p62: Friend or Foe? Evidences for OncoJanus and NeuroJanus Roles


 ,
,  , ,
, , 
Abstract
:1. Introduction
2. p62 Structure, Post-translational Modifications, and Conformational Changes
2.1. PB1 Domain and p62 Oligomerization
2.2. The Zinc Finger ZZ Domain, Nuclear Localization, and Export Sequences
2.3. The TRAF binding TB Domain
2.4. The LC3-Interacting Region (LIR) Domain and Autophagy
2.5. The Keap1-Interacting Region (KIR) Domain and Nrf2 Signalling Activation
2.6. The Ubiquitin Associated (UBA) Domain, Phosphorylation, and Acetylation of p62
3. p62 Functional Interplay with Key Factors Regulating Cell Death and Survival
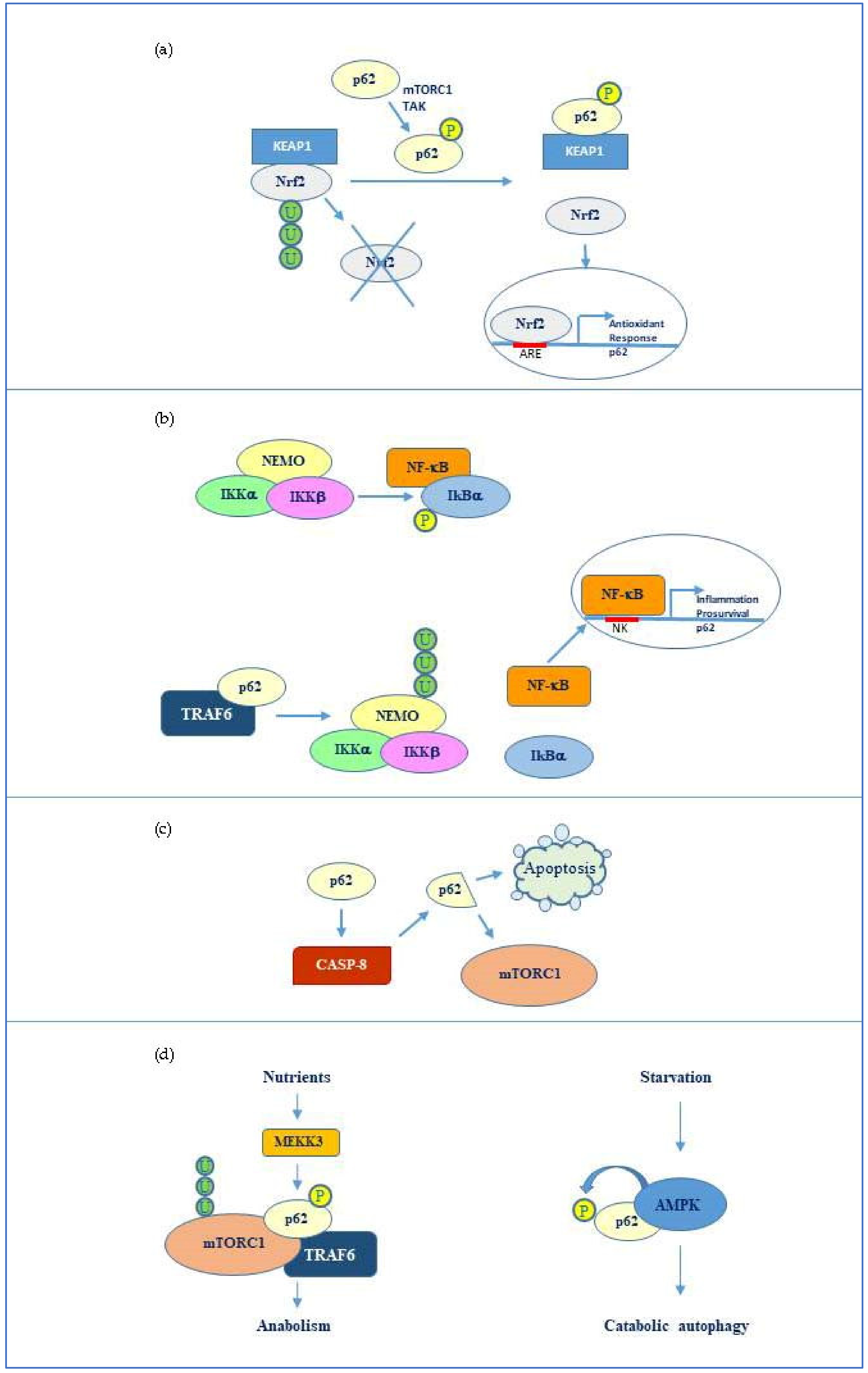
3.1. Interplay p62-Nrf2/Keap1
3.2. p62 Relationship with NF-κB and Caspase 8
3.3. p62 Interaction with mTORC1
4. OncoJanus Role of p62
4.1. Pro-Tumor Role of p62
4.2. Antitumor Role of p62
5. NeuroJanus Role of p62
5.1. p62 in Alzheimer Disease
5.2. p62 in Parkinson Disease
5.3. p62 in Amyotrophic Lateral Sclerosis
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shin, J. P62 and the sequestosome, a novel mechanism for protein metabolism. Arch. Pharm. Res. 1998, 21, 629–633. [Google Scholar] [CrossRef] [PubMed]
- Thompson, H.G.R. Identification and confirmation of a module of coexpressed genes. Genome Res. 2002, 12, 1517–1522. [Google Scholar] [CrossRef] [PubMed]
- Lamark, T.; Svenning, S.; Johansen, T. Regulation of selective autophagy: The p62/SQSTM1 paradigm. Essays Biochem. 2017, 61, 609–624. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Biological functions of autophagy genes: A disease perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef]
- Yun, H.R.; Jo, Y.H.; Kim, J.; Shin, Y.; Kim, S.S.; Choi, T.G. Roles of autophagy in oxidative stress. Int. J. Mol. Sci. 2020, 21, 3289. [Google Scholar] [CrossRef]
- Sánchez-Martín, P.; Komatsu, M. Physiological stress response by selective autophagy. J. Mol. Biol. 2020, 432, 53–62. [Google Scholar] [CrossRef]
- Zaffagnini, G.; Martens, S. Mechanisms of selective autophagy. J. Mol. Biol. 2016, 428, 1714–1724. [Google Scholar] [CrossRef] [Green Version]
- Rogov, V.; Dötsch, V.; Johansen, T.; Kirkin, V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol. Cell 2014, 53, 167–178. [Google Scholar] [CrossRef] [Green Version]
- Danieli, A.; Martens, S. p62-mediated phase separation at the intersection of the ubiquitin-proteasome system and autophagy. J. Cell Sci. 2018, 131, jcs214304. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.; Lachenmayer, M.L.; Wu, S.; Liu, W.; Kundu, M.; Wang, R.; Komatsu, M.; Oh, Y.J.; Zhao, Y.; Yue, Z. Proteotoxic stress induces phosphorylation of p62/SQSTM1 by ULK1 to regulate selective autophagic clearance of protein aggregates. PLoS Genet. 2015, 11, e1004987. [Google Scholar] [CrossRef]
- Sun, D.; Wu, R.; Zheng, J.; Li, P.; Yu, L. Polyubiquitin chain-induced p62 phase separation drives autophagic cargo segregation. Cell Res. 2018, 28, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Ciuffa, R.; Lamark, T.; Tarafder, A.K.; Guesdon, A.; Rybina, S.; Hagen, W.J.H.; Johansen, T.; Sachse, C. The selective autophagy receptor p62 forms a flexible filamentous helical scaffold. Cell Rep. 2015, 11, 748–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Min, Z.; Ting, Y.; Mingtao, G.; Xiaofei, T.; Dong, Y.; Chenguang, Z.; Wei, D. Monitoring autophagic flux using p62/SQSTM1 based luciferase reporters in glioma cells. Exp. Cell Res. 2018, 363, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Pellerito, C.; Emanuele, S.; Ferrante, F.; Celesia, A.; Giuliano, M.; Fiore, T. Tributyltin(IV) ferulate, a novel synthetic ferulic acid derivative, induces autophagic cell death in colon cancer cells: From chemical synthesis to biochemical effects. J. Inorg. Biochem. 2020, 205, 110999. [Google Scholar] [CrossRef]
- Cernigliaro, C.; D’Anneo, A.; Carlisi, D.; Giuliano, M.; Marino Gammazza, A.; Barone, R.; Longhitano, L.; Cappello, F.; Emanuele, S.; Distefano, A.; et al. Ethanol-mediated stress promotes autophagic survival and aggressiveness of colon cancer cells via activation of Nrf2/HO-1 pathway. Cancers 2019, 11, 505. [Google Scholar] [CrossRef] [Green Version]
- Emanuele, S.; Notaro, A.; Palumbo Piccionello, A.; Maggio, A.; Lauricella, M.; D’Anneo, A.; Cernigliaro, C.; Calvaruso, G.; Giuliano, M. Sicilian litchi fruit extracts induce autophagy versus apoptosis switch in human colon cancer cells. Nutrients 2018, 10, 1490. [Google Scholar] [CrossRef]
