Role of Flavonoids in the Prevention of AhR-Dependent Resistance During Treatment with BRAF Inhibitors

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
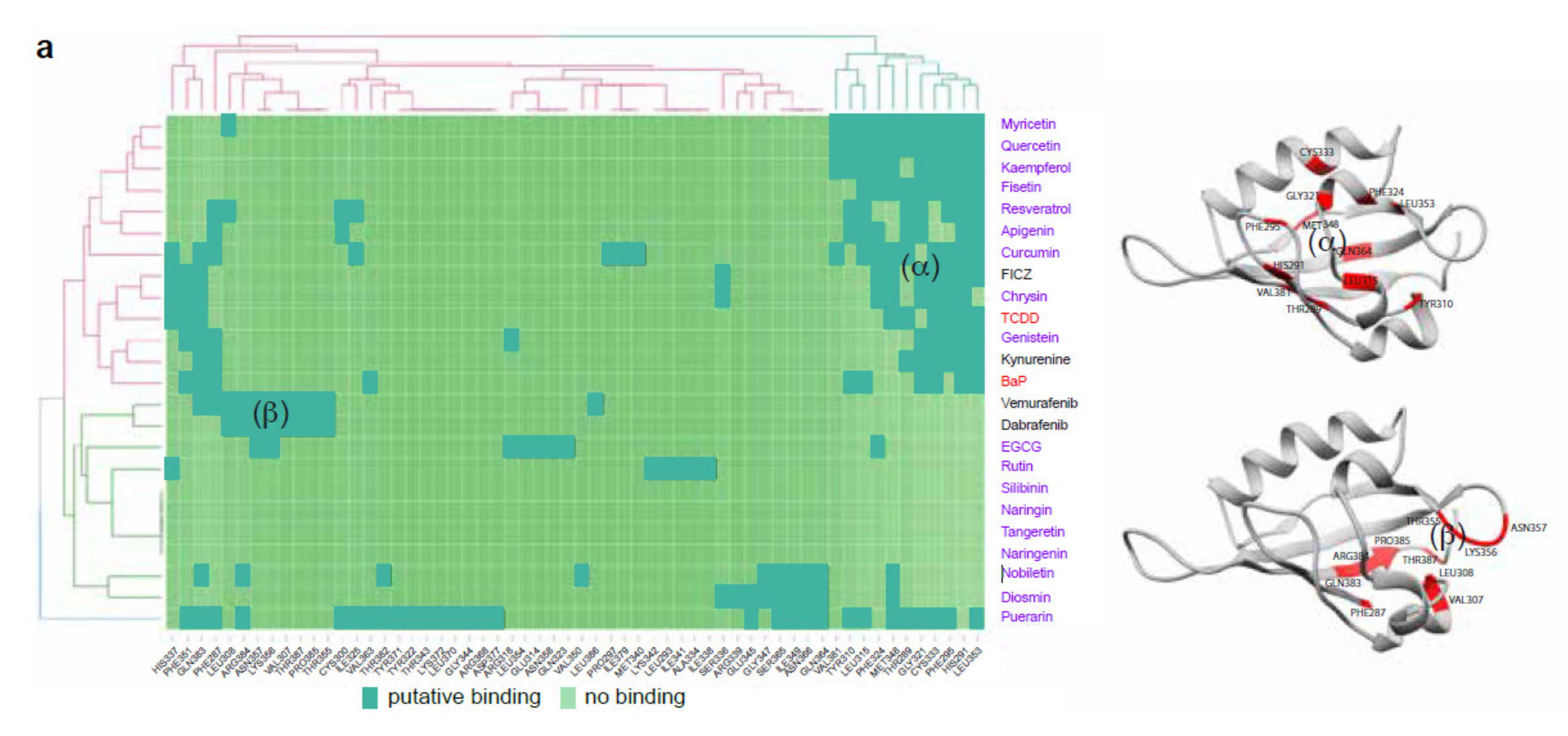
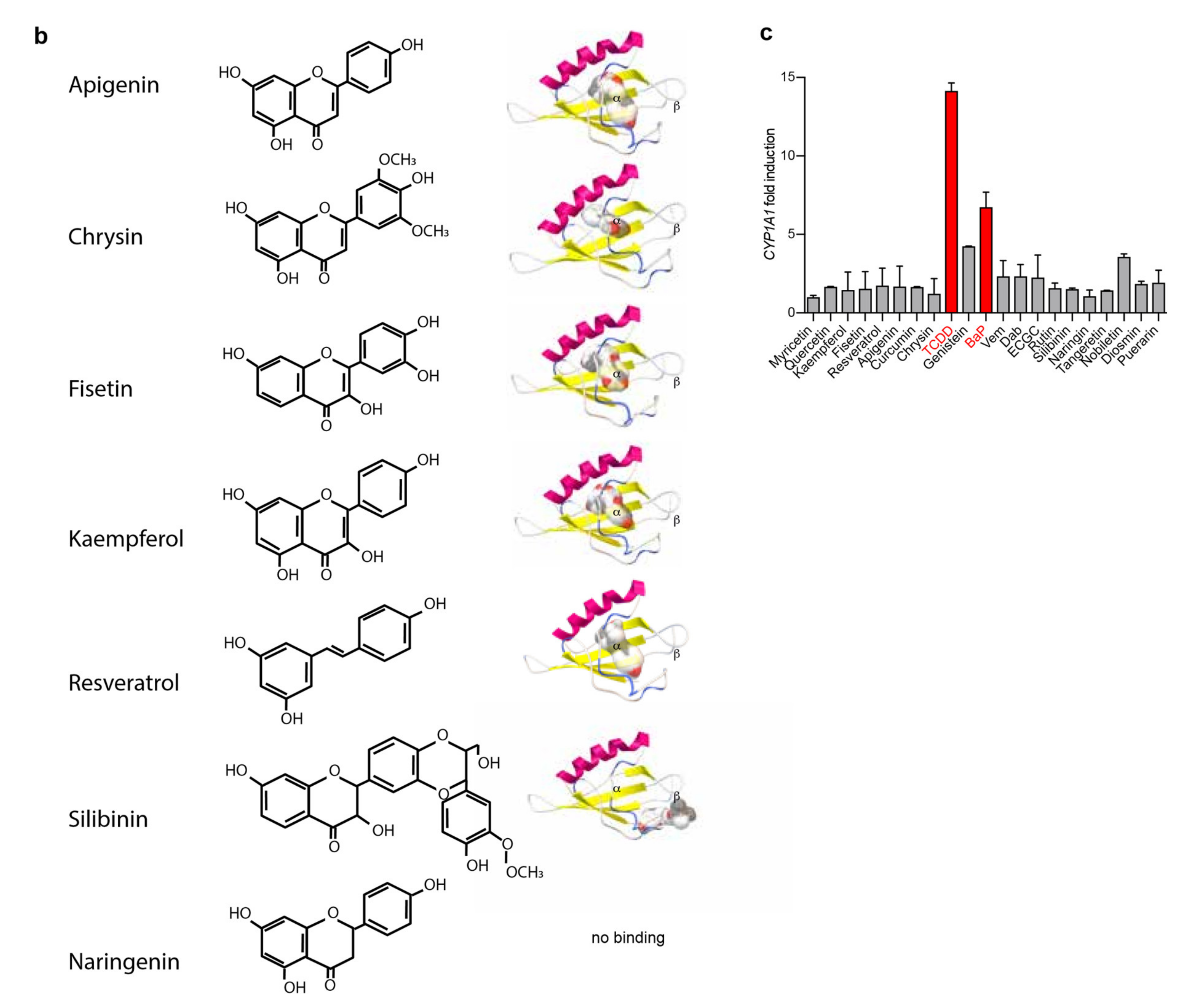
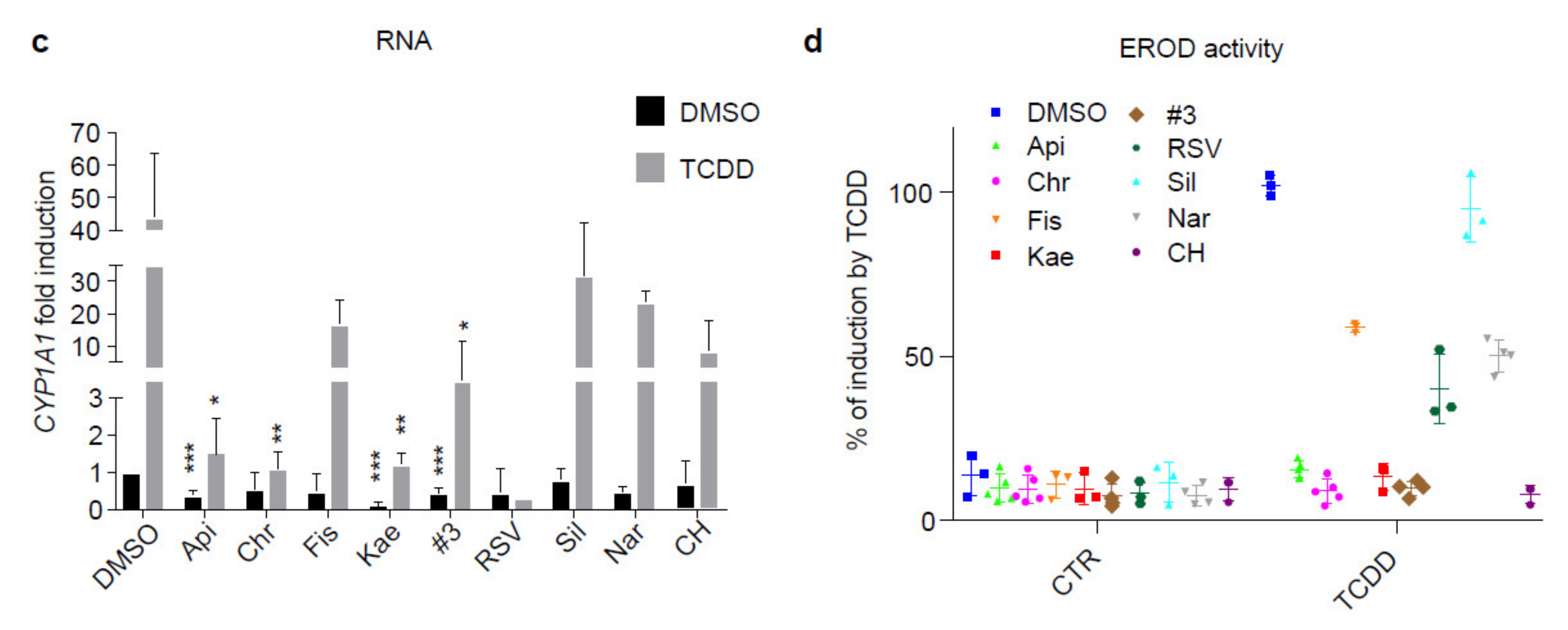
2.1. Flavonoids Are AhR Ligands That Control Its Transcriptional Activity
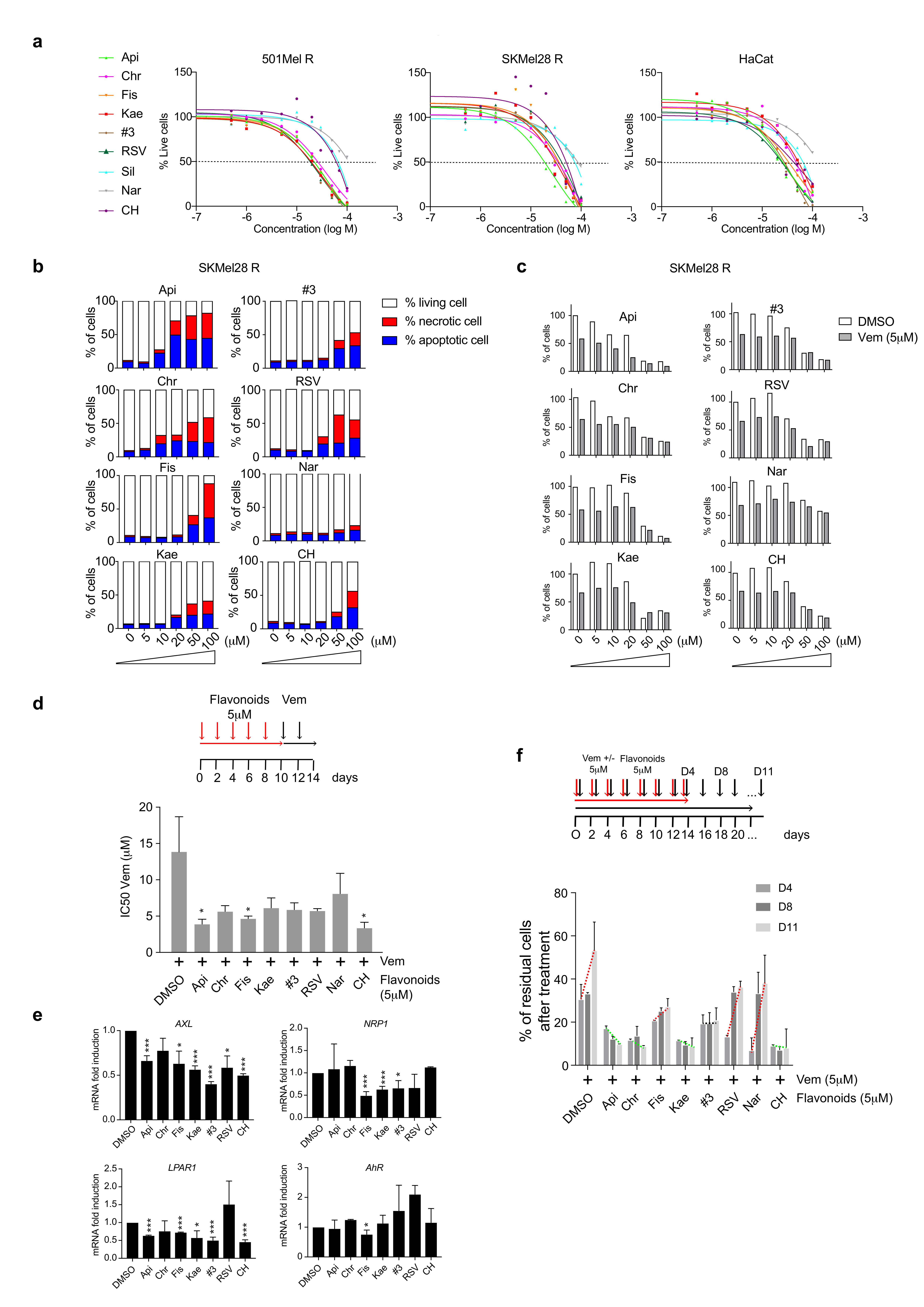
2.2. Flavonoids Increase BRAFi Sensitivity and Diminish the Pool of Persister Cells
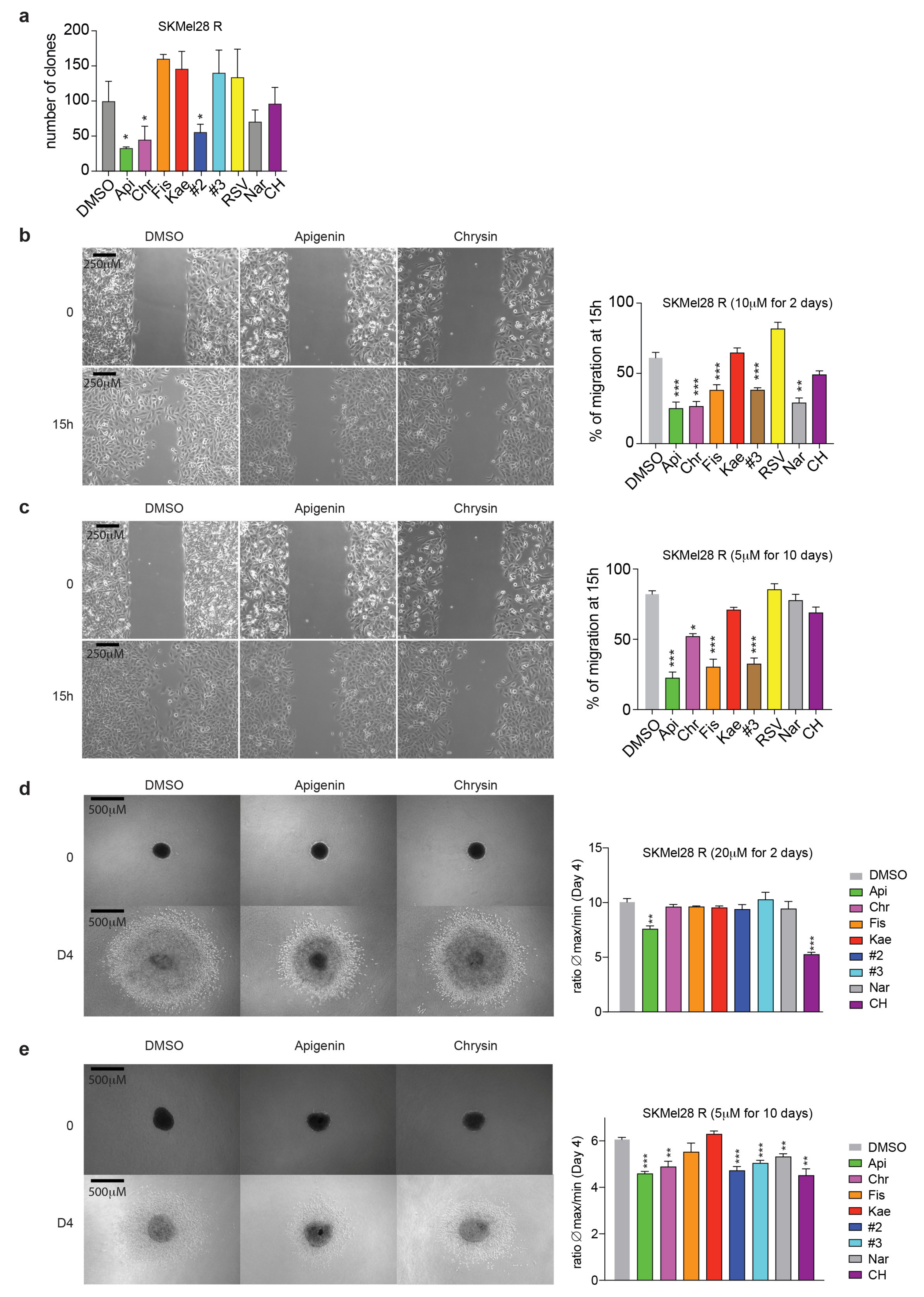
2.3. Flavonoids Prevent Migration and Invasion of Resistant Melanoma Cells
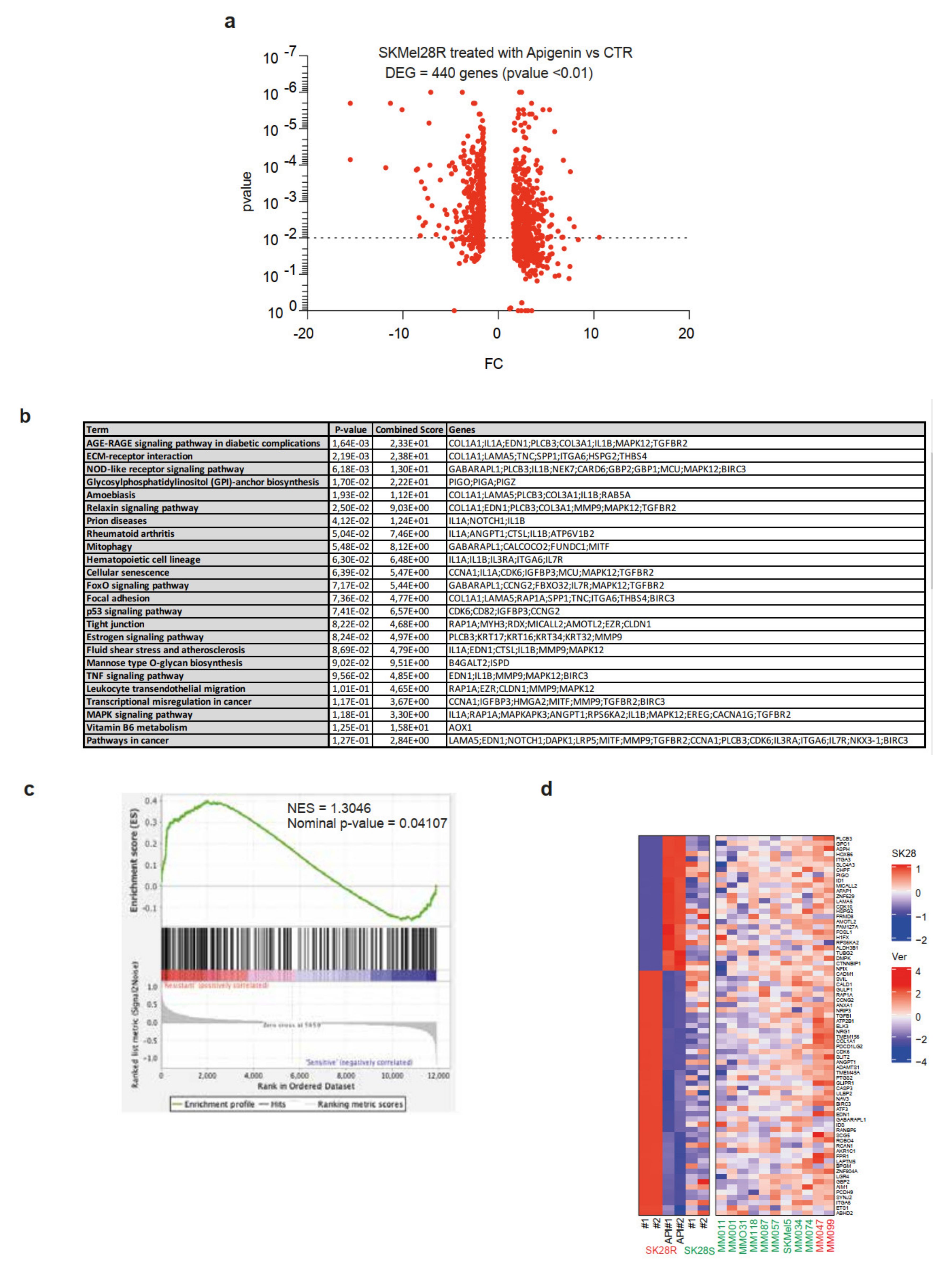
2.4. Treatment of SKMel28-Resistant Cell Lines by Apigenin Leads to a Decrease in the Expression of Genes Associated with the Resistance Phenotype
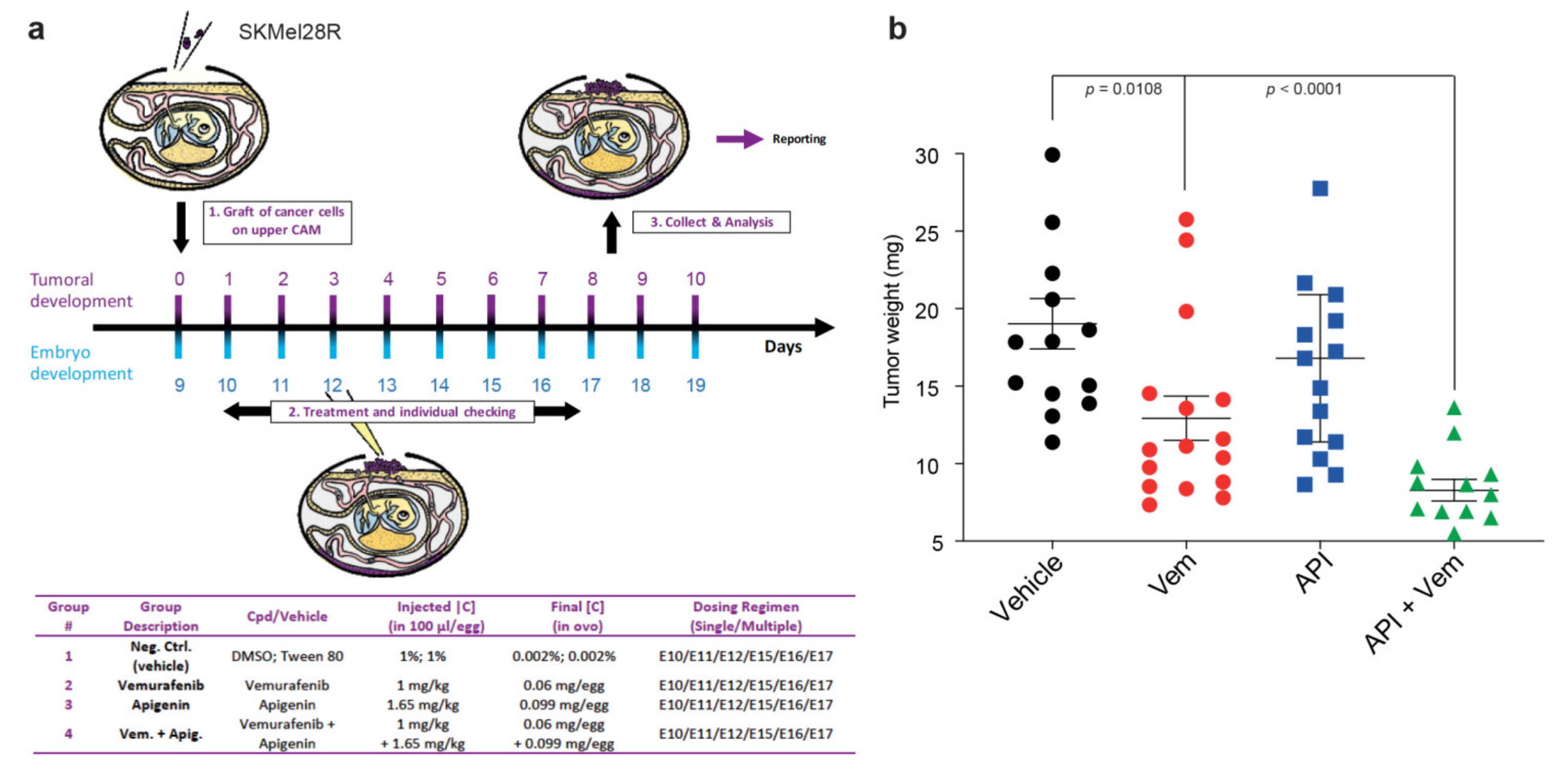
2.5. Apigenin Increases BRAFi Sensitivity of Melanoma Xenografts in a Chick Embryo Model
3. Discussion
4. Methods
4.1. Reagents (AhR Ligands)
4.2. Cell Lines and Culture Conditions
4.3. Molecular Modelling
4.4. Evaluation of Cell Density
4.5. Wound-Healing Migration Assay
4.6. Spheroid-Formation Assay
4.7. Colony-Formation Assay
4.8. Apoptosis and Necrosis Assays
4.9. Ethoxyresorufin O-Deethylase Activity Assay
4.10. Luciferase Activity
4.11. RNA Extraction and RT-qPCR Expression
4.12. RNA-Seq
4.13. Data Mining
4.14. Tumor Grafts in the Chicken CAM Model
4.14.1. Preparation of Chicken Embryos
4.14.2. Amplification and Grafting of Tumor Cells
4.14.3. Treatments
4.14.4. Quantitative Evaluation of Tumor Growth
4.15. Statistics
4.16. Data Availability
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MDPI | Multidisciplinary Digital Publishing Institute |
| DOAJ | Directory of open access journals |
| AhR | Aryl hydrocarbon Receptor |
| Api | Apigenin |
| ARNT | AhR Nuclear Translocator |
| AXL | AXL tyrosine kinase |
| BaP | Benzo (a) pyrene |
| BRAF | serine/threonine-protein kinase B-Raf |
| BRAFi | BRAF inhibitor |
| Chr | Chrysin |
| CYP1A1 | Cytochrome P450, family 1, subfamily A |
| DMSO | DiMethylSulfOxide |
| EGFR | Epidermal Growth Factor Receptor |
| FICZ | 6-formylindolo[3 ,2-b]carbazole |
| Fis | Fisetin |
| FOXO | Forkhead box |
| HIF | Hypoxia Inducible Factor |
| Kae | Kaempferol |
| KO | Knockout |
| MAPK | Mitogen Activated Protein Kinase |
| MEK | Mitogen-activated protein kinase kinase |
| MITF | Microphtalmia-associated Transcription |
| Nar | Naringenin |
| NGFR | Nerve Growth Factor Receptor |
| PAS | Per-ARNT-Sim family |
| PDGFRB | Platelet Derived Growth Factor Receptor Beta |
| RSV | Resveratrol |
| Sil | Silibin |
| TCDD | 2,3,7,8-tétrachlorodibenzo-p-dioxine |
| TEAD | Transcriptional enhancer factor TEF-3 |
| Vem | Vemurafenib |
| XRE | Xenobiotic Response Elements |
References
