EPAC in Vascular Smooth Muscle Cells

 , ,
, , 

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. EPAC
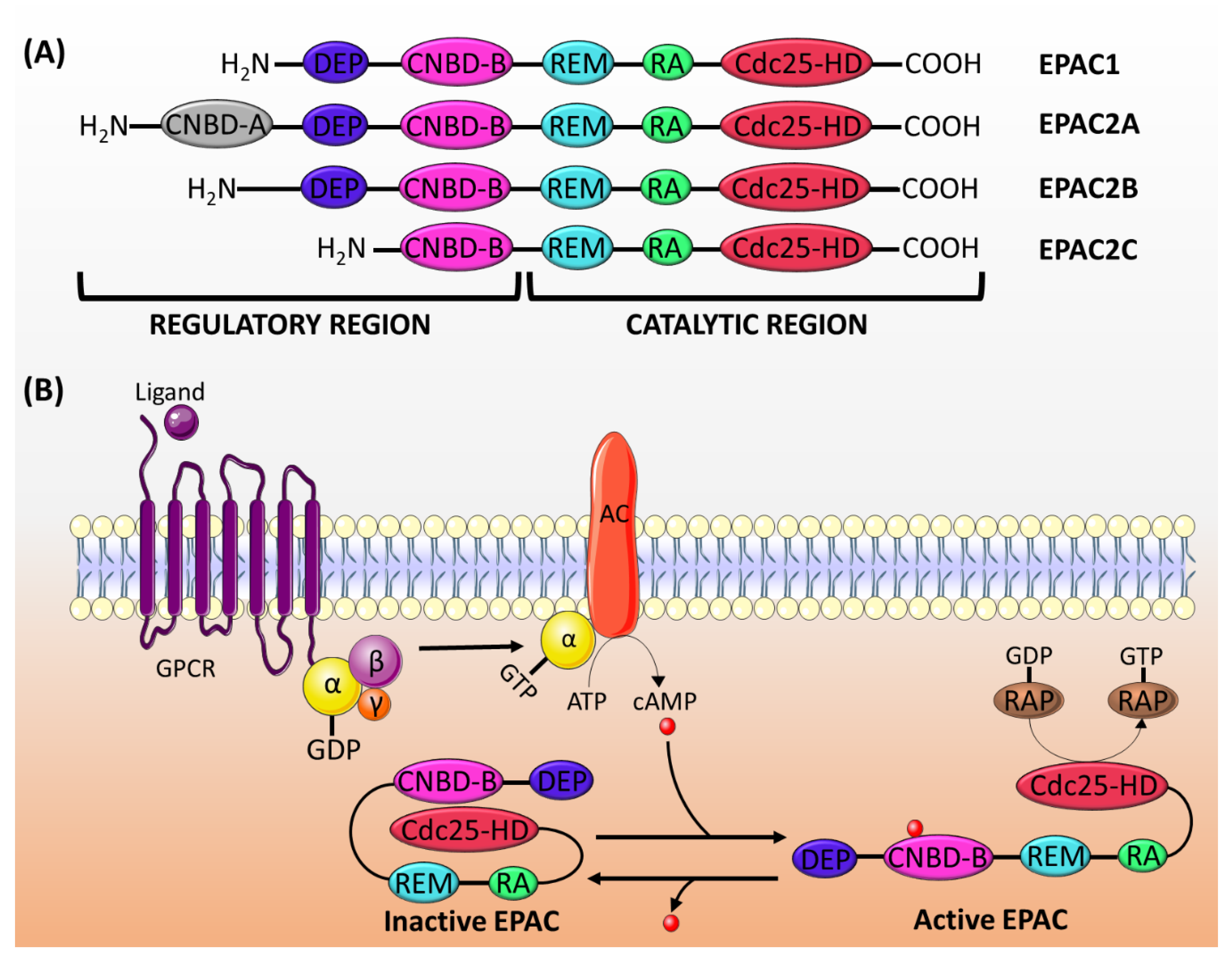
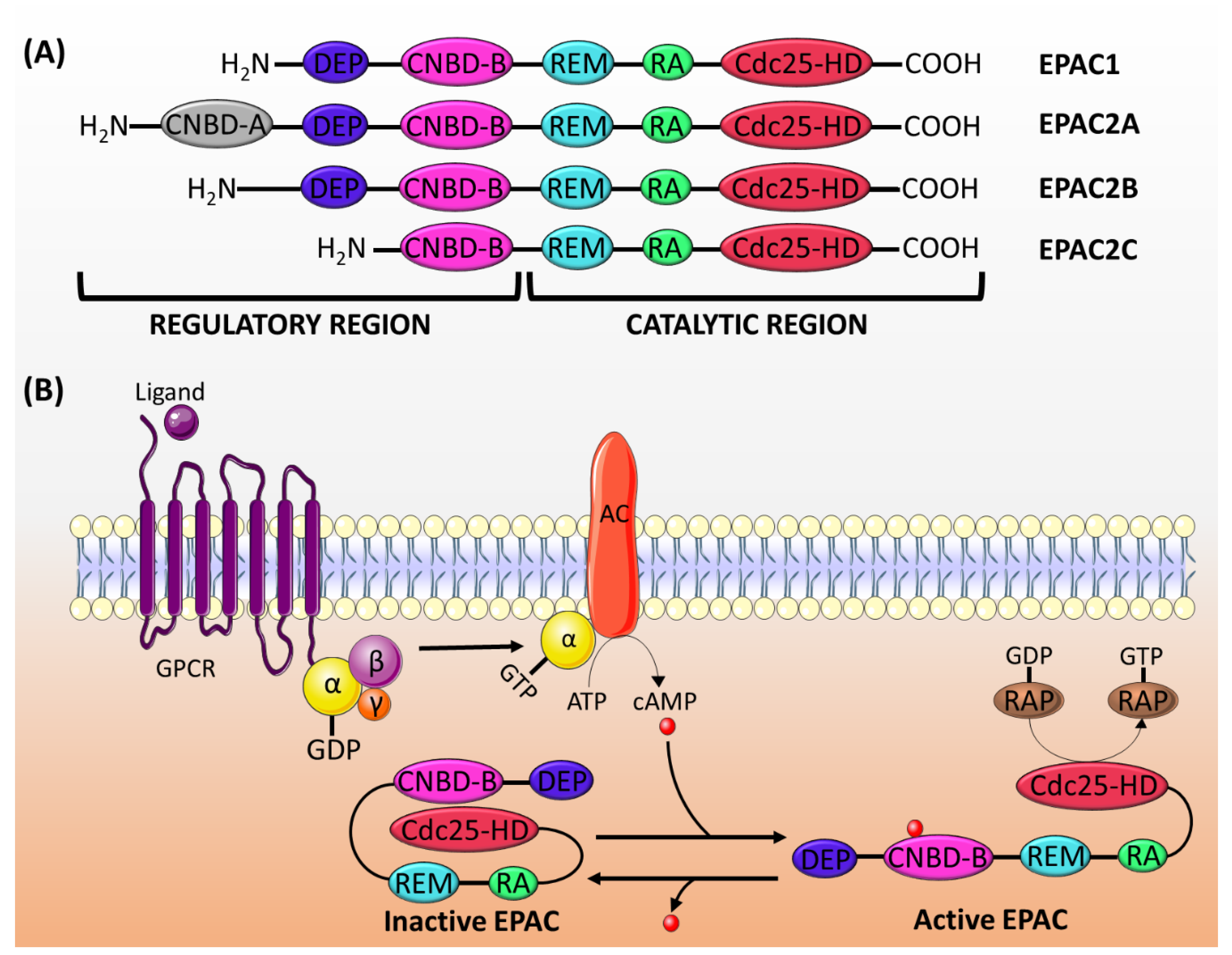
2.1. Genes and Expression
2.2. Structure and Activation
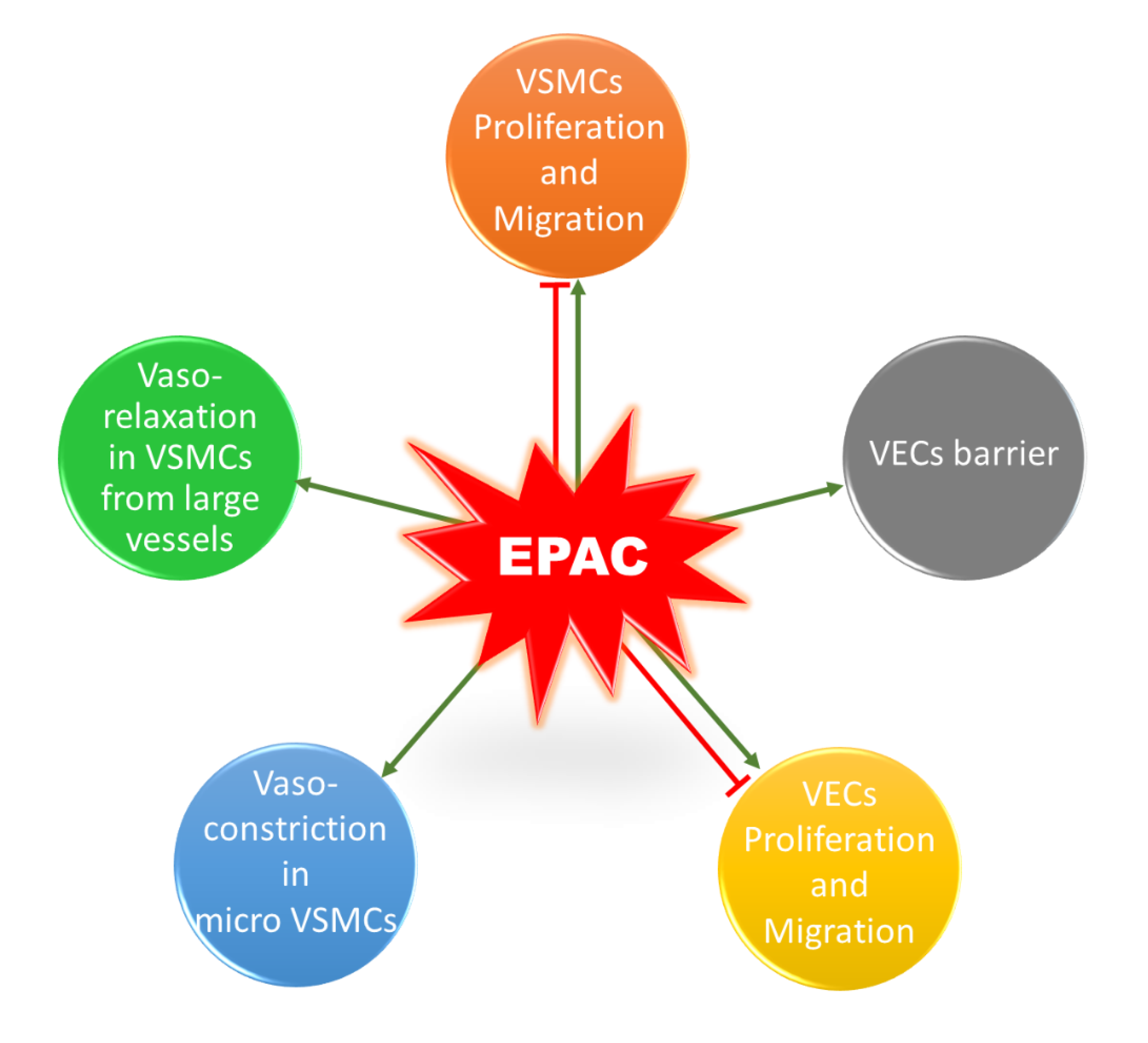
3. The Role of EPAC in VSMCs
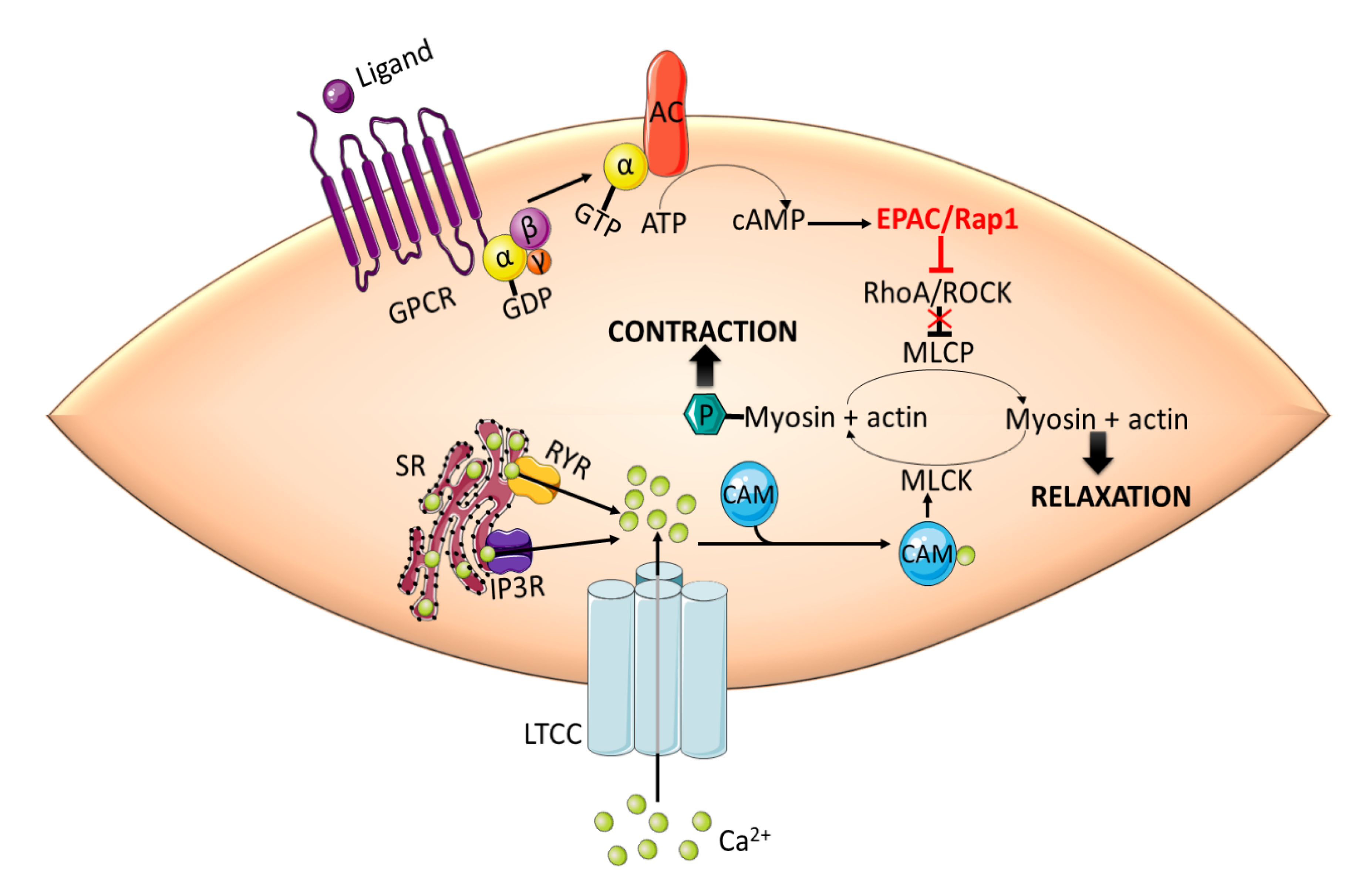
3.1. Vascular Tone
3.2. Phenotypic Switching: VSMCs Proliferation and Migration
4. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Fardoun, M.M.; Issa, K.; Maaliki, D.; Nasser, S.A.; Baydoun, E.; Eid, A.H. Estrogen increases expression of vascular alpha 2C adrenoceptor through the cAMP/Epac/JNK/AP-1 pathway and potentiates cold-induced vasoconstriction. Vasc. Pharmacol. 2020, 131, 106690. [Google Scholar] [CrossRef] [PubMed]
- Anwar, M.A.; Samaha, A.A.; Baydoun, S.; Iratni, R.; Eid, A.H. Rhus coriaria L.(Sumac) Evokes Endothelium-Dependent Vasorelaxation of Rat Aorta: Involvement of the cAMP and cGMP Pathways. Front. Pharmacol. [CrossRef] [PubMed]
- Eid, A.H. cAMP induces adhesion of microvascular smooth muscle cells to fibronectin via an Epac-mediated but PKA-independent mechanism. Cell. Physiol. Biochem. 2012, 30, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Chotani, M.A.; Mitra, S.; Eid, A.H.; Han, S.A.; Flavahan, N.A. Distinct cAMP signaling pathways differentially regulate alpha2C-adrenoceptor expression: Role in serum induction in human arteriolar smooth muscle cells. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H69–H76. [Google Scholar] [CrossRef] [Green Version]
- Motawea, H.K.; Jeyaraj, S.C.; Eid, A.H.; Mitra, S.; Unger, N.T.; Ahmed, A.A.; Flavahan, N.A.; Chotani, M.A. Cyclic AMP-Rap1A signaling mediates cell surface translocation of microvascular smooth muscle alpha2C-adrenoceptors through the actin-binding protein filamin-2. Am. J. Physiol. Cell Physiol. 2013, 305, C829–C845. [Google Scholar] [CrossRef] [Green Version]
- Jeyaraj, S.C.; Unger, N.T.; Eid, A.H.; Mitra, S.; Paul El-Dahdah, N.; Quilliam, L.A.; Flavahan, N.A.; Chotani, M.A. Cyclic AMP-Rap1A signaling activates RhoA to induce alpha(2c)-adrenoceptor translocation to the cell surface of microvascular smooth muscle cells. Am. J. Physiol. Cell Physiol. 2012, 303, C499–C511. [Google Scholar] [CrossRef] [Green Version]
- Eid, A.H.; Chotani, M.A.; Mitra, S.; Miller, T.J.; Flavahan, N.A. Cyclic AMP acts through Rap1 and JNK signaling to increase expression of cutaneous smooth muscle alpha2C-adrenoceptors. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H266–H272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wehbe, Z.; Nasser, S.A.; El-Yazbi, A.; Nasreddine, S.; Eid, A.H. Estrogen and Bisphenol A in Hypertension. Curr. Hypertens. Rep. 2020, 22, 23. [Google Scholar] [CrossRef] [PubMed]
- Fardoun, M.; Dehaini, H.; Shaito, A.; Mesmar, J.; El-Yazbi, A.; Badran, A.; Beydoun, E.; Eid, A.H. The hypertensive potential of estrogen: An untold story. Vasc. Pharmacol. 2020, 124, 106600. [Google Scholar] [CrossRef]
- Dehaini, H.; Fardoun, M.; Abou-Saleh, H.; El-Yazbi, A.; Eid, A.A.; Eid, A.H. Estrogen in vascular smooth muscle cells: A friend or a foe? Vasc. Pharmacol. 2018, 111, 15–21. [Google Scholar] [CrossRef]
- Eid, A.H.; Maiti, K.; Mitra, S.; Chotani, M.A.; Flavahan, S.; Bailey, S.R.; Thompson-Torgerson, C.S.; Flavahan, N.A. Estrogen increases smooth muscle expression of alpha2C-adrenoceptors and cold-induced constriction of cutaneous arteries. Am. J. Physiol. 2007, 293, H1955–H1961. [Google Scholar] [CrossRef] [Green Version]
- Tresguerres, M.; Levin, L.R.; Buck, J. Intracellular cAMP signaling by soluble adenylyl cyclase. Kidney Int. 2011, 79, 1277–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, D.M. Regulation and organization of adenylyl cyclases and cAMP. Biochem. J. 2003, 375, 517–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mika, D.; Leroy, J.; Vandecasteele, G.; Fischmeister, R. PDEs create local domains of cAMP signaling. J. Mol. Cell. Cardiol. 2012, 52, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Kritzer, M.D.; Li, J.; Dodge-Kafka, K.; Kapiloff, M.S. AKAPs: The architectural underpinnings of local cAMP signaling. J. Mol. Cell. Cardiol. 2012, 52, 351–358. [Google Scholar] [CrossRef] [Green Version]
