Application of Coordination Compounds with Transition Metal Ions in the Chemical Industry—A Review

 , ,
, ,
Abstract
:
1. Introduction
2. Oxidation Processes
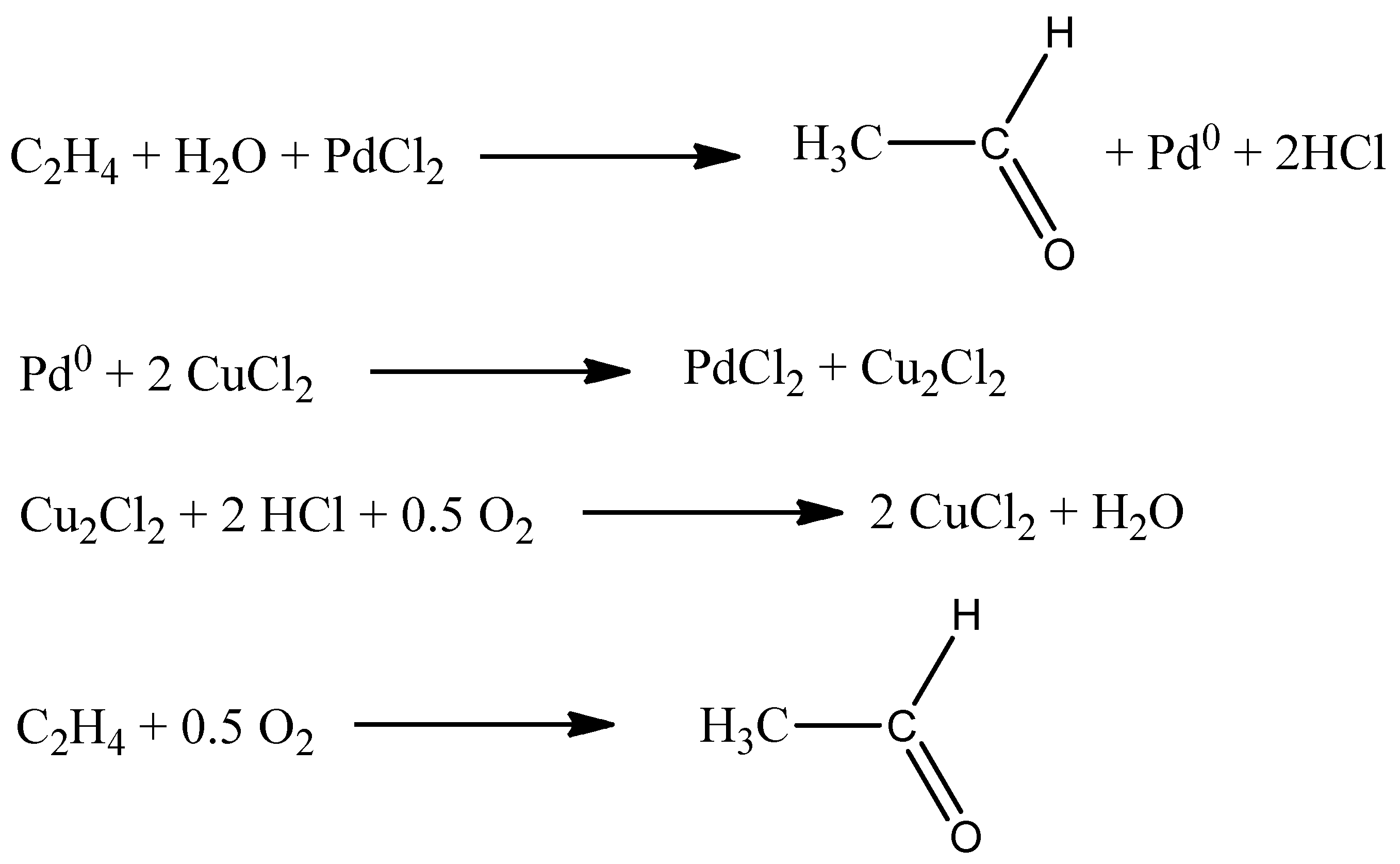
2.1. Wacker Process
2.2. Hydroperoxide Epoxidation Reaction of Olefins Catalyzed by Mo(VI) Complex
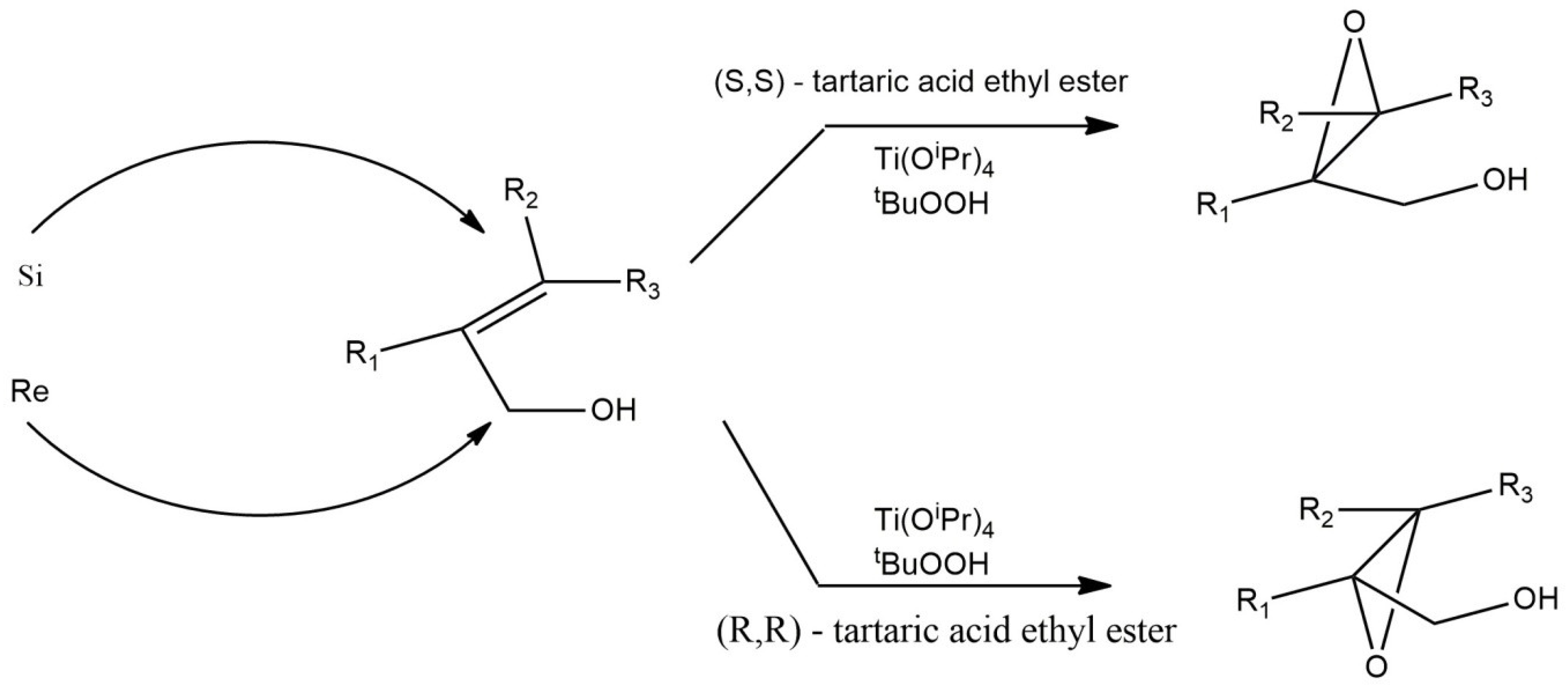
2.3. Sharpless System for Asymmetric Epoxidation of Olefins with Hydroperoxide with Dimeric Titanium Complex as a Catalytically Active Form
2.4. Cyclohexyl Hydroperoxide Decomposition Reaction Catalyzed by Cobalt(II) and Cobalt(III) Complex Compounds
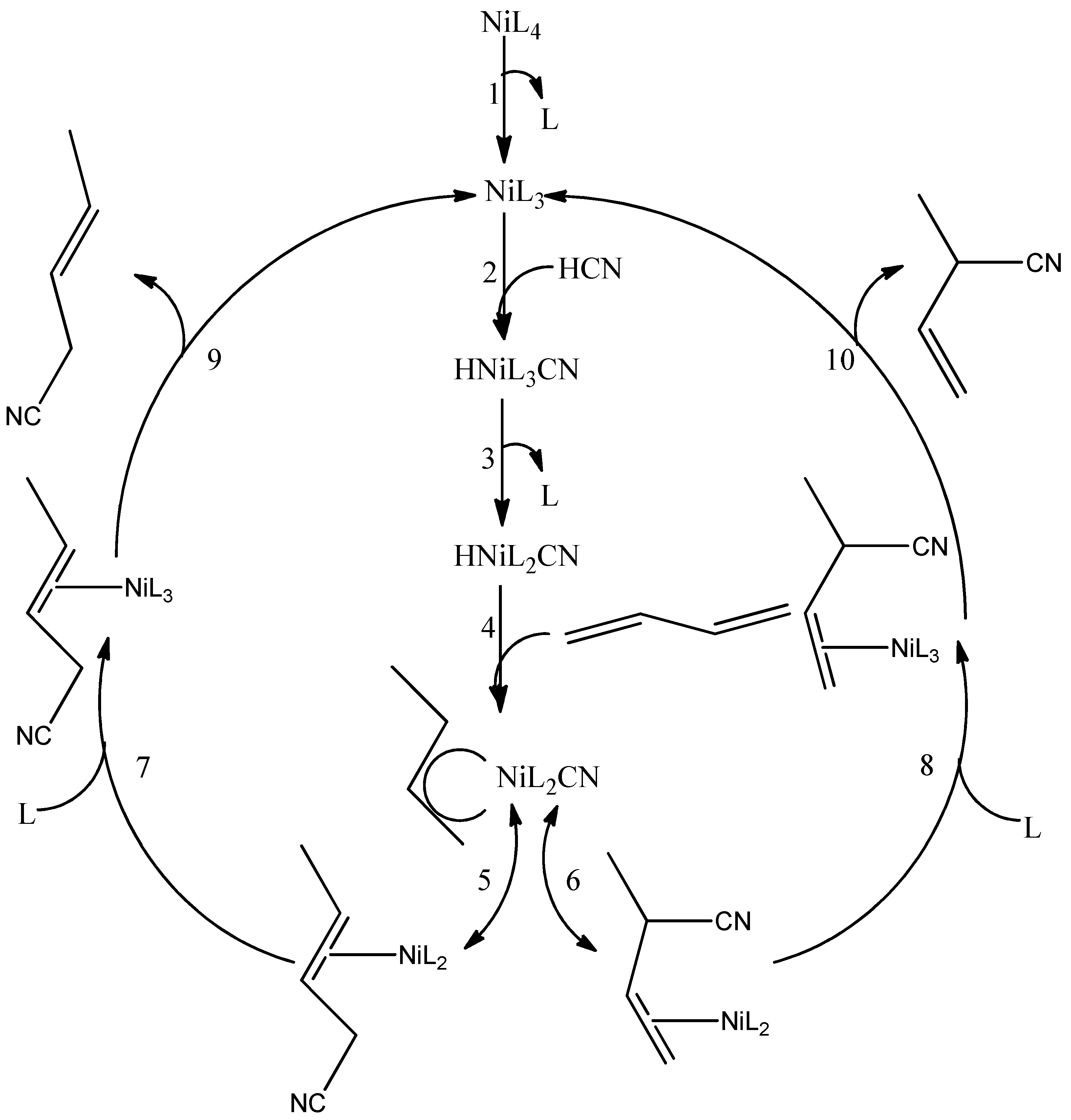
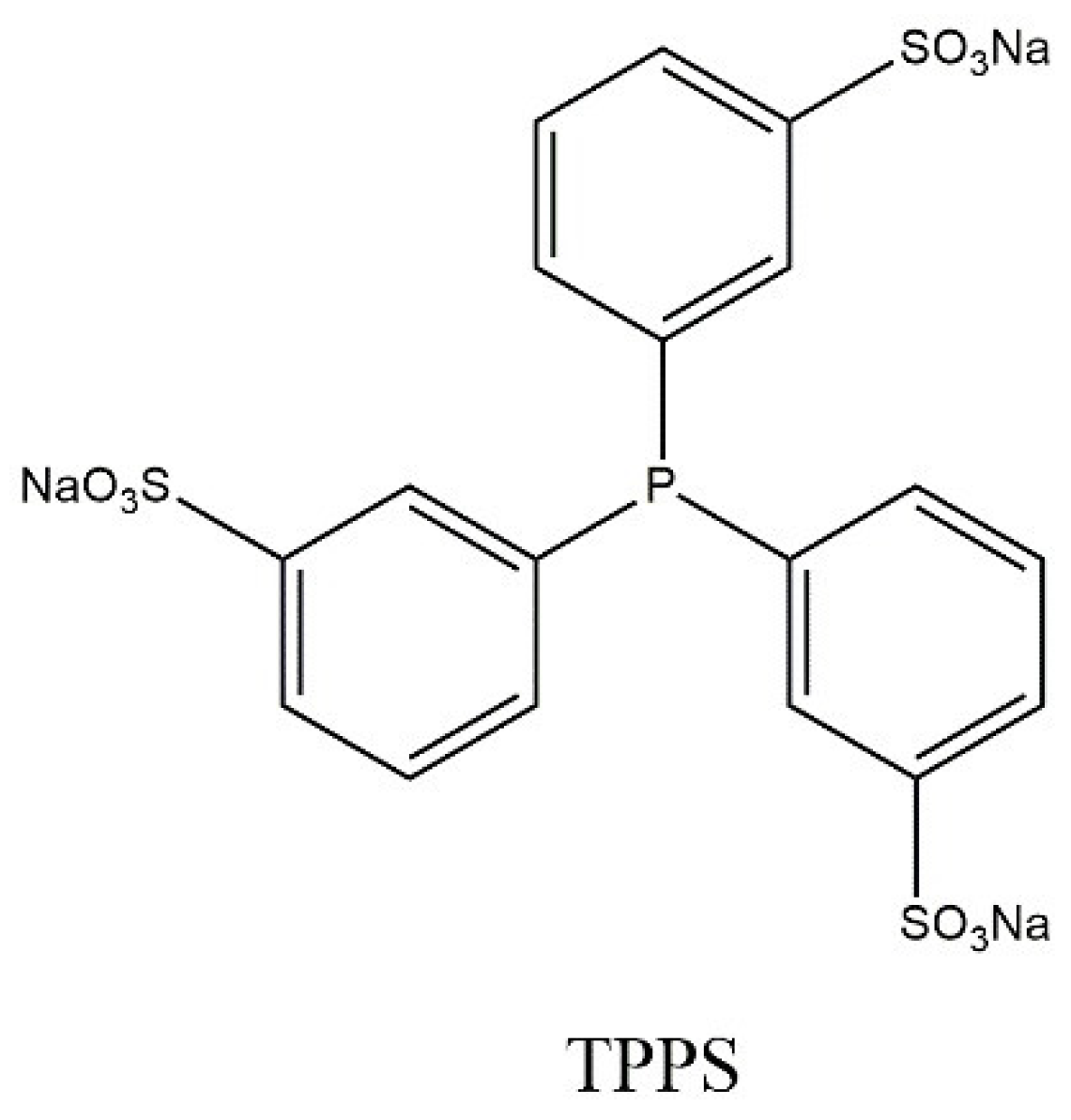
3. The Hydrocyanation Reaction
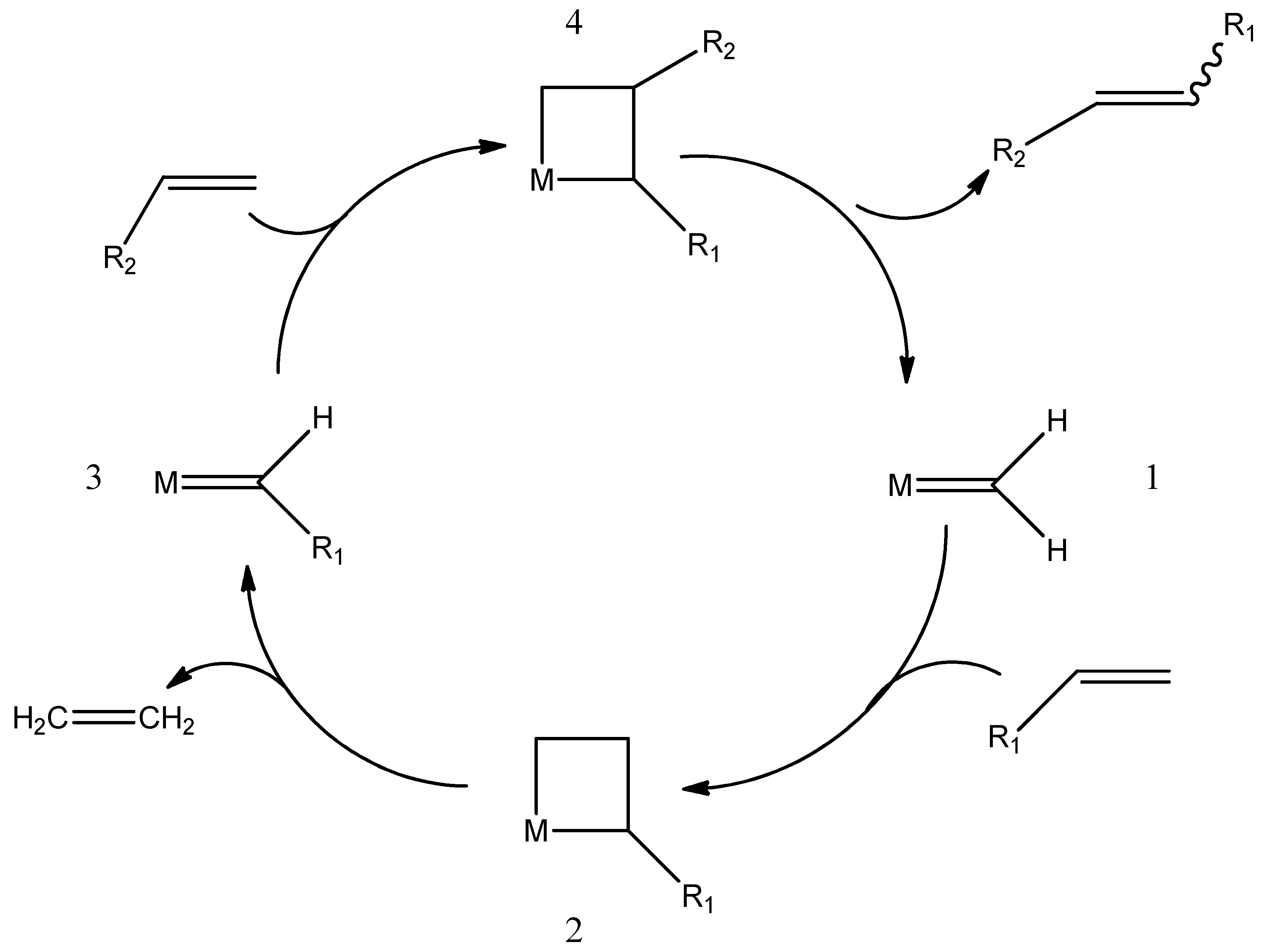
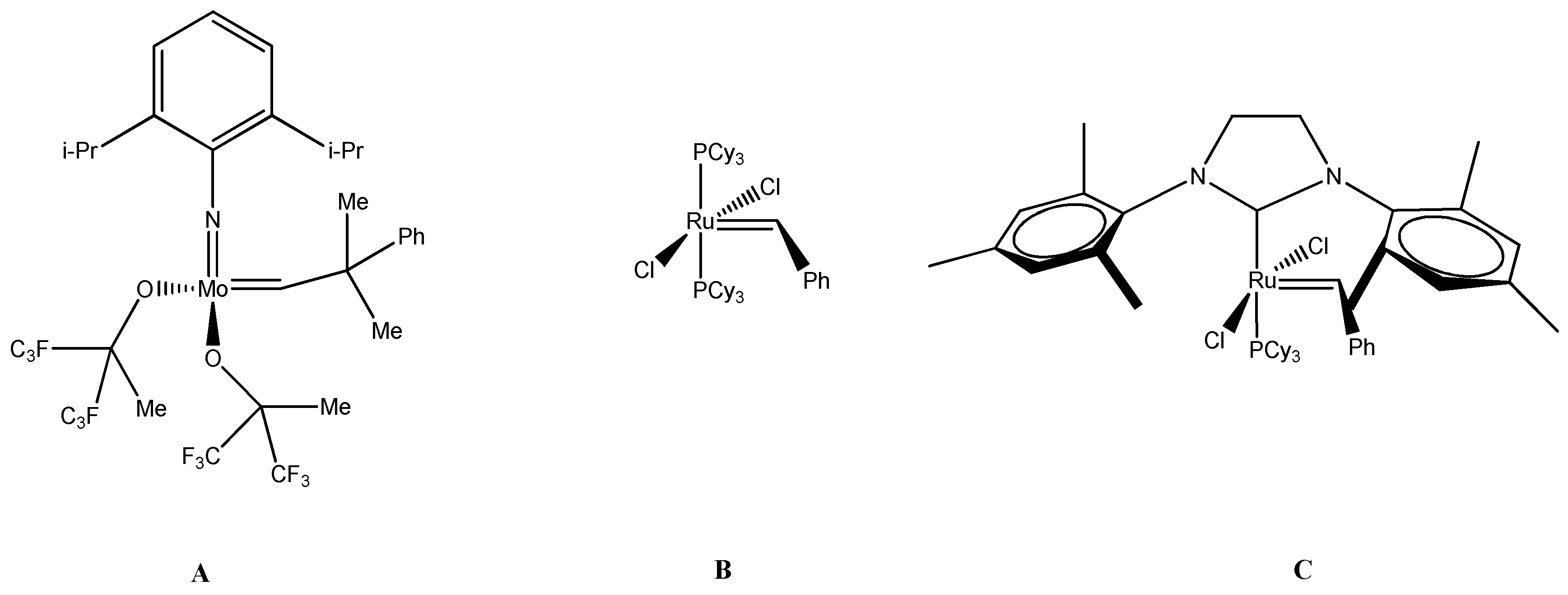
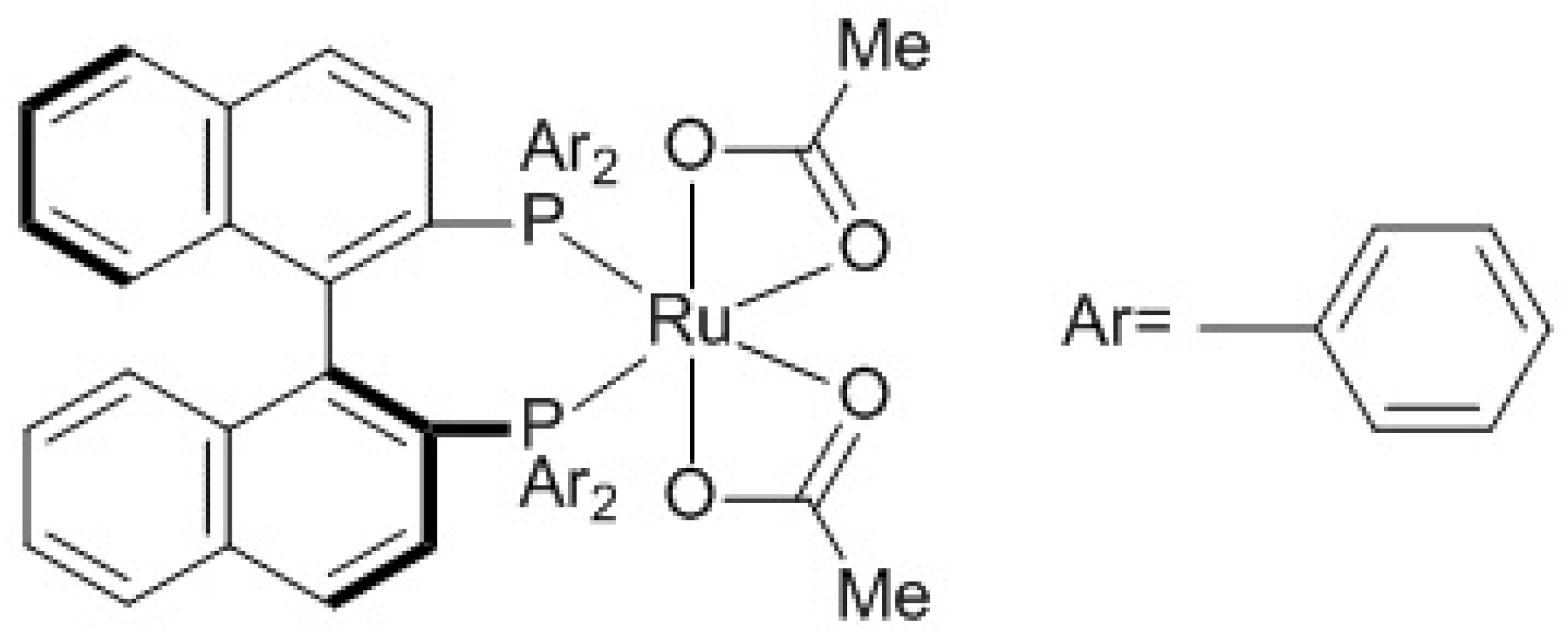
4. Metathesis Reaction
- Cross Metathesis—CM
![Ijms 21 05443 i001]()

- Ring Closing Metathesis—RCM
- Ring Opening Metathesis—ROM
![Ijms 21 05443 i002]()

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
- Ring Opening Metathesis Polymerization—ROMP
- Acyclic Diene Metathesis—ADMET
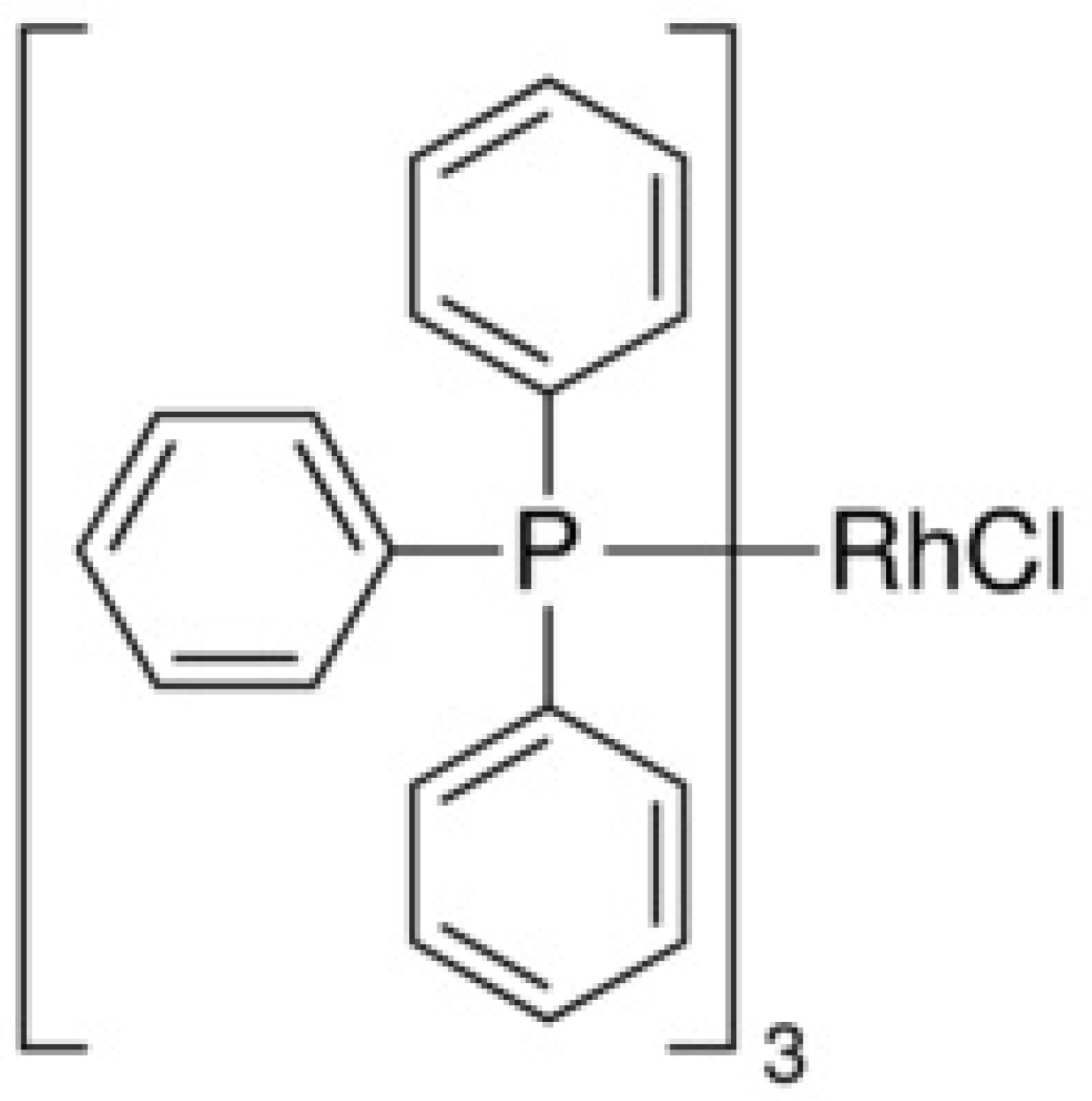
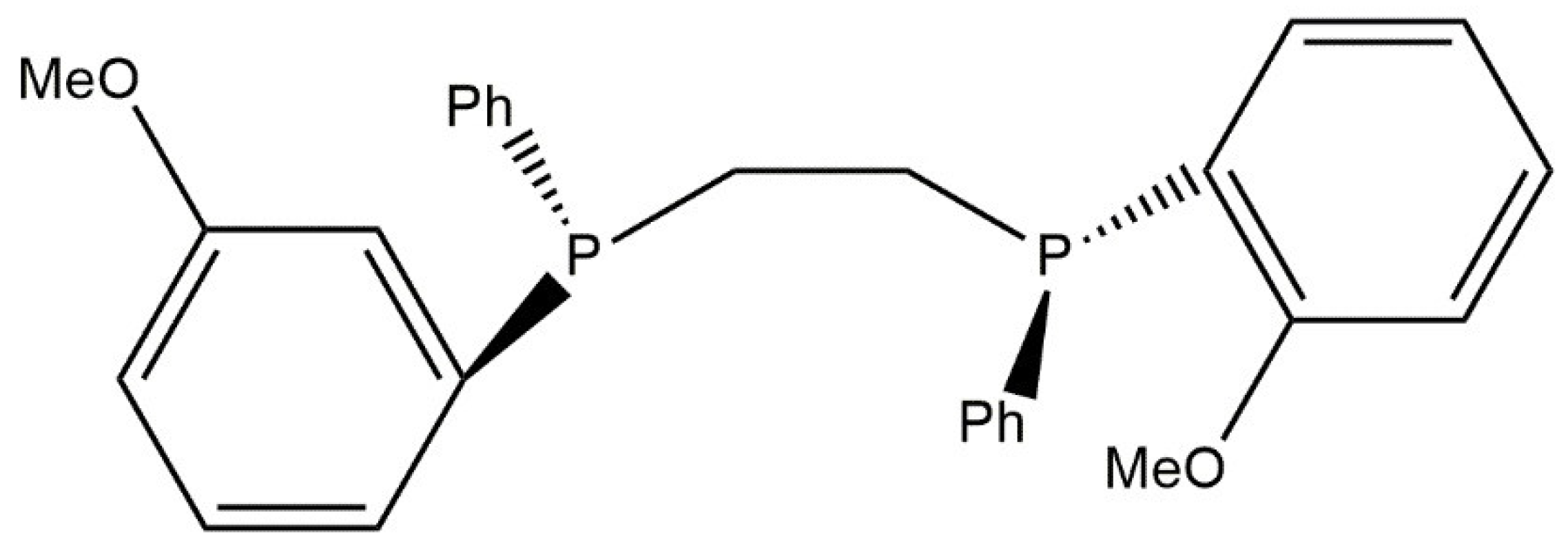
5. Hydrosilylation Reaction
5.1. Reduction Pt(II) to Pt(0) in Reaction with Silane HSi(OEt)3
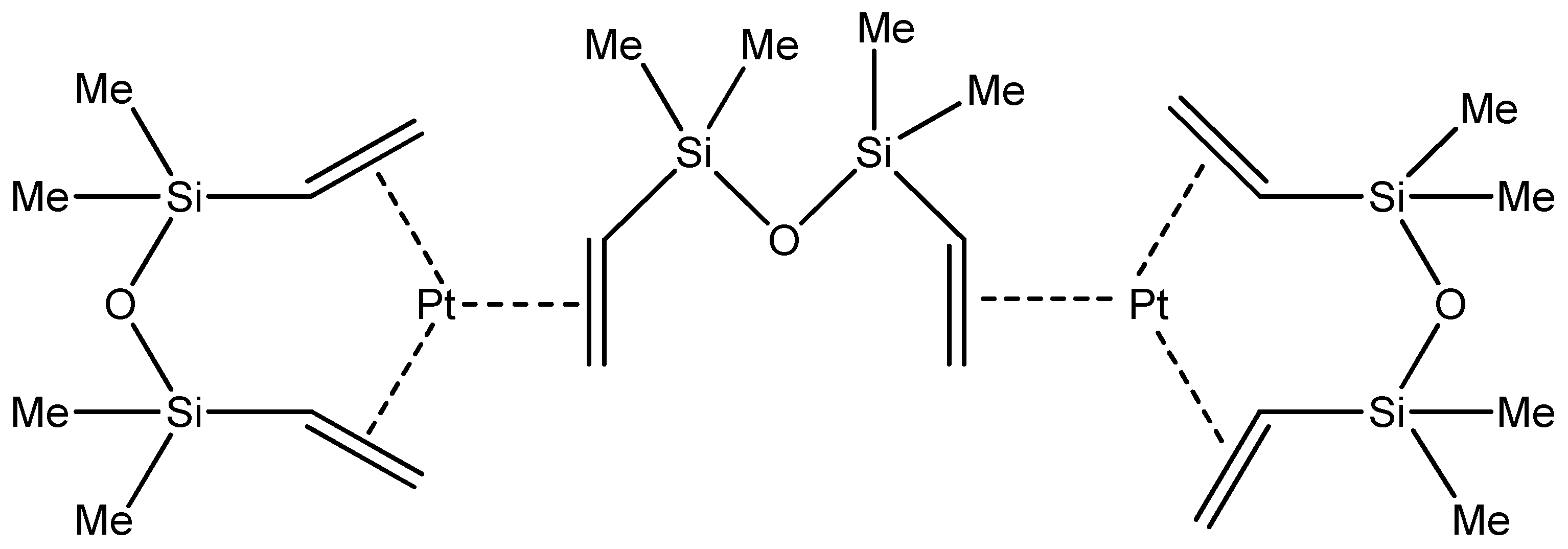
5.2. Karstedt’s Catalyst
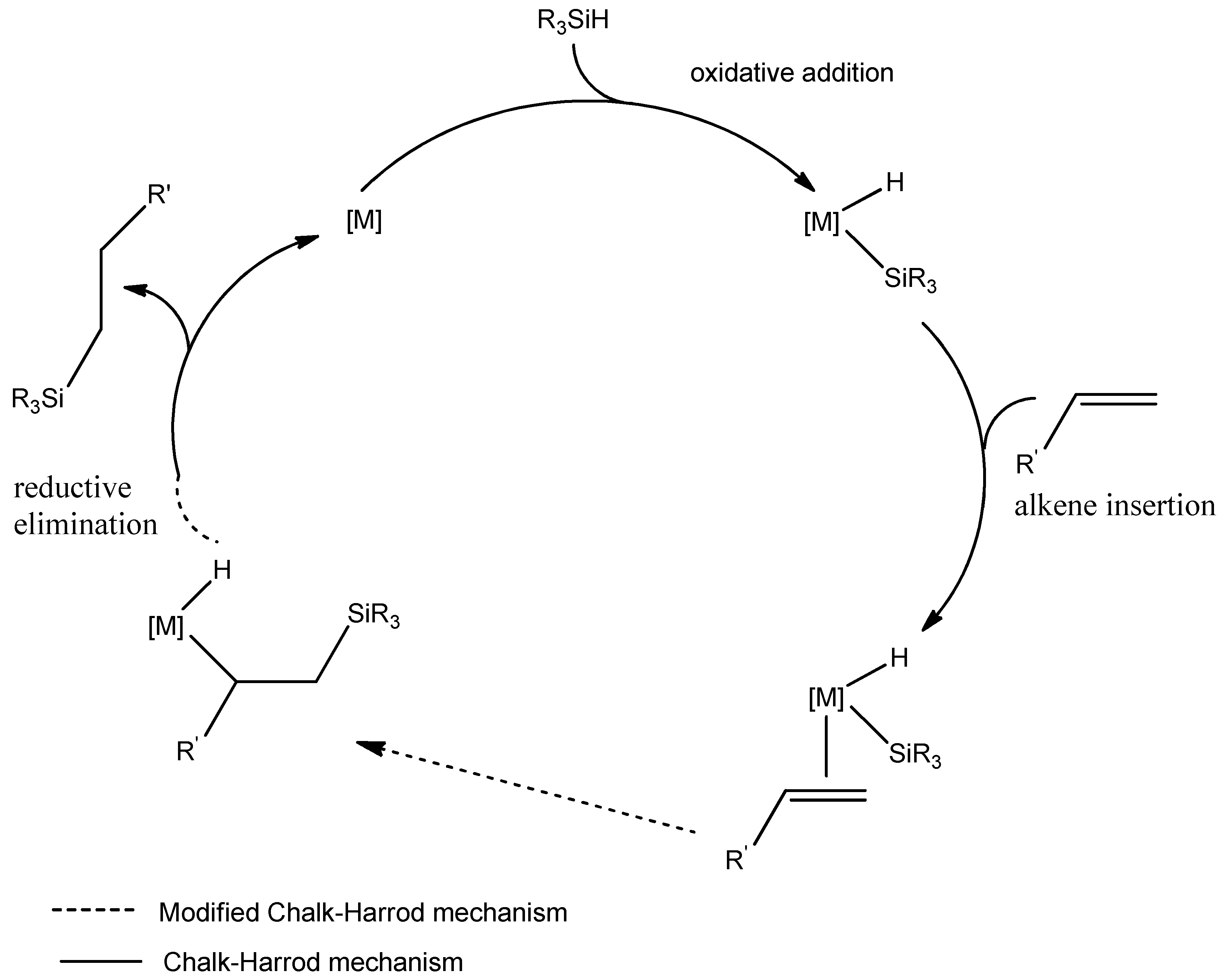
5.3. Hydrosilylation Mechanism Proposed by Chalk and Harrod and Modified Mechanism of Chal–Harrod—Catalyzed by Rhodium Complex Compounds
6. Hydroformylation Reaction
7. Carbonylation Reaction
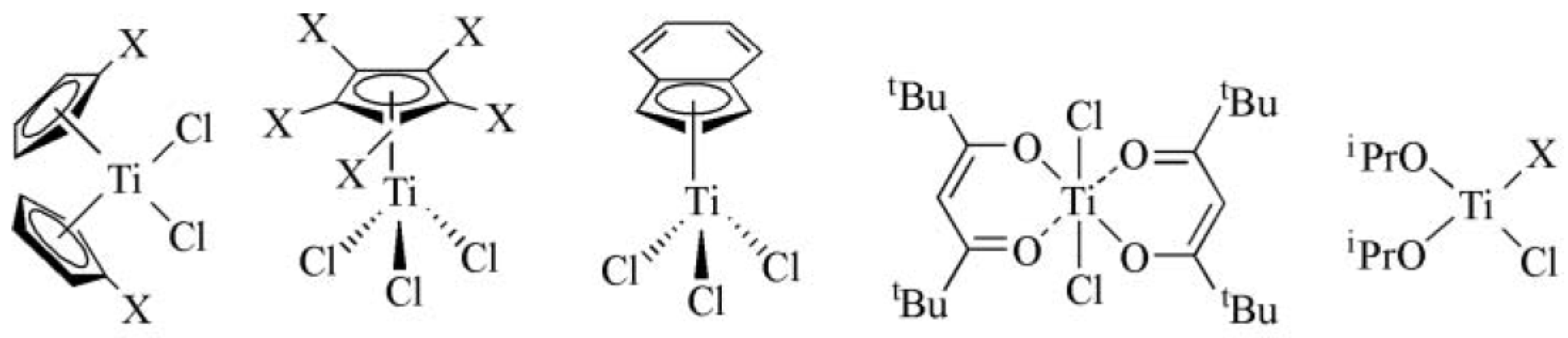
8. Olefin Polymerization
9. Hydrogenation Reaction
10. Catalysis and Green Chemistry
10.1. Ionic Liquids—Catalytic Reactions
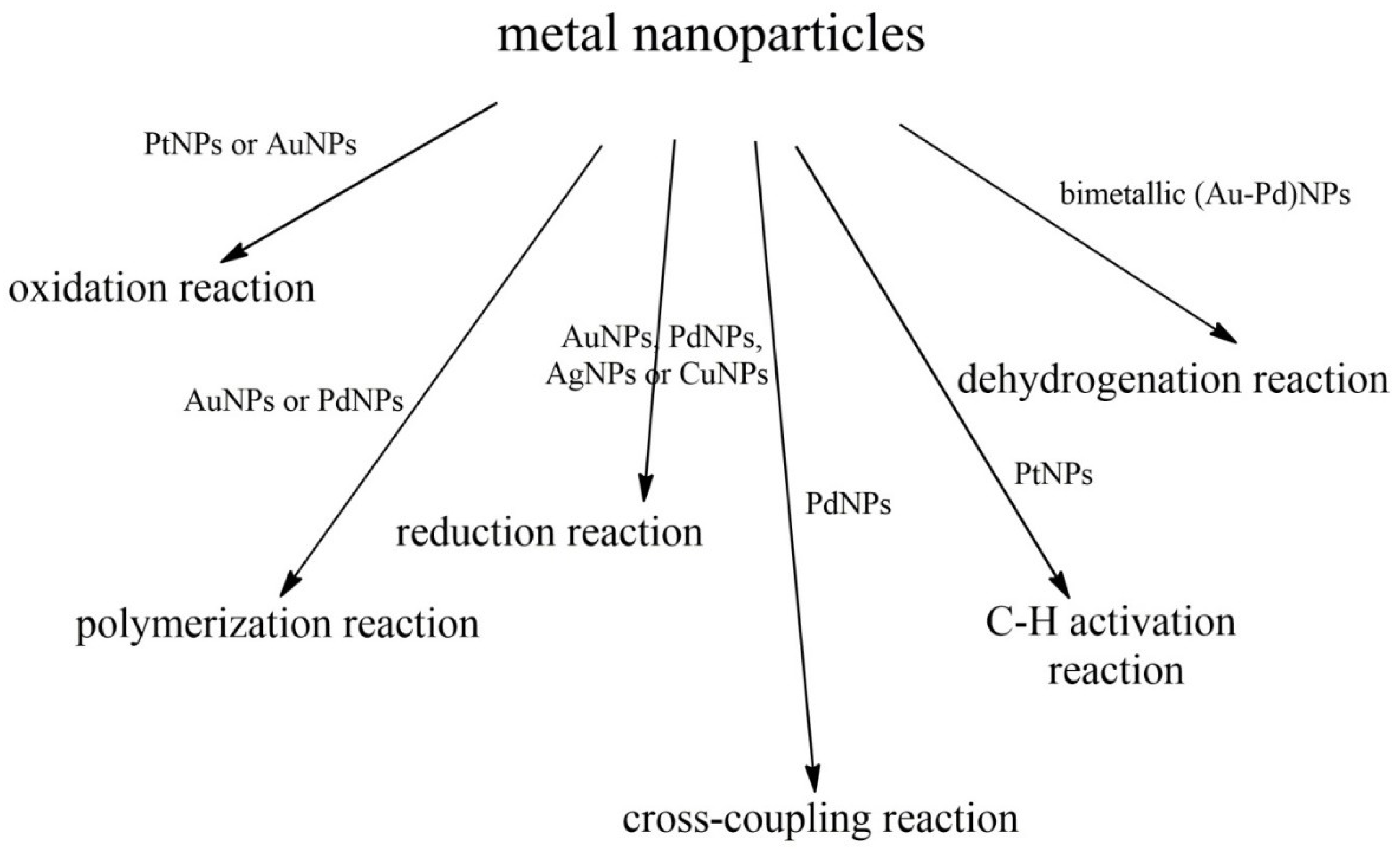
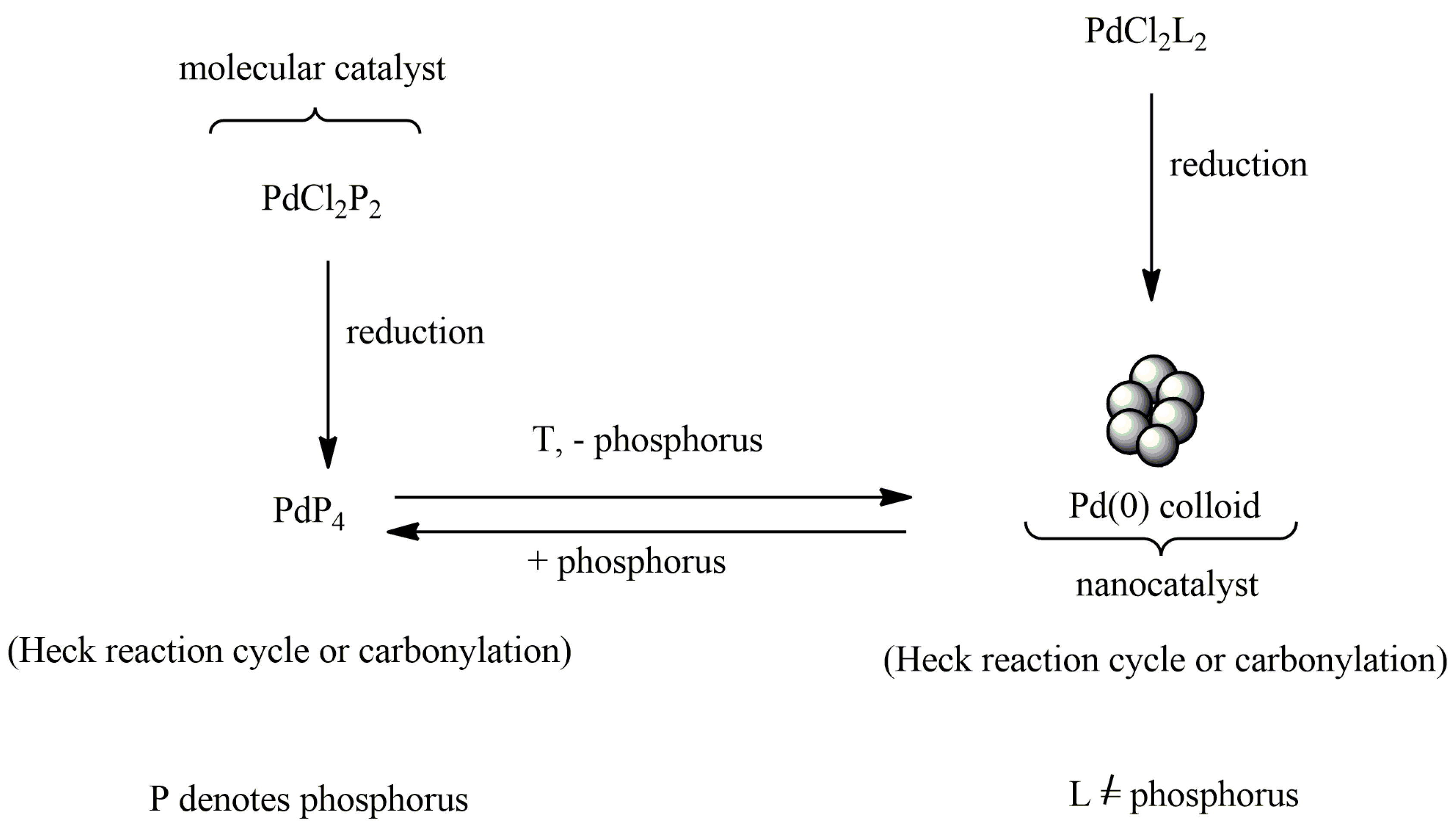
10.2. Catalysts of Organic Reactions—Metal Nanoparticles
11. Conclusions
- PdCl2 taking part in the Wacker process forming a stoichiometric amount of acetaldehyde;
- Modernized CYCLOPOL-bis technology for caprolactam production allows for production costs to be minimized and the quality of products to be improved;
- Ni complexes with bidentate chelate ligands applied as catalysts in SHOP allow a separated C6–C18 fraction to be obtained, which is used for the production of plasticizers and detergents;
- [PtCl2(cod)], [Pt(CH2=CHSiMe2)2O3], [RhCl(PPh3)3], [Pd2(dba)3] are the hydrosilylation catalysts;
- Cu(I) and Fe(II) complexes with ligands such as halides, 2,2′-bipyridine, or tris(2-pyridylmethyl)amine are the catalysts in ATRP;
- [PdCl2(cod)] and Pd(0)/PVP are catalysts for the methoxycarbonylation reaction;
- K2[Fe(CO)4] catalyse carbonylation;
- [Cu(2,3-pydc)(bpp)]·2.5H2O, [Zn(2,3-pydc)(bpp)]·2.5H2O, and [Cd(2,3-pydc) (bpp)(H2O)]·3H2O are metal–organic frameworks which are applied in heterogeneous catalysis;
- [IL]2[PdX4] complex is more active than [PdCl2(cod)] in the Suzuki reaction.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Adam, R.; Mon, M.; Greco, R.; Kalinke, L.H.; Vidal-Moya, A.; Fernandez, A.; Winpenny, R.E.P.; Doménech-Carbó, A.; Leyva-Pérez, A.; Armentano, D.; et al. Self-Assembly of Catalytically Active Supramolecular Coordination Compounds within Metal–Organic Frameworks. J. Am. Chem. Soc. 2019, 141, 10350–10360. [Google Scholar] [CrossRef] [PubMed]
- Glaser, F.; Wenger, O.S. Recent progress in the development of transition-metal based photoredox catalysts. Coordin. Chem. Rev. 2020, 405, 213129. [Google Scholar] [CrossRef]
- Pessoa, J.C.; Correia, I. Salan vs. salen metal complexes in catalysis and medicinal applications: Virtues and pitfalls. Coordin. Chem. Rev. 2019, 388, 227–247. [Google Scholar] [CrossRef]