- Davies, H. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Nazarian, R.; Shi, H.; Wang, Q.; Kong, X.; Koya, R.C.; Lee, H.; Chen, Z.; Lee, M.-K.; Attar, N.; Sazegar, H.; et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 2010, 468, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Wagle, N.; Van Allen, E.M.; Treacy, D.J.; Frederick, D.T.; Cooper, Z.A.; Taylor-Weiner, A.; Rosenberg, M.; Goetz, E.M.; Sullivan, R.J.; Farlow, D.N.; et al. MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Discov. 2014, 4, 61–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriceau, G.; Hugo, W.; Hong, A.; Shi, H.; Kong, X.; Yu, C.C.; Koya, R.C.; Samatar, A.A.; Khanlou, N.; Braun, J.; et al. Tunable-combinatorial Mechanisms of Acquired Resistance Limit the Efficacy of BRAF/MEK Co-targeting but Result in Melanoma Drug Addiction. Cancer Cell 2015, 27, 240–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heidorn, S.J. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell 2010, 140, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Paluncic, J.; Kovacevic, Z.; Jansson, P.J.; Kalinowski, D.; Merlot, A.M.; Huang, M.L.-H.; Lok, H.C.; Sahni, S.; Lane, D.J.R.; Richardson, D.R. Roads to melanoma: Key pathways and emerging players in melanoma progression and oncogenic signaling. Biochim. Biophys. Acta Mol. Cell Res. 2016, 1863, 770–784. [Google Scholar] [CrossRef]
- Chan, X.Y.; Singh, A.; Osman, N.; Piva, T.J. Role Played by Signalling Pathways in Overcoming BRAF Inhibitor Resistance in Melanoma. Int. J. Mol. Sci. 2017, 18, 1527. [Google Scholar] [CrossRef]
- Shi, H.; Hugo, W.; Kong, X.; Hong, A.; Koya, R.C.; Moriceau, G.; Chodon, T.; Guo, R.; Johnson, D.B.; Dahlman, K.B.; et al. Acquired Resistance and Clonal Evolution in Melanoma during BRAF Inhibitor Therapy. Cancer Discov. 2014, 4, 80–93. [Google Scholar] [CrossRef] [Green Version]
- Da Silva-Diz, V.; Simón-Extremera, P.; Bernat-Peguera, A.; de Sostoa, J.; Urpí, M.; Penín, R.M.; Sidelnikova, D.P.; Bermejo, O.; Viñals, J.M.; Rodolosse, A.; et al. Cancer Stem-like Cells Act via Distinct Signaling Pathways in Promoting Late Stages of Malignant Progression. Cancer Res. 2016, 76, 1245–1259. [Google Scholar] [CrossRef] [Green Version]
- Girotti, M.R.; Pedersen, M.; Sanchez-Laorden, B.; Viros, A.; Turajlic, S.; Niculescu-Duvaz, D.; Zambon, A.; Sinclair, J.; Hayes, A.; Gore, M.; et al. Inhibiting EGF receptor or SRC family kinase signaling overcomes BRAF inhibitor resistance in melanoma. Cancer Discov. 2013, 3, 158–167. [Google Scholar] [CrossRef] [Green Version]
- Arozarena, I.; Wellbrock, C. Targeting invasive properties of melanoma cells. FEBS J. 2017, 284, 2148–2162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoek, K.S.; Goding, C.R. Cancer stem cells versus phenotype-switching in melanoma. Pigment. Cell Melanoma Res. 2010, 23, 746–759. [Google Scholar] [CrossRef]
