Dietary Gluten and Neurodegeneration: A Case for Preclinical Studies




{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Preclinical Evidence
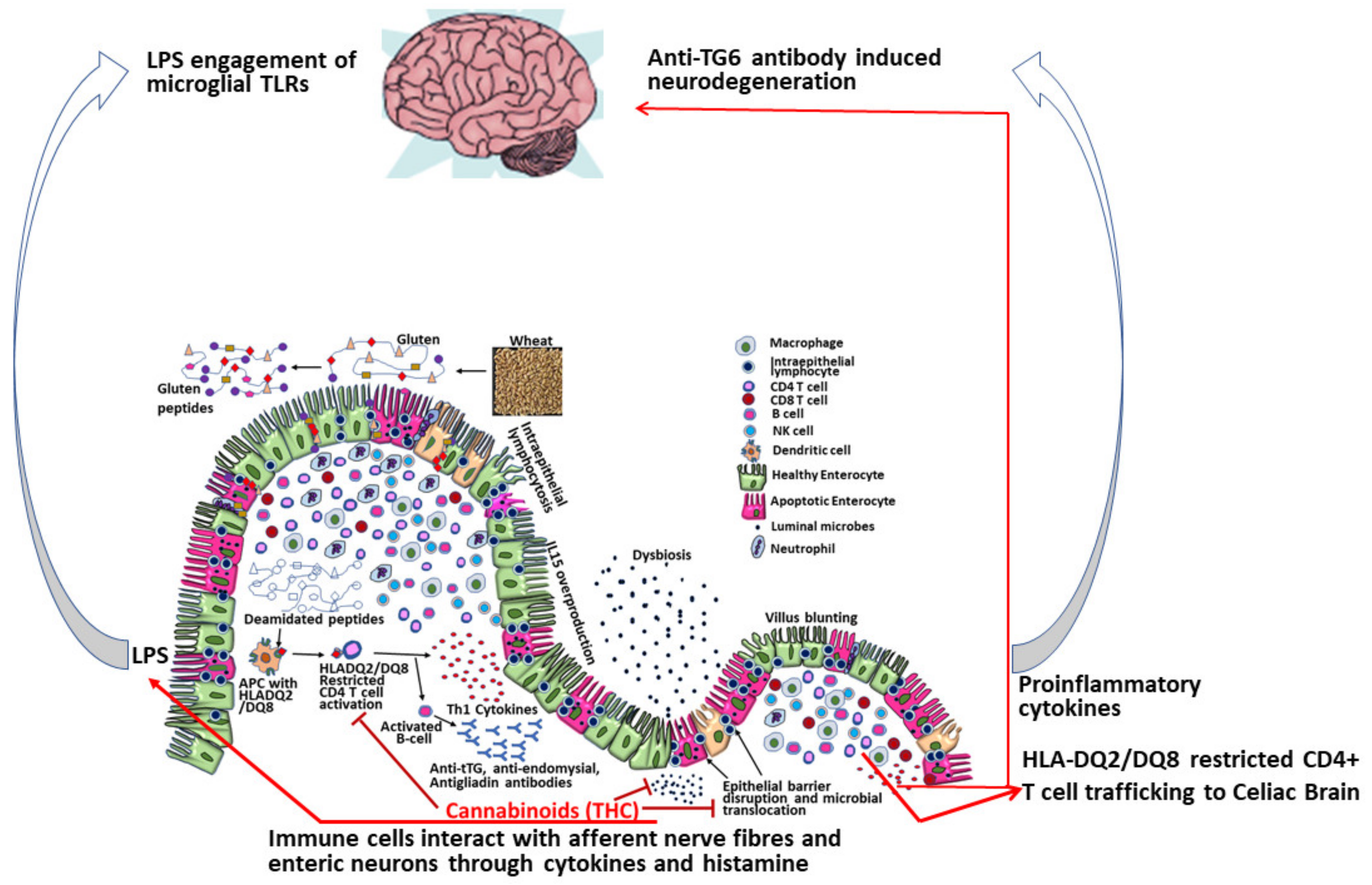
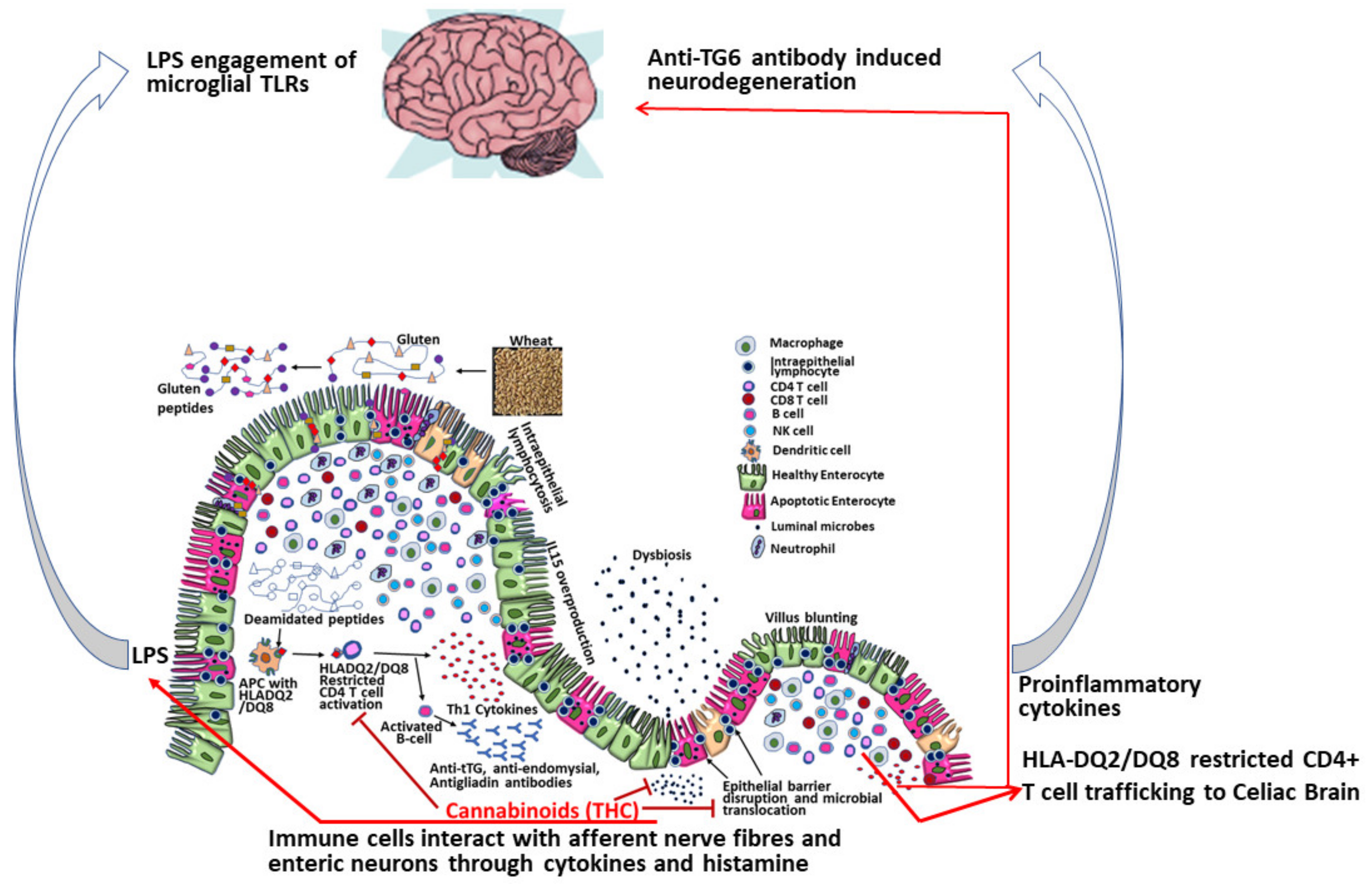
2.2. Mechanisms of Dietary Gluten-Induced Neuropathy
2.3. MicroRNA Evidence
2.4. Gut Dysbiosis-Neurodegeneration Link
2.5. Potential Role of Extracellular Structures—Biomolecular Condensates and Extracellular Vesicles in Pathogenesis of CD-Associated Neurodegeneration
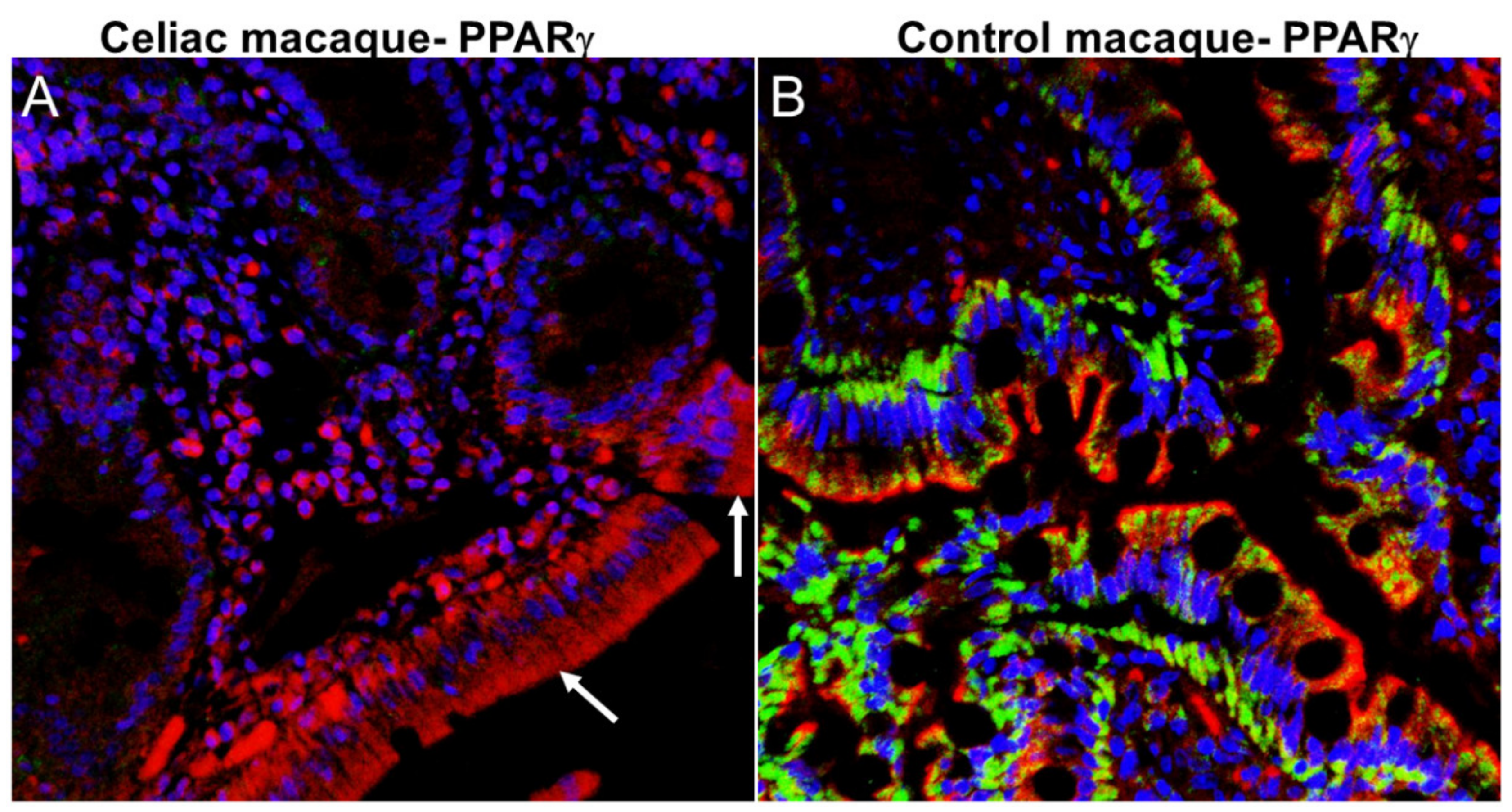
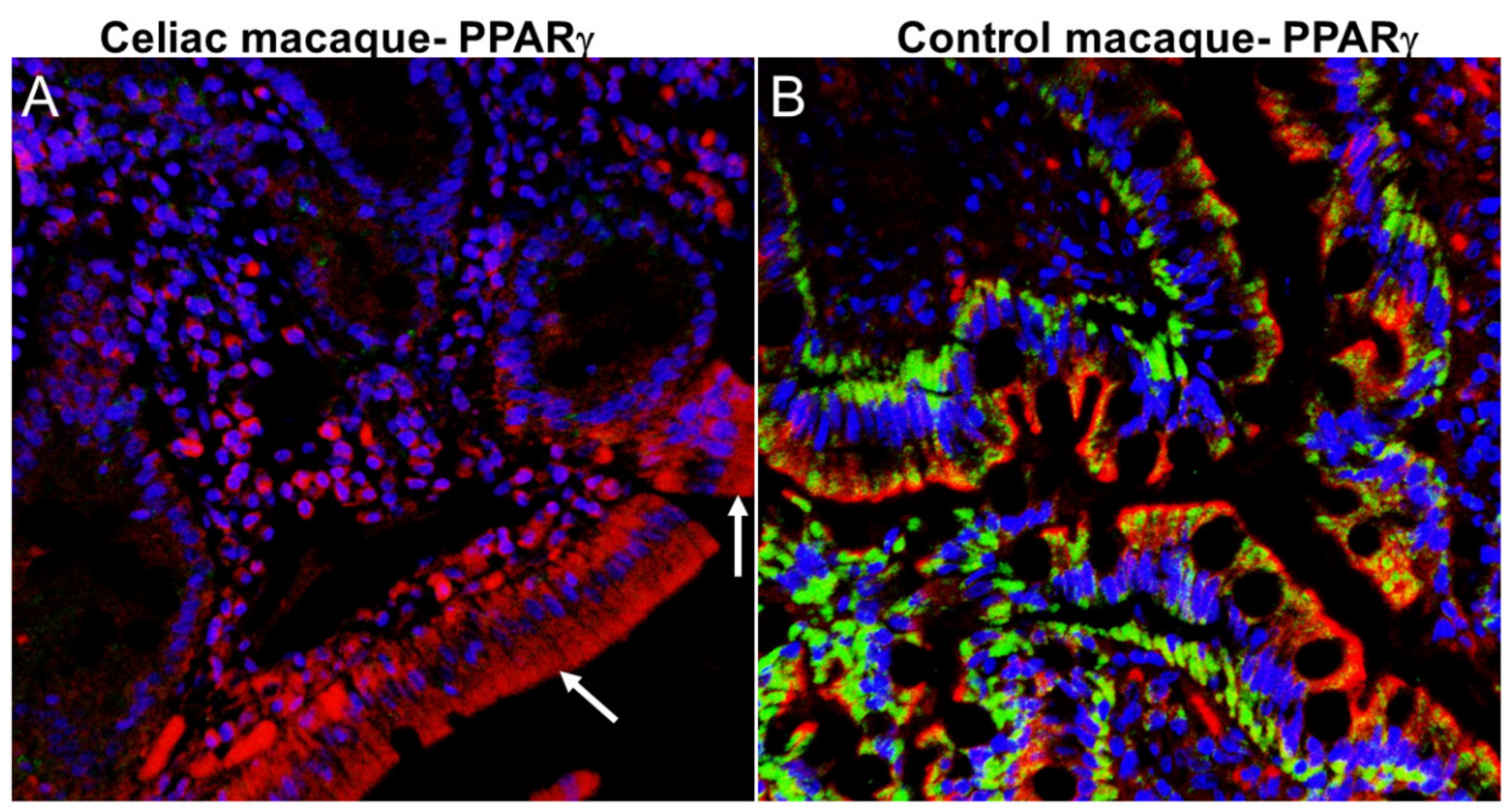
2.6. ES and Neurodegeneration
2.7. ES and the Gut-Brain Barrier
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CD | Celiac Disease |
| GFD | Gluten-Free Diet |
| tTG2 | Intestinal Tissue Transglutaminase |
| tTG6 | Neural Tissue Transglutaminase |
| NCGS | Non-Celiac Gluten Sensitivity |
| CNS | Central Nervous System |
| BBB | Blood-Brain Barrier |
| HLA | Human leukocyte antigen |
| mRNA | messenger RNA |
| miRNA | micro RNA |
| tRNA | transfer RNA |
| dsDNA | double-stranded DNA |
| SIV | Simian Immunodeficiency Virus |
| AD | Alzheimer’s Disease |
| PD | Parkinson’s Disease |
| HD | Huntington’s Disease |
| ASD | Autism Spectrum Disorder |
| DS | Down’s Syndrome |
| IBD | Inflammatory Bowel Disease |
| IBS | Irritable Bowel Syndrome |
| ZO1 | Zona Occludens Protein 1 |
| PPARγ | Peroxisome proliferator activated receptor gamma |
| STAT3 | Signal transducer and activator of transcription 3 |
| c-MYC | MYC proto-oncogene BHLH transcription factor |
| NOTCH1 | Notch receptor 1 |
| KLF4 | Kruppel like factor 4 |
| NFκB | Nuclear factor kappa B |
| NOS2 | Nitric oxide synthase 2 |
| THC | Delta-9-Tetra-Hydrocannabinol |
| ES | Extracellular Structure |
| BMS | Biomolecular Condensate |
| EV | Extracellular Vesicle |
| FTD | Frontotemporal Dementia |
| ALS | Amyotrophic Lateral Sclerosis |
| MAPT | Microtubule-Associated Protein Tau |
References
- Cooke, W.T.; Smith, W.T. Neurological disorders associated with adult coeliac disease. Brain 1966, 89, 683–722. [Google Scholar] [CrossRef] [PubMed]
- Cooke, W.T.; Johnson, A.G.; Woolf, A.L. Vital staining and electron microscopy of the intramuscular nerve endings in the neuropathy of adult coeliac disease. Brain 1966, 89, 663–682. [Google Scholar] [CrossRef] [PubMed]
- Kinney, H.C.; Burger, P.C.; Hurwitz, B.J.; Hijmans, J.C.; Grant, J.P. Degeneration of the central nervous system associated with celiac disease. J. Neurol. Sci. 1982, 53, 9–22. [Google Scholar] [CrossRef]
- Ward, M.E.; Murphy, J.T.; Greenberg, G.R. Celiac disease and spinocerebellar degeneration with normal vitamin E status. Neurology 1985, 35, 1199. [Google Scholar] [CrossRef] [PubMed]
- Dohan, F.C.; Harper, E.H.; Clark, M.H.; Rodrigue, R.B.; Zigas, V. Is schizophrenia rare if grain is rare? Boil. Psychiatry 1984, 19, 385–399. [Google Scholar]
- Ashe, P.C.; Berry, M.; Boulton, A. Schizophrenia, a neurodegenerative disorder with neurodevelopmental antecedents. Prog. Neuro-Psychopharmacol. Boil. Psychiatry 2001, 25, 691–707. [Google Scholar] [CrossRef]
- Rao, J.; Chiappelli, J.; Kochunov, P.; Regenold, W.T.; Rapoport, S.I.; Hong, L.E. Is schizophrenia a neurodegenerative disease? Evidence from age-related decline of brain-derived neurotrophic factor in the brains of schizophrenia patients and matched nonpsychiatric controls. Neurodegener. Dis. 2014, 15, 38–44. [Google Scholar] [CrossRef] [Green Version]
- Caccamo, D.; Currò, M.; Ientile, R. Potential of transglutaminase 2 as a therapeutic target. Expert Opin. Ther. Targets 2010, 14, 989–1003. [Google Scholar] [CrossRef]
- Sestak, K.; Fortgang, I. Celiac and Non-Celiac Forms of Gluten Sensitivity: Shifting Paradigms of an Old Disease. Br. Microbiol. Res. J. 2013, 3, 585–589. [Google Scholar] [CrossRef]
- Griffin, M.; Casadio, R.; Bergamini, C.M. Transglutaminases: Nature’s biological glues. Biochem. J. 2002, 368, 377–396. [Google Scholar] [CrossRef] [Green Version]
- Baizabal-Carvallo, J.F.; Jankovic, J. Movement disorders in autoimmune diseases. Mov. Disord. 2012, 27, 935–946. [Google Scholar] [CrossRef] [PubMed]
- Hadjivassiliou, M.; Aeschlimann, P.; Sanders, D.S.; Mäki, M.; Kaukinen, K.; Grünewald, R.; Bandmann, O.; Woodroofe, N.; Haddock, G.; Aeschlimann, D. Transglutaminase 6 antibodies in the diagnosis of gluten ataxia. Neurology 2013, 80, 1740–1745. [Google Scholar] [CrossRef] [PubMed]
