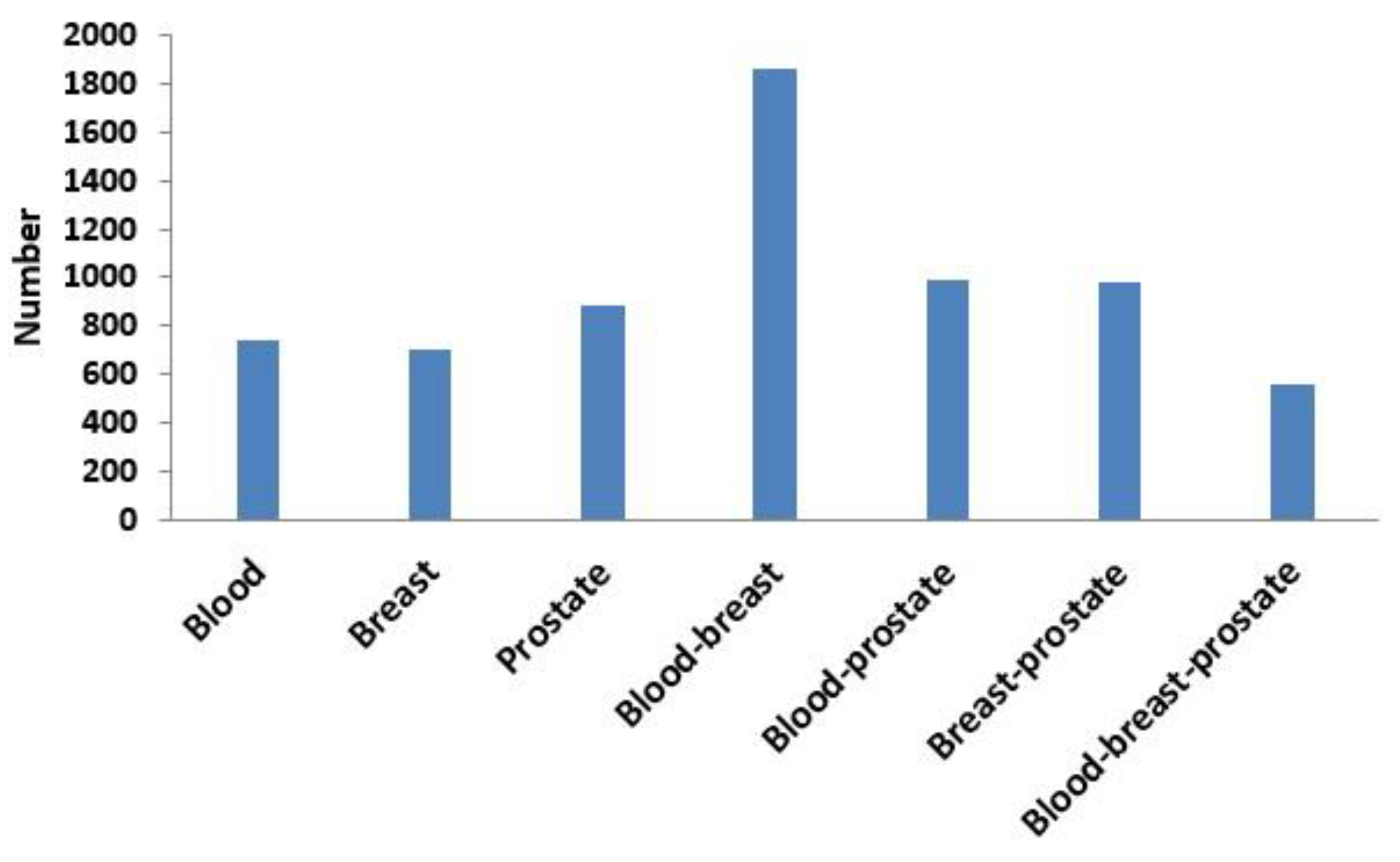
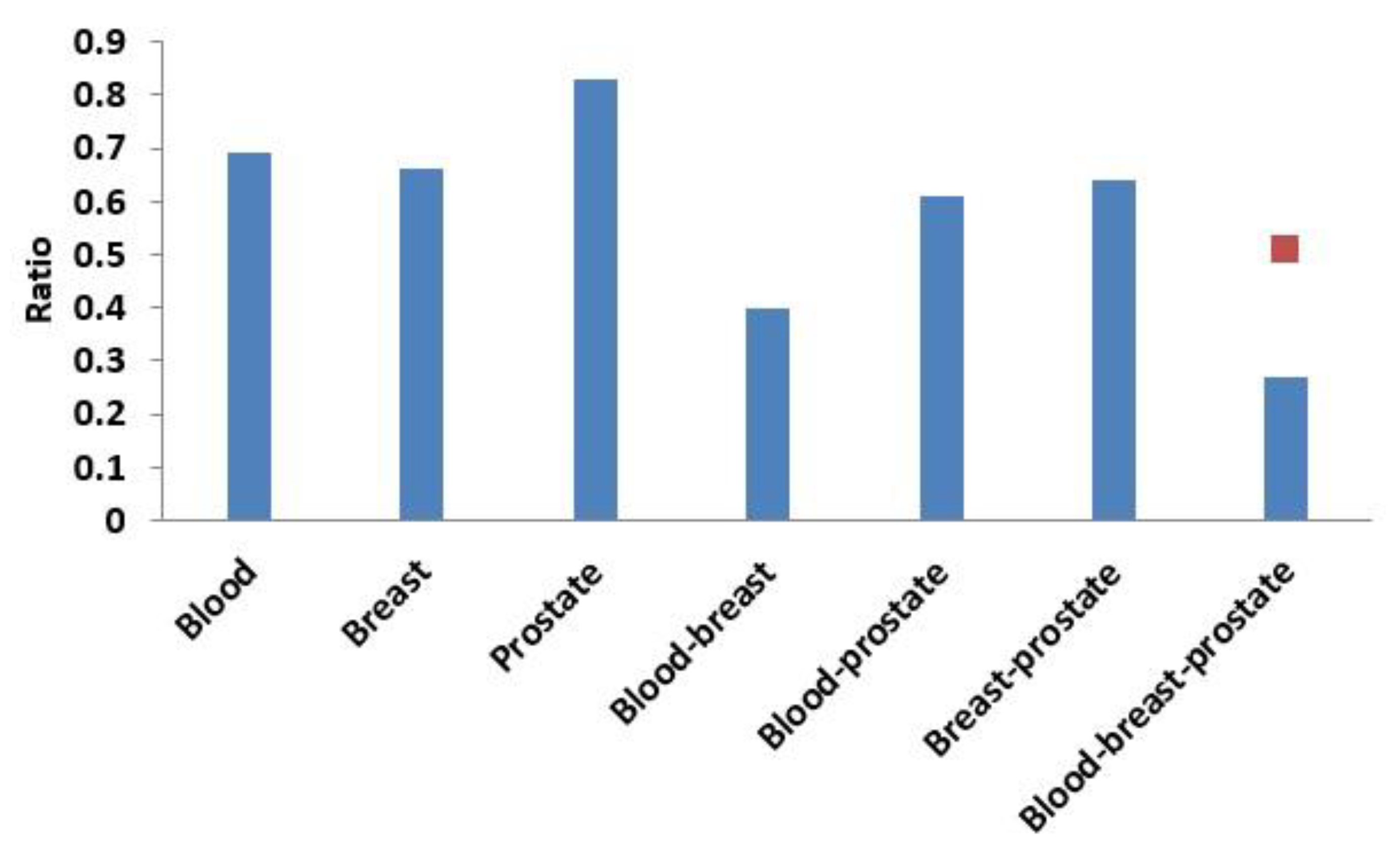
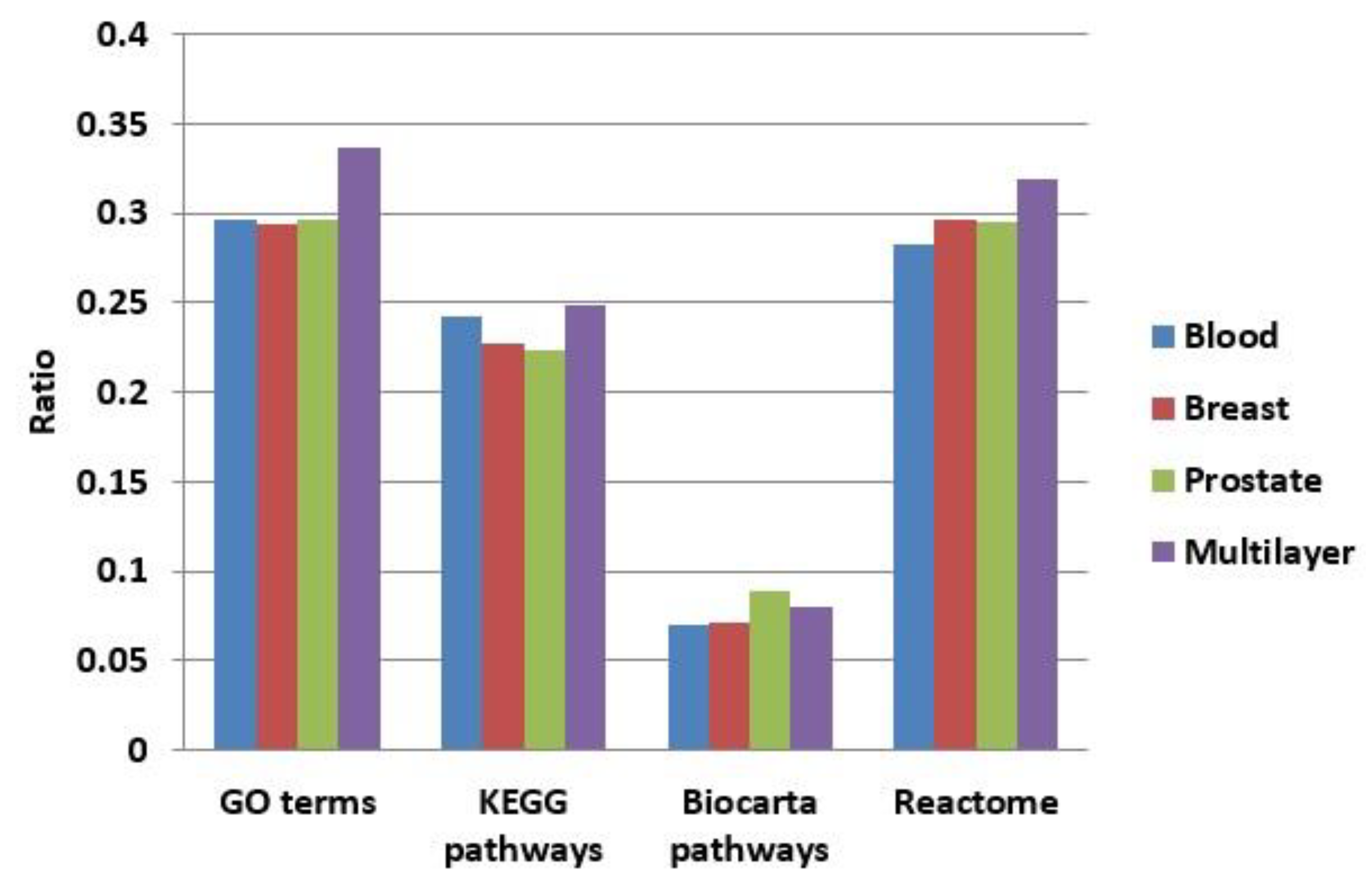
3.3.1. Comparison of Overlap between Modules
3.3.2. Functional Enrichment Comparison
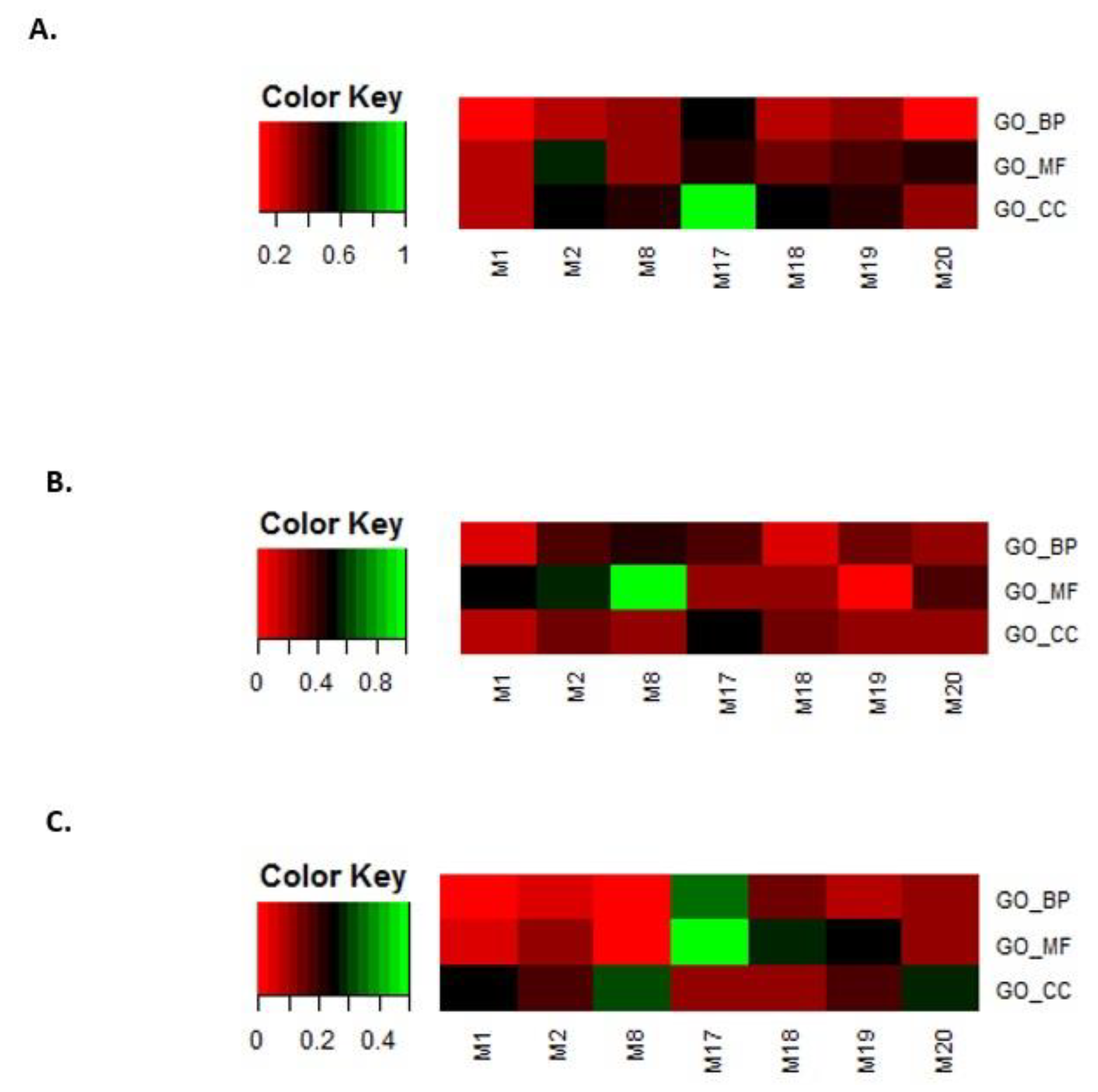
3.4. Filtering Extracted Modules in the Multilayer Network
3.4.1. Analysis Based on TSA Activity
3.4.2. Analysis Based on GO Terms
3.4.3. Analysis Based on KEGG Pathways
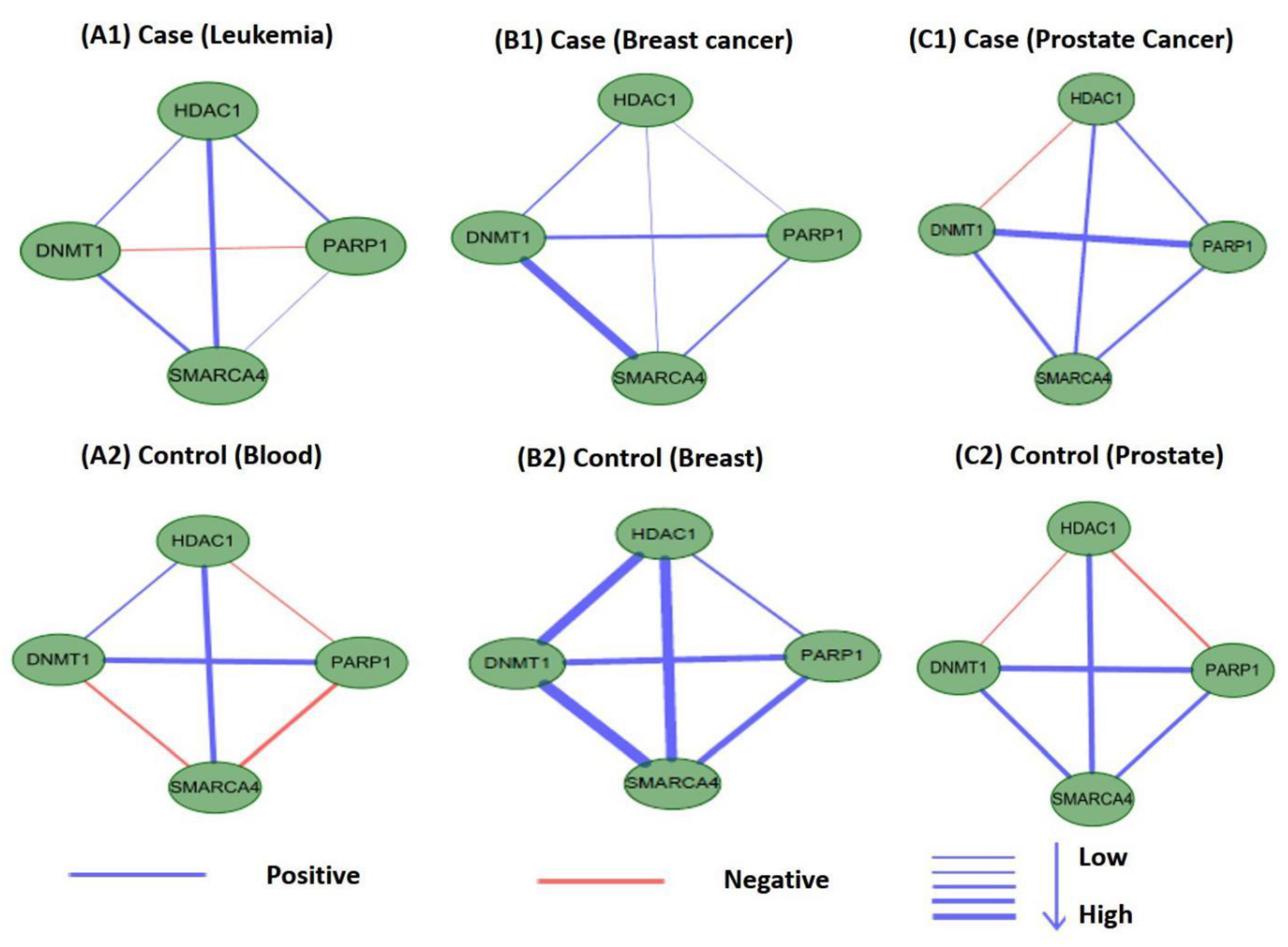
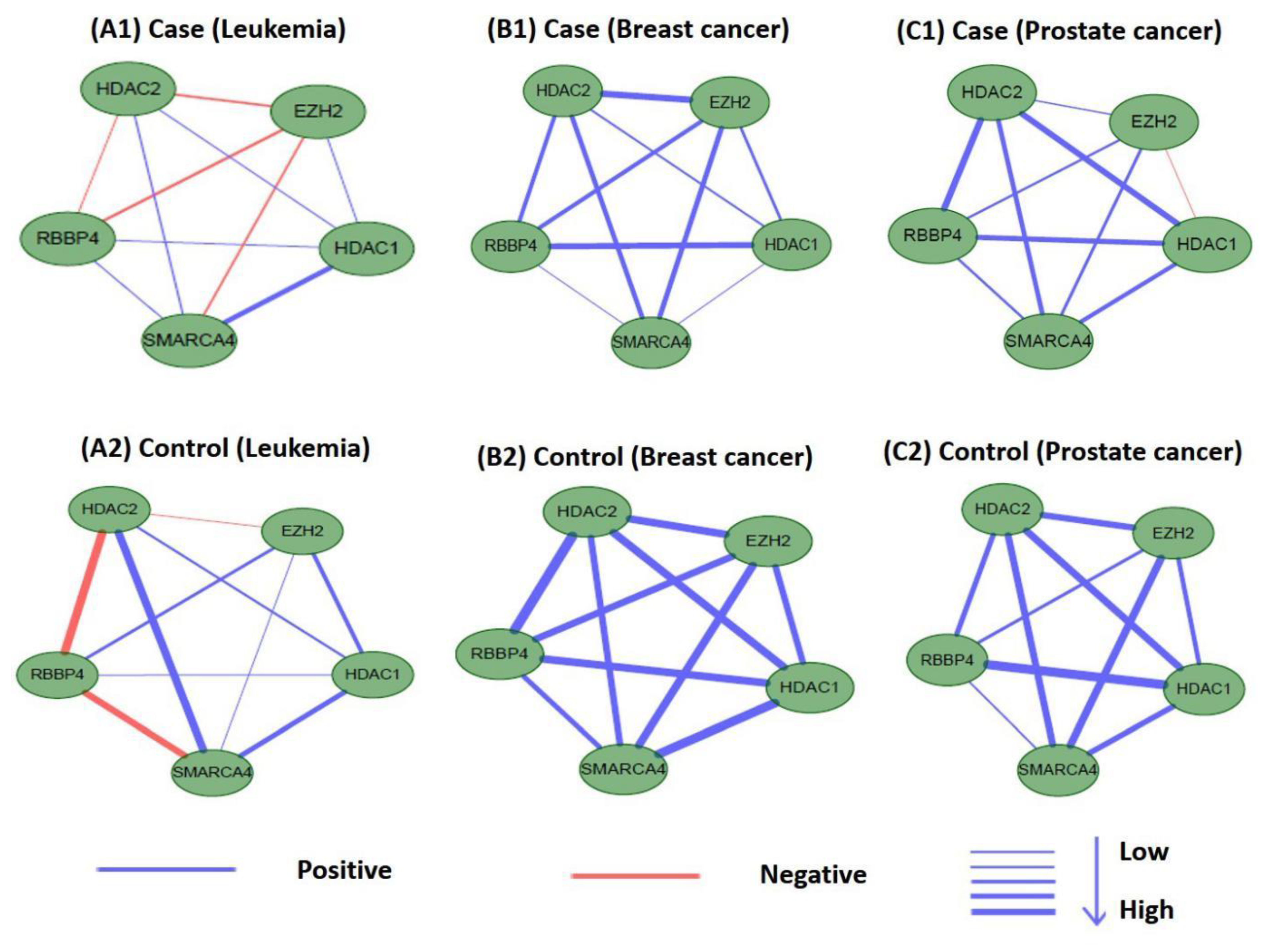
3.5. Validating and Analyzing the Significance of M17 and M18
3.5.1. Statistical Significance
3.5.2. Significance of Other TSA-Related TSPPI Networks
3.6. Differential Analysis of Internal Connections in M17 and M18 for Co-Expression Networks
3.7. PubMed Literature Validation of Genes in Modules M17 and M18
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Isik, Z.; Baldow, C.; Cannistraci, C.V.; Schroeder, M. Drug target prioritization by perturbed gene expression and network information. Sci. Rep. 2015, 5, 17417. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Iyengar, R. Systems pharmacology: Network analysis to identify multiscale mechanisms of drug action. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 505–521. [Google Scholar] [CrossRef] [PubMed]
- Rahmoune, H.; Guest, P.C. Application of Multiplex Biomarker Approaches to Accelerate Drug Discovery and Development. Methods Mol. Biol. 2017, 1546, 3–17. [Google Scholar] [PubMed]
- Suhara, T.; Chaki, S.; Kimura, H.; Furusawa, M.; Matsumoto, M.; Ogura, H.; Negishi, T.; Saijo, T.; Higuchi, M.; Omura, T.; et al. Strategies for Utilizing Neuroimaging Biomarkers in CNS Drug Discovery and Development: CINP/JSNP Working Group Report. Int. J. Neuropsychopharmacol. 2017, 20, 285–294. [Google Scholar] [CrossRef]
- Yu, L.; Huang, J.; Ma, Z.; Zhang, J.; Zou, Y.; Gao, L. Inferring drug-disease associations based on known protein complexes. BMC Med Genom. 2015, 8, 13. [Google Scholar] [CrossRef]
- Yu, L.; Wang, B.; Ma, X.; Gao, L. The extraction of drug-disease correlations based on module distance in incomplete human interactome. BMC Syst. Biol. 2016, 10, 111. [Google Scholar] [CrossRef]
- Koch, L. Pathogen genetics: Evolutionary dynamics driving drug resistance. Nat. Rev. Genet. 2017, 18, 578–579. [Google Scholar] [CrossRef]