- Müller, J.; Krijgsman, O.; Tsoi, J.; Robert, L.; Hugo, W.; Song, C.; Kong, X.; Possik, P.A.; Cornelissen-Steijger, P.D.M.; Foppen, M.H.G.; et al. Low MITF/AXL ratio predicts early resistance to multiple targeted drugs in melanoma. Nat. Commun. 2014, 5, 5712. [Google Scholar] [CrossRef] [PubMed]
- Konieczkowski, D.J.; Johannessen, C.M.; Abudayyeh, O.; Kim, J.W.; Cooper, Z.A.; Piris, A.; Frederick, D.T.; Barzily-Rokni, M.; Straussman, R.; Haq, R.; et al. A melanoma cell state distinction influences sensitivity to MAPK pathway inhibitors. Cancer Discov. 2014, 4, 816–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaffer, S.M.; Dunagin, M.C.; Torborg, S.R.; Torre, E.A.; Emert, B.; Krepler, C.; Beqiri, M.; Sproesser, K.; Brafford, P.A.; Xiao, M.; et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 2017, 546, 431–435. [Google Scholar] [CrossRef] [Green Version]
- Verfaillie, A.; Imrichova, H.; Atak, Z.K.; Dewaele, M.; Rambow, F.; Hulselmans, G.; Christiaens, V.; Svetlichnyy, D.; Luciani, F.; Van den Mooter, L.; et al. Decoding the regulatory landscape of melanoma reveals TEADS as regulators of the invasive cell state. Nat. Commun. 2015, 6, 6683. [Google Scholar] [CrossRef] [Green Version]
- Tsoi, J.; Robert, L.; Paraiso, K.; Galvan, C.; Sheu, K.M.; Lay, J.; Wong, D.J.L.L.; Atefi, M.; Shirazi, R.; Wang, X.; et al. Multi-stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell 2018, 33, 890–904. [Google Scholar] [CrossRef] [Green Version]
- Rambow, F.; Rogiers, A.; Marin-Bejar, O.; Aibar, S.; Femel, J.; Dewaele, M.; Karras, P.; Brown, D.; Chang, Y.H.; Debiec-Rychter, M.; et al. Toward Minimal Residual Disease-Directed Therapy in Melanoma. Cell 2018, 174, 843–855. [Google Scholar] [CrossRef] [Green Version]
- Rose, A.A.N.; Annis, M.G.; Frederick, D.T.; Biondini, M.; Dong, Z.; Kwong, L.; Chin, L.; Keler, T.; Hawthorne, T.; Watson, I.R.; et al. MAPK Pathway Inhibitors Sensitize BRAF-Mutant Melanoma to an Antibody-Drug Conjugate Targeting GPNMB. Clin. Cancer Res. 2016, 22, 6088–6098. [Google Scholar] [CrossRef] [Green Version]
- Corre, S.; Tardif, N.; Mouchet, N.; Leclair, H.M.; Boussemart, L.; Gautron, A.; Bachelot, L.; Perrot, A.; Rambow, F.; Soshilov, A.; et al. Sustained activation of the Aryl hydrocarbon Receptor (AhR) transcription factor promotes the resistance to BRAF inhibitors in melanoma. Nat. Commun. 2018, 9, 4775. [Google Scholar] [CrossRef]
- Bersten, D.C.; Sullivan, A.E.; Peet, D.J.; Whitelaw, M.L. bHLH–PAS proteins in cancer. Nat. Rev. Cancer 2013, 13, 827. [Google Scholar] [CrossRef] [PubMed]
- Veldhoen, M.; Duarte, J.H. The aryl hydrocarbon receptor: Fine-tuning the immune-response. Curr. Opin. Immunol. 2010, 22, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Denison, M.S.; Pandini, A.; Nagy, S.R.; Baldwin, E.P.; Bonati, L. Ligand binding and activation of the Ah receptor. Chem. Biol. Interact. 2002, 141, 3–24. [Google Scholar] [CrossRef] [Green Version]