- Lee, D.H.; Park, J.S.; Lee, Y.S.; Han, J.; Lee, D.-K.; Kwon, S.W.; Han, D.H.; Lee, Y.-H.; Bae, S.H. SQSTM1/p62 activates NFE2L2/NRF2 via ULK1-mediated autophagic KEAP1 degradation and protects mouse liver from lipotoxicity. Autophagy 2020, 1–25. [Google Scholar] [CrossRef]
- Schwob, A.; Teruel, E.; Dubuisson, L.; Lormières, F.; Verlhac, P.; Abudu, Y.P.; Gauthier, J.; Naoumenko, M.; Cloarec-Ung, F.-M.; Faure, M.; et al. SQSTM-1/p62 potentiates HTLV-1 Tax-mediated NF-κB activation through its ubiquitin binding function. Sci. Rep. 2019, 9, 16014. [Google Scholar] [CrossRef] [Green Version]
- Linares, J.F.; Duran, A.; Yajima, T.; Pasparakis, M.; Moscat, J.; Diaz-Meco, M.T. K63 Polyubiquitination and activation of mTOR by the p62-TRAF6 complex in nutrient-activated cells. Mol. Cell 2013, 51, 283–296. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.; Zhong, X.; Yu, S.; Zhang, L.; Liu, Y.; Zhang, Y.; Sun, L.; Su, J. p62 aggregates mediated Caspase 8 activation is responsible for progression of ovarian cancer. J. Cell. Mol. Med. 2019, 23, 4030–4042. [Google Scholar] [CrossRef]
- Islam, M.; Sooro, M.; Zhang, P. Autophagic regulation of p62 is critical for cancer therapy. Int. J. Mol. Sci. 2018, 19, 1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, S.; Attarwala, I.Y.; Xie, X.-Q. SQSTM1/p62: A potential target for neurodegenerative disease. ACS Chem. Neurosci. 2019, 10, 2094–2114. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Martín, P.; Saito, T.; Komatsu, M. p62/SQSTM1: “Jack of all trades” in health and cancer. FEBS J. 2019, 286, 8–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Z.; Umemura, A.; Sanchez-Lopez, E.; Liang, S.; Shalapour, S.; Wong, J.; He, F.; Boassa, D.; Perkins, G.; Ali, S.R.; et al. NF-κB restricts inflammasome activation via elimination of damaged mitochondria. Cell 2016, 164, 896–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.; Liu, E.; Tang, L.; Lei, Y.; Sun, X.; Hu, J.; Dong, H.; Yang, S.-M.; Gao, M.; Tang, B. Emerging roles and regulation of MiT/TFE transcriptional factors. Cell. Commun. Signal. 2018, 16, 31. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Hanawa, N.; Katsumata, S.-I.; Katsumata-Tsuboi, R.; Takahashi, N.; Uehara, M. Iron deficiency induces autophagy and activates Nrf2 signal through modulating p62/SQSTM. Biomed. Res. 2017, 38, 343–350. [Google Scholar] [CrossRef] [Green Version]
- Gong, J. Differential stimulation of PKC phosphorylation of potassium channels by ZIP1 and ZIP2. Science 1999, 285, 1565–1569. [Google Scholar] [CrossRef]
- Kageyama, S.; Saito, T.; Obata, M.; Koide, R.; Ichimura, Y.; Komatsu, M. Negative regulation of the Keap1-Nrf2 pathway by a p62/Sqstm1 splicing variant. Mol. Cell. Biol. 2018, 38, e00642-17. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Cano, M.; Handa, J.T. p62 provides dual cytoprotection against oxidative stress in the retinal pigment epithelium. Biochem. Biophys. Acta BBA Mol. Cell. Res. 2014, 1843, 1248–1258. [Google Scholar] [CrossRef] [Green Version]
- Kwon, D.H.; Park, O.H.; Kim, L.; Jung, Y.O.; Park, Y.; Jeong, H.; Hyun, J.; Kim, Y.K.; Song, H.K. Insights into degradation mechanism of N-end rule substrates by p62/SQSTM1 autophagy adapter. Nat. Commun. 2018, 9, 3291. [Google Scholar] [CrossRef]
- Sánchez-Martín, P.; Sou, Y.-S.; Kageyama, S.; Koike, M.; Waguri, S.; Komatsu, M. NBR1-mediated p62-liquid droplets enhance the Keap1-Nrf2 system. EMBO Rep. 2020, 21, e48902. [Google Scholar] [CrossRef] [PubMed]
- Jakobi, A.J.; Huber, S.T.; Mortensen, S.A.; Schultz, S.W.; Palara, A.; Kuhm, T.; Shrestha, B.K.; Lamark, T.; Hagen, W.J.H.; Wilmanns, M.; et al. Structural basis of p62/SQSTM1 helical filaments and their role in cellular cargo uptake. Nat. Commun. 2020, 11, 440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Willis, T.L.; Button, R.W.; Strang, C.J.; Fu, Y.; Wen, X.; Grayson, P.R.C.; Evans, T.; Sipthorpe, R.J.; Roberts, S.L.; et al. Cytoplasmic DAXX drives SQSTM1/p62 phase condensation to activate Nrf2-mediated stress response. Nat. Commun. 2019, 10, 3759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moscat, J.; Diaz-Meco, M.T.; Albert, A.; Campuzano, S. Cell signaling and function organized by PB1 domain interactions. Mol. Cell 2006, 23, 631–640. [Google Scholar] [CrossRef]
- Lin, Q.; Dai, Q.; Meng, H.; Sun, A.; Wei, J.; Peng, K.; Childress, C.; Chen, M.; Shao, G.; Yang, W. The HECT E3 ubiquitin ligase NEDD4 interacts with and ubiquitylates SQSTM1 for inclusion body autophagy. J. Cell. Sci. 2017, 130, 3839–3850. [Google Scholar] [CrossRef]