- Gopinath, K.; Karthikeyan, M. Understanding the Evolutionary Relationship of M2 Channel Protein of Influenza A Virus and its Structural Variation and Drug Resistance. Curr. Bioinform. 2017, 12, 265–274. [Google Scholar] [CrossRef]
- Wong, Y.H.; Lin, C.L.; Chen, T.S.; Chen, C.A.; Jiang, P.S.; Lai, Y.H.; Chu, L.J.; Li, C.W.; Chen, J.J.W.; Chen, B.S. Multiple target drug cocktail design for attacking the core network markers of four cancers using ligand-based and structure-based virtual screening methods. BMC Med. Genomics 2015, 8, S4. [Google Scholar] [CrossRef]
- Vilar, S.; Quezada, E.; Uriarte, E.; Costanzi, S.; Borges, F.; Vina, D.; Hripcsak, G. Computational Drug Target Screening through Protein Interaction Profiles. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- McCormick, F. K-Ras protein as a drug target. J. Mol. Med. 2016, 94, 253–258. [Google Scholar] [CrossRef]
- Kaltdorf, M.; Srivastava, M.; Gupta, S.K.; Liang, C.; Binder, J.; Dietl, A.M.; Meir, Z.; Haas, H.; Osherov, N.; Krappmann, S.; et al. Systematic Identification of Anti-Fungal Drug Targets by a Metabolic Network Approach. Front. Mol. Biosci. 2016, 3, 22. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Ding, Y.; Tang, J.; Xu, X.; Guo, F. An Ameliorated Prediction of Drug–Target Interactions Based on Multi-Scale Discrete Wavelet Transform and Network Features. Int. J. Mol. Sci. 2017, 18, 1781. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Tang, J.; Guo, F. Identification of Protein–Ligand Binding Sites by Sequence Information and Ensemble Classifier. J. Chem. Inf. Modeling 2017, 57, 3149–3161. [Google Scholar] [CrossRef]
- Ding, Y.; Tang, J.; Guo, F. Identification of drug-target interactions via multiple information integration. Inf. Sci. 2017, 418, 546–560. [Google Scholar] [CrossRef]
- Huang, L.; Li, F.H.; Sheng, J.T.; Xia, X.F.; Ma, J.W.; Zhan, M.; Wong, S.T.C. DrugComboRanker: Drug combination discovery based on target network analysis. Bioinformatics 2014, 30, 228–236. [Google Scholar] [CrossRef]
- Yue, Z.L.; Arora, I.; Zhang, E.Y.; Laufer, V.; Bridges, S.L.; Chen, J.Y. Repositioning drugs by targeting network modules: A Parkinson’s disease case study. BMC Bioinform. 2017, 18, 532. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Zhao, J.; Gao, L. Predicting Potential Drugs for Breast Cancer based on miRNA and Tissue Specificity. Int. J. Biol. Sci. 2018, 14, 971–980. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Guo, J.; Winnenburg, R.; Baumbach, J. Drug repurposing by integrated literature mining and drug-gene-disease triangulation. Drug Discov. Today 2017, 22, 615–619. [Google Scholar] [CrossRef]