- Safe, S.; Lee, S.-O.; Jin, U.-H. Role of the aryl hydrocarbon receptor in carcinogenesis and potential as a drug target. Toxicol. Sci. 2013, 135, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, I.A.; Patterson, A.D.; Perdew, G.H. Aryl hydrocarbon receptor ligands in cancer: Friend and foe. Nat. Rev. Cancer 2014, 14, 801–814. [Google Scholar] [CrossRef]
- Abotaleb, M.; Samuel, S.M.; Varghese, E.; Varghese, S.; Kubatka, P.; Liskova, A.; Büsselberg, D. Flavonoids in Cancer and Apoptosis. Cancers 2018, 11, 28. [Google Scholar] [CrossRef] [Green Version]
- Chahar, M.K.; Sharma, N.; Dobhal, M.P.; Joshi, Y.C. Flavonoids: A versatile source of anticancer drugs. Pharmacogn. Rev. 2011, 5, 1–12. [Google Scholar]
- Panche, A.N.; Diwan, A.D.; Chandra, S.R. Flavonoids: An overview. J. Nutr. Sci. 2016, 5, e47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodek, P.; Trefil, P.; Stiborová, M. Flavonoids-potent and versatile biologically active compounds interacting with cytochromes P450. Chem. Biol. Interact. 2002, 139, 1–21. [Google Scholar] [CrossRef]
- Neuhouser, M.L. Review: Dietary Flavonoids and Cancer Risk: Evidence From Human Population Studies. Nutr. Cancer 2004, 50, 1–7. [Google Scholar] [CrossRef]
- Amawi, H.; Ashby, C.R., Jr.; Tiwari, A.K. Cancer chemoprevention through dietary flavonoids: What’s limiting? Chin. J. Cancer 2017, 36, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Qin, C.; Safe, S.H. Flavonoids as aryl hydrocarbon receptor agonists/antagonists: Effects of structure and cell context. Environ. Health Perspect. 2003, 111, 1877–1882. [Google Scholar] [CrossRef] [PubMed]
- Kolluri, S.K.; Jin, U.-H.; Safe, S. Role of the aryl hydrocarbon receptor in carcinogenesis and potential as an anti-cancer drug target. Arch. Toxicol. 2017, 91, 2497–2513. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Solal, K.A.; Kaufman, H.L.; Lasfar, A. Transcription Factors as Critical Players in Melanoma Invasiveness, Drug Resistance and Opportunities for Therapeutic Drug Development. Pigment. Cell Melanoma Res. 2017, 31, 241–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Straussman, R.; Morikawa, T.; Shee, K.; Barzily-Rokni, M.; Qian, Z.R.; Du, J.; Davis, A.; Mongare, M.M.; Gould, J.; Frederick, D.T.; et al. Tumor microenvironment induces innate RAF-inhibitor resistance through HGF secretion. Nature 2012, 487, 500–504. [Google Scholar] [CrossRef] [Green Version]
- Villanueva, J.; Vultur, A.; Lee, J.T.; Somasundaram, R.; Fukunaga-Kalabis, M.; Cipolla, A.K.; Wubbenhorst, B.; Xu, X.; Gimotty, P.A.; Kee, D.; et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by co-targeting MEK and IGF-1R/PI3K. Cancer Cell 2010, 18, 683–695. [Google Scholar] [CrossRef] [Green Version]
- Paraiso, K.H.T.; Xiang, Y.; Rebecca, V.W.; Abel, E.V.; Chen, Y.A.; Munko, A.C.; Wood, E.; Fedorenko, I.V.; Sondak, V.K.; Anderson, A.R.A.; et al. PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res. 2011, 71, 2750–2760. [Google Scholar] [CrossRef] [Green Version]