- Yetra, S.R.; Rogge, T.; Warratz, S.; Struwe, J.; Peng, W.; Vana, P.; Ackermann, L. Micellar Catalysis for Ruthenium(II)-Catalyzed C−H Arylation: Weak-Coordination-Enabled C−H Activation in H2O. Angew. Chem. Int. Ed. 2019, 58, 7490–7494. [Google Scholar] [CrossRef]
- Böhm, L.L. The Ethylene Polymerization with Ziegler catalysts: Fifty years after the discovery. Angew. Chem. Int. Ed. 2003, 42, 5010–5030. [Google Scholar] [CrossRef]
- Ziegler, K.; Holzkamp, E.; Breil, H.; Martin, H. Das Mülheimer normaldruck-polyäthylen-verfahren. Angew. Chem. 1955, 67, 541–547. [Google Scholar] [CrossRef]
- Natta, G.; Pino, P.; Corradini, P.; Danusso, F.; Mantica, E.; Mazzanti, G.; Moraglio, G. Crystalline high polymers of α-olefins. J. Am. Chem. Soc. 1955, 77, 1708–1710. [Google Scholar] [CrossRef]
- Natta, G. Stereospecific catalysis and isotactic polymers. Angew. Chem. 1956, 168, 393–403. [Google Scholar] [CrossRef]
- Cornils, B.; Herrmann, W.A. Aqueous-Phase Organometallic Catalysis: Concepts and Applications; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar]
- James, D.E.; Stille, J.K. The palladium(II) catalyzed olefin carbonylation reaction. Mechanisms and synthetic utility. J. Am. Chem. Soc. 1976, 98, 1810–1823. [Google Scholar] [CrossRef]
- Crabtree, R.H. The Organometallic Chemistry of the Transition Metals; John Wiley & Sons, Inc.: New York, NY, USA, 2001. [Google Scholar]
- Chiusoli, G.P.; Maitlis, P.M. Metal.-Catalysis. Mechanism and Industrial Organic Processes; RSC Publishing: Cambride, UK, 2006. [Google Scholar]
- Beyramabadi, S.A.; Eshtiagh-Hosseini, H.; Housaindokht, M.R.; Morsali, A. Mechanism and kinetics of the Wacker process: A quantum mechanical approach. Organometallics 2008, 27, 72–79. [Google Scholar] [CrossRef]
- Keith, J.A.; Nielsen, R.J.; Oxgaard, J.; Goddard III, W.A.; Henry, P.M. Comment on “Mechanism and Kinetics of the Wacker Process: A Quantum Mechanical Approach”. Organometallics 2009, 28, 1618–1619. [Google Scholar] [CrossRef] [Green Version]
- Peili, T.; Wickens, Z.K.; Dong, G.; Grubbs, R.H. Efficient and highly aldehyde selective Wacker oxidation. Am. Chem. Soc. 2012, 14, 3237–3239. [Google Scholar] [CrossRef] [Green Version]
- Wickens, Z.K.; Skakuj, K.; Morandi, B.; Grubbs, R.H. Catalyst-controlled Wacker-type oxidation: Facile access to functionalized aldehydes. J. Am. Chem. Soc. 2014, 136, 890–893. [Google Scholar] [CrossRef] [Green Version]
- Hu, K.F.; Ning, X.S.; Qu, J.P.; Kang, Y.B. Tuning regioselectivity of Wacker oxidation in one catalytic system: Small change makes big step. J. Org. Chem. 2018, 83, 11327–11332. [Google Scholar] [CrossRef] [PubMed]
- Corless, V.B.; Holownia, A.; Foy, H.; Mendoza-Sanchez, R.; Adachi, S.; Dudding, T.; Yudin, A.K. Synthesis of α-borylated ketones by regioselective Wacker oxidation of alkenylboronates. Org. Lett. 2018, 20, 5300–5303. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Xie, X.; Wang, Y.; Wen, X.; Zhao, Y.; Ding, C. Highly selective Wacker reaction of styrene derivatives: A green and efficient aerobic oxidative process promoted by benzoquinone/NaNO2/HClO4 under mild conditions. Org. Biomol. Chem. 2013, 11, 2947–2950. [Google Scholar] [CrossRef]
- Kulkarni, M.G.; Shaikh, Y.B.; Borhade, A.S.; Chavhan, S.W.; Dhondge, A.P.; Gaikwad, D.D.; Dhatrak, N.R. Greening the Wacker process. Tetrahedron Lett. 2013, 54, 2293–2295. [Google Scholar] [CrossRef]
- Zawadzki, P.; Czardybon, W.; Chrobok, A. The oxidation reactions of selected functional groups using oxone® as a source of molecular oxygen. Wiad. Chem. 2016, 70, 5–6. [Google Scholar]
- Yudin, A.K. Aziridines and Epoxides in Organic Synthesis; Wiley-WCH: Weinheim, Germany, 2006. [Google Scholar]
- Itsuno, S. Polymeric Chiral Catalyst Design and Chiral Polymer Synthesis; Wiley-WCH: Weinheim, Germany, 2011. [Google Scholar]
