DNA Mismatch Repair Gene Variants in Sporadic Solid Cancers

 , and
, and 
Abstract
:1. Introduction
1.1. DNA Mismatch Repair System and Its Role in Tumorigenesis
1.2. Function of DNA Mismatch Repair System
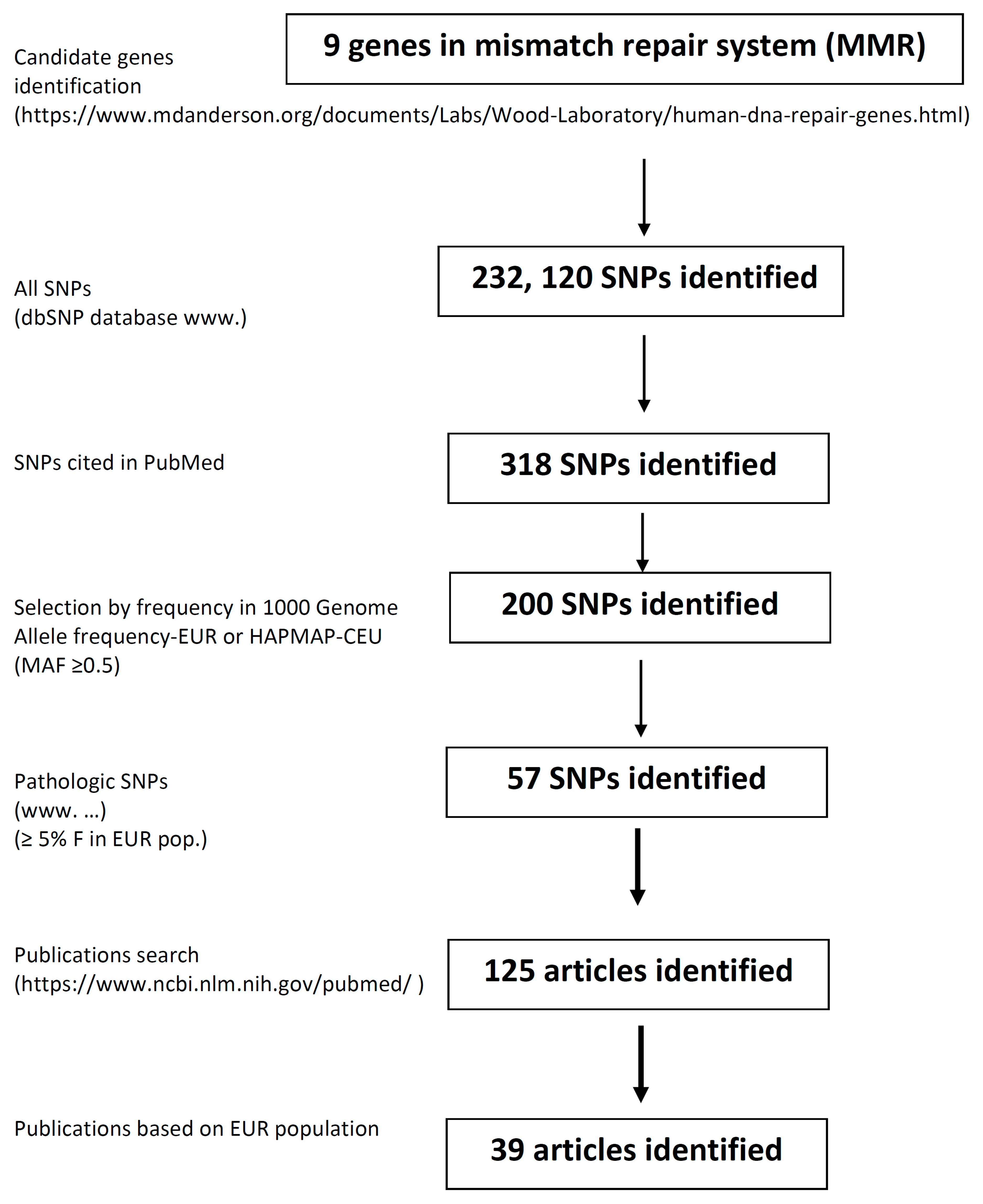
2. Materials and Methods
3. Results on MMR Gene Variants
3.1. MLH1 Gene (Mutl Homolog 1)
3.2. MLH3 Gene (Mutl Homolog 3)
3.3. MSH2 Gene (MutS Homolog 2)
3.4. MSH3 Gene (MutS Homolog 3)
3.5. MSH4 Gene (MutS Homolog 4)
3.6. MSH5 Gene (MutS Homolog 5)
3.7. MSH6 Gene (MutS Homolog 6)
3.8. PMS1 Gene (Postmeiotic Segregation Increased 1)
3.9. PMS2 (Postmeiotic Segregation Increased 1, Homolog 2) Gene
3.10. PMS2P1 (Postmeiotic Segregation Increased 1 Homolog 2, Pseudogene 1) Gene
3.11. PMS2P2 (Postmeiotic Segregation Increased 1 Homolog 2, Pseudogene 2) Gene
4. Role of MMR Variants in Sporadic Cancer
5. SNPs in MMR Genes and Therapy
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Baretti, M.; Le, D.T. DNA mismatch repair in cancer. Pharmacol. Ther. 2018, 189, 45–62. [Google Scholar] [CrossRef] [PubMed]
- Peters, U.; Bien, S.; Zubair, N. Genetic architecture of colorectal cancer. Gut 2015, 64, 1623–1636. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Pearlman, A.H.; Hsieh, P. DNA mismatch repair and the DNA damage response. DNA Repair 2016, 38, 94–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearl, L.H.; Schierz, A.C.; Ward, S.E.; Al-Lazikani, B.; Pearl, F. Therapeutic opportunities within the DNA damage response. Nat. Rev. Cancer 2015, 15, 166–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carethers, J.M.; Jung, B.H. Genetics and Genetic Biomarkers in Sporadic Colorectal Cancer. Gastroenterology 2015, 149, 1177–1190.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grady, W.M.; Markowitz, S.D. The molecular pathogenesis of colorectal cancer and its potential application to colorectal cancer screening. Dig. Dis. Sci. 2014, 60, 762–772. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, F.C.; Hansen, T.V.O.; Sørensen, C.S. Hereditary breast and ovarian cancer: New genes in confined pathways. Nat. Rev. Cancer 2016, 16, 599–612. [Google Scholar] [CrossRef]
- Niskakoski, A.; Pasanen, A.; Lassus, H.; Renkonen-Sinisalo, L.; Kaur, S.; Mecklin, J.-P.; Bützow, R.; Peltomäki, P. Molecular changes preceding endometrial and ovarian cancer: A study of consecutive endometrial specimens from Lynch syndrome surveillance. Mod. Pathol. 2018, 31, 1291–1301. [Google Scholar] [CrossRef]