- Magger, O.; Waldman, Y.Y.; Ruppin, E.; Sharan, R. Enhancing the Prioritization of Disease-Causing Genes through Tissue Specific Protein Interaction Networks. PLoS Comput. Biol. 2012, 8, e1002690. [Google Scholar] [CrossRef]
- Guan, Y.F.; Gorenshteyn, D.; Burmeister, M.; Wong, A.K.; Schimenti, J.C.; Handel, M.A.; Bult, C.J.; Hibbs, M.A.; Troyanskaya, O.G. Tissue-Specific Functional Networks for Prioritizing Phenotype and Disease Genes. PLoS Comput. Biol. 2012, 8, e1002694. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Zhao, J.; Gao, L. Drug repositioning based on triangularly balanced structure for tissue-specific diseases in incomplete interactome. Artif. Intell. Med. 2017, 77, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, S.; Takami, T.; Murakami, Y. Porous PLGA microparticles formed by "one-step" emulsification for pulmonary drug delivery: The surface morphology and the aerodynamic properties. Colloid Surf. B 2017, 159, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chu, C.; Zhang, Y.H.; Zheng, M.Y.; Zhu, L.C.; Kong, X.Y.; Huang, T. Identification of Drug-Drug Interactions Using Chemical Interactions. Curr. Bioinform. 2017, 12, 526–534. [Google Scholar] [CrossRef]
- Yu, L.; Ma, X.; Zhang, L.; Zhang, J.; Gao, L. Prediction of new drug indications based on clinical data and network modularity. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Cichonska, A.; Ravikumar, B.; Parri, E.; Timonen, S.; Pahikkala, T.; Airola, A.; Wennerberg, K.; Rousu, J.; Aittokallio, T. Computational-experimental approach to drug-target interaction mapping: A case study on kinase inhibitors. PLoS Comput. Biol. 2017, 13, e1005678. [Google Scholar] [CrossRef]
- Greene, C.S.; Krishnan, A.; Wong, A.K.; Ricciotti, E.; Zelaya, R.A.; Himmelstein, D.S.; Zhang, R.; Hartmann, B.M.; Zaslavsky, E.; Sealfon, S.C.; et al. Understanding multicellular function and disease with human tissue-specific networks. Nat. Genet. 2015, 47, 569–576. [Google Scholar] [CrossRef]