- Feng, Y.; Klionsky, D.J. Autophagy regulates DNA repair through SQSTM1/p62. Autophagy 2017, 13, 995–996. [Google Scholar] [CrossRef]
- Salmina, K.; Bojko, A.; Inashkina, I.; Staniak, K.; Dudkowska, M.; Podlesniy, P.; Rumnieks, F.; Vainshelbaum, N.M.; Pjanova, D.; Sikora, E.; et al. “Mitotic slippage” and extranuclear DNA in cancer chemoresistance: A focus on telomeres. Int. J. Mol.Sci. 2020, 21, 2779. [Google Scholar] [CrossRef]
- Noguchi, T.; Suzuki, M.; Mutoh, N.; Hirata, Y.; Tsuchida, M.; Miyagawa, S.; Hwang, G.-W.; Aoki, J.; Matsuzawa, A. Nuclear-accumulated SQSTM1/p62-based ALIS act as microdomains sensing cellular stresses and triggering oxidative stress-induced parthanatos. Cell Death Dis. 2018, 9, 1193. [Google Scholar] [CrossRef]
- Yan, C.; Dodd, T.; He, Y.; Tainer, J.A.; Tsutakawa, S.E.; Ivanov, I. Transcription preinitiation complex structure and dynamics provide insight into genetic diseases. Nat. Struct. Mol. Biol. 2019, 26, 397–406. [Google Scholar] [CrossRef]
- Greber, B.J.; Toso, D.B.; Fang, J.; Nogales, E. The complete structure of the human TFIIH core complex. eLife 2019, 8, e44771. [Google Scholar] [CrossRef]
- Schimmack, G.; Schorpp, K.; Kutzner, K.; Gehring, T.; Brenke, J.K.; Hadian, K.; Krappmann, D. YOD1/TRAF6 association balances p62-dependent IL-1 signaling to NF-κB. eLife 2017, 6, e22416. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Lee, D.-H.; Dilly, A.-K.; Lee, Y.-S.; Choudry, H.A.; Kwon, Y.T.; Bartlett, D.L.; Lee, Y.J. Crosstalk between apoptosis and autophagy is regulated by the arginylated BiP/Beclin-1/p62 complex. Mol Cancer Res. 2018, 16, 1077–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Godar, R.J.; Liu, H.; Diwan, A. Enhancing lysosome biogenesis attenuates BNIP3-induced cardiomyocyte death. Autophagy 2012, 8, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Harder, B.; Rojo de la Vega, M.; Wong, P.K.; Chapman, E.; Zhang, D.D. p62 links autophagy and Nrf2 signaling. Free Rad. Biol. Med. 2015, 88, 199–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichimura, Y.; Komatsu, M. Activation of p62/SQSTM1–Keap1–nuclear factor erythroid 2-related factor 2 pathway in cancer. Front. Oncol. 2018, 8, 210. [Google Scholar] [CrossRef]
- You, Z.; Jiang, W.-X.; Qin, L.-Y.; Gong, Z.; Wan, W.; Li, J.; Wang, Y.; Zhang, H.; Peng, C.; Zhou, T.; et al. Requirement for p62 acetylation in the aggregation of ubiquitylated proteins under nutrient stress. Nat. Commun. 2019, 10, 5792. [Google Scholar] [CrossRef]
- Yun, C.; Lee, S. The roles of autophagy in cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef] [Green Version]
- Guo, F.; Liu, X.; Cai, H.; Le, W. Autophagy in neurodegenerative diseases: Pathogenesis and therapy: Autophagy in neurodegenerative diseases. Brain Pathol. 2018, 28, 3–13. [Google Scholar] [CrossRef]
- Denton, D.; Kumar, S. Autophagy-dependent cell death. Cell Death Differ. 2019, 26, 605–616. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.; Lamark, T.; Sjøttem, E.; Larsen, K.B.; Awuh, J.A.; Øvervatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, K.; Fujikawa, N.; Komatsu, M.; Ishii, T.; Unno, M.; Akaike, T.; Motohashi, H.; Yamamoto, M. Keap1 degradation by autophagy for the maintenance of redox homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 13561–13566. [Google Scholar] [CrossRef]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 Years of NF-κB: A blossoming of relevance to human pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.X.; Sibon, O.C.M.; Dijkers, P.F. Inhibition of NF-κB in astrocytes is sufficient to delay neurodegeneration induced by proteotoxicity in neurons. J. Neuroinflam. 2018, 15, 261. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Moreno-Villanueva, M.; Krieger, S.; Ramesh, G.; Neelam, S.; Wu, H. Transcriptomics, NF-κB pathway, and their potential spaceflight-related health consequences. Int. J. Mol. Sci. 2017, 18, 1166. [Google Scholar] [CrossRef] [PubMed]
- Zotti, T.; Scudiero, I.; Settembre, P.; Ferravante, A.; Mazzone, P.; D’Andrea, L.; Reale, C.; Vito, P.; Stilo, R. TRAF6-mediated ubiquitination of NEMO requires p62/sequestosome-1. Mol. Immun. 2014, 58, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Ozato, K. The sequestosome 1/p62 attenuates cytokine gene expression in activated macrophages by inhibiting IFN regulatory factor 8 and TNF receptor-associated factor 6/NF-κB activity. J. Immunol. 2009, 182, 2131–2140. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Kageyama, S.; Ichimura, Y. p62/SQSTM1/A170: Physiology and pathology. Pharmacol. Res. 2012, 66, 457–462. [Google Scholar] [CrossRef]