- Vörös, P.; Sziksz, E.; Himer, L.; Ónody, A.; Pap, D.; Frivolt, K.; Szebeni, B.; Lippai, R.; Győrffy, H.; Fekete, A.; et al. Expression of PARK7 is increased in celiac disease. Virchows Archiv 2013, 463, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Cristofanilli, M.; Gratch, D.; Pagano, B.; McDermott, K.; Huang, J.; Jian, J.; Bates, D.; A Sadiq, S. Transglutaminase-6 is an autoantigen in progressive multiple sclerosis and is upregulated in reactive astrocytes. Mult. Scler. J. 2016, 23, 1707–1715. [Google Scholar] [CrossRef] [PubMed]
- Cascella, N.; Kryszak, D.; Gregory, P.; Fasano, D.L.K.A.; Eaton, W.W. Increased Prevalence of Transglutaminase 6 Antibodies in Sera From Schizophrenia Patients. Schizophr. Res. 2012, 136, S12. [Google Scholar] [CrossRef] [Green Version]
- Bavykina, I.A.; Zvyagin, A.A.; Petrova, I.V.; Nastausheva, T.L. Markers of gluten intolerance in children with autism spectrum disorders and Down’syndrome. Zhurnal Nevrol. Psikhiatrii Im. SS Korsakova 2018, 118, 64–68. [Google Scholar] [CrossRef]
- Bennabi, M.; Gaman, A.; Delorme, R.; Boukouaci, W.; Manier, C.; Scheid, I.; Mohammed, N.S.; Bengoufa, D.; Charron, D.; Krishnamoorthy, R.; et al. HLA-class II haplotypes and Autism Spectrum Disorders. Sci. Rep. 2018, 8, 7639. [Google Scholar] [CrossRef] [Green Version]
- Rahmoune, H.; Boutrid, N. Autism & Gluten: The Proof By Regression. Pediatr. Neurol. Briefs 2018, 32, 9. [Google Scholar] [CrossRef]
- Dickerson, F.B.; Stallings, C.; Origoni, A.; Vaughan, C.; Khushalani, S.; Alaedini, A.; Yolken, R. Markers of gluten sensitivity and celiac disease in bipolar disorder. Bipolar Disord. 2011, 13, 52–58. [Google Scholar] [CrossRef]
- Hadjivassiliou, M.; Aeschlimann, D.; A Grünewald, R.; Sanders, D.S.; Sharrack, B.; Woodroofe, M.N. GAD antibody-associated neurological illness and its relationship to gluten sensitivity. Acta Neurol. Scand. 2011, 123, 175–180. [Google Scholar] [CrossRef]
- Alaedini, A.; Okamoto, H.; Briani, C.; Wollenberg, K.; Shill, H.A.; Bushara, K.O.; Sander, H.W.; Green, P.H.R.; Hallett, M.; Latov, N. Immune cross-reactivity in celiac disease: anti-gliadin antibodies bind to neuronal synapsin I. J. Immunol. 2007, 178, 6590–6595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadjivassiliou, M.; Boscolo, S.; Davies–Jones, G.A.; Grünewald, R.A.; Not, T.; Sanders, D.S.; Simpson, J.E.; Tongiorgi, E.; Williamson, C.A.; Woodroofe, N. The humoral response in the pathogenesis of gluten ataxia. Neurology 2002, 58, 1221–1226. [Google Scholar] [CrossRef]
- Hu, W.T.; Murray, J.A.; Greenaway, M.C.; Parisi, J.E.; Josephs, K.A. Cognitive Impairment and Celiac Disease. Arch. Neurol. 2006, 63, 1440–1446. [Google Scholar] [CrossRef] [Green Version]
- Zis, P.; Rao, D.G.; Sarrigiannis, P.G.; Aeschlimann, P.; Aeschlimann, D.; Rigby, R.; Grünewald, R.; Hadjivassiliou, M. Transglutaminase 6 antibodies in gluten neuropathy. Dig. Liver Dis. 2017, 49, 1196–1200. [Google Scholar] [CrossRef]
- Aziz, I.; Hadjivassiliou, M.; Sanders, D.S. The spectrum of noncoeliac gluten sensitivity. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Daulatzai, M.A. Non-celiac gluten sensitivity triggers gut dysbiosis, neuroinflammation, gut-brain axis dysfunction, and vulnerability for dementia. CNS Neurol. Disord. Drug Targets 2015, 14, 110–131. [Google Scholar] [CrossRef] [PubMed]
- Hadjivassiliou, M.; Sanders, D.S.; A Grünewald, R.; Woodroofe, N.; Boscolo, S.; Aeschlimann, D. Gluten sensitivity: from gut to brain. Lancet Neurol. 2010, 9, 318–330. [Google Scholar] [CrossRef]
- Lionetti, E.; Leonardi, S.; Franzonello, C.; Mancardi, M.; Ruggieri, M.; Catassi, C. Gluten Psychosis: Confirmation of a New Clinical Entity. Nutrients 2015, 7, 5532–5539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boscolo, S.; Sarich, A.; Lorenzon, A.; Passoni, M.; Rui, V.; Stebel, M.; Sblattero, D.; Marzari, R.; Hadjivassiliou, M.; Tongiorgi, E. Gluten Ataxia: Passive Transfer in a Mouse Model. Ann. N.Y. Acad. Sci. 2007, 1107, 319–328. [Google Scholar] [CrossRef]
- Tarlac, V.; Kelly, L.; Nag, N.; Allen-Graham, J.; Anderson, R.P.; Storey, E. HLA-DR3-DQ2 Mice Do Not Develop Ataxia in the Presence of High Titre Anti-gliadin Antibodies. Cerebellum 2012, 12, 370–376. [Google Scholar] [CrossRef]