- Liu, X.; Hong, Z.; Liu, J.; Lin, Y.; Alfonso, R.-P.; Zou, Q.; Zeng, X. Computational methods for identifying the critical nodes in biological networks. Brief. Bioinform. 2019, 21, 486–497. [Google Scholar] [CrossRef]
- Yu, L.; Su, R.; Wang, B.; Zhang, L.; Zou, Y.; Zhang, J.; Gao, L. Prediction of Novel Drugs for Hepatocellular Carcinoma Based on Multi-Source Random Walk. IEEE/ACM Trans. Comput. Biol. Bioinform. 2017, 14, 966–977. [Google Scholar] [CrossRef]
- Boccaletti, S.; Bianconi, G.; Criado, R.; del Genio, C.I.; Gomez-Gardenes, J.; Romance, M.; Sendina-Nadal, I.; Wang, Z.; Zanin, M. The structure and dynamics of multilayer networks. Phys. Rep. 2014, 544, 1–122. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.P.; Grondin, C.J.; Johnson, R.J.; Sciaky, D.; King, B.L.; McMorran, R.; Wiegers, J.; Wiegers, T.C.; Mattingly, C.J. The Comparative Toxicogenomics Database: Update 2017. Nucleic Acids Res. 2017, 45, D972–D978. [Google Scholar] [CrossRef] [PubMed]
- Lamb, J. Innovation—The Connectivity Map: A new tool for biomedical research. Nat. Rev. Cancer 2007, 7, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.P.; Subramanian, A.; Ross, K.N.; et al. The connectivity map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef]
- Piao, J.J.; Chen, L.Y.; Quan, T.H.; Li, L.S.; Quan, C.J.; Piao, Y.S.; Jin, T.F.; Lin, Z.H. Superior efficacy of co-treatment with the dual PI3K/mTOR inhibitor BEZ235 and histone deacetylase inhibitor Trichostatin A against NSCLC. Oncotarget 2016, 7, 60169–60180. [Google Scholar] [CrossRef]
- Makishima, H.; Yoshizato, T.; Yoshida, K.; Sekeres, M.A.; Radivoyevitch, T.; Suzuki, H.; Przychodzen, B.; Nagata, Y.; Meggendorfer, M.; Sanada, M.; et al. Dynamics of clonal evolution in myelodysplastic syndromes. Nat. Genet. 2017, 49, 204–212. [Google Scholar] [CrossRef]
- Vigushin, D.M.; Ali, S.; Pace, P.E.; Mirsaidi, N.; Ito, K.; Adcock, I.; Coombes, R.C. Trichostatin A is a histone deacetylase inhibitor with potent antitumor activity against breast cancer in vivo. Clin. Cancer Res. 2001, 7, 971–976. [Google Scholar]
- Keil, K.P.; Altmann, H.M.; Abler, L.L.; Hernandez, L.L.; Vezina, C.M. Histone acetylation regulates prostate ductal morphogenesis through a bone morphogenetic protein-dependent mechanism. Dev. Dynam. 2015, 244, 1404–1414. [Google Scholar] [CrossRef]