- Robichaux, W.G., 3rd; Cheng, X. Intracellular cAMP Sensor EPAC: Physiology, Pathophysiology, and Therapeutics Development. Physiol. Rev. 2018, 98, 919–1053. [Google Scholar] [CrossRef]
- Arora, K.; Sinha, C.; Zhang, W.; Ren, A.; Moon, C.S.; Yarlagadda, S.; Naren, A.P. Compartmentalization of cyclic nucleotide signaling: A question of when, where, and why? Pflug. Arch. 2013, 465, 1397–1407. [Google Scholar] [CrossRef] [Green Version]
- Walsh, D.A.; Perkins, J.P.; Krebs, E.G. An adenosine 3’,5’-monophosphate-dependant protein kinase from rabbit skeletal muscle. J. Biol. Chem. 1968, 243, 3763–3765. [Google Scholar]
- De Rooij, J.; Zwartkruis, F.J.; Verheijen, M.H.; Cool, R.H.; Nijman, S.M.; Wittinghofer, A.; Bos, J.L. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 1998, 396, 474–477. [Google Scholar] [CrossRef]
- Kawasaki, H.; Springett, G.M.; Mochizuki, N.; Toki, S.; Nakaya, M.; Matsuda, M.; Housman, D.E.; Graybiel, A.M. A family of cAMP-binding proteins that directly activate Rap1. Science 1998, 282, 2275–2279. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, M.; Dekker, F.J.; Maarsingh, H. Exchange protein directly activated by cAMP (epac): A multidomain cAMP mediator in the regulation of diverse biological functions. Pharmacol. Rev. 2013, 65, 670–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lezoualc’h, F.; Fazal, L.; Laudette, M.; Conte, C. Cyclic AMP Sensor EPAC Proteins and Their Role in Cardiovascular Function and Disease. Circ. Res. 2016, 118, 881–897. [Google Scholar] [CrossRef] [PubMed]
- Fardoun, M.M.; Nassif, J.; Issa, K.; Baydoun, E.; Eid, A.H. Raynaud’s Phenomenon: A Brief Review of the Underlying Mechanisms. Front. Pharmacol. 2016, 7, 438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopkins, P.N. Molecular biology of atherosclerosis. Physiol. Rev. 2013, 93, 1317–1542. [Google Scholar] [CrossRef]
- Basatemur, G.L.; Jorgensen, H.F.; Clarke, M.C.H.; Bennett, M.R.; Mallat, Z. Vascular smooth muscle cells in atherosclerosis. Nat. Rev. Cardiol. 2019, 16, 727–744. [Google Scholar] [CrossRef]
- Newby, A.C.; Zaltsman, A.B. Molecular mechanisms in intimal hyperplasia. J. Pathol. 2000, 190, 300–309. [Google Scholar] [CrossRef]
- Hoivik, E.A.; Witsoe, S.L.; Bergheim, I.R.; Xu, Y.; Jakobsson, I.; Tengholm, A.; Doskeland, S.O.; Bakke, M. DNA methylation of alternative promoters directs tissue specific expression of Epac2 isoforms. PLoS ONE 2013, 8, e67925. [Google Scholar] [CrossRef] [Green Version]
- Aumo, L.; Rusten, M.; Mellgren, G.; Bakke, M.; Lewis, A.E. Functional roles of protein kinase A (PKA) and exchange protein directly activated by 3’,5’-cyclic adenosine 5’-monophosphate (cAMP) 2 (EPAC2) in cAMP-mediated actions in adrenocortical cells. Endocrinology 2010, 151, 2151–2161. [Google Scholar] [CrossRef] [Green Version]
- Ueno, H.; Shibasaki, T.; Iwanaga, T.; Takahashi, K.; Yokoyama, Y.; Liu, L.M.; Yokoi, N.; Ozaki, N.; Matsukura, S.; Yano, H.; et al. Characterization of the gene EPAC2: Structure, chromosomal localization, tissue expression, and identification of the liver-specific isoform. Genomics 2001, 78, 91–98. [Google Scholar] [CrossRef]
- Boriack-Sjodin, P.A.; Margarit, S.M.; Bar-Sagi, D.; Kuriyan, J. The structural basis of the activation of Ras by Sos. Nature 1998, 394, 337–343. [Google Scholar] [CrossRef]
- Liu, C.; Takahashi, M.; Li, Y.; Song, S.; Dillon, T.J.; Shinde, U.; Stork, P.J. Ras is required for the cyclic AMP-dependent activation of Rap1 via Epac2. Mol. Cell. Biol. 2008, 28, 7109–7125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popovic, M.; Rensen-de Leeuw, M.; Rehmann, H. Selectivity of CDC25 homology domain-containing guanine nucleotide exchange factors. J. Mol. Biol. 2013, 425, 2782–2794. [Google Scholar] [CrossRef] [PubMed]