- Ramus, S.J.; Song, H.; Dicks, E.; Tyrer, J.P.; Rosenthal, A.N.; Intermaggio, M.P.; Fraser, L.; Gentry-Maharaj, A.; Hayward, J.; Philpott, S.; et al. Germline Mutations in the BRIP1, BARD1, PALB2, and NBN Genes in Women With Ovarian Cancer. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef]
- Chubb, D.; Broderick, P.; Frampton, M.; Kinnersley, B.; Sherborne, A.; Penegar, S.; Lloyd, A.; Ma, Y.P.; Dobbins, S.E.; Houlston, R. Genetic Diagnosis of High-Penetrance Susceptibility for Colorectal Cancer (CRC) Is Achievable for a High Proportion of Familial CRC by Exome Sequencing. J. Clin. Oncol. 2015, 33, 426–432. [Google Scholar] [CrossRef]
- Peltomäki, P. Role of DNA Mismatch Repair Defects in the Pathogenesis of Human Cancer. J. Clin. Oncol. 2003, 21, 1174–1179. [Google Scholar] [CrossRef] [PubMed]
- Valle, L.; De Voer, R.M.; Goldberg, Y.; Sjursen, W.; Försti, A.; Ruiz-Ponte, C.; Caldés, T.; Garré, P.; Olsen, M.F.; Nordling, M.; et al. Update on genetic predisposition to colorectal cancer and polyposis. Mol. Asp. Med. 2019, 69, 10–26. [Google Scholar] [CrossRef] [PubMed]
- Lavebratt, C.; Şengül, S. Single nucleotide polymorphism (SNP) allele frequency estimation in DNA pools using Pyrosequencing™. Nat. Protoc. 2006, 1, 2573–2582. [Google Scholar] [CrossRef]
- Mohrenweiser, H.W.; Xi, T.; Vázquez-Matías, J.; Jones, I.M. Identification of 127 amino acid substitution variants in screening 37 DNA repair genes in humans. Cancer Epidemiol. Biomark. Prev. 2002, 11, 1054–1064. [Google Scholar]
- Vymetalkova, V.; Pardini, B.; Rosa, F.; Di Gaetano, C.; Novotny, J.; Levy, M.; Büchler, T.; Slyskova, J.; Vodickova, L.; Naccarati, A.; et al. Variations in mismatch repair genes and colorectal cancer risk and clinical outcome. Mutagenesis 2014, 29, 259–265. [Google Scholar] [CrossRef] [Green Version]
- Han, W.; Kim, K.-Y.; Yang, S.-J.; Noh, D.-Y.; Kang, D.; Kwack, K. SNP-SNP interactions between DNA repair genes were associated with breast cancer risk in a Korean population. Cancer 2011, 118, 594–602. [Google Scholar] [CrossRef]
- Schwender, H.R.; Selinski, S.; Blaszkewicz, M.; Marchan, R.; Ickstadt, K.; Golka, K.; Hengstler, J.G. Distinct SNP Combinations Confer Susceptibility to Urinary Bladder Cancer in Smokers and Non-Smokers. PLoS ONE 2012, 7, e51880. [Google Scholar] [CrossRef] [Green Version]
- Ito, H.; Sueta, A.; Iwata, H.; Hosono, S.; Oze, I.; Watanabe, M.; Iwase, H.; Tanaka, H.; Matsuo, K. Abstract 06: A genetic risk predictor for breast cancer using a combination of low-penetrance polymorphisms in a Japanese population. Cancer Epidemiol. Biomark. Prev. 2012, 21, 6. [Google Scholar] [CrossRef]