- Mazumdar, K.; Álvarez, X.; Borda, J.T.; Dufour, J.; Martin, E.; Bethune, M.T.; Khosla, C.; Sestak, K. Visualization of Transepithelial Passage of the Immunogenic 33-Residue Peptide from α-2 Gliadin in Gluten-Sensitive Macaques. PLoS ONE 2010, 5, e10228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohan, M.; Chow, C.-E.T.; Ryan, C.N.; Chan, L.S.; Dufour, J.; Aye, P.P.; Blanchard, J.; Moehs, C.P.; Sestak, K. Dietary Gluten-Induced Gut Dysbiosis Is Accompanied by Selective Upregulation of microRNAs with Intestinal Tight Junction and Bacteria-Binding Motifs in Rhesus Macaque Model of Celiac Disease. Nutrients 2016, 8, 684. [Google Scholar] [CrossRef] [PubMed]
- Sestak, K.; Conroy, L.; Aye, P.P.; Mehra, S.; Doxiadis, G.G.; Kaushal, D. Improved Xenobiotic Metabolism and Reduced Susceptibility to Cancer in Gluten-Sensitive Macaques upon Introduction of a Gluten-Free Diet. PLoS ONE 2011, 6, e18648. [Google Scholar] [CrossRef] [PubMed]
- Du Pré, M.F.; Blazevski, J.; Dewan, A.E.; Stamnaes, J.; Kanduri, C.; Sandve, G.K.; Johannesen, M.K.; Lindstad, C.B.; Hnida, K.; Fugger, L.; et al. B cell tolerance and antibody production to the celiac disease autoantigen transglutaminase 2. J. Exp. Med. 2019, 217. [Google Scholar] [CrossRef]
- Klöck, C.; DiRaimondo, T.R.; Khosla, C. Role of transglutaminase 2 in celiac disease pathogenesis. Semin. Immunopathol. 2012, 34, 513–522. [Google Scholar] [CrossRef] [Green Version]
- Sulic, A.-M.; Kurppa, K.; Rauhavirta, T.; Kaukinen, K.; Lindfors, K. Transglutaminase as a therapeutic target for celiac disease. Expert Opin. Ther. Targets 2014, 19, 335–348. [Google Scholar] [CrossRef]
- Gentile, V.; Cooper, A.J.L. Transglutaminases—Possible drug targets in human diseases. Curr. Drug Target -CNS Neurol. Disord. 2004, 3, 99–104. [Google Scholar] [CrossRef]
- Schmid, A.W.; Condemi, E.; Tuchscherer, G.; Chiappe, D.; Mutter, M.; Vogel, H.; Moniatte, M.; Tsybin, Y.O. Tissue Transglutaminase-mediated Glutamine Deamidation of β-Amyloid Peptide Increases Peptide Solubility, Whereas Enzymatic Cross-linking and Peptide Fragmentation May Serve as Molecular Triggers for Rapid Peptide Aggregation. J. Boil. Chem. 2011, 286, 12172–12188. [Google Scholar] [CrossRef] [Green Version]
- Wilhelmus, M.M.; de Jager, M.; Bakker, E.N.; Drukarch, B. Tissue transglutaminase in Alzheimer’s disease: involvement in pathogenesis and its potential as a therapeutic target. J. Alzheimers Dis. 2014, 42, S289–S303. [Google Scholar] [CrossRef]
- Junn, E.; Ronchetti, R.D.; Quezado, M.M.; Kim, S.Y.; Mouradian, M.M. Tissue transglutaminase-induced aggregation of alpha-synuclein: Implications for Lewy body formation in Parkinson’s disease and dementia with Lewy bodies. Proc. Natl. Acad. Sci. USA 2003, 100, 2047–2052. [Google Scholar] [CrossRef] [Green Version]
- Wilhelmus, M.M.; Verhaar, R.; Andringa, G.; Bol, J.G.; Cras, P.; Shan, L.; Hoozemans, J.J.; Drukarch, B. Presence of tissue transglutaminase in granular endoplasmic reticulum is characteristic of melanized neurons in Parkinson’s disease brain. Brain Pathol. 2011, 21, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Min, B.; Chung, K.C. New insight into transglutaminase 2 and link to neurodegenerative diseases. BMB Rep. 2018, 51, 5–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agnihotri, N.; Mehta, K. Transglutaminase-2: evolution from pedestrian protein to a promising therapeutic target. Amino Acids 2016, 49, 425–439. [Google Scholar] [CrossRef] [PubMed]
- DiRaimondo, T.R.; Klöck, C.; Khosla, C. Interferon-γ activates transglutaminase 2 via a phosphatidylinositol-3-kinase-dependent pathway: implications for celiac sprue therapy. J. Pharmacol. Exp. Ther. 2012, 341, 104–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, R.; Khosla, C. Substrates, inhibitors, and probes of mammalian transglutaminase 2. Anal. Biochem. 2020, 591, 113560. [Google Scholar] [CrossRef]
- Aaron, L.; Torsten, M.; Patricia, W. Autoimmunity in celiac disease: Extra-intestinal manifestations. Autoimmun. Rev. 2019, 18, 241–246. [Google Scholar] [CrossRef]
- Lerner, A.; Neidhöfer, S.; Matthias, T. The Gut Microbiome Feelings of the Brain: A Perspective for Non-Microbiologists. Microorganisms 2017, 5, 66. [Google Scholar] [CrossRef]
- Magni, S.; Comani, G.B.; Elli, L.; Vanessi, S.; Ballarini, E.; Nicolini, G.; Rusconi, M.; Castoldi, M.; Meneveri, R.; Muckenthaler, M.U.; et al. miRNAs Affect the Expression of Innate and Adaptive Immunity Proteins in Celiac Disease. Am. J. Gastroenterol. 2014, 109, 1662–1674. [Google Scholar] [CrossRef]
- Vaira, V.; Roncoroni, L.; Barisani, D.; Gaudioso, G.; Bosari, S.; Bulfamante, G.; Doneda, L.; Conte, D.; Tomba, C.; Bardella, M.T.; et al. microRNA profiles in coeliac patients distinguish different clinical phenotypes and are modulated by gliadin peptides in primary duodenal fibroblasts. Clin. Sci. 2013, 126, 417–423. [Google Scholar] [CrossRef]