- Young, M.M.; Takahashi, Y.; Khan, O.; Park, S.; Hori, T.; Yun, J.; Sharma, A.K.; Amin, S.; Hu, C.-D.; Zhang, J.; et al. Autophagosomal membrane serves as platform for intracellular death-inducing signaling complex (iDISC)-mediated Caspase-8 activation and apoptosis. J. Biol. Chem. 2012, 287, 12455–12468. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Zhu, Q.; Bu, X.; Zhou, Y.; Bai, D.; Guo, Q.; Gao, Y.; Lu, N. Triggering apoptosis by oroxylin A through caspase-8 activation and p62/SQSTM1 proteolysis. Redox Biol. 2020, 29, 101392. [Google Scholar] [CrossRef]
- Sanchez-Garrido, J.; Sancho-Shimizu, V.; Shenoy, A.R. Regulated proteolysis of p62/SQSTM1 enables differential control of autophagy and nutrient sensing. Sci. Signal. 2018, 11, eaat6903. [Google Scholar] [CrossRef] [Green Version]
- Rabanal-Ruiz, Y.; Korolchuk, V. mTORC1 and nutrient homeostasis: The central role of the lysosome. Int. J. Mol. Sci. 2018, 19, 818. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Duran, A.; Amanchy, R.; Linares, J.F.; Joshi, J.; Abu-Baker, S.; Porollo, A.; Hansen, M.; Moscat, J.; Diaz-Meco, M.T. p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol. Cell 2011, 44, 134–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linares, J.F.; Duran, A.; Reina-Campos, M.; Aza-Blanc, P.; Campos, A.; Moscat, J.; Diaz-Meco, M.T. Amino acid activation of mTORC1 by a PB1-domain-driven kinase complex cascade. Cell Rep. 2015, 12, 1339–1352. [Google Scholar] [CrossRef] [PubMed]
- Howell, J.J.; Hellberg, K.; Turner, M.; Talbott, G.; Kolar, M.J.; Ross, D.S.; Hoxhaj, G.; Saghatelian, A.; Shaw, R.J.; Manning, B.D. Metformin inhibits hepatic mTORC1 signaling via dose-dependent mechanisms involving AMPK and the TSC complex. Cell Metab. 2017, 25, 463–471. [Google Scholar] [CrossRef]
- Ha, S.; Jeong, S.-H.; Yi, K.; Chung, K.M.; Hong, C.J.; Kim, S.W.; Kim, E.-K.; Yu, S.-W. Phosphorylation of p62 by AMP-activated protein kinase mediates autophagic cell death in adult hippocampal neural stem cells. J. Biol. Chem. 2017, 292, 13795–13808. [Google Scholar] [CrossRef] [Green Version]
- Umemura, A.; He, F.; Taniguchi, K.; Nakagawa, H.; Yamachika, S.; Font-Burgada, J.; Zhong, Z.; Subramaniam, S.; Raghunandan, S.; Duran, A.; et al. p62, Upregulated during preneoplasia, induces hepatocellular carcinogenesis by maintaining survival of stressed HCC-initiating cells. Cancer Cell 2016, 29, 935–948. [Google Scholar] [CrossRef]
- Ryoo, I.; Choi, B.; Ku, S.-K.; Kwak, M.-K. High CD44 expression mediates p62-associated NFE2L2/NRF2 activation in breast cancer stem cell-like cells: Implications for cancer stem cell resistance. Redox Biol. 2018, 17, 246–258. [Google Scholar] [CrossRef]
- Schläfli, A.M.; Adams, O.; Galván, J.A.; Gugger, M.; Savic, S.; Bubendorf, L.; Schmid, R.A.; Becker, K.-F.; Tschan, M.P.; Langer, R.; et al. Prognostic value of the autophagy markers LC3 and p62/SQSTM1 in early-stage non-small cell lung cancer. Oncotarget 2016, 7, 39544–39555. [Google Scholar] [CrossRef]
- Masuda, G.O.; Yashiro, M.; Kitayama, K.; Miki, Y.; Kasashima, H.; Kinoshita, H.; Morisaki, T.; Fukuoka, T.; Hasegawa, T.; Sakurai, K.; et al. Clinicopathological correlations of autophagy-related proteins LC3, Beclin 1 and p62 in gastric cancer. Anticancer Res. 2016, 36, 129–136. [Google Scholar]
- Lei, C.; Zhao, B.; Liu, L.; Zeng, X.; Yu, Z.; Wang, X. Expression and clinical significance of p62 protein in colon cancer. Medicine 2020, 99, e18791. [Google Scholar] [CrossRef]
- Lam, H.C.; Baglini, C.V.; Lope, A.L.; Parkhitko, A.A.; Liu, H.-J.; Alesi, N.; Malinowska, I.A.; Ebrahimi-Fakhari, D.; Saffari, A.; Yu, J.J.; et al. p62/SQSTM1 cooperates with hyperactive mTORC1 to regulate glutathione production, maintain mitochondrial integrity, and promote tumorigenesis. Cancer Res. 2017, 77, 3255–3267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, H.; Wang, C.; Croce, C.M.; Guan, J.-L. p62/SQSTM1 synergizes with autophagy for tumor growth in vivo. Genes Dev. 2014, 28, 1204–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Jiang, D.; Wu, K. p62 promotes bladder cancer cell growth by activating KEAP1/NRF2-dependent antioxidative response. Cancer Sci. 2020, 111, 1156–1164. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, D.; Ohe, T.; Takahashi, K.; Imamura, R.; Kojima, H.; Okabe, T.; Ichimura, Y.; Komatsu, M.; Yamamoto, M.; Nagano, T.; et al. Inhibitors of the protein–protein interaction between phosphorylated p62 and Keap1 attenuate chemoresistance in a human hepatocellular carcinoma cell line. Free Rad. Res. 2020, 1–13. [Google Scholar] [CrossRef]