- Katsuki, T.; Sharpless, K.B. The first practical method for asymmetric epoxidation. J. Am. Chem. Soc. 1980, 102, 5974–5976. [Google Scholar] [CrossRef]
- Irie, R.; Noda, K.; Ito, Y.; Matsumoto, N.; Katsuki, T. Catalytic asymmetric epoxidation of unfunctionalized olefins. Tetrahedron Lett. 1990, 31, 7345–7348. [Google Scholar] [CrossRef]
- Woodard, S.; Finn, M.G.; Sharpless, K.B. Mechanism of asymmetric epoxidation. 1. Kinetics. J. Am. Chem. Soc. 1991, 113, 106–113. [Google Scholar] [CrossRef]
- Kozieł, S.J.; Luksa, A. Economic conditions for the utilization of chemical synthesis by-products—Caprolactam production. JCEEA 2017, 4, 511–519. [Google Scholar]
- Gruszka, M.; Malinowski, T.; Rygiel, S.; Wais, J. CYCLOPOL and CYCLOPOL-bis; cyclohexanone technology in India. CHEMIK 2012, 66, 1083–1094. [Google Scholar]
- Bhaduri, S.; Mukesh, D. Homogenous Catalysi: Mechanism and Industrial Appplications A.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2000. [Google Scholar]
- Parshall, G.W.; Ittel, S.D. Homogenous Catalysis: The Aplications and Chemistry of Catalysis by Soluble Transition Metal Complexes; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1992. [Google Scholar]
- Tolman, C.A. Phosphorus ligand exchange equilibriums on zerovalent nickel. Dominant role for steric effects. J. Am. Chem. Soc. 1970, 92, 2956–2965. [Google Scholar] [CrossRef]
- Rahman, M.M.; Liu, H.Y.; Eriks, K.; Prock, A.; Giering, W.P. Quantitative analysis of ligand effects. Part 3. Separation of phosphorus(III) ligands into pure sigma-donors and sigma.-donor/.pi.-acceptors. Comparison of basicity and sigma-donicity. Organometallics 1989, 8, 1–7. [Google Scholar] [CrossRef]
- Gosser, L.W.; Tolman, C.A. New three-coordinate complex of nickel(0). Tris(tri-o-tolyl phosphite)nickel. Inorg. Chem. 1970, 9, 2350–2353. [Google Scholar] [CrossRef]
- Seidel, W.C.; Tolman, C.A. Ethylene [bis(tri-o-tolyl phosphite)] nickel(0). Inorg. Chem. 1970, 9, 2354–2357. [Google Scholar] [CrossRef]
- Tolman, C.A. The 16 and 18 electron rule in organometallic chemistry and homogeneous catalysis. Chem. Soc. Rev. 1972, 1, 337–353. [Google Scholar] [CrossRef]
- Tolman, C.A. Chemistry of tetrakis(triethyl phosphite) nickel hydride, HNi[P(OEt)3]4+. I. Nickel hydride formation and decay. J. Am. Chem. Soc. 1970, 92, 4217–4222. [Google Scholar] [CrossRef]
- Goertz, W.; Keim, W.; Vogt, D.; Englert, U.; Boele, M.D.K.; van de Veen, L.A.; Kamer, P.C.J.; van Leeuwen, P.W.N.M. Electronic effects in the nickel-catalysed hydrocyanation of styrene applying chelating phosphorus ligands with large bite angles. J. Chem. Soc. Dalton Trans. 1998, 2981–2988. [Google Scholar] [CrossRef]
- Tolman, C.A.; Mckinney, R.J.; Seidel, W.C.; Druliner, J.D.; Stevens, W.R. Homogeneous nickel-catalyzed olefin hydrocyanation. Adv. Catal. 1985, 33, 1–46. [Google Scholar] [CrossRef]
- Schuppe, A.W.; Borrajo-Calleja, G.M.; Buchwald, S.L. Enantioselective olefin hydrocyanation without cyanide. J. Am. Chem. Soc. 2019, 141, 18668–18672. [Google Scholar] [CrossRef] [PubMed]
- Bini, L.; Müller, C.; Vogt, D. Ligand development in the Ni-catalyzed hydrocyanation of alkenes. Chem. Commun. 2010, 46, 8325–8334. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Chen, J.; Ritter, T. Rh-catalyzed anti-markovnikov hydrocyanation of terminal alkynes. J. Am. Chem. Soc. 2017, 139, 7184–7187. [Google Scholar] [CrossRef] [PubMed]
- Michel, N.W.M.; Jeanneret, A.D.M.; Kim, H.; Rousseaux, S.A.L. Nickel-catalyzed cyanation of benzylic and allylic pivalate esters. J. Org. Chem. 2018, 83, 11860–11872. [Google Scholar] [CrossRef] [PubMed]
- Zahrt, A.F.; Athavale, S.V.; Denmark, S.E. Quantitative structure–selectivity relationships in enantioselective catalysis: Past, present, and future. Chem. Rev. 2020, 120, 1620–1689. [Google Scholar] [CrossRef] [Green Version]