- Wu, F.; Zikusoka, M.; Trindade, A.; Dassopoulos, T.; Harris, M.L.; Bayless, T.M.; Brant, S.R.; Chakravarti, S.; Kwon, J.H. MicroRNAs Are Differentially Expressed in Ulcerative Colitis and Alter Expression of Macrophage Inflammatory Peptide-2α. Gastroenterology 2008, 135, 1624–1635. [Google Scholar] [CrossRef]
- Vaira, V.; Gaudioso, G.; Laginestra, M.A.; Terrasi, A.; Agostinelli, C.; Bosari, S.; Di Sabatino, A.; Vanoli, A.; Paulli, M.; Ferrero, S.; et al. Deregulation of miRNAs-cMYC circuits is a key event in refractory celiac disease type-2 lymphomagenesis. Clin. Sci. 2020, 134, 1151–1166. [Google Scholar] [CrossRef] [PubMed]
- Capuano, M.; Iaffaldano, L.; Tinto, N.; Montanaro, D.; Capobianco, V.; Izzo, V.; Tucci, F.; Troncone, G.; Greco, L.; Sacchetti, L. MicroRNA-449a Overexpression, Reduced NOTCH1 Signals and Scarce Goblet Cells Characterize the Small Intestine of Celiac Patients. PLoS ONE 2011, 6, e29094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amr, K.; Bayoumi, F.; Eissa, E.; Abu-Zekry, M. Circulating microRNAs as potential non-invasive biomarkers in pediatric patients with celiac disease. Eur. Ann. Allergy Clin. Immunol. 2019, 51, 159–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandra, L.C.; Kumar, V.; Torben, W.; Stouwe, C.V.; Winsauer, P.; Amedee, A.; Molina, P.E.; Mohan, M. Chronic Administration of Δ9-Tetrahydrocannabinol Induces Intestinal Anti-Inflammatory MicroRNA Expression during Acute Simian Immunodeficiency Virus Infection of Rhesus Macaques. J. Virol. 2014, 89, 1168–1181. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Torben, W.; Mansfield, J.; Alvarez, X.; Stouwe, C.V.; Li, J.; Byrareddy, S.N.; Didier, P.J.; Pahar, B.; Molina, P.E.; et al. Cannabinoid Attenuation of Intestinal Inflammation in Chronic SIV-Infected Rhesus Macaques Involves T Cell Modulation and Differential Expression of Micro-RNAs and Pro-inflammatory Genes. Front. Immunol. 2019, 10, 914. [Google Scholar] [CrossRef]
- De Palma, G.; Cinova, J.; Stepankova, R.; Tuckova, L.; Sanz, Y. Pivotal Advance: Bifidobacteria and Gram-negative bacteria differentially influence immune responses in the proinflammatory milieu of celiac disease. J. Leukoc. Boil. 2009, 87, 765–778. [Google Scholar] [CrossRef]
- Scheperjans, F.; Aho, V.; Pereira, P.; Koskinen, K.; Paulin, L.; Pekkonen, E.; Haapaniemi, E.; Kaakkola, S.; Eerola-Rautio, J.; Pohja, M.; et al. Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov. Disord. 2014, 30, 350–358. [Google Scholar] [CrossRef]
- Villapol, S. Roles of Peroxisome Proliferator-Activated Receptor Gamma on Brain and Peripheral Inflammation. Cell. Mol. Neurobiol. 2017, 38, 121–132. [Google Scholar] [CrossRef]
- Byndloss, M.X.; Olsan, E.E.; Rivera-Chávez, F.; Tiffany, C.R.; Cevallos, S.A.; Lokken, K.L.; Torres, T.P.; Byndloss, A.J.; Faber, F.; Gao, Y.; et al. Microbiota-activated PPAR-γ signaling inhibits dysbiotic Enterobacteriaceae expansion. Science 2017, 357, 570–575. [Google Scholar] [CrossRef]
- Vetuschi, A.; Pompili, S.; Gaudio, E.; Latella, G.; Sferra, R. PPAR-γ with its anti-inflammatory and anti-fibrotic action could be an effective therapeutic target in IBD. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 8839–8848. [Google Scholar]
- Luciani, A.; Villella, V.R.; Vasaturo, A.; Giardino, I.; Pettoello-Mantovani, M.; Guido, S.; Cexus, O.N.; Peake, N.; Londei, M.; Quaratino, S.; et al. Lysosomal accumulation of gliadin p31-43 peptide induces oxidative stress and tissue transglutaminase-mediated PPARgamma downregulation in intestinal epithelial cells and coeliac mucosa. Gut 2010, 59, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Soares, F.L.P.; Matoso, R.D.O.; Teixeira, L.G.; Menezes, Z.; Pereira, S.S.; Alves, A.C.; Batista, N.V.; Faria, A.C.; Cara, D.C.; Ferreira, A.V.M.; et al. Gluten-free diet reduces adiposity, inflammation and insulin resistance associated with the induction of PPAR-alpha and PPAR-gamma expression. J. Nutr. Biochem. 2013, 24, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Sziksz, E.; Molnár, K.; Lippai, R.; Pap, D.; Ónody, A.; Veres-Székely, A.; Vörös, P.; Szabo, D.; Győrffy, H.; Veres, G.; et al. Peroxisome proliferator-activated receptor-γ and thymic stromal lymphopoietin are involved in the pathophysiology of childhood coeliac disease. Virchows Archiv 2014, 465, 385–393. [Google Scholar] [CrossRef]