- Hwang, S.-K.; Jeong, Y.-J.; Chang, Y.-C. PDCD4 inhibits lung tumorigenesis by the suppressing p62-Nrf2 signaling pathway and upregulating Keap1 expression. Am. J. Cancer Res. 2020, 10, 424–439. [Google Scholar]
- Manirujjaman, M.; Ozaki, I.; Murata, Y.; Guo, J.; Xia, J.; Nishioka, K.; Perveen, R.; Takahashi, H.; Anzai, K.; Matsuhashi, S. Degradation of the tumor suppressor PDCD4 is impaired by the suppression of p62/SQSTM1 and autophagy. Cells 2020, 9, 218. [Google Scholar] [CrossRef]
- Sanchez-Lopez, E.; Ghia, E.M.; Antonucci, L.; Sharma, N.; Rassenti, L.Z.; Xu, J.; Sun, B.; Kipps, T.J.; Karin, M. NF-κB-p62-NRF2 survival signaling is associated with high ROR1 expression in chronic lymphocytic leukemia. Cell Death Differ. 2020. [Google Scholar] [CrossRef]
- Tamura, K.; Watanabe, K.; Matsushita, Y.; Watanabe, H.; Motoyama, D.; Ito, T.; Sugiyama, T.; Otsuka, A.; Miyake, H. Enhanced sensitivity to NVP-BEZ235 by inhibition of p62/SQSTM1 in human bladder cancer KoTCC-1 cells both in vitro and in vivo. In Vivo 2020, 34, 1001–1008. [Google Scholar] [CrossRef]
- Xu, L.; Xu, F.; Kong, Q.; Yang, T.; Tan, D.; Zhang, X.; Li, N.; Zhao, S.; Zhao, J.; Li, M. Inhibition of p62/SQSTM1 sensitizes small-cell lung cancer cells to cisplatin-induced cytotoxicity by targeting NEDD9 expression. Mol. Carcinog. 2020, mc.23215. [Google Scholar] [CrossRef]
- Valencia, T.; Kim, J.Y.; Abu-Baker, S.; Moscat-Pardos, J.; Ahn, C.S.; Reina-Campos, M.; Duran, A.; Castilla, E.A.; Metallo, C.M.; Diaz-Meco, M.T.; et al. Metabolic reprogramming of stromal fibroblasts through p62-mTORC1 signaling promotes inflammation and tumorigenesis. Cancer Cell 2014, 26, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Diaz-Meco, M.T.; Moscat, J. The macroenviromental control of cancer metabolism by p62. Cell Cycle 2018, 17, 2110–2121. [Google Scholar] [CrossRef]
- Linares, J.F.; Cordes, T.; Duran, A.; Reina-Campos, M.; Valencia, T.; Ahn, C.S.; Castilla, E.A.; Moscat, J.; Metallo, C.M.; Diaz-Meco, M.T. ATF4-induced metabolic reprograming is a synthetic vulnerability of the p62-deficient tumor stroma. Cell Metabolism 2017, 26, 817–829.e6. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Wang, Y.; Zhu, W.-G.; Zhao, Y. Autophagy substrate SQSTM1/p62 regulates chromatin ubiquitination during the DNA damage response. Autophagy 2017, 13, 212–213. [Google Scholar] [CrossRef]
- Venanzi, F.M.; Gabai, V.; Mariotti, F.; Magi, G.E.; Vullo, C.; Sufianov, A.A.; Kolesnikov, S.I.; Shneider, A. p62-DNA-encoding plasmid reverts tumor grade, changes tumor stroma, and enhances anticancer immunity. Aging 2019, 11, 10711–10722. [Google Scholar] [CrossRef]
- Choi, Y.K.; Kang, J.-I.; Han, S.; Kim, Y.R.; Jo, J.; Kang, Y.W.; Choo, D.R.; Hyun, J.W.; Koh, Y.S.; Yoo, E.-S.; et al. L-ascorbic acid inhibits breast cancer growth by inducing IRE/JNK/CHOP-related endoplasmic reticulum stress-mediated p62/SQSTM1 accumulation in the nucleus. Nutrients 2020, 12, 1351. [Google Scholar] [CrossRef]
- Liu, J.-L.; Chen, F.-F.; Lung, J.; Lo, C.-H.; Lee, F.-H.; Lu, Y.-C.; Hung, C.-H. Prognostic significance of p62/SQSTM1 subcellular localization and LC3B in oral squamous cell carcinoma. Br. J. Cancer 2014, 111, 944–954. [Google Scholar] [CrossRef] [Green Version]
- Ponomarenko, D.M.; Gabai, V.L.; Sufianov, A.A.; Kolesnikov, S.I.; Shneider, A.M. Response of a chemo-resistant triple-negative breast cancer patient to a combination of p62-encoding plasmid, Elenagen, and CMF chemotherapy. Oncotarget 2020, 11, 294–299. [Google Scholar] [CrossRef]
- Lou, J.-S.; Bi, W.-C.; Chan, G.K.L.; Jin, Y.; Wong, C.-W.; Zhou, Z.-Y.; Wang, H.-Y.; Yao, P.; Dong, T.T.X.; Tsim, K.W.K. Ginkgetin induces autophagic cell death through p62/SQSTM1-mediated autolysosome formation and redox setting in non-small cell lung cancer. Oncotarget 2017, 8, 93131–93148. [Google Scholar] [CrossRef]
- Li, S.-S.; Xu, L.-Z.; Zhou, W.; Yao, S.; Wang, C.-L.; Xia, J.-L.; Wang, H.-F.; Kamran, M.; Xue, X.-Y.; Dong, L.; et al. p62/SQSTM1 interacts with vimentin to enhance breast cancer metastasis. Carcinogenesis 2017, 38, 1092–1103. [Google Scholar] [CrossRef]
- Xu, L.-Z.; Li, S.-S.; Zhou, W.; Kang, Z.-J.; Zhang, Q.-X.; Kamran, M.; Xu, J.; Liang, D.-P.; Wang, C.-L.; Hou, Z.-J.; et al. p62/SQSTM1 enhances breast cancer stem-like properties by stabilizing MYC mRNA. Oncogene 2017, 36, 304–317. [Google Scholar] [CrossRef]