- De Rooij, J.; Rehmann, H.; van Triest, M.; Cool, R.H.; Wittinghofer, A.; Bos, J.L. Mechanism of regulation of the Epac family of cAMP-dependent RapGEFs. J Biol Chem 2000, 275, 20829–20836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niimura, M.; Miki, T.; Shibasaki, T.; Fujimoto, W.; Iwanaga, T.; Seino, S. Critical role of the N-terminal cyclic AMP-binding domain of Epac2 in its subcellular localization and function. J. Cell. Physiol. 2009, 219, 652–658. [Google Scholar] [CrossRef]
- Rembold, C.M. Regulation of contraction and relaxation in arterial smooth muscle. Hypertension 1992, 20, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Raeymaekers, L.; Eggermont, J.A.; Wuytack, F.; Casteels, R. Effects of cyclic nucleotide dependent protein kinases on the endoplasmic reticulum Ca2+ pump of bovine pulmonary artery. Cell Calcium 1990, 11, 261–268. [Google Scholar] [CrossRef]
- Oishi, A.; Makita, N.; Sato, J.; Iiri, T. Regulation of RhoA signaling by the cAMP-dependent phosphorylation of RhoGDIalpha. J. Biol. Chem. 2012, 287, 38705–38715. [Google Scholar] [CrossRef] [Green Version]
- Sauzeau, V.; Le Jeune, H.; Cario-Toumaniantz, C.; Smolenski, A.; Lohmann, S.M.; Bertoglio, J.; Chardin, P.; Pacaud, P.; Loirand, G. Cyclic GMP-dependent protein kinase signaling pathway inhibits RhoA-induced Ca2+ sensitization of contraction in vascular smooth muscle. J. Biol. Chem. 2000, 275, 21722–21729. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Haystead, T.A.; Nakamoto, R.K.; Somlyo, A.V.; Somlyo, A.P. Acceleration of myosin light chain dephosphorylation and relaxation of smooth muscle by telokin. Synergism with cyclic nucleotide-activated kinase. J. Biol. Chem. 1998, 273, 11362–11369. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Somlyo, A.V.; Somlyo, A.P. Cyclic GMP-dependent stimulation reverses G-protein-coupled inhibition of smooth muscle myosin light chain phosphate. Biochem. Biophys. Res. Commun. 1996, 220, 658–663. [Google Scholar] [CrossRef]
- Lubomirov, L.T.; Reimann, K.; Metzler, D.; Hasse, V.; Stehle, R.; Ito, M.; Hartshorne, D.J.; Gagov, H.; Pfitzer, G.; Schubert, R. Urocortin-induced decrease in Ca2+ sensitivity of contraction in mouse tail arteries is attributable to cAMP-dependent dephosphorylation of MYPT1 and activation of myosin light chain phosphatase. Circ. Res. 2006, 98, 1159–1167. [Google Scholar] [CrossRef] [Green Version]
- Wooldridge, A.A.; MacDonald, J.A.; Erdodi, F.; Ma, C.; Borman, M.A.; Hartshorne, D.J.; Haystead, T.A. Smooth muscle phosphatase is regulated in vivo by exclusion of phosphorylation of threonine 696 of MYPT1 by phosphorylation of Serine 695 in response to cyclic nucleotides. J. Biol. Chem. 2004, 279, 34496–34504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seko, T.; Ito, M.; Kureishi, Y.; Okamoto, R.; Moriki, N.; Onishi, K.; Isaka, N.; Hartshorne, D.J.; Nakano, T. Activation of RhoA and inhibition of myosin phosphatase as important components in hypertension in vascular smooth muscle. Circ. Res. 2003, 92, 411–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sward, K.; Mita, M.; Wilson, D.P.; Deng, J.T.; Susnjar, M.; Walsh, M.P. The role of RhoA and Rho-associated kinase in vascular smooth muscle contraction. Curr. Hypertens. Rep. 2003, 5, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.A.; Zou, H.; Potocnik, S.J.; Meininger, G.A.; Davis, M.J. Arteriolar smooth muscle mechanotransduction: Ca(2+) signaling pathways underlying myogenic reactivity. J. Appl. Physiol. 