- Liao, Z.J.; Li, D.P.; Wang, X.R.; Li, L.S.; Zou, Q. Cancer Diagnosis Through IsomiR Expression with Machine Learning Method. Curr. Bioinform. 2018, 13, 57–63. [Google Scholar] [CrossRef]
- Tang, W.; Wan, S.; Yang, Z.; Teschendorff, A.E.; Zou, Q. Tumor origin detection with tissue-specific miRNA and DNA methylation markers. Bioinformatics 2018, 34, 398–406. [Google Scholar] [CrossRef]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M.; Network, C.G.A.R. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Smyth, G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3, 3. [Google Scholar] [CrossRef]
- Li, W.Y.; Liu, C.C.; Zhang, T.; Li, H.F.; Waterman, M.S.; Zhou, X.H.J. Integrative Analysis of Many Weighted Co-Expression Networks Using Tensor Computation. PLoS Comput. Biol. 2011, 7, e1001106. [Google Scholar] [CrossRef] [PubMed]
- Barshir, R.; Shwartz, O.; Smoly, I.Y.; Yeger-Lotem, E. Comparative Analysis of Human Tissue Interactomes Reveals Factors Leading to Tissue-Specific Manifestation of Hereditary Diseases. PLoS Comput. Biol. 2014, 10, e1003632. [Google Scholar] [CrossRef] [PubMed]
- Barabasi, A.L.; Albert, R. Emergence of scaling in random networks. Science 1999, 286, 509–512. [Google Scholar] [CrossRef]
- Nicosia, V.; Bianconi, G.; Latora, V.; Barthelemy, M. Growing Multiplex Networks. Phys. Rev. Lett. 2013, 111, 058701. [Google Scholar] [CrossRef] [PubMed]
- Mucha, P.J.; Richardson, T.; Macon, K.; Porter, M.A.; Onnela, J.P. Community Structure in Time-Dependent, Multiscale, and Multiplex Networks. Science 2010, 328, 876–878. [Google Scholar] [CrossRef]
- Berardini, T.Z.; Li, D.H.; Huala, E.; Bridges, S.; Burgess, S.; McCarthy, F.; Carbon, S.; Lewis, S.E.; Mungall, C.J.; Abdulla, A.; et al. The Gene Ontology in 2010: Extensions and refinements The Gene Ontology Consortium. Nucleic Acids Res. 2010, 38, D331–D335. [Google Scholar]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef]
- Hecker, N.; Ahmed, J.; Eichborn, J.; Dunkel, M.; Macha, K.; Eckert, A.; Gilson, M.K.; Bourne, P.E.; Preissner, R. SuperTarget goes quantitative: Update on drug-target interactions. Nucleic Acids Res. 2012, 40, D1113–D1117. [Google Scholar] [CrossRef]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hamosh, A.; Scott, A.F.; Amberger, J.S.; Bocchini, C.A.; McKusick, V.A. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005, 33, D514–D517. [Google Scholar] [CrossRef]