- Webster, M.R.; Fane, M.E.; Alicea, G.M.; Basu, S.; Kossenkov, A.V.; Marino, G.E.; Douglass, S.M.; Kaur, A.; Ecker, B.L.; Gnanapradeepan, K.; et al. Paradoxical Role for Wild-Type p53 in Driving Therapy Resistance in Melanoma. Mol. Cell 2019, 77, 633–644. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modeling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Hugo, W.; Shi, H.; Sun, L.; Piva, M.; Song, C.; Kong, X.; Moriceau, G.; Hong, A.; Dahlman, K.B.B.; Johnson, D.B.B.; et al. Non-genomic and Immune Evolution of Melanoma Acquiring MAPKi Resistance. Cell 2015, 162, 1271–1285. [Google Scholar] [CrossRef] [Green Version]
- Song, C.; Piva, M.; Sun, L.; Hong, A.; Moriceau, G.; Kong, X.; Zhang, H.; Lomeli, S.; Qian, J.; Yu, C.C.; et al. Recurrent Tumor Cell–Intrinsic and –Extrinsic Alterations during MAPKi-Induced Melanoma Regression and Early Adaptation. Cancer Discov. 2017, 7, 1248–1265. [Google Scholar] [CrossRef] [Green Version]
- Brun, S.; Bassissi, F.; Serdjebi, C.; Novello, M.; Tracz, J.; Autelitano, F.; Guillemot, M.; Fabre, P.; Courcambeck, J.; Ansaldi, C.; et al. GNS561, a new lysosomotropic small molecule, for the treatment of intrahepatic cholangiocarcinoma. Investig. New Drugs 2019, 37, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Sonego, S.; Gyengesi, E.; Rangel, A.; Niedermayer, G.; Karl, T.; Münch, G. OP-25–Anti-Inflammatory and Neuroprotective Effect of Apigenin: Studies in the GFAP-IL6 Mouse Model of Chronic Neuroinflammation. Free Radic. Biol. Med. 2017, 108, S10. [Google Scholar] [CrossRef]
- Yi, L.-T.; Li, J.-M.; Li, Y.-C.; Pan, Y.; Xu, Q.; Kong, L.-D. Antidepressant-like behavioral and neurochemical effects of the citrus-associated chemical apigenin. Life Sci. 2008, 82, 741–751. [Google Scholar] [CrossRef]
- Mao, J.J.; Xie, S.X.; Keefe, J.R.; Soeller, I.; Li, Q.S.; Amsterdam, J.D. Long-term chamomile (Matricaria chamomilla L.) treatment for generalized anxiety disorder: A randomized clinical trial. Phytomedicine 2016, 23, 1735–1742. [Google Scholar] [CrossRef] [Green Version]
- Shoara, R.; Hashempur, M.H.; Ashraf, A.; Salehi, A.; Dehshahri, S.; Habibagahi, Z. Efficacy and safety of topical Matricaria chamomilla L. (chamomile) oil for knee osteoarthritis: A randomized controlled clinical trial. Complement. Ther. Clin. Pract. 2015, 21, 181–187. [Google Scholar] [CrossRef]
- Ugurel, S.; Röhmel, J.; Ascierto, P.A.; Flaherty, K.T.; Grob, J.J.; Hauschild, A.; Larkin, J.; Long, G.V.; Lorigan, P.; McArthur, G.A.; et al. Survival of patients with advanced metastatic melanoma: The impact of novel therapies–update 2017. Eur. J. Cancer 2017, 83, 247–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kemper, K.; de Goeje, P.L.; Peeper, D.S.; van Amerongen, R. Phenotype Switching: Tumor Cell Plasticity as a Resistance Mechanism and Target for Therapy. Cancer Res. 2014, 74, 5937–5941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, F.; Haass, N.K. Microenvironment-Driven Dynamic Heterogeneity and Phenotypic Plasticity as a Mechanism of Melanoma Therapy Resistance. Front. Oncol. 2018, 8, 173. [Google Scholar] [CrossRef]