- Schneider, V.; Frolich, P.K. Mechanism of formation of aromatics from lower paraffins. J. Ind. Eng. Chem. 1931, 23, 1405–1410. [Google Scholar] [CrossRef]
- Banks, R.L.; Bailey, G.C. Olefin disproportionation. A new catalytic process. Ind. Eng. Chem. Prod. Res. Dev. 1964, 3, 170–173. [Google Scholar] [CrossRef]
- Dall’Asta, G.; Mazzanti, G.; Natta, G.; Porri, L. Anionic-coordinated polymerization of cyclobutene. Makromol. Chem. 1962, 56, 244–256. [Google Scholar] [CrossRef]
- Natta, G.; Dall’Asta, G.; Mazzanti, G. Stereospecific homopolymerization of cyclopentene. Angew. Chem. Int. Ed. 1964, 3, 723–729. [Google Scholar] [CrossRef]
- Calderon, N. Olefin metathesis reaction. Acc. Chem. Res. 1972, 5, 127–132. [Google Scholar] [CrossRef]
- Calderon, N.; Chen, H.Y.; Scott, K.W. Olefin metathesis—A novel reaction for skeletal transformations of unsaturated hydrocarbons. Tetrahedron Lett. 1967, 34, 3327–3329. [Google Scholar] [CrossRef]
- Trnka, T.M.; Grubbs, R.H. The Development of L2X2RuCHR olefin metathesis catalysts: an organometallic success story. Acc. Chem. Res. 2001, 34, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Sanford, M.S.; Love, J.A.; Grubbs, R.H. Mechanism and activity of ruthenium olefin metathesis catalysts. J. Am. Chem. Soc. 2001, 123, 6543–6554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herisson, J.L.; Chauvin, Y. Catalyse de transformation des oléfines par les complexes du tungstène. II. Télomérisation des oléfines cycliques en présence d’oléfines acycliques. Makromol. Chem. 1971, 141, 161–176. [Google Scholar] [CrossRef]
- Schrock, R.R.; Hoveyda, A.H. Molybdenum and tungsten imido alkylidene complexes as efficient olefin-metathesis catalysts. Angew. Chem. Int. Ed. 2003, 42, 4592–4633. [Google Scholar] [CrossRef]
- Grubbs, R.H. Handbook of Metathesis; Wiley-VCH: Weinheim, Germany, 2003. [Google Scholar]
- Lutz, E.F. Shell higher olefins process. J. Chem. Ed. 1986, 63, 202. [Google Scholar] [CrossRef]
- Vougioukalakis, G.C.; Grubbs, R.H. Ruthenium-based heterocyclic carbene-coordinated olefin metathesis catalysts. Chem. Rev. 2010, 110, 1746–1787. [Google Scholar] [CrossRef]
- Li, X.Y.; Kang, Q.P.; Liu, L.Z.; Ma, J.C.; Dong, W.K. Trinuclear Co(II) and mononuclear Ni(II) salamo-type bisoxime coordination compounds. Crystals 2018, 8, 43. [Google Scholar] [CrossRef] [Green Version]
- Assi, H.; Mouchaham, G.; Steunou, N.; Devic, T.; Serre, C. Titanium coordination compounds: From discrete metal complexes to metal–organic frameworks. Chem. Soc. Rev. 2017, 46, 3431–3452. [Google Scholar] [CrossRef]
- Ludwig, J.R.; Zimmerman, P.M.; Gianino, J.; Corinna, B.; Schindler, S. Iron(III)-catalysed carbonyl–olefin metathesis. Nature 2016, 533, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Ravindar, L.; Lekkala, R.; Rakesh, K.P.; Asiri, A.M.; Marwani, H.M.; Qin, H.L. Carbonyl–olefin metathesis: A key review. Org. Chem. Front. 2018, 5, 1381–1391. [Google Scholar] [CrossRef]
- Marcinec, B. Hydrosililation. A Comprehensive Review on Recent Advances. Advances in Silicon Science; Springer: Basel, Switzerland, 2009. [Google Scholar]
- Lewis, L.N.; Stein, J.; Gao, Y.; Colborn, R.E.; Hutchins, G. Platinum Catalysts Used in the Silicones Industry Their Synthesis and Activity in Hydrosilylation; GE Corporate Research & Development Center, General Electric Company, Schenectady: New York, NY, USA, 1997. [Google Scholar]
- D’Amelia, R.P.; Mancuso, J.; Nirode, W.F.; Singh, S. Characterization and synthesis of functionalized polysilalkylene siloxane monomers by hydrosilylation reaction. World J. Org. Chem. 2019, 7, 5–13. [Google Scholar] [CrossRef]
- Nakajima, Y.; Shimada, S. Hydrosilylation reaction of olefins: Recent advances and perspectives. RSC Adv. 2015, 5, 20603–20616. [Google Scholar] [CrossRef]