- Bhushan, S.; McLeod, H.; Walko, C.M. Role of Pharmacogenetics as Predictive Biomarkers of Response and/or Toxicity in the Treatment of Colorectal Cancer. Clin. Color. Cancer 2009, 8, 15–21. [Google Scholar] [CrossRef]
- Kunkel, T.A.; Erie, D.A. Eukaryotic Mismatch Repair in Relation to DNA Replication. Annu. Rev. Genet. 2015, 49, 291–313. [Google Scholar] [CrossRef] [Green Version]
- Lahue, R.S.; Au, K.G.; Modrich, P. DNA mismatch correction in a defined system. Science 1989, 245, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Modrich, P.; Lahue, R. Mismatch repair in replication fidelity, genetic recombination, and cancer biology. Annu. Rev. Biochem. 1996, 65, 101–133. [Google Scholar] [CrossRef] [PubMed]
- Nevers, P.; Spatz, H.C. Escherichia coli mutants uvr D and uvr E deficient in gene conversion of lambda-heteroduplexes. Mol. Gen. Genet. 1975, 139, 233–243. [Google Scholar] [CrossRef] [PubMed]
- Ban, C.; Junop, M.; Yang, W. Transformation of MutL by ATP binding and hydrolysis: A switch in DNA mismatch repair. Cell 1999, 97, 85–97. [Google Scholar] [CrossRef] [Green Version]
- Längle-Rouault, F.; Maenhaut-Michel, G.; Radman, M. GATC sequences, DNA nicks and the MutH function in Escherichia coli mismatch repair. EMBO J. 1987, 6, 1121–1127. [Google Scholar] [CrossRef]
- Kadyrov, F.A.; Dzantiev, L.; Constantin, N.; Modrich, P. Endonucleolytic Function of MutLα in Human Mismatch Repair. Cell 2006, 126, 297–308. [Google Scholar] [CrossRef] [Green Version]
- Genschel, J.; Littman, S.J.; Drummond, J.T.; Modrich, P. Isolation of MutSbeta from human cells and comparison of the mismatch repair specificities of MutSbeta and MutSalpha. J. Biol. Chem. 1998, 273, 19895–19901. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.; Predeus, A.V.; Kovacs, N.; Feig, M. Differential Mismatch Recognition Specificities of Eukaryotic MutS Homologs, MutSα and MutSβ. Biophys. J. 2014, 106, 2483–2492. [Google Scholar] [CrossRef] [Green Version]
- Marsischky, G.T.; Filosi, N.; Kane, M.F.; Kolodner, R. Redundancy of Saccharomyces cerevisiae MSH3 and MSH6 in MSH2-dependent mismatch repair. Genes Dev. 1996, 10, 407–420. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Gellert, M.; Yang, W. Mechanism of mismatch recognition revealed by human MutSbeta bound to unpaired DNA loops. Nat. Struct. Mol. Biol. 2011, 19, 8–72. [Google Scholar]
- Kondo, E. The interacting domains of three MutL heterodimers in man: hMLH1 interacts with 36 homologous amino acid residues within hMLH3, hPMS1 and hPMS. Nucleic Acids Res. 2001, 29, 1695–1702. [Google Scholar] [CrossRef] [PubMed]