- Mottaz, A.; Yip, Y.L.; Ruch, P.; Veuthey, A.L. Mapping proteins to disease terminologies: From UniProt to MeSH. BMC Bioinform. 2008, 9, S3. [Google Scholar] [CrossRef]
- Ramos, E.M.; Hoffman, D.; Junkins, H.A.; Maglott, D.; Phan, L.; Sherry, S.T.; Feolo, M.; Hindorff, L.A. Phenotype-Genotype Integrator (PheGenI): Synthesizing genome- wide association study (GWAS) data with existing genomic resources. Eur. J. Hum. Genet. 2014, 22, 144–147. [Google Scholar] [CrossRef] [PubMed]
- van Dam, S.; Vosa, U.; van der Graaf, A.; Franke, L.; de Magalhaes, J.P. Gene co-expression analysis for functional classification and gene-disease predictions. Brief Bioinform 2018, 19, 575–592. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Marischuk, K.; Castelvecchi, G.D.; Bashirullah, A. HDAC Inhibitors Disrupt Programmed Resistance to Apoptosis During Drosophila Development. G3 (Bethesda) 2017, 7, 1985–1993. [Google Scholar] [CrossRef]
- Robertson, K.D.; Ait-Si-Ali, S.; Yokochi, T.; Wade, P.A.; Jones, P.L.; Wolffe, A.P. DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters. Nat. Genet. 2000, 25, 338–342. [Google Scholar] [CrossRef]
- Furumai, R.; Komatsu, Y.; Nishino, N.; Khochbin, S.; Yoshida, M.; Horinouchi, S. Potent histone deacetylase inhibitors built from trichostatin A and cyclic tetrapeptide antibiotics including trapoxin. Proc. Natl. Acad. Sci. USA 2001, 98, 87–92. [Google Scholar] [CrossRef]
- Qiu, Z.; Ghosh, A. A calcium-dependent switch in a CREST-BRG1 complex regulates activity-dependent gene expression. Neuron 2008, 60, 775–787. [Google Scholar] [CrossRef]
- King, H.A.; Trotter, K.W.; Archer, T.K. Chromatin remodeling during glucocorticoid receptor regulated transactivation. BBA 2012, 1819, 716–726. [Google Scholar] [CrossRef] [PubMed]
- Barutcu, A.R.; Lajoie, B.R.; Fritz, A.J.; McCord, R.P.; Nickerson, J.A.; van Wijnen, A.J.; Lian, J.B.; Stein, J.L.; Dekker, J.; Stein, G.S.; et al. SMARCA4 regulates gene expression and higher order chromatin structure in proliferating mammary epithelial cells. Genome Res. 2016, 26, 1188–1201. [Google Scholar] [CrossRef] [PubMed]
- Mackmull, M.T.; Iskar, M.; Parca, L.; Singer, S.; Bork, P.; Ori, A.; Beck, M. Histone Deacetylase Inhibitors (HDACi) Cause the Selective Depletion of Bromodomain Containing Proteins (BCPs). Mol. Cell Proteom. 2015, 14, 1350–1360. [Google Scholar] [CrossRef] [PubMed]