2001, 91, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Ratz, P.H.; Hill, M.A. Temporal aspects of Ca(2+) and myosin phosphorylation during myogenic and norepinephrine-induced arteriolar constriction. J. Vasc. Res. 2000, 37, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Domínguez, A.; Colinas, O.; El-Yazbi, A.; Walsh, E.J.; Hill, M.A.; Walsh, M.P.; Cole, W.C. Ca 2+ sensitization due to myosin light chain phosphatase inhibition and cytoskeletal reorganization in the myogenic response of skeletal muscle resistance arteries. J. Physiol. 2013, 591, 1235–1250. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Dominguez, A.; El-Yazbi, A.F.; Zhu, H.L.; Colinas, O.; Zhong, X.Z.; Walsh, E.J.; Cole, D.M.; Kargacin, G.J.; Walsh, M.P.; Cole, W.C. Cytoskeletal reorganization evoked by Rho-associated kinase- and protein kinase C-catalyzed phosphorylation of cofilin and heat shock protein 27, respectively, contributes to myogenic constriction of rat cerebral arteries. J. Biol. Chem. 2014, 289, 20939–20952. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.P.; El-Yazbi, A.F.; Takeya, K.; Walsh, E.J.; Walsh, M.P.; Cole, W.C. Ca2+ sensitization via phosphorylation of myosin phosphatase targeting subunit at threonine-855 by Rho kinase contributes to the arterial myogenic response. J. Physiol. 2009, 587, 2537–2553. [Google Scholar] [CrossRef]
- El-Yazbi, A.F.; Johnson, R.P.; Walsh, E.J.; Takeya, K.; Walsh, M.P.; Cole, W.C. Pressure-dependent contribution of Rho kinase-mediated calcium sensitization in serotonin-evoked vasoconstriction of rat cerebral arteries. J. Physiol. 2010, 588, 1747–1762. [Google Scholar] [CrossRef]
- El-Yazbi, A.F.; Abd-Elrahman, K.S.; Moreno-Dominguez, A. PKC-mediated cerebral vasoconstriction: Role of myosin light chain phosphorylation versus actin cytoskeleton reorganization. Biochem. Pharmacol. 2015, 95, 263–278. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, C.; Walsh, M.P. Myosin regulatory light chain diphosphorylation slows relaxation of arterial smooth muscle. J. Biol. Chem. 2012, 287, 24064–24076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khromov, A.; Choudhury, N.; Stevenson, A.S.; Somlyo, A.V.; Eto, M. Phosphorylation-dependent autoinhibition of myosin light chain phosphatase accounts for Ca2+ sensitization force of smooth muscle contraction. J. Biol. Chem. 2009, 284, 21569–21579. [Google Scholar] [CrossRef] [Green Version]
- Zieba, B.J.; Artamonov, M.V.; Jin, L.; Momotani, K.; Ho, R.; Franke, A.S.; Neppl, R.L.; Stevenson, A.S.; Khromov, A.S.; Chrzanowska-Wodnicka, M.; et al. The cAMP-responsive Rap1 guanine nucleotide exchange factor, Epac, induces smooth muscle relaxation by down-regulation of RhoA activity. J. Biol. Chem. 2011, 286, 16681–16692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, O.L.; Kamishima, T.; Barrett-Jolley, R.; Quayle, J.M.; Dart, C. Exchange protein activated by cAMP (Epac) induces vascular relaxation by activating Ca2+-sensitive K+ channels in rat mesenteric artery. J. Physiol. 2013, 591, 5107–5123. [Google Scholar] [CrossRef]
- Hill, M.A.; Yang, Y.; Ella, S.R.; Davis, M.J.; Braun, A.P. Large conductance, Ca2+-activated K+ channels (BKCa) and arteriolar myogenic signaling. Febs Lett. 2010, 584, 2033–2042. [Google Scholar] [CrossRef] [Green Version]
- Nelson, M.T.; Patlak, J.B.; Worley, J.F.; Standen, N.B. Calcium channels, potassium channels, and voltage dependence of arterial smooth muscle tone. Am. J. Physiol. 1990, 259, C3–C18. [Google Scholar] [CrossRef]
- Purves, G.I.; Kamishima, T.; Davies, L.M.; Quayle, J.M.; Dart, C. Exchange protein activated by cAMP (Epac) mediates cAMP-dependent but protein kinase A-insensitive modulation of vascular ATP-sensitive potassium channels. J. Physiol. 2009, 587, 3639–3650. [Google Scholar] [CrossRef]
- Mufti, R.E.; Brett, S.E.; Tran, C.H.T.; Abd El-Rahman, R.; Anfinogenova, Y.; El-Yazbi, A.; Cole, W.C.; Jones, P.P.; Chen, S.R.W.; Welsh, D.G. Intravascular pressure augments cerebral arterial constriction by inducing voltage-insensitive Ca2+ waves. J. Physiol. 2010, 588, 3983–4005. [Google Scholar] [CrossRef]