- Banasik, M.; Sachadyn, P. Conserved motifs of MutL proteins. Mutat. Res. Mol. Mech. Mutagenesis 2014, 769, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Vodicka, P.; Musak, L.; Frank, C.; Kazimirova, A.; Vymetalkova, V.; Barancokova, M.; Smolkova, B.; Dzupinkova, Z.; Jiraskova, K.; Vodenkova, S.; et al. Interactions of DNA repair gene variants modulate chromosomal aberrations in healthy subjects. Carcinogenesis 2015, 36, 1299–1306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillotin, D.; Martin, S.A. Exploiting DNA mismatch repair deficiency as a therapeutic strategy. Exp. Cell Res. 2014, 329, 110–115. [Google Scholar] [CrossRef]
- Larrea, A.A.; Lujan, S.A.; Kunkel, T.A. SnapShot: DNA Mismatch Repair. Cell 2010, 141, 730–730.e1. [Google Scholar] [CrossRef] [Green Version]
- Peltomäki, P.; Vasen, H. Mutations predisposing to hereditary nonpolyposis colorectal cancer: Database and results of a collaborative study. The International Collaborative Group on Hereditary Nonpolyposis Colorectal Cancer. Gastroenterology 1997, 113, 1146–1158. [Google Scholar] [CrossRef]
- Mrkonjic, M.; Roslin, N.M.; Greenwood, C.M.T.; Raptis, S.; Pollett, A.; Laird, P.W.; Pethe, V.V.; Chiang, T.; Daftary, D.; Dicks, E.; et al. Specific Variants in the MLH1 Gene Region May Drive DNA Methylation, Loss of Protein Expression, and MSI-H Colorectal Cancer. PLoS ONE 2010, 5, e13314. [Google Scholar] [CrossRef]
- Nizam, Z.M.; Aziz, A.A.A.; Kaur, G.; Abu Hassan, M.R.; Sidek, A.S.M.; Lee, Y.Y.; Mazuwin, M.; Ankathil, R. Contribution of the MLH1 −93G>A Promoter Polymorphism in Modulating Susceptibility Risk in Malaysian Colorectal Cancer Patients. Asian Pac. J. Cancer Prev. 2013, 14, 619–624. [Google Scholar] [CrossRef] [Green Version]
- Beiner, M.E.; Rosen, B.; Harley, I.; Siminovitch, K.; Zhang, S.; Fyles, A.; Pal, T.; Sun, P.; Narod, S.A. Endometrial Cancer Risk Is Associated with Variants of the Mismatch Repair Genes MLH1 and MSH. Cancer Epidemiol. Biomark. Prev. 2006, 15, 1636–1640. [Google Scholar] [CrossRef] [Green Version]
- Whiffin, N.; Broderick, P.; Lubbe, S.J.; Pittman, A.; Penegar, S.; Chandler, I.; Houlston, R.S. MLH1-93G > A is a risk factor for MSI colorectal cancer. Carcinogenesis 2011, 32, 1157–1161. [Google Scholar] [CrossRef] [Green Version]