- Kim, J.S.; Bae, G.E.; Kim, K.-H.; Lee, S.-I.; Chung, C.; Lee, D.; Lee, T.H.; Kwon, I.S.; Yeo, M.-K. Prognostic significance of LC3B and p62/SQSTM1 expression in gastric adenocarcinoma. Anticancer Res. 2019, 39, 6711–6722. [Google Scholar] [CrossRef]
- Yan, X.-Y.; Zhang, Y.; Zhang, J.-J.; Zhang, L.-C.; Liu, Y.-N.; Wu, Y.; Xue, Y.-N.; Lu, S.-Y.; Su, J.; Sun, L.-K. p62/SQSTM1 as an oncotarget mediates cisplatin resistance through activating RIP1-NF-κB pathway in human ovarian cancer cells. Cancer Sci. 2017, 108, 1405–1413. [Google Scholar] [CrossRef]
- Wu, Q.; Xiang, M.; Wang, K.; Chen, Z.; Long, L.; Tao, Y.; Liang, Y.; Yan, Y.; Xiao, Z.; Qiu, S.; et al. Overexpression of p62 induces autophagy and promotes proliferation, migration and invasion of nasopharyngeal carcinoma cells through promoting ERK signaling pathway. Curr. Cancer Drug Targets 2020, 20. [Google Scholar] [CrossRef]
- Saito, T.; Ichimura, Y.; Taguchi, K.; Suzuki, T.; Mizushima, T.; Takagi, K.; Hirose, Y.; Nagahashi, M.; Iso, T.; Fukutomi, T.; et al. p62/Sqstm1 promotes malignancy of HCV-positive hepatocellular carcinoma through Nrf2-dependent metabolic reprogramming. Nat. Commun. 2016, 7, 12030. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, I. p62 manipulation affects chlorin e6-mediated photodynamic therapy efficacy in colorectal cancer cell lines. Oncol. Lett. 2020. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Dai, C.; Fan, Y.; Guo, B.; Ren, K.; Sun, T.; Wang, W. From autophagy to mitophagy: The roles of P62 in neurodegenerative diseases. J. Bioenerg. Biomembr. 2017, 49, 413–422. [Google Scholar] [CrossRef]
- Khan, S.; Barve, K.H.; Kumar, M.S. Recent advancements in pathogenesis, diagnostics and treatment of Alzheimer’s disease. Curr. Neuropharmacol. 2020, 18. [Google Scholar] [CrossRef]
- Nilsson, P.; Sekiguchi, M.; Akagi, T.; Izumi, S.; Komori, T.; Hui, K.; Sörgjerd, K.; Tanaka, M.; Saito, T.; Iwata, N.; et al. Autophagy-related protein 7 deficiency in amyloid β (Aβ) precursor protein transgenic mice decreases Aβ in the multivesicular bodies and induces Aβ accumulation in the Golgi. Am. J. Pathol. 2015, 185, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Wang, W.; Liu, R.; Huang, H.; Zhang, R.; Sun, L. Effect of p62 on tau hyperphosphorylation in a rat model of Alzheimer’s disease. Neural Regen. Res. 2012, 7, 1304–1311. [Google Scholar] [CrossRef] [PubMed]
- Geetha, T.; Zheng, C.; McGregor, W.C.; Douglas White, B.; Diaz-Meco, M.T.; Moscat, J.; Babu, J.R. TRAF6 and p62 inhibit amyloid β-induced neuronal death through p75 neurotrophin receptor. Neurochem. Intern. 2012, 61, 1289–1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanji, K.; Miki, Y.; Ozaki, T.; Maruyama, A.; Yoshida, H.; Mimura, J.; Matsumiya, T.; Mori, F.; Imaizumi, T.; Itoh, K.; et al. Phosphorylation of serine 349 of p62 in Alzheimer’s disease brain. Acta Neuropathol. Commun. 2014, 2, 50. [Google Scholar] [CrossRef] [PubMed]
- Fão, L.; Mota, S.I.; Rego, A.C. Shaping the Nrf2-ARE-related pathways in Alzheimer’s and Parkinson’s diseases. Ageing Res. Rev. 2019, 54, 100942. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Siu, W.; Li, L.; Jin, Y.; Liang, S.; Cao, M.; Ma, M.; Wu, Z. Autophagy in Alzheimer’s disease and promising modulatory effects of herbal medicine. Exp. Geront. 2019, 119, 100–110. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, T.; Ge, X.; Chen, J.; Zhao, Y.; Fu, J. Parkin overexpression attenuates Aβ-induced mitochondrial dysfunction in HEK293 cells by restoring impaired mitophagy. Life Sci. 2020, 244, 117322. [Google Scholar] [CrossRef]
- Deng, M.; Huang, L.; Zhong, X. β-asarone modulates Beclin-1, LC3 and p62 expression to attenuate Aβ40 and Aβ42 levels in APP/PS1 transgenic mice with Alzheimer’s disease. Mol. Med. Rep. 2020. [Google Scholar] [CrossRef]
- Cacabelos, R. Parkinson’s disease: From pathogenesis to pharmacogenomics. Int. J. Mol. Sci. 2017, 18, 551. [Google Scholar] [CrossRef]
- Raza, C.; Anjum, R.; Shakeel, N.U.A. Parkinson’s disease: Mechanisms, translational models and management strategies. Life Sci. 2019, 226, 77–90. [Google Scholar] [CrossRef]
- Sato, S.; Uchihara, T.; Fukuda, T.; Noda, S.; Kondo, H.; Saiki, S.; Komatsu, M.; Uchiyama, Y.; Tanaka, K.; Hattori, N. Loss of autophagy in dopaminergic neurons causes Lewy pathology and motor dysfunction in aged mice. Sci. Rep. 2018, 8, 2813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, Y.; Tatebe, H.; Taguchi, K.; Endo, Y.; Tokuda, T.; Mizuno, T.; Nakagawa, M.; Tanaka, M. p62/SQSTM1-dependent autophagy of Lewy body-like α-synuclein inclusions. PLoS ONE 2012, 7, e52868. [Google Scholar] [CrossRef]