- Kang, D.-W.; Park, J.G.; Ilhan, Z.E.; Wallstrom, G.; LaBaer, J.; Adams, J.B.; Krajmalnik-Brown, R. Reduced Incidence of Prevotella and Other Fermenters in Intestinal Microflora of Autistic Children. PLoS ONE 2013, 8, e68322. [Google Scholar] [CrossRef] [Green Version]
- Ning, L.; Lou, X.; Zhang, F.; Xu, G. Nuclear Receptors in the Pathogenesis and Management of Inflammatory Bowel Disease. Mediat. Inflamm. 2019, 2019, 2624941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benedetti, E.; Viscido, A.; Castelli, V.; Maggiani, C.; D’Angelo, M.; Di Giacomo, E.; Antonosante, A.; Picarelli, A.; Frieri, G. Mesalazine treatment in organotypic culture of celiac patients: Comparative study with gluten free diet. J. Cell. Physiol. 2018, 233, 4383–4390. [Google Scholar] [CrossRef]
- Sehgal, P.; Colombel, J.-F.; Aboubakr, A.; Narula, N. Systematic review: safety of mesalazine in ulcerative colitis. Aliment. Pharmacol. Ther. 2018, 47, 1597–1609. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Mansfield, J.; Fan, R.; MacLean, A.G.; Li, J.; Mohan, M. miR-130a and miR-212 Disrupt the Intestinal Epithelial Barrier through Modulation of PPARγ and Occludin Expression in Chronic Simian Immunodeficiency Virus–Infected Rhesus Macaques. J. Immunol. 2018, 200, 2677–2689. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, X.; Sestak, K.; Byrareddy, S.N.; Mohan, M. Long Term Delta-9-tetrahydrocannabinol Administration Inhibits Proinflammatory Responses in Minor Salivary Glands of Chronically Simian Immunodeficieny Virus Infected Rhesus Macaques. Viruses 2020, 12, 713. [Google Scholar] [CrossRef]
- Toretsky, J.A.; Wright, P.E. Assemblages: Functional units formed by cellular phase separation. J. Cell Boil. 2014, 206, 579–588. [Google Scholar] [CrossRef]
- Navarro, M.G.-J.; Kashida, S.; Chouaib, R.; Souquere, S.; Pierron, G.; Weil, D.; Gueroui, Z. RNA is a critical element for the sizing and the composition of phase-separated RNA-protein condensates. Nat. Commun. 2019, 10, 3230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittag, T.; Parker, R. Multiple Modes of Protein-Protein Interactions Promote RNP Granule Assembly. J. Mol. Boil. 2018, 430, 4636–4649. [Google Scholar] [CrossRef] [PubMed]
- Atkin-Smith, G.K.; Tixeira, R.; Paone, S.; Mathivanan, S.; Collins, C.; Liem, M.; Goodall, K.J.; Ravichandran, K.S.; Hulett, M.D.; Poon, I.K. A novel mechanism of generating extracellular vesicles during apoptosis via a beads-on-a-string membrane structure. Nat. Commun. 2015, 6, 7439. [Google Scholar] [CrossRef]
- Akers, J.C.; Gonda, D.; Kim, R.; Carter, B.S.; Chen, C.C. Biogenesis of extracellular vesicles (EV): Exosomes, microvesicles, retrovirus-like vesicles, and apoptotic bodies. J. Neuro-Oncol. 2013, 113, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ihara, T.; Yamamoto, T.; Sugamata, M.; Okumura, H.; Ueno, Y. The process of ultrastructural changes from nuclei to apoptotic body. Virchows. Archiv. 1998, 433, 443–447. [Google Scholar] [CrossRef]
- Hristov, M.; Erl, W.; Linder, S.; Weber, P.C. Apoptotic bodies from endothelial cells enhance the number and initiate the differentiation of human endothelial progenitor cells in vitro. Blood 2004, 104, 2761–2766. [Google Scholar] [CrossRef]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Boil. 2013, 200, 373–383. [Google Scholar] [CrossRef] [Green Version]
- Thakur, B.K.; Zhang, H.; Becker, A.; Matei, I.; Huang, Y.; Costa-Silva, B.; Zheng, Y.; Hoshino, A.; Brazier, H.; Xiang, J.; et al. Double-stranded DNA in exosomes: a novel biomarker in cancer detection. Cell Res. 2014, 24, 766–769. [Google Scholar] [CrossRef] [Green Version]
- Welch, J.L.; Kaddour, H.; Winchester, L.; Fletcher, C.V.; Stapleton, J.T.; Okeoma, C.M. Semen Extracellular Vesicles From HIV-1–Infected Individuals Inhibit HIV-1 Replication In Vitro, and Extracellular Vesicles Carry Antiretroviral Drugs In Vivo. JAIDS J. Acquir. Immune Defic. Syndr. 2020, 83, 90–98. [Google Scholar] [CrossRef]
- Balaj, L.; Lessard, R.; Dai, L.; Cho, Y.-J.; Pomeroy, S.L.; Breakefield, X.O.; Skog, J. Tumour microvesicles contain retrotransposon elements and amplified oncogene sequences. Nat. Commun. 2011, 2, 1–9. [Google Scholar] [CrossRef]
- Hnisz, D.; Shrinivas, K.; Young, R.A.; Chakraborty, A.K.; Sharp, P.A. A Phase Separation Model for Transcriptional Control. Cell 2017, 169, 13–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabari, B.R.; Dall’Agnese, A.; Boija, A.; Klein, I.A.; Coffey, E.L.; Shrinivas, K.; Abraham, B.J.; Hannett, N.M.; Zamudio, A.V.; Manteiga, J.C.; et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science 2018, 361, eaar3958. [Google Scholar] [CrossRef] [Green Version]