- Liu, Y.; Deng, L. Mode of activation of cobalt(II) amides for catalytic hydrosilylation of alkenes with tertiary silanes. J. Am. Chem. Soc. 2017, 139, 1798–1801. [Google Scholar] [CrossRef]
- Du, X.; Huang, Z. Advances in base-metal-catalyzed alkene hydrosilylation. ACS Catal. 2017, 7, 1227–1243. [Google Scholar] [CrossRef]
- Naganawa, Y.; Inomata, K.; Sato, K.; Nakajima, Y. Hydrosilylation reactions of functionalized alkenes. Tetrahedron Lett. 2020, 61, 151513. [Google Scholar] [CrossRef]
- Obligacion, J.V.; Chirik, P.J. Earth-abundant transition metal catalysts for alkene hydrosilylation and hydroboration. Nat. Rev. Chem. 2018, 15–34. [Google Scholar] [CrossRef]
- Wilkinson, G.; Gordon, F.; Stone, A.; Abel, E.W. Comprehensive Organometallic Chemistry; Elsevier Science Ltd.: Amsterdam, The Netherlands, 1982. [Google Scholar]
- Trzeciak, A.M.; Ziółkowski, J.J. Perspectives of rhodium organometallic catalysis. Fundamental and applied aspects of hydroformylation. Coord. Chem. Rev. 1999, 190–192, 883–900. [Google Scholar] [CrossRef]
- Frohling, C.D.; Kohlpaitner, C.W. Applied homogeneous Catalysis. Carbon Monoxide and Synthesis Gas Chemistry. In Applied Homogeneous Catalysis with Organometallic Compounds: A Comprehensive Handbook in Two Volumes; Wiley–VCH: Weinheim, Germany, 1996. [Google Scholar]
- Evans, D.; Osborn, J.A.; Wilkinson, G. Hydroformylation of alkenes by use of rhodium complex catalysts. J. Chem. Soc. A 1968, 0, 3133–3142. [Google Scholar] [CrossRef]
- Yagupsky, G.; Brown, C.K.; Wilkinson, G. Further studies on hydridocarbonyltris (triphenylphosphine) hodium (I); intermediate species in hydroformylation; rhodium and iridium analogues. J. Chem. Soc. A 1970, 1392–1401. [Google Scholar] [CrossRef]
- Kuntz, E.G.; Godard, G.; Basset, J.M.; Vittori, O.M. Nickel(0)-TPPTS-cyanide complex in water. An efficient and flexible catalyst for the isomerisation of olefinic compounds at room temperature. Oil Gas. Sci. Technol. 2007, 62, 781–785. [Google Scholar] [CrossRef]
- Cornils, B.; Herrmann, W.A. Aqueous–Phase catalysis, Multiphase Homogeneous Catalysis, B. Cornil; Wiley–VCH Verlag GmbH & Co. KgaA: Weinheim, Germany, 2005. [Google Scholar]
- Trzeciak, A.M.; Ziółkowski, J.J. Carbonylation of benzyl bromide to benzeneacetic acid and its esters catalysed by water-soluble palladium complexes. J. Mol. Catal. A. Chem. 2000, 154, 93–101. [Google Scholar] [CrossRef]
- Tolman, C.A. Steric effects of phosphorus ligands in organometallic chemistry and homogeneous catalysis. Chem. Rev. 1977, 77, 313–348. [Google Scholar] [CrossRef]
- Van Leeuven, P.W.N.M. Rhodium Catalyzed Hydroformylation; Kluver Academic Publisher: Dordrecht, The Netherlands, 2000. [Google Scholar]
- Behr, A.; Neubert, P. Applied Homogeneous Catalysis; Wiley–VCH: Weinheim, Germany, 2012. [Google Scholar]
- Casey, C.P.; Paulsen, E.L.; Beuttenmueller, E.W.; Proft, B.R.; Petrovich, L.M.; Matter, B.A.; Powell, D.R. Electron withdrawing substituents on equatorial and apical phosphines have opposite effects on the regioselectivity of rhodium catalyzed hydroformylation. J. Am. Chem. Soc. 1997, 119, 11817–11825. [Google Scholar] [CrossRef]
- Gil, W.; Trzeciak, A.M.; Ziółkowski, J.J. Rhodium(I) N-heterocyclic carbene complexes as highly selective catalysts for 1-hexene hydroformylation. Organometallics 2008, 27, 4131–4138. [Google Scholar] [CrossRef]
- Gil, W.; Trzeciak, A.M. N-Heterocyclic carbene–rhodium complexes as catalysts for hydroformylation and related. Coord. Chem. Rev. 2011, 255, 473–483. [Google Scholar] [CrossRef]
- Sakai, N.; Nozaki, K.; Takaya, H. Asymmetric hydroformylation of 1,2-disubstituted olefins catalysed by chiral phosphinephosphite–rhodium(I) complexes. J. Chem. Soc. Chem. Comm. 1994, 4, 395–396. [Google Scholar] [CrossRef]