- Wu, F.; Xu, H.-D.; Guan, J.-J.; Hou, Y.-S.; Gu, J.-H.; Zhen, X.-C.; Qin, Z.-H. Rotenone impairs autophagic flux and lysosomal functions in Parkinson’s disease. Neuroscience 2015, 284, 900–911. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Li, S.; Wu, H.; Gao, R.; Rao, G.; Wang, D.; Chen, Z.; Ma, B.; Wang, H.; Sui, N.; et al. Parkin promotes proteasomal degradation of p62: Implication of selective vulnerability of neuronal cells in the pathogenesis of Parkinson’s disease. Protein Cell 2016, 7, 114–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ajroud-Driss, S.; Siddique, T. Sporadic and hereditary amyotrophic lateral sclerosis (ALS). Biochem. Biophys. Acta BBA Mol. Basis Dis. 2015, 1852, 679–684. [Google Scholar] [CrossRef]
- Nowicka, N.; Juranek, J.; Juranek, J.K.; Wojtkiewicz, J. Risk factors and emerging therapies in amyotrophic lateral sclerosis. Int. J. Mol. Sci. 2019, 20, 2616. [Google Scholar] [CrossRef] [Green Version]
- Pansarasa, O.; Bordoni, M.; Diamanti, L.; Sproviero, D.; Gagliardi, S.; Cereda, C. SOD1 in amyotrophic lateral sclerosis: “Ambivalent” behavior connected to the disease. Int. J. Mol. Sci. 2018, 19, 1345. [Google Scholar] [CrossRef] [PubMed]
- Kwok, C.T.; Morris, A.; de Belleroche, J.S. Sequestosome-1 (SQSTM1) sequence variants in ALS cases in the UK: Prevalence and coexistence of SQSTM1 mutations in ALS kindred with PDB. Eur. J. Hum. Genet. 2014, 22, 492–496. [Google Scholar] [CrossRef]
- Teyssou, E.; Takeda, T.; Lebon, V.; Boillée, S.; Doukouré, B.; Bataillon, G.; Sazdovitch, V.; Cazeneuve, C.; Meininger, V.; LeGuern, E.; et al. Mutations in SQSTM1 encoding p62 in amyotrophic lateral sclerosis: Genetics and neuropathology. Acta Neuropathol. 2013, 125, 511–522. [Google Scholar] [CrossRef]
- Yilmaz, R.; Müller, K.; Brenner, D.; Volk, A.E.; Borck, G.; Hermann, A.; Meitinger, T.; Strom, T.M.; Danzer, K.M.; Ludolph, A.C.; et al. SQSTM1/p62 variants in 486 patients with familial ALS from Germany and Sweden. Neurobiol. Aging 2020, 87, 139.e9–139.e15. [Google Scholar] [CrossRef]
- Gal, J.; Ström, A.-L.; Kwinter, D.M.; Kilty, R.; Zhang, J.; Shi, P.; Fu, W.; Wooten, M.W.; Zhu, H. Sequestosome 1/p62 links familial ALS mutant SOD1 to LC3 via an ubiquitin-independent mechanism. J. Neurochem. 2009, 111, 1062–1073. [Google Scholar] [CrossRef] [Green Version]
- Vicencio, E.; Beltrán, S.; Labrador, L.; Manque, P.; Nassif, M.; Woehlbier, U. Implications of selective autophagy dysfunction for ALS pathology. Cells 2020, 9, 381. [Google Scholar] [CrossRef]
- Deng, Z.; Lim, J.; Wang, Q.; Purtell, K.; Wu, S.; Palomo, G.M.; Tan, H.; Manfredi, G.; Zhao, Y.; Peng, J.; et al. ALS-FTLD-linked mutations of SQSTM1/p62 disrupt selective autophagy and NFE2L2/NRF2 anti-oxidative stress pathway. Autophagy 2020, 16, 917–931. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.K.H.; Thombre, R.; Wang, J. Autophagy as a common pathway in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 697, 34–48. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Jeon, Y.-M.; Cha, S.J.; Kim, S.; Kwon, Y.; Jo, M.; Jang, Y.-N.; Lee, S.; Kim, J.; Kim, S.R.; et al. PTK2/FAK regulates UPS impairment via SQSTM1/p62 phosphorylation in TARDBP/TDP-43 proteinopathies. Autophagy 2019, 1–17. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| p62 pro-tumor activity | |||
| Tumor Type | Supposed Role | Identified Intermediate | References |
| Lung cancer | - Promotion of cisplatin resistance - Resistance to Autophagic Cell Death - Promotion of tumorigenesis | Positive NEDD9 regulation Increased Nrf2/ARE activity and ROS production Increased Nrf2 Signalling and Upregulating Keap1 | [81] [91] [77] |
| Breast cancer | - Association with invasive phenotypes - Tumour growth and progression, drug resistance. - Enhancement of breast cancer stem-like properties | Vimentin upregulation CD44-mediated Nrf2 activation MYC mRNA stabilization | [92] [69] [93] |
| Gastric cancer | - Correlation with lymph node metastasis, vessel invasion and hepatic metastasis - High cytoplasmic and low nuclear p62 levels associated with poor prognosis | Autophagy promotion Not determined | [71] [94] |
| Colon cancer | - Independent risk factor for poor prognosis | Not determined | [72] |
| Ovarian carcinoma | - Involvement in cisplatin resistance | NF-κB activation and Lys63-linked RIP1 ubiquitination | [95] |