- Aguzzi, A.; Altmeyer, M. Phase Separation: Linking Cellular Compartmentalization to Disease. Trends Cell Boil. 2016, 26, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Dobra, I.; Pankivskyi, S.; Samsonova, A.; Pastré, D.; Hamon, L. Relation Between Stress Granules and Cytoplasmic Protein Aggregates Linked to Neurodegenerative Diseases. Curr. Neurol. Neurosci. Rep. 2018, 18, 107. [Google Scholar] [CrossRef] [PubMed]
- Ryan, V.H.; Fawzi, N.L. Physiological, Pathological, and Targetable Membraneless Organelles in Neurons. Trends Neurosci. 2019, 42, 693–708. [Google Scholar] [CrossRef] [PubMed]
- Bobrie, A.; Colombo, M.; Raposo, G.; Théry, C. Exosome Secretion: Molecular Mechanisms and Roles in Immune Responses. Traffic 2011, 12, 1659–1668. [Google Scholar] [CrossRef]
- Simons, M.; Raposo, G. Exosomes—Vesicular carriers for intercellular communication. Curr. Opin. Cell Boil. 2009, 21, 575–581. [Google Scholar] [CrossRef]
- Théry, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef]
- Buzas, E.; György, B.; Nagy, G.; Falus, A.; Gay, S. Emerging role of extracellular vesicles in inflammatory diseases. Nat. Rev. Rheumatol. 2014, 10, 356–364. [Google Scholar] [CrossRef]
- Kulp, A.; Kuehn, M.J. Biological Functions and Biogenesis of Secreted Bacterial Outer Membrane Vesicles. Annu. Rev. Microbiol. 2010, 64, 163–184. [Google Scholar] [CrossRef] [Green Version]
- Avila-Calderón, E.D.; Villanueva, M.G.A.; Cancino-Diaz, J.C.; López-Villegas, E.O.; Sriranganathan, N.; Boyle, S.M.; Contreras-Rodríguez, A. Roles of bacterial membrane vesicles. Arch. Microbiol. 2014, 197, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Nordgreen, J.; Munsterhjelm, C.; Aae, F.; Popova, A.; Boysen, P.; Ranheim, B.; Heinonen, M.; Raszplewicz, J.; Piepponen, T.P.; Lervik, A.; et al. The effect of lipopolysaccharide (LPS) on inflammatory markers in blood and brain and on behavior in individually-housed pigs. Physiol. Behav. 2018, 195, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Costinean, S.; Croce, C.M.; Brasier, A.R.; Merwat, S.; Larson, S.A.; Basra, S.; Verne, G.N. MicroRNA 29 targets nuclear factor-κB-repressing factor and Claudin 1 to increase intestinal permeability. Gastroenterology 2014, 148, 158–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerner, A.; Arleevskaya, M.; Schmiedl, A.; Matthias, T. Microbes and Viruses Are Bugging the Gut in Celiac Disease. Are They Friends or Foes? Front. Microbiol. 2017, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.J.; Hardy, J.; Fischbeck, K.H. Toxic Proteins in Neurodegenerative Disease. Science 2002, 296, 1991–1995. [Google Scholar] [CrossRef]
- Gan, L.; Cookson, M.R.; Petrucelli, L.; La Spada, A.R. Converging pathways in neurodegeneration, from genetics to mechanisms. Nat. Neurosci. 2018, 21, 1300–1309. [Google Scholar] [CrossRef]
- Jucker, M.; Walker, L.C. Propagation and spread of pathogenic protein assemblies in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1341–1349. [Google Scholar] [CrossRef]
- Davis, D.; Yuan, H.; Liang, F.-X.; Yang, Y.-M.; Westley, J.; Petzold, C.; Dancel-Manning, K.; Deng, Y.; Sall, J.; Sehgal, P.B. Human Antiviral Protein MxA Forms Novel Metastable Membraneless Cytoplasmic Condensates Exhibiting Rapid Reversible Tonicity-Driven Phase Transitions. J. Virol. 2019, 93, 22. [Google Scholar] [CrossRef]
- Heinrich, B.S.; Maliga, Z.; Stein, D.A.; Hyman, A.; Whelan, P.J. Phase Transitions Drive the Formation of Vesicular Stomatitis Virus Replication Compartments. mBio 2018, 9, e02290-17. [Google Scholar] [CrossRef] [Green Version]
- Molliex, A.; Temirov, J.; Lee, J.; Coughlin, M.; Kanagaraj, A.P.; Kim, H.J.; Mittag, T.; Taylor, J.P. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 2015, 163, 123–133. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.; Lee, H.O.; Jawerth, L.; Maharana, S.; Jahnel, M.; Hein, M.Y.; Stoynov, S.; Mahamid, J.; Saha, S.; Franzmann, T.M.; et al. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 2015, 162, 1066–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozelle, D.K.; Filone, C.M.; Kedersha, N.; Connor, J.H. Activation of Stress Response Pathways Promotes Formation of Antiviral Granules and Restricts Virus Replication. Mol. Cell. Boil. 2014, 34, 2003–2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.-J.; Wang, B.; Kodali, M.C.; Chen, C.; Kim, E.; Patters, B.J.; Lan, L.; Kumar, S.; Wang, X.; Yue, J.; et al. In Vivo evidence for the contribution of peripheral circulating inflammatory exosomes to neuroinflammation. J. Neuroinflam. 2018, 15, 8. [Google Scholar] [CrossRef] [PubMed]