- Ginwala, R.; Bhavsar, R.; Chigbu, D.I.; Jain, P.; Khan, Z.K. Potential Role of Flavonoids in Treating Chronic Inflammatory Diseases with a Special Focus on the Anti-Inflammatory Activity of Apigenin. Antioxidants 2019, 8, 35. [Google Scholar] [CrossRef] [Green Version]
- Jin, U.-H.; Park, H.; Li, X.; Davidson, L.A.; Allred, C.; Patil, B.; Jayaprakasha, G.; Orr, A.A.; Mao, L.; Chapkin, R.S.; et al. Structure-Dependent Modulation of Aryl Hydrocarbon Receptor-Mediated Activities by Flavonoids. Toxicol. Sci. 2018, 164, 205–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukai, R.; Shirai, Y.; Saito, N.; Fukuda, I.; Nishiumi, S.; Yoshida, K.I.; Ashida, H. Suppression mechanisms of flavonoids on aryl hydrocarbon receptor-mediated signal transduction. Arch. Biochem. Biophys. 2010, 501, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Gautron, A.; Bachelot, L.; Quemener, A.; Corre, S.; Aubry, M.; Rambow, F.; Paris, A.; Tardif, N.; Leclair, H.; Coulouarn, C.; et al. Gain-of-function CRISPR screens define tumor driver genes conferring melanoma cell plasticity and therapy-resistance. Nat. Commun. 2019, 51, 66. [Google Scholar]
- Yan, X.; Qi, M.; Li, P.; Zhan, Y.; Shao, H. Apigenin in cancer therapy: Anti-cancer effects and mechanisms of action. Cell Biosci. 2017, 7, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, G.; Han, X.; Cheng, W.; Ni, J.; Zhang, Y.; Lin, J.S. Apigenin inhibits proliferation and invasion, and induces apoptosis and cell cycle arrest in human melanoma cells. Oncol. Rep. 2017, 37, 2277–2285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, M.; Shiao, C.; Lee, Y.; Shih, Y. Apigenin, a dietary flavonoid, inhibits proliferation of human bladder cancer T-24 cells via blocking cell cycle progression and inducing apoptosis. Cancer Cell Int. 2015, 15, 33. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Zhang, Y.; Tian, K.; Chen, X.; Zhang, R.; Mu, X.; Wu, Y.; Wang, D.; Wang, S.; Liu, F.; et al. Apigenin suppresses PD-L1 expression in melanoma and host dendritic cells to elicit synergistic therapeutic effects. J. Exp. Clin. Cancer Res. 2018, 37, 261. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.-W.; Xu, J.; Zhu, G.-Y.; Huang, Z.-J.; Lu, Y.; Li, X.-Q.; Wang, N.; Zhang, F.-X. Apigenin suppresses the stem cell-like properties of triple-negative breast cancer cells by inhibiting YAP/TAZ activity. Cell Death Discov. 2018, 4, 105. [Google Scholar] [CrossRef]
- Ketkaew, Y.; Osathanon, T.; Pavasant, P.; Sooampon, S. Apigenin inhibited hypoxia induced stem cell marker expression in a head and neck squamous cell carcinoma cell line. Arch. Oral Biol. 2017, 74, 69–74. [Google Scholar] [CrossRef]