- Li, C.; Wang, W.; Yan, L. A mini review on strategies for heterogenization of rhodium-based hydroformylation catalysts. Front. Chem. Sci. Eng. 2018, 12, 113–123. [Google Scholar] [CrossRef]
- Lang, R.; Li, T.; Matsumura, D.; Miao, S.; Ren, Y.; Cui, Y.T.; Tan, Y.; Qiao, B.; Li, L.; Wang, A.; et al. Hydroformylation of olefins by a rhodium single-atom catalyst with activity comparable to RhCl(PPh3)3. Angew. Chem. Int. Ed. 2016, 55, 16054–16058. [Google Scholar] [CrossRef]
- Ndolomingo, M.J.; Bingwa, N.; Meijboom, R. Review of supported metal nanoparticles: Synthesis methodologies, advantages and application as catalysts. J Mater Sci. 2020, 55, 6195–6241. [Google Scholar] [CrossRef]
- Liu, S.; Dai, X.; Wang, H.; Wang, X.; Shi, F. Organic ligand-free hydroformylation with Rh particles as catalyst. Chin. J. Chem. 2020, 38, 139–143. [Google Scholar] [CrossRef]
- Pandey, S.; Raj, K.V.; Shinde, D.R.; Vanka, K.; Kashyap, V.; Kurungot, S.; Vinod, C.P.; Chikkali, S.H. Iron catalyzed hydroformylation of alkenes under mild conditions: Evidence of an Fe(II) catalyzed process. J. Am. Chem. Soc. 2018, 140, 4430–4439. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, J. Palladium Reagents and Catalysts: New Perspectives for the 21th Century; John Wiley&Sons Ltd.: Weinheim, Germany, 2004. [Google Scholar]
- Beller, M.; Bolm, C. Transition Metals for Organic Synthesis: Building Blocks and Fine Chemicals; WILEY-VCH Verlag GmbH & Co.: Weinheim, Germany, 2004. [Google Scholar]
- Beller, M.; Cornils, B.; Frohning, C.D.; Kohlpaintner, C.W. Progress in hydroformylation and carbonylation. J. Mol. Catal. A Chem. 1995, 10, 417–485. [Google Scholar] [CrossRef]
- Maitlis, P.M.; Haynes, A.; James, B.R.; Catellani, M.; Chiusoli, G.P. Iodide effects in transition metal catalyzed reactions. Dalton Trans. 2004, 21, 3409–3419. [Google Scholar] [CrossRef] [PubMed]
- Skoda-Foldes, R.; Kollar, L. Iodide effects in transition metal catalyzed reactions. Curr. Org. Chem. 2002, 6, 1097–1119. [Google Scholar] [CrossRef]
- Zhao, S.; Mankad, N.P. Metal-catalysed radical carbonylation reactions. Catal. Sci. Technol. 2019, 9, 3603–3613. [Google Scholar] [CrossRef]
- Liu, S.; Wang, H.; Daiab, X.; Shi, F. Organic ligand-free carbonylation reactions with unsupported bulk Pd as catalyst. Green Chem. 2018, 20, 3457–3462. [Google Scholar] [CrossRef]
- Kalck, P.; Le Berre, C.; Serp, P. Recent advances in the methanol carbonylation reaction into acetic acid. Coord. Chem. Rev. 2020, 402, 213078. [Google Scholar] [CrossRef]
- Galiano, F.; Castro-Muñoz, R.; Mancuso, R.; Gabriele, B.; Figoli, A. Membrane technology in catalytic carbonylation reactions. Catalysts 2019, 9, 614. [Google Scholar] [CrossRef] [Green Version]
- Allah, T.N.; Savourey, S.; Berthet, J.C.; Nicolas, E.; Cantat, T. Carbonylation of C-N bonds in tertiary amines catalyzed by low valent iron catalysts. Angew. Chem. 2019, 131, 11000–11003. [Google Scholar] [CrossRef]
- Kaminsky, W.; Arndt, M. Polymerization, Oligomerization and Copolymerization of Olefins. In Applied Homogeneous Catalysis with Organometallic Compounds: A Comprehensive Handbook in Two Volumes; Wiley–VCH: Weinheim, Germany, 1996. [Google Scholar]
- Mathey, F.; Sevin, A. Molecular Chemistry of the Transition Elements: An Introductory Course; John Wiley&Sons: Chichester, UK, 1996. [Google Scholar]
- Matyjaszewski, K. Atom transfer radical polymerization (ATRP): Current status and future perspectives. Macromolecules 2012, 45, 4015–4039. [Google Scholar] [CrossRef]
- Schroder, K.; Konkolewicz, D.; Poli, R.; Matyjaszewski, K. Formation and possible reactions of organometallic intermediates with active copper(I) catalysts in ATRP. Organometallics 2012, 31, 7994–7999. [Google Scholar] [CrossRef]