- Quinn, K.V.; Giblin, J.P.; Tinker, A. Multisite phosphorylation mechanism for protein kinase A activation of the smooth muscle ATP-sensitive K+ channel. Circ. Res. 2004, 94, 1359–1366. [Google Scholar] [CrossRef] [Green Version]
- Cuinas, A.; Garcia-Morales, V.; Vina, D.; Gil-Longo, J.; Campos-Toimil, M. Activation of PKA and Epac proteins by cyclic AMP depletes intracellular calcium stores and reduces calcium availability for vasoconstriction. Life Sci. 2016, 155, 102–109. [Google Scholar] [CrossRef]
- Garcia-Morales, V.; Luaces-Regueira, M.; Campos-Toimil, M. The cAMP effectors PKA and Epac activate endothelial NO synthase through PI3K/Akt pathway in human endothelial cells. Biochem. Pharmacol. 2017, 145, 94–101. [Google Scholar] [CrossRef]
- Garcia-Morales, V.; Cuinas, A.; Elies, J.; Campos-Toimil, M. PKA and Epac activation mediates cAMP-induced vasorelaxation by increasing endothelial NO production. Vasc. Pharmacol. 2014, 60, 95–101. [Google Scholar] [CrossRef]
- Lakshmikanthan, S.; Zieba, B.J.; Ge, Z.D.; Momotani, K.; Zheng, X.; Lund, H.; Artamonov, M.V.; Maas, J.E.; Szabo, A.; Zhang, D.X.; et al. Rap1b in smooth muscle and endothelium is required for maintenance of vascular tone and normal blood pressure. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1486–1494. [Google Scholar] [CrossRef] [Green Version]
- Daunt, D.A.; Hurt, C.; Hein, L.; Kallio, J.; Feng, F.; Kobilka, B.K. Subtype-specific intracellular trafficking of alpha2-adrenergic receptors. Mol. Pharmacol. 1997, 51, 711–720. [Google Scholar] [CrossRef] [PubMed]
- Chotani, M.A.; Flavahan, S.; Mitra, S.; Daunt, D.; Flavahan, N.A. Silent alpha(2C)-adrenergic receptors enable cold-induced vasoconstriction in cutaneous arteries. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H1075–H1083. [Google Scholar] [CrossRef]
- Bailey, S.R.; Eid, A.H.; Mitra, S.; Flavahan, S.; Flavahan, N.A. Rho kinase mediates cold-induced constriction of cutaneous arteries: Role of alpha2C-adrenoceptor translocation. Circ. Res. 2004, 94, 1367–1374. [Google Scholar] [CrossRef] [Green Version]
- Kulinskii, V.I.; Zobova, N.V. Submitochondrial distribution of cAMP during incubation with rat liver mitochondria. Biokhimiia 1985, 50, 1546–1552. [Google Scholar]
- Qiao, J.; Mei, F.C.; Popov, V.L.; Vergara, L.A.; Cheng, X. Cell cycle-dependent subcellular localization of exchange factor directly activated by cAMP. J. Biol. Chem. 2002, 277, 26581–26586. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Robichaux, W.G.; Wang, Z.; Mei, F.C.; Cai, M.; Du, G.; Chen, J.; Cheng, X. Inhibition of Epac1 suppresses mitochondrial fission and reduces neointima formation induced by vascular injury. Sci. Rep. 2016, 6, 36552. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.; Huang, Y.; Li, L. Drp1-Dependent Mitochondrial Fission Plays Critical Roles in Physiological and Pathological Progresses in Mammals. Int. J. Mol. Sci. 2017, 18, 144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, T.; Sheu, S.S.; Robotham, J.L.; Yoon, Y. Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovasc. Res. 2008, 79, 341–351. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Ying, Z.; Webb, R.C. Activation of Rho/Rho kinase signaling pathway by reactive oxygen species in rat aorta. Am. J. Physiol. 2004, 287, H1495–H1500. [Google Scholar] [CrossRef]
- Bailey, S.R.; Mitra, S.; Flavahan, S.; Flavahan, N.A. Reactive oxygen species from smooth muscle mitochondria initiate cold-induced constriction of cutaneous arteries. Am. J. Physiol. 2005, 289, H243–H250. [Google Scholar] [CrossRef] [PubMed]
- Chotani, M.A.; Mitra, S.; Su, B.Y.; Flavahan, S.; Eid, A.H.; Clark, K.R.; Montague, C.R.; Paris, H.; Handy, D.E.; Flavahan, N.A. Regulation of alpha(2)-adrenoceptors in human vascular smooth muscle cells. Am. J. Physiol. 2004, 286, H59–H67. [Google Scholar] [CrossRef] [Green Version]