- Ogino, H.; Nakayama, R.; Sakamoto, H.; Yoshida, T.; Sugimura, T.; Masutani, M. Analysis of poly(ADP-ribose) polymerase-1 (PARP1) gene alteration in human germ cell tumor cell lines. Cancer Genet. Cytogen. 2010, 197, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Nagaria, P.K.; Pawar, N.; Adewuyi, A.; Gojo, I.; Meyers, D.J.; Cole, P.A.; Rassool, F.V. Histone deacetylase inhibitors decrease NHEJ both by acetylation of repair factors and trapping of PARP1 at DNA double-strand breaks in chromatin. Leuk. Res. 2016, 45, 14–23. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Module ID | Entrez IDs of Genes in the Modules |
|---|---|
| M1 | 890, 7153, 4085, 6241, 701, 22974, 6790, 3161, 11130, 10403, 6240, 10051, 51203, 1434, 1719, 3832, 7298, 5984, 10592, 4173, 891, 9319, 2237, 3838, 990, 47, 90, 87 |
| M2 | 7520, 142, 1019, 5111, 5591, 6749, 2237, 5036, 4522, 6241, 4175, 10606, 5982, 1736 |
| M3 | 22948, 10213, 10969, 471, 1434, 3329, 5686, 1503, 9221, 908, 5901, 5036, 3838, 7371 |
| M4 | 5901, 7334, 7520, 7443, 10576, 7153, 10213, 26135, 6636, 6427, 5902, 6428, 6240 |
| M5 | 22948, 7203, 6950, 10574, 11222, 1164, 4830, 7334 |
| M6 | 4172, 6627, 1503, 10528, 11130, 2237, 7398, 9521, 5985 |
| M7 | 6426, 4436, 10772, 10236, 3838, 26135, 1665, 23165, 10576, 7520 |
| M8 | 7153, 5557, 6790, 672, 8317, 10733, 4001, 1736 |
| M9 | 6426, 9221, 6434, 7334, 3015, 1736, 2237, 3184, 2956, 6427 |
| M10 | 10574, 158, 7965, 142, 1503, 7411, 4176, 1736, 8607, 7203, 5901, 5902 |
| M11 | 6637, 5111, 3148, 3182, 6434 |
| M12 | 1434, 3308, 908, 4869, 6950, 7203, 3336, 3838 |
| M13 | 10492, 1503, 3182 |
| M14 | 3276, 5725, 3609, 6597, 4176, 6627 |
| M15 | 6194, 6124, 6201, 6137, 11224, 6143, 6193, 6217, 6152, 6139, 6136, 6161, 23521, 6133, 6175, 4736, 6207, 6218, 6135, 6128, 6146, 3646, 1933, 47, 87, 39, 29, 90, 95 |
| M16 | 3014, 84823, 6597, 5036 |
| M17 | 3065, 142, 1786, 6597 |
| M18 | 3066, 3065, 5928, 2146, 6597 |
| M19 | 86, 6597, 10856 |
| M20 | 6597, 6599, 5591, 4173, 4172 |
| M21 | 6597, 23246, 8662 |
| M22 | 5036, 10574, 3182 |
| M23 | 3329, 7203, 6428 |
| M24 | 10606, 6950, 4691, 3183, 6741, 3843, 5901 |
| M25 | 890, 7371, 3251, 1665 |
| M26 | 5557, 990, 9493, 9833, 1060 |
| Module | Threshold of Score | Total Number of Genes | Number of KEGG Pathways |