- Drzeżdżon, J.; Malinowski, J.; Sikorski, A.; Gawdzik, B.; Rybiński, P.; Chmurzyński, L.; Jacewicz, D. Iminodiacetate complex of cobalt(II)—Structure, physicochemical characteristics, biological properties and catalytic activity for 2-chloro-2-propen-1-ol oligomerization. Polyhedron 2020, 175, 114168. [Google Scholar] [CrossRef]
- Drzeżdżon, J.; Malinowski, J.; Chmurzyński, L.; Jacewicz, D. The oxydiacetate and iminodiacetate complexes of oxidovanadium(IV) as the new series of the catalysts for the oligomerization of beta-olefin derivatives. Polyhedron 2020, 180, 114409. [Google Scholar] [CrossRef]
- Drzeżdżon, J.; Zych, D.; Malinowski, J.; Sikorski, A.; Chmurzyński, L.; Jacewicz, D. Formation of 2-chloroallyl alcohol oligomers using a new crystalline dipicolinate complex of Cr(III) as a catalyst. J. Catal. 2019, 375, 287–293. [Google Scholar] [CrossRef]
- Drzeżdżon, J.; Sikorski, A.; Chmurzyński, L.; Jacewicz, D. New type of highly active chromium(III) catalysts containing both organic cations and anions designed for polymerization of beta-olefin derivatives. Sci. Rep. 2018, 8, 2315. [Google Scholar] [CrossRef]
- Drzeżdżon, J.; Sikorski, A.; Chmurzyński, L.; Jacewicz, D. Oligomerization of 2-chloroallyl alcohol by 2-pyridinecarboxylate complex of chromium(III) - new highly active and selective catalyst. Sci. Rep. 2018, 8, 8632. [Google Scholar] [CrossRef]
- Drzeżdżon, J.; Chmurzyński, L.; Jacewicz, D. Geometric isomerism effect on catalytic activities of bis(oxalato)diaquochromates(III) for 2-chloroallyl alcohol oligomerization. J. Chem. Sci. 2018, 130, 116. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Liu, W.; Chen, C. Late transition metal catalyzed α-olefin polymerization and copolymerization with polar monomers. Mater. Chem. Front. 2017, 1, 2487–2494. [Google Scholar] [CrossRef]
- Kaminsky, W. The discovery and evolution of metallocene-based olefin polymerization catalysts. Rend. Fis. Acc. Lincei 2017, 28, 87–95. [Google Scholar] [CrossRef]
- Remya, V.R.; Kurian, M. Synthesis and catalytic applications of metal–organic frameworks: A review on recent literature. Int. Nano. Lett. 2019, 9, 17–29. [Google Scholar] [CrossRef] [Green Version]
- Bariashirab, C.; Huangab, C.; Solanac, G.A.; Sun, W.H. Recent advances in homogeneous chromium catalyst design for ethylene tri-, tetra-, oligo- and polymerization. Coord. Chem. Rev. 2019, 385, 208–229. [Google Scholar] [CrossRef] [Green Version]
- Drzeżdżon, J.; Jacewicz, D.; Sielicka, A.; Chmurzyński, L. A review of new approaches to analytical methods to determine the structure and morphology of polymers. Trac–Trend Anal. Chem. 2019, 118, 470–476. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Arends, I.; Henefeld, U. Green Chemistry and Catalysis; Wiley–VCH Verlag GmbH & Co. KgaA: Weinheim, Germany, 2007. [Google Scholar]
- Pruchnik, F. Kataliza homogeniczna; PWN: Warsaw, Poland, 1993. [Google Scholar]
- Osborn, J.A.; Jardine, F.H.; Young, J.F.; Wilkinson, G. The preparation and properties of tris(triphenylphosphine)halogenorhodium(I) and some reactions thereof including catalytic homogeneous hydrogenation of olefins and acetylenes and their derivatives. J. Chem. Soc. A 1966, 0, 1711–1732. [Google Scholar] [CrossRef]
- Plechkova, N.V.; Seddon, K.R. Applications of ionic liquids in the chemical industry. Chem. Soc. Rev. 2008, 37, 123–150. [Google Scholar] [CrossRef]
- Stojanovic, A.; Keppler, B.K. Ionic liquids as extracting agents for heavy metals. Sep. Sci. Technol. 2012, 47, 189–203. [Google Scholar] [CrossRef]
- Kollár, L. Modern Carbonylation Methods; John Wiley & Sons: Weinheim, Germany, 2008. [Google Scholar]
- Sans, V.; Trzeciak, A.M.; Luis, S.; Ziółkowski, J.J. PdCl2(P(OPh)3)2 catalyzed coupling and carbonylative coupling of phenylacetylenes with aryl iodides in organic solvents and in ionic iiquids. Catal. Lett. 2006, 109, 37–41. [Google Scholar] [CrossRef]