- Xu, M.; Wang, S.; Song, Y.U.; Yao, J.; Huang, K.; Zhu, X. Apigenin suppresses colorectal cancer cell proliferation, migration and invasion via inhibition of the Wnt/β-catenin signaling pathway. Oncol. Lett. 2016, 11, 3075–3080. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Wei, Y.-X.; Shen, M.-C.; Tu, Y.-H.; Wang, C.-C.; Huang, H.-C. Chrysin, Abundant in Morinda citrifolia Fruit Water–EtOAc Extracts, Combined with Apigenin Synergistically Induced Apoptosis and Inhibited Migration in Human Breast and Liver Cancer Cells. J. Agric. Food Chem. 2016, 64, 4235–4245. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Afaq, F.; Mukhtar, H. Involvement of nuclear factor-kappa B, Bax and Bcl-2 in induction of cell cycle arrest and apoptosis by apigenin in human prostate carcinoma cells. Oncogene 2002, 21, 3727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angulo, P.; Kaushik, G.; Subramaniam, D.; Dandawate, P.; Neville, K.; Chastain, K.; Anant, S. Natural compounds targeting major cell signaling pathways: A novel paradigm for osteosarcoma therapy. J. Hematol. Oncol. 2017, 10, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, S.G.M. Bioavailability Challenges Associated with Development of Anti-Cancer Phenolics. Mini Rev. Med. Chem. 2010, 10, 550–567. [Google Scholar]
- Zhang, J.; Liu, D.; Huang, Y.; Gao, Y.; Qian, S. Biopharmaceutics classification and intestinal absorption study of apigenin. Int. J. Pharm. 2012, 436, 311–317. [Google Scholar] [CrossRef]
- Bunkar, N.; Shandilya, R.; Bhargava, A.; Samarth, R.M.; Tiwari, R.; Mishra, D.K.; Srivastava, R.K.; Sharma, R.S.; Lohiya, N.K. Nano-engineered flavonoids for cancer protection. Front. Biosci. 2019, 24, 1097–1157. [Google Scholar]
- Karim, R.; Palazzo, C.; Laloy, J.; Delvigne, A.-S.; Vanslambrouck, S.; Jerome, C.; Lepeltier, E.; Orange, F.; Dogne, J.-M.; Evrard, B.; et al. Development and evaluation of injectable nanosized drug delivery systems for apigenin. Int. J. Pharm. 2017, 532, 757–768. [Google Scholar] [CrossRef] [Green Version]
- Santini, A.; Cammarata, S.M.; Capone, G.; Ianaro, A.; Tenore, G.C.; Pani, L.; Novellino, E. Nutraceuticals: Opening the debate for a regulatory framework. Br. J. Clin. Pharmacol. 2018, 84, 659–672. [Google Scholar] [CrossRef] [Green Version]
- Salehi, B.; Venditti, A.; Sharifi-Rad, M.; Kręgiel, D.; Sharifi-Rad, J.; Durazzo, A.; Lucarini, M.; Santini, A.; Souto, E.B.; Novellino, E.; et al. The therapeutic potential of Apigenin. Int. J. Mol. Sci. 2019, 20, 1305. [Google Scholar] [CrossRef] [Green Version]
- Thompson, K.G.; Kim, N. Dietary supplements in dermatology: A review of the evidence for zinc, biotin, vitamin D, nicotinamide, and polypodium. J. Am. Acad. Dermatol. 2020. [Google Scholar] [CrossRef]