|---|---|---|---|
| M2 | 7.0 | 46 | 12 |
| M17 | 2.0 | 21 | 11 |
| M18 | 2.1 | 18 | 6 |
| M20 | 2.3 | 35 | 4 |
| Module | Threshold of Score | Number of Total Genes | Number of KEGG Pathways |
|---|---|---|---|
| M2 | 7.2 | 59 | 13 |
| M17 | 2.0 | 29 | 10 |
| M18 | 2.0 | 28 | 6 |
| M20 | 2.5 | 42 | 5 |
| Module | Threshold of Score | Number of Total Genes | Number of KEGG Pathways |
|---|---|---|---|
| M2 | 4.5 | 65 | 12 |
| M17 | 1.2 | 28 | 15 |
| M18 | 1.4 | 21 | 14 |
| M20 | 1.8 | 30 | 5 |
| Tissue | M2 | M17 | M18 | M20 |
|---|---|---|---|---|
| Blood | 0 | 3 | 1 | 0 |
| Breast | 1 | 4 | 1 | 0 |
| Prostate | 1 | 4 | 4 | 1 |
| Cancer | M17 | M18 |
|---|---|---|
| Leukemia | 6 | 1 |
| Breast cancer | 3 | 1 |
| Prostate cancer | 1 | 1 |
| Tissue | Value for M17 | Value for M18 |
|---|---|---|
| Blood | 6.27 × 104 | 6.36 × 106 |
| Breast | 3.24 × 104 | 0 |
| Prostate | 1.64 × 104 | 0 |
| Tissue | Number of Edges | Minimal Edge Weight | Value for M17 | Value for M18 | |
|---|---|---|---|---|---|
| Lung | 149,495 | 0.374935 | 2.12 × 104 | 0 | |
| Colon | 163,180 | 0.317351 | 6.91 × 104 | 0 | |
| Ovarian | 161,487 | 0.37902 | 2.67 × 104 | 0 | |
| Pancreas | 161,147 | 0.312249 | 6.58 × 104 | 8.43 × 105 | |
| Marrow | 154,621 | 0.391356 | 0.0242 | 9.23 × 104 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, L.; Shi, Y.; Zou, Q.; Wang, S.; Zheng, L.; Gao, L. Exploring Drug Treatment Patterns Based on the Action of Drug and Multilayer Network Model. Int. J. Mol. Sci. 2020, 21, 5014. https://doi.org/10.3390/ijms21145014
Yu L, Shi Y, Zou Q, Wang S, Zheng L, Gao L. Exploring Drug Treatment Patterns Based on the Action of Drug and Multilayer Network Model. International Journal of Molecular Sciences. 2020; 21(14):5014. https://doi.org/10.3390/ijms21145014
Chicago/Turabian StyleYu, Liang, Yayong Shi, Quan Zou, Shuhang Wang, Liping Zheng, and Lin Gao. 2020. "Exploring Drug Treatment Patterns Based on the Action of Drug and Multilayer Network Model" International Journal of Molecular Sciences 21, no. 14: 5014. https://doi.org/10.3390/ijms21145014
APA StyleYu, L., Shi, Y., Zou, Q., Wang, S., Zheng, L., & Gao, L. (2020). Exploring Drug Treatment Patterns Based on the Action of Drug and Multilayer Network Model. International Journal of Molecular Sciences, 21(14), 5014. https://doi.org/10.3390/ijms21145014