- Mathews, C.J.; Smith, P.J.; Welton, T.; White, A.J.; Williams, D.J. In situ formation of mixed phosphine−imidazolylidene palladium complexes in room-temperature ionic liquids. Organometallics 2001, 20, 3848–3850. [Google Scholar] [CrossRef]
- Herrmann, W.A.; Elison, M.; Fischer, J.; Koecher, C.; Artus, G.R.J. Metal complexes of N-heterocyclic carbenes—A new structural principle for catalysts in homogeneous catalysis. Angew. Chem. Int. Ed. 1995, 34, 2371–2374. [Google Scholar] [CrossRef]
- Zawartka, W.; Trzeciak, A.M.; Ziółkowski, J.J.; Lis, T.; Ciunik, Z.; Pernak, J. Methoxycarbonylation of iodobenzene in ionic liquids. A case of inhibiting effect of imidazolium halides. Adv. Synth. Catal. 2006, 348, 1689–1698. [Google Scholar] [CrossRef]
- Zawartka, W.; Gniewek, A.; Trzeciak, A.M.; Ziółkowski, J.J.; Pernak, J. PdII square planar complexes of the type [IL]2[PdX4] as catalyst precursors for the Suzuki–Miyaura cross-coupling reaction. The first in situ ESI-MS evidence of [(IL)xPd3] clusters formation. J. Mol. Catal. A Chem. 2009, 304, 8–15. [Google Scholar] [CrossRef]
- Silarska, E.; Trzeciak, A.M.; Pernak, J.; Skrzypczak, A. [IL]2[PdCl4] complexes (IL=imidazolium cation) as efficient catalysts for Suzuki–Miyaura cross-coupling of aryl bromides and aryl chlorides. Appl. Catal. A-Gen. 2013, 466, 216–223. [Google Scholar] [CrossRef]
- Cotugno, P.; Monopoli, A.; Ciminale, F.; Cioffi, N.; Nacci, A. Pd nanoparticle catalysed one-pot sequential Heck and Suzuki couplings of bromo-chloroarenes in ionic liquids and water. Org. Biomol. Chem. 2012, 10, 808–813. [Google Scholar] [CrossRef] [PubMed]
- Astruc, D.; Lu, F.; Aranzaes, J.R. Nanoparticles as recyclable catalysts: The frontier between homogeneous and heterogeneous catalysis. Angew. Chem. Int. Ed. 2005, 44, 7852–7872. [Google Scholar] [CrossRef] [PubMed]
- Ananikov, V.P.; Beletskaya, I.P. Toward the ideal catalyst: From atomic centers to a “cocktail” of catalysts. Organometallics 2012, 31, 1595–1604. [Google Scholar] [CrossRef]
- Trzeciak, A.M.; Ziółkowski, J.J. Structural and mechanistic studies of Pd-catalyzed CC bond formation: The case of carbonylation and Heck reaction. Coordin. Chem. Rev. 2005, 249, 2308–2322. [Google Scholar] [CrossRef]
- Jun, Y.W.; Choi, J.S.; Cheon, J. Heterostructured magnetic nanoparticles: Their versatility and high performance capabilities. Chem. Commun. 2007, 12, 1203–1214. [Google Scholar] [CrossRef]
- Ranu, B.C.; Chattopadhyay, K.; Adak, L.; Saha, A.; Bhadra, S.; Dey, R.; Saha, D. Metal nanoparticles as efficient catalysts for organic reactions. Pure Applied Chem. 2009, 81, 2337–2354. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.X.; Zhu, M.; Zuo, M.; Chu, S.Q.; Zhang, J.; Wu, Y.; Liang, H.W.; Feng, X. Identification of catalytic sites for oxygen reduction in metal/nitrogen-doped carbons with encapsulated metal nanoparticles. Angew. Chem. Int. Ed. 2020, 59, 1627–1633. [Google Scholar] [CrossRef]

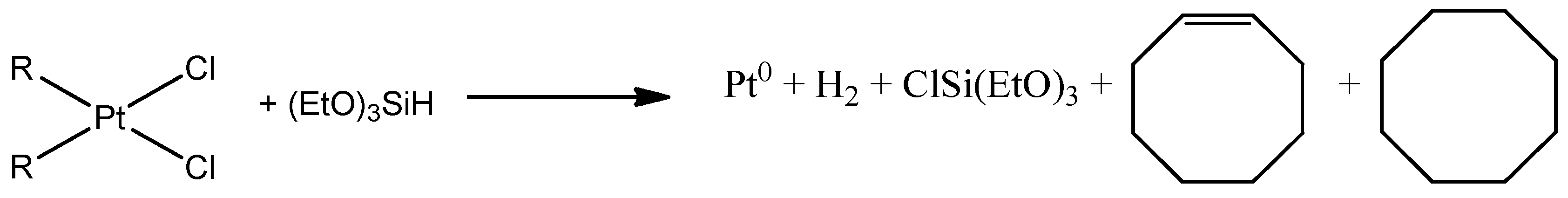


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malinowski, J.; Zych, D.; Jacewicz, D.; Gawdzik, B.; Drzeżdżon, J. Application of Coordination Compounds with Transition Metal Ions in the Chemical Industry—A Review. Int. J. Mol. Sci. 2020, 21, 5443. https://doi.org/10.3390/ijms21155443
Malinowski J, Zych D, Jacewicz D, Gawdzik B, Drzeżdżon J. Application of Coordination Compounds with Transition Metal Ions in the Chemical Industry—A Review. International Journal of Molecular Sciences. 2020; 21(15):5443. https://doi.org/10.3390/ijms21155443
Chicago/Turabian StyleMalinowski, Jacek, Dominika Zych, Dagmara Jacewicz, Barbara Gawdzik, and Joanna Drzeżdżon. 2020. "Application of Coordination Compounds with Transition Metal Ions in the Chemical Industry—A Review" International Journal of Molecular Sciences 21, no. 15: 5443. https://doi.org/10.3390/ijms21155443