| Bladder Cancer Cells | - Promotion of Cell Growth - Resistance to apoptotic cell death | Keap1/Nrf2-dependent antioxidant response activation Resistance to PI3K/mTOR inhibitor effect. | [75] [80] |
| Nasopharyngeal Carcinoma Cells | - Induction of pro-survival autophagy, proliferation, migration and invasion | Stimulation of ERK Signaling Pathway | [96] |
| Chronic lymphocytic leukemia | - Promotion of cell survival | Enhancement of mTORC1 signalling and activation of Nrf2 | [79] |
| Hepatocellular carcinoma | - Protection from oxidative stress-induced death - Tolerance to anti-cancer drugs, metabolic reprogramming and promotion of malignancy | Activation of Nrf2 and mTORC1, and c-Myc induction Nrf2 activation | [68] [97] |
| Renal carcinoma | - Stimulation of TSC2-driven tumorigenesis | Interaction with mTORC1 complex | [73] |
| p62 anti-tumor activity | |||
| Tumor Type | Supposed Role | Identified Intermediate | References |
| Prostate cancer | - p62 loss in the tumor microenvironment and increased inflammation and tumorigenesis - Metabolic reprogramming of tumor associated stroma lacking p62 and tumor growth | Regulation of mTORC1/c-Myc pathway Upregulation of stromal ATF4 by p62 deficiency | [82] [84] |
| Ovarian cancer | - Autophagic flux blockage | Caspase-8 Activation | [20] |
| Cervical cancer | - Sensitization to radiation by nuclear p62 | Inhibition of Histone H2A ubiquitination | [86] |
| Colorectal cancer | - Sensitization to photodynamic therapy - Induction of autophagic cell death | Activation of autophagic cell death pathway | [14,98] |
| Breast cancer | - Mediation of ascorbic acid anti-proliferative effect. - Chemoresistance attenuation by p62 encoding plasmid | Induction of IRE/JNK/CHOP -Related ER Stress Not determined | [88] [90] |
| Mammary carcinoma | - Reversion of tumor grade and anticancer immunity stimulation by ectopic expression of p62 | Increase in intra-tumor T cells | [87] |
| Melanoma | - Tumor growth reversion by p62 encoding plasmid | Not determined | [87] |
| Oral squamous cell carcinoma | - Association of low nuclear p62 expression with aggressive clinicopathologic features | Not determined | [89] |
| Neurodegenerative Disease | p62 Status | Supposed Mechanism or Identified Interactor | References |
|---|---|---|---|
| p62 Neurotoxic Function | |||
| Alzheimer | - Increased p62 (Ser349) phosphorylation - Increased levels | Nrf2 signalling aberrant increase Excessive mitophagy | [104] [99] |
| Parkinson | - Aberrant expression - Accumulation and aggregation in dopamingeric neurons | Increased α-synuclein in pathological inclusions Parkin deficiency | [113] [114] |
| Lateral Amyotrophic Sclerosis | - Mutation and accumulation - TDP43 overexpression dependent p62 (Ser403) phosphorylation | Protein aggregates in motor neurons Poly-ubiquitinated protein accumulation | [120] [118,125] |
| p62 neuroprotective function | |||
| Alzheimer | - Low expression or loss of function - Induced expression of p62 | Autophagy failure, misfolded protein aggregation, Aβ accumulation, Tau hyperphsphorylation Pro-survival autophagy, Aβ accumulation and toxicity TRAF6-mediated p75 ubiquitination | [22,99] [101,102,103,108] |
| Parkinson | - Normal levels - Loss of p62 | Autophagic clearance of α-Synuclein inclusions Parkin/Pink1-mediated mitophagy Lewy pathology and motor dysfunction | [112] [114] [111] |
| Amyotrophic Lateral Sclerosis | - Normal levels | Targeting mutant SOD1 to selective autophagy. Stimulation of Nrf2-mediated antioxidant response Autophagic elimination of TDP43/FUS positive stress granules | [121] [123,124] [125] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Emanuele, S.; Lauricella, M.; D’Anneo, A.; Carlisi, D.; De Blasio, A.; Di Liberto, D.; Giuliano, M. p62: Friend or Foe? Evidences for OncoJanus and NeuroJanus Roles. Int. J. Mol. Sci. 2020, 21, 5029. https://doi.org/10.3390/ijms21145029
Emanuele S, Lauricella M, D’Anneo A, Carlisi D, De Blasio A, Di Liberto D, Giuliano M. p62: Friend or Foe? Evidences for OncoJanus and NeuroJanus Roles. International Journal of Molecular Sciences. 2020; 21(14):5029. https://doi.org/10.3390/ijms21145029
Chicago/Turabian StyleEmanuele, Sonia, Marianna Lauricella, Antonella D’Anneo, Daniela Carlisi, Anna De Blasio, Diana Di Liberto, and Michela Giuliano. 2020. "p62: Friend or Foe? Evidences for OncoJanus and NeuroJanus Roles" International Journal of Molecular Sciences 21, no. 14: 5029. https://doi.org/10.3390/ijms21145029