- Leech, C.J.; Faber, J.E. Different alpha-adrenoceptor subtypes mediate constriction of arterioles and venules. Am. J. Physiol. 1996, 270, H710–H722. [Google Scholar] [CrossRef]
- Dong, H.; Claffey, K.P.; Brocke, S.; Epstein, P.M. Inhibition of breast cancer cell migration by activation of cAMP signaling. Breast Cancer Res. Treat. 2015, 152, 17–28. [Google Scholar] [CrossRef]
- Lee, J.W.; Lee, J.; Moon, E.Y. HeLa human cervical cancer cell migration is inhibited by treatment with dibutyryl-cAMP. Anticancer Res. 2014, 34, 3447–3455. [Google Scholar]
- Schmitt, J.M.; Stork, P.J. Cyclic AMP-mediated inhibition of cell growth requires the small G protein Rap1. Mol. Cell. Biol. 2001, 21, 3671–3683. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.O.; Ryu, J.M.; Suh, H.N.; Park, S.H.; Oh, Y.M.; Lee, S.H.; Han, H.J. cAMP Promotes Cell Migration Through Cell Junctional Complex Dynamics and Actin Cytoskeleton Remodeling: Implications in Skin Wound Healing. Stem Cells Dev. 2015, 24, 2513–2524. [Google Scholar] [CrossRef]
- Misra, U.K.; Pizzo, S.V. Epac1-induced cellular proliferation in prostate cancer cells is mediated by B-Raf/ERK and mTOR signaling cascades. J. Cell. Biochem. 2009, 108, 998–1011. [Google Scholar] [CrossRef] [Green Version]
- Hochbaum, D.; Hong, K.; Barila, G.; Ribeiro-Neto, F.; Altschuler, D.L. Epac, in synergy with cAMP-dependent protein kinase (PKA), is required for cAMP-mediated mitogenesis. J. Biol. Chem. 2008, 283, 4464–4468. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.A.; Newby, A.C.; Bond, M. Ending Restenosis: Inhibition of Vascular Smooth Muscle Cell Proliferation by cAM. Cells 2019, 8, 1447. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.J.; Bond, M.; Sala-Newby, G.B.; Newby, A.C. Altered S-phase kinase-associated protein-2 levels are a major mediator of cyclic nucleotide-induced inhibition of vascular smooth muscle cell proliferation. Circ. Res. 2006, 98, 1141–1150. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, S.; Morishita, R.; Matsushita, H.; Nakagami, H.; Taniyama, Y.; Nakamura, T.; Aoki, M.; Yamamoto, K.; Higaki, J.; Ogihara, T. Cyclic AMP inhibited proliferation of human aortic vascular smooth muscle cells, accompanied by induction of p53 and p21. Hypertension 2000, 35, 237–243. [Google Scholar] [CrossRef] [Green Version]
- Indolfi, C.; Avvedimento, E.V.; Di Lorenzo, E.; Esposito, G.; Rapacciuolo, A.; Giuliano, P.; Grieco, D.; Cavuto, L.; Stingone, A.M.; Ciullo, I.; et al. Activation of cAMP-PKA signaling in vivo inhibits smooth muscle cell proliferation induced by vascular injury. Nat. Med. 1997, 3, 775–779. [Google Scholar] [CrossRef]
- Hewer, R.C.; Sala-Newby, G.B.; Wu, Y.J.; Newby, A.C.; Bond, M. PKA and Epac synergistically inhibit smooth muscle cell proliferation. J. Mol. Cell. Cardiol. 2011, 50, 87–98. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Teng, Y.; Wang, J.; Yu, M.; Li, Y.; Zheng, H. Rap1 promotes proliferation and migration of vascular smooth muscle cell via the ERK pathway. Pathol. Res. Pract. 2018, 214, 1045–1050. [Google Scholar] [CrossRef]
- Kimura, T.E.; Duggirala, A.; Hindmarch, C.C.; Hewer, R.C.; Cui, M.Z.; Newby, A.C.; Bond, M. Inhibition of Egr1 expression underlies the anti-mitogenic effects of cAMP in vascular smooth muscle cells. J. Mol. Cell. Cardiol. 2014, 72, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Min, I.M.; Pietramaggiori, G.; Kim, F.S.; Passegue, E.; Stevenson, K.E.; Wagers, A.J. The transcription factor EGR1 controls both the proliferation and localization of hematopoietic stem cells. Cell Stem Cell 2008, 2, 380–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fahmy, R.G.; Khachigian, L.M. Locked nucleic acid modified DNA enzymes targeting early growth response-1 inhibit human vascular smooth muscle cell growth. Nucleic Acids Res. 2004, 32, 2281–2285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, P.; Hinze, A.V.; Harst, A.; von Kugelgen, I. A(2)B receptors mediate the induction of early genes and inhibition of arterial smooth muscle cell proliferation via Epac. Cardiovasc. Res. 2011, 90, 148–156. [Google Scholar] [CrossRef]