- Hoensch, H.; Groh, B.; Edler, L.; Kirch, W. Prospective cohort comparison of flavonoid treatment in patients with resected colorectal cancer to prevent recurrence. World J. Gastroenterol. 2008, 14, 2187–2193. [Google Scholar] [CrossRef] [PubMed]
- Samavat, H.; Ursin, G.; Emory, T.H.; Lee, E.; Wang, R.; Torkelson, C.J.; Dostal, A.M.; Swenson, K.; Le, C.T.; Yang, C.S.; et al. A Randomized Controlled Trial of Green Tea Extract Supplementation and Mammographic Density in Postmenopausal Women at Increased Risk of Breast Cancer. Cancer Prev. Res. 2017, 10, 710–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesinski, G.B.; Reville, P.K.; Mace, T.A.; Young, G.S.; Ahn-Jarvis, J.; Thomas-Ahner, J.; Vodovotz, Y.; Ameen, Z.; Grainger, E.; Riedl, K.; et al. Consumption of soy isoflavone enriched bread in men with prostate cancer is associated with reduced proinflammatory cytokines and immunosuppressive cells. Cancer Prev. Res. 2015, 8, 1036–1044. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandini, A.; Denison, M.S.; Song, Y.; Soshilov, A.A.; Bonati, L. Structural and Functional Characterization of the Aryl Hydrocarbon Receptor Ligand Binding Domain by Homology Modeling and Mutational Analysis. Biochemistry 2007, 46, 696–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilot, D.; Le Meur, N.; Giudicelli, F.; Le Vée, M.; Lagadic-Gossmann, D.; Théret, N.; Fardel, O. Rnai-based screening identifies kinases interfering with dioxin-mediated up-regulation of CYP1A1 activity. PLoS ONE 2011, 6, e18261. [Google Scholar] [CrossRef]
- Monteiro, P.; Gilot, D.; Langouet, S.; Fardel, O. Activation of the aryl hydrocarbon receptor by the calcium/calmodulin-dependent protein kinase kinase inhibitor 7-oxo-7H-benzimidazo[2,1-a]benz[de]isoquinoline-3-carboxylic acid (STO-609). Drug. Metab. Dispos. 2008, 36, 2556–2563. [Google Scholar] [CrossRef]
- Bouafia, A.; Corre, S.; Gilot, D.; Mouchet, N.; Prince, S.; Galibert, M.-D.D. p53 Requires the Stress Sensor USF1 to Direct Appropriate Cell Fate Decision. PLoS Genet. 2014, 10, e1004309. [Google Scholar] [CrossRef] [Green Version]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leclair, H.M.; Tardif, N.; Paris, A.; Galibert, M.-D.; Corre, S. Role of Flavonoids in the Prevention of AhR-Dependent Resistance During Treatment with BRAF Inhibitors. Int. J. Mol. Sci. 2020, 21, 5025. https://doi.org/10.3390/ijms21145025
Leclair HM, Tardif N, Paris A, Galibert M-D, Corre S. Role of Flavonoids in the Prevention of AhR-Dependent Resistance During Treatment with BRAF Inhibitors. International Journal of Molecular Sciences. 2020; 21(14):5025. https://doi.org/10.3390/ijms21145025
Chicago/Turabian StyleLeclair, Héloïse M., Nina Tardif, Anaïs Paris, Marie-Dominique Galibert, and Sébastien Corre. 2020. "Role of Flavonoids in the Prevention of AhR-Dependent Resistance During Treatment with BRAF Inhibitors" International Journal of Molecular Sciences 21, no. 14: 5025. https://doi.org/10.3390/ijms21145025