- Sinha, M.S.; Ansell-Schultz, A.; Civitelli, L.; Hildesjö, C.; Larsson, M.; Lannfelt, L.; Ingelsson, M.; Hallbeck, M. Alzheimer’s disease pathology propagation by exosomes containing toxic amyloid-beta oligomers. Acta Neuropathol. 2018, 136, 41–56. [Google Scholar] [CrossRef] [Green Version]
- Joshi, P.; Turola, E.; Ruiz, A.; Bergami, A.; Libera, D.D.; Benussi, L.; Giussani, P.; Magnani, G.; Comi, G.; Legname, G.; et al. Microglia convert aggregated amyloid-β into neurotoxic forms through the shedding of microvesicles. Cell Death Differ. 2013, 21, 582–593. [Google Scholar] [CrossRef] [Green Version]
- Halverson, K.; Fraser, P.E.; Kirschner, D.A.; Lansbury, P.T., Jr. Molecular determinants of amyloid deposition in Alzheimer’s disease: conformational studies of synthetic beta-protein fragments. Biochemistry 1990, 29, 2639–2644. [Google Scholar] [CrossRef]
- Saman, S.; Kim, W.; Raya, M.; Visnick, Y.; Miro, S.; Saman, S.; Jackson, B.; McKee, A.C.; Alvarez, V.E.; Lee, N.C.Y.; et al. Exosome-associated Tau Is Secreted in Tauopathy Models and Is Selectively Phosphorylated in Cerebrospinal Fluid in Early Alzheimer Disease. J. Boil. Chem. 2011, 287, 3842–3849. [Google Scholar] [CrossRef] [Green Version]
- Willing, B.P.; Russell, S.L.; Finlay, B.B. Shifting the balance: antibiotic effects on host–microbiota mutualism. Nat. Rev. Genet. 2011, 9, 233–243. [Google Scholar] [CrossRef]
- Everard, A.; Cani, P.D. Diabetes, obesity and gut microbiota. Best Pr. Res. Clin. Gastroenterol. 2013, 27, 73–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, C.-S.; Ban, M.; Choi, E.-J.; Moon, H.-G.; Jeon, J.-S.; Kim, D.-K.; Park, S.-K.; Jeon, S.G.; Roh, T.-Y.; Myung, S.-J.; et al. Extracellular Vesicles Derived from Gut Microbiota, Especially Akkermansia muciniphila, Protect the Progression of Dextran Sulfate Sodium-Induced Colitis. PLoS ONE 2013, 8, e76520. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Torchia, M.L.G.; Lawson, G.W.; Karp, C.; Ashwell, J.D.; Mazmanian, S.K. Outer membrane vesicles of a human commensal mediate immune regulation and disease protection. Cell Host Microbe 2012, 12, 509–520. [Google Scholar] [CrossRef] [Green Version]
- Elsen, L.W.J.V.D.; Poyntz, H.C.; Weyrich, L.S.; Young, W.; E Forbes-Blom, E. Embracing the gut microbiota: the new frontier for inflammatory and infectious diseases. Clin. Transl. Immunol. 2017, 6, e125. [Google Scholar] [CrossRef] [PubMed]
- Kapogiannis, D.; Boxer, A.; Schwartz, J.B.; Abner, E.L.; Biragyn, A.; Masharani, U.; Frassetto, L.; Petersen, R.C.; Miller, B.L.; Goetzl, E.J. Dysfunctionally phosphorylated type 1 insulin receptor substrate in neural-derived blood exosomes of preclinical Alzheimer’s disease. FASEB J. 2015, 29, 589–596. [Google Scholar] [CrossRef] [Green Version]
- Vella, L.J.; Hill, A.F.; Cheng, L. Focus on Extracellular Vesicles: Exosomes and Their Role in Protein Trafficking and Biomarker Potential in Alzheimer’s and Parkinson’s Disease. Int. J. Mol. Sci. 2016, 17, 173. [Google Scholar] [CrossRef]
- Xu, J.; Yang, M.; Kosterin, P.; Salzberg, B.M.; Milovanova, T.N.; Bhopale, V.M.; Thom, S.R. Carbon monoxide inhalation increases microparticles causing vascular and CNS dysfunction. Toxicol. Appl. Pharmacol. 2013, 273, 410–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.; Milovanova, T.N.; Bogush, M.; Uzun, G.; Bhopale, V.M.; Thom, S.R. Microparticle enlargement and altered surface proteins after air decompression are associated with inflammatory vascular injuries. J. Appl. Physiol. 2011, 112, 204–211. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Kosterin, P.; Salzberg, B.M.; Milovanova, T.N.; Bhopale, V.M.; Thom, S.R. Microparticles generated by decompression stress cause central nervous system injury manifested as neurohypophysial terminal action potential broadening. J. Appl. Physiol. 2013, 115, 1481–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Stoica, B.; Loane, D.J.; Yang, M.; Abulwerdi, G.; Khan, N.; Kumar, A.; Thom, S.R.; Faden, A. Microglial-derived microparticles mediate neuroinflammation after traumatic brain injury. J. Neuroinflam. 2017, 14, 47. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Shan, A.; Wei, Z.; Xu, B. Intravenous mesenchymal stem cell-derived exosomes ameliorate myocardial inflammation in the dilated cardiomyopathy. Biochem. Biophys. Res. Commun. 2018, 503, 2611–2618. [Google Scholar] [CrossRef]