- Yokoyama, U.; Minamisawa, S.; Quan, H.; Ghatak, S.; Akaike, T.; Segi-Nishida, E.; Iwasaki, S.; Iwamoto, M.; Misra, S.; Tamura, K.; et al. Chronic activation of the prostaglandin receptor EP4 promotes hyaluronan-mediated neointimal formation in the ductus arteriosus. J. Clin. Investig. 2006, 116, 3026–3034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoyama, U.; Minamisawa, S.; Quan, H.; Akaike, T.; Suzuki, S.; Jin, M.; Jiao, Q.; Watanabe, M.; Otsu, K.; Iwasaki, S.; et al. Prostaglandin E2-activated Epac promotes neointimal formation of the rat ductus arteriosus by a process distinct from that of cAMP-dependent protein kinase A. J. Biol. Chem. 2008, 283, 28702–28709. [Google Scholar] [CrossRef] [Green Version]
- Kimura, T.E.; Duggirala, A.; Smith, M.C.; White, S.; Sala-Newby, G.B.; Newby, A.C.; Bond, M. The Hippo pathway mediates inhibition of vascular smooth muscle cell proliferation by cAMP. J. Mol. Cell. Cardiol. 2016, 90, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Stone, J.D.; Narine, A.; Tulis, D.A. Inhibition of vascular smooth muscle growth via signaling crosstalk between AMP-activated protein kinase and cAMP-dependent protein kinase. Front. Physiol. 2012, 3, 409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, D.; Tsoi, K.; Maurice, D.H. Synergistic inhibition of vascular smooth muscle cell migration by phosphodiesterase 3 and phosphodiesterase 4 inhibitors. Circ. Res. 1998, 82, 852–861. [Google Scholar] [CrossRef]
- McKean, J.S.; Murray, F.; Gibson, G.; Shewan, D.A.; Tucker, S.J.; Nixon, G.F. The cAMP-producing agonist beraprost inhibits human vascular smooth muscle cell migration via exchange protein directly activated by cAMP. Cardiovasc. Res. 2015, 107, 546–555. [Google Scholar] [CrossRef] [Green Version]
- Min, J.; Reznichenko, M.; Poythress, R.H.; Gallant, C.M.; Vetterkind, S.; Li, Y.; Morgan, K.G. Src modulates contractile vascular smooth muscle function via regulation of focal adhesions. J. Cell. Physiol. 2012, 227, 3585–3592. [Google Scholar] [CrossRef] [Green Version]
- Turner, C.E. Paxillin and focal adhesion signalling. Nat. Cell Biol. 2000, 2, E231–E236. [Google Scholar] [CrossRef]
- Yokoyama, U.; Minamisawa, S.; Quan, H.; Akaike, T.; Jin, M.; Otsu, K.; Ulucan, C.; Wang, X.; Baljinnyam, E.; Takaoka, M.; et al. Epac1 is upregulated during neointima formation and promotes vascular smooth muscle cell migration. Am. J. Physiol. 2008, 295, H1547–H1555. [Google Scholar] [CrossRef] [Green Version]
- Jawien, A.; Bowen-Pope, D.F.; Lindner, V.; Schwartz, S.M.; Clowes, A.W. Platelet-derived growth factor promotes smooth muscle migration and intimal thickening in a rat model of balloon angioplasty. J. Clin. Investig. 1992, 89, 507–511. [Google Scholar] [CrossRef]
- Chen, P.Y.; Qin, L.; Li, G.; Tellides, G.; Simons, M. Fibroblast growth factor (FGF) signaling regulates transforming growth factor beta (TGFbeta)-dependent smooth muscle cell phenotype modulation. Sci. Rep. 2016, 6, 33407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, Y.; Yokoyama, U.; Yanai, C.; Ishige, R.; Kurotaki, D.; Umemura, M.; Fujita, T.; Kubota, T.; Okumura, S.; Sata, M.; et al. Epac1 Deficiency Attenuated Vascular Smooth Muscle Cell Migration and Neointimal Formation. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2617–2625. [Google Scholar] [CrossRef] [Green Version]
- Kato, Y.; Yokoyama, U.; Fujita, T.; Umemura, M.; Kubota, T.; Ishikawa, Y. Epac1 deficiency inhibits basic fibroblast growth factor-mediated vascular smooth muscle cell migration. J. Physiol. Sci. 2019, 69, 175–184. [Google Scholar] [CrossRef]