- Cosenza, S.; Toupet, K.; Maumus, M.; Luz-Crawford, P.; Blanc-Brude, O.; Jorgensen, C.; Noël, D. Mesenchymal stem cells-derived exosomes are more immunosuppressive than microparticles in inflammatory arthritis. Theranostics 2018, 8, 1399–1410. [Google Scholar] [CrossRef]
- Fujita, Y.; Kadota, T.; Araya, J.; Ochiya, T.; Kuwano, K. Clinical Application of Mesenchymal Stem Cell-Derived Extracellular Vesicle-Based Therapeutics for Inflammatory Lung Diseases. J. Clin. Med. 2018, 7, 355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, B.S.; Kim, J.O.; Ha, D.H.; Yi, Y.W. Exosomes derived from human adipose tissue-derived mesenchymal stem cells alleviate atopic dermatitis. Stem Cell Res. Ther. 2018, 9, 187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Chopp, M.; Meng, Y.; Katakowski, M.; Xin, H.; Mahmood, A.; Xiong, Y. Effect of exosomes derived from multipluripotent mesenchymal stromal cells on functional recovery and neurovascular plasticity in rats after traumatic brain injury. J. Neurosurg. 2015, 122, 856–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Ye, Y.; Su, X.; He, J.; Bai, W.; He, X. MSCs-Derived Exosomes and Neuroinflammation, Neurogenesis and Therapy of Traumatic Brain Injury. Front. Cell. Neurosci. 2017, 11, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xin, H.; Li, Y.; Cui, Y.; Yang, J.J.; Zhang, Z.G.; Chopp, M. Systemic Administration of Exosomes Released from Mesenchymal Stromal Cells Promote Functional Recovery and Neurovascular Plasticity After Stroke in Rats. Br. J. Pharmacol. 2013, 33, 1711–1715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomi, G.; Surbek, D.; Haesler, V.; Joerger-Messerli, M.; Schoeberlein, A. Exosomes derived from umbilical cord mesenchymal stem cells reduce microglia-mediated neuroinflammation in perinatal brain injury. Stem Cell Res. Ther. 2019, 10, 105. [Google Scholar] [CrossRef]
- Long, Q.; Upadhya, D.; Hattiangady, B.; Kim, D.-K.; An, S.Y.; Shuai, B.; Prockop, D.J.; Shetty, A.K. Intranasal MSC-derived A1-exosomes ease inflammation, and prevent abnormal neurogenesis and memory dysfunction after status epilepticus. Proc. Natl. Acad. Sci. USA 2017, 114, E3536–E3545. [Google Scholar] [CrossRef] [Green Version]
- Bui, T.M.; Mascarenhas, L.A.; Sumagin, R. Extracellular vesicles regulate immune responses and cellular function in intestinal inflammation and repair. Tissue Barriers 2018, 6, e1431038. [Google Scholar] [CrossRef]
- Mitsuhashi, S.; Feldbrügge, L.; Csizmadia, E.; Mitsuhashi, M.; Robson, S.C.; Moss, A.C. Luminal Extracellular Vesicles (EVs) in Inflammatory Bowel Disease (IBD) Exhibit Proinflammatory Effects on Epithelial Cells and Macrophages. Inflamm. Bowel Dis. 2016, 22, 1587–1595. [Google Scholar] [CrossRef]
- Kanninen, K.M.; Bister, N.; Koistinaho, J.; Malm, T. Exosomes as new diagnostic tools in CNS diseases. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2016, 1862, 403–410. [Google Scholar] [CrossRef]
- Gilbert, J.A.; Krajmalnik-Brown, R.; Porazinska, R.L.; Weiss, S.J.; Knight, R. Toward effective probiotics for autism and other neurodevelopmental disorders. Cell 2013, 155, 1446–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gagliardi, A.; Totino, V.; Cacciotti, F.; Iebba, V.; Neroni, B.; Bonfiglio, G.; Trancassini, M.; Passariello, C.; Pantanella, F.; Schippa, S. Rebuilding the Gut Microbiota Ecosystem. Int. J. Environ. Res. Public Health 2018, 15, 1679. [Google Scholar] [CrossRef] [PubMed] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohan, M.; Okeoma, C.M.; Sestak, K. Dietary Gluten and Neurodegeneration: A Case for Preclinical Studies. Int. J. Mol. Sci. 2020, 21, 5407. https://doi.org/10.3390/ijms21155407
Mohan M, Okeoma CM, Sestak K. Dietary Gluten and Neurodegeneration: A Case for Preclinical Studies. International Journal of Molecular Sciences. 2020; 21(15):5407. https://doi.org/10.3390/ijms21155407
Chicago/Turabian StyleMohan, Mahesh, Chioma M. Okeoma, and Karol Sestak. 2020. "Dietary Gluten and Neurodegeneration: A Case for Preclinical Studies" International Journal of Molecular Sciences 21, no. 15: 5407. https://doi.org/10.3390/ijms21155407
APA StyleMohan, M., Okeoma, C. M., & Sestak, K. (2020). Dietary Gluten and Neurodegeneration: A Case for Preclinical Studies. International Journal of Molecular Sciences, 21(15), 5407. https://doi.org/10.3390/ijms21155407