- Shigematsu, K.; Koyama, H.; Olson, N.E.; Cho, A.; Reidy, M.A. Phosphatidylinositol 3-kinase signaling is important for smooth muscle cell replication after arterial injury. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 2373–2378. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Yu, T.; Lee, H.; O’Brien, D.K.; Sesaki, H.; Yoon, Y. Decreasing mitochondrial fission diminishes vascular smooth muscle cell migration and ameliorates intimal hyperplasia. Cardiovasc. Res. 2015, 106, 272–283. [Google Scholar] [CrossRef] [Green Version]
- Clempus, R.E.; Griendling, K.K. Reactive oxygen species signaling in vascular smooth muscle cells. Cardiovasc. Res. 2006, 71, 216–225. [Google Scholar] [CrossRef]
- Netherton, S.J.; Sutton, J.A.; Wilson, L.S.; Carter, R.L.; Maurice, D.H. Both protein kinase A and exchange protein activated by cAMP coordinate adhesion of human vascular endothelial cells. Circ. Res. 2007, 101, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Cullere, X.; Shaw, S.K.; Andersson, L.; Hirahashi, J.; Luscinskas, F.W.; Mayadas, T.N. Regulation of vascular endothelial barrier function by Epac, a cAMP-activated exchange factor for Rap GTPase. Blood 2005, 105, 1950–1955. [Google Scholar] [CrossRef] [Green Version]
- Fukuhara, S.; Sakurai, A.; Sano, H.; Yamagishi, A.; Somekawa, S.; Takakura, N.; Saito, Y.; Kangawa, K.; Mochizuki, N. Cyclic AMP potentiates vascular endothelial cadherin-mediated cell-cell contact to enhance endothelial barrier function through an Epac-Rap1 signaling pathway. Mol. Cell. Biol. 2005, 25, 136–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Mei, F.C.; Yang, W.; Wang, H.; Wong, E.; Cai, J.; Toth, E.; Luo, P.; Li, Y.M.; Zhang, W.; et al. Epac1 inhibition ameliorates pathological angiogenesis through coordinated activation of Notch and suppression of VEGF signaling. Sci. Adv. 2020, 6, eaay3566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doebele, R.C.; Schulze-Hoepfner, F.T.; Hong, J.; Chlenski, A.; Zeitlin, B.D.; Goel, K.; Gomes, S.; Liu, Y.; Abe, M.K.; Nor, J.E.; et al. A novel interplay between Epac/Rap1 and mitogen-activated protein kinase kinase 5/extracellular signal-regulated kinase 5 (MEK5/ERK5) regulates thrombospondin to control angiogenesis. Blood 2009, 114, 4592–4600. [Google Scholar] [CrossRef] [Green Version]
- Namkoong, S.; Kim, C.K.; Cho, Y.L.; Kim, J.H.; Lee, H.; Ha, K.S.; Choe, J.; Kim, P.H.; Won, M.H.; Kwon, Y.G.; et al. Forskolin increases angiogenesis through the coordinated cross-talk of PKA-dependent VEGF expression and Epac-mediated PI3K/Akt/eNOS signaling. Cell. Signal. 2009, 21, 906–915. [Google Scholar] [CrossRef] [PubMed]
- Roberts, O.L.; Dart, C. cAMP signalling in the vasculature: The role of Epac (exchange protein directly activated by cAMP). Biochem. Soc. Trans 2014, 42, 89–97. [Google Scholar] [CrossRef] [PubMed]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wehbe, N.; Nasser, S.A.; Al-Dhaheri, Y.; Iratni, R.; Bitto, A.; El-Yazbi, A.F.; Badran, A.; Kobeissy, F.; Baydoun, E.; Eid, A.H. EPAC in Vascular Smooth Muscle Cells. Int. J. Mol. Sci. 2020, 21, 5160. https://doi.org/10.3390/ijms21145160
Wehbe N, Nasser SA, Al-Dhaheri Y, Iratni R, Bitto A, El-Yazbi AF, Badran A, Kobeissy F, Baydoun E, Eid AH. EPAC in Vascular Smooth Muscle Cells. International Journal of Molecular Sciences. 2020; 21(14):5160. https://doi.org/10.3390/ijms21145160
Chicago/Turabian StyleWehbe, Nadine, Suzanne Awni Nasser, Yusra Al-Dhaheri, Rabah Iratni, Alessandra Bitto, Ahmed F. El-Yazbi, Adnan Badran, Firas Kobeissy, Elias Baydoun, and Ali H. Eid. 2020. "EPAC in Vascular Smooth Muscle Cells" International Journal of Molecular Sciences 21, no. 14: 5160. https://doi.org/10.3390/ijms21145160
APA StyleWehbe, N., Nasser, S. A., Al-Dhaheri, Y., Iratni, R., Bitto, A., El-Yazbi, A. F., Badran, A., Kobeissy, F., Baydoun, E., & Eid, A. H. (2020). EPAC in Vascular Smooth Muscle Cells. International Journal of Molecular Sciences, 21(14), 5160. https://doi.org/10.3390/ijms21145160