- Koref, M.F.S.; Wilson, V.; Cartwright, N.; Cunnington, M.S.; Mathers, J.C.; Bishop, D.; Curtis, A.; Dunlop, M.G.; Burn, J. MLH1 Differential Allelic Expression in Mutation Carriers and Controls. Ann. Hum. Genet. 2010, 74, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, I.; Houlston, R.S.; Montgomery, G.W.; Sieber, O.; Dunlop, M.G. Investigation of the effects of DNA repair gene polymorphisms on the risk of colorectal cancer. Mutagenesis 2012, 27, 219–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allan, J.M.; Shorto, J.; Adlard, J.; Bury, J.; Coggins, R.; George, R.; Katory, M.; Quirke, P.; Richman, S.; Scott, D.; et al. MLH1 −93G>A promoter polymorphism and risk of mismatch repair deficient colorectal cancer. Int. J. Cancer 2008, 123, 2456–2459. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Hernandez, I.; Perdomo, S.; Santos-Briz, A.; Hernández, J.L.G.; Gómez-Moreta, J.A.; Cruz, J.J.; González-Sarmiento, R. Analysis of DNA repair gene polymorphisms in glioblastoma. Gene 2014, 536, 79–83. [Google Scholar] [CrossRef]
- Lo, Y.-L.; Hsiao, C.-F.; Jou, Y.-S.; Chang, G.-C.; Tsai, Y.-H.; Su, W.-C.; Chen, K.-Y.; Chen, Y.-M.; Huang, M.-S.; Hsieh, W.-S.; et al. Polymorphisms of MLH1 and MSH2 genes and the risk of lung cancer among never smokers. Lung Cancer 2011, 72, 280–286. [Google Scholar] [CrossRef]
- Shih, C.-M.; Chen, C.-Y.; Lee, I.-H.; Kao, W.-T.; Wang, Y.-C. A polymorphism in the hMLH1 gene (−93G-->A) associated with lung cancer susceptibility and prognosis. Int. J. Mol. Med. 2010, 25, 165–170. [Google Scholar]
- Park, S.H.; Lee, G.Y.; Jeon, H.-S.; Lee, S.J.; Kim, K.M.; Jang, S.S.; Kim, C.H.; Lee, W.K.; Kam, S.; Park, R.W.; et al. −93G→A polymorphism ofhMLH1 and risk of primary lung cancer. Int. J. Cancer 2004, 112, 678–682. [Google Scholar] [CrossRef]
- Halasova, E.; Matáková, T.; Skerenova, M.; Krutakova, M.; Slovakova, P.; Dzian, A.; Javorkova, S.; Pec, M.; Kypusova, K.; Hamzik, J. Polymorphisms of Selected DNA Repair Genes and Lung Cancer in Chromium Exposure. Retin. Degener. Dis. 2016, 911, 17–22. [Google Scholar] [CrossRef]
- Wang, J.; Liu, Q.; Yuan, S.; Xie, W.; Liu, Y.; Xiang, Y.; Wu, N.; Wu, L.; Ma, X.; Cai, T.; et al. Genetic predisposition to lung cancer: Comprehensive literature integration, meta-analysis, and multiple evidence assessment of candidate-gene association studies. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Raptis, S.; Mrkonjic, M.; Green, R.C.; Pethe, V.V.; Monga, N.; Chan, Y.M.; Daftary, D.; Dicks, E.; Younghusband, B.H.; Parfrey, P.S.; et al. MLH1 −93G>A Promoter Polymorphism and the Risk of Microsatellite-Unstable Colorectal Cancer. J. Natl. Cancer Inst. 2007, 99, 463–474. [Google Scholar] [CrossRef]
- Liu, N.Q.; Ter Huurne, M.; Nguyen, L.N.; Peng, T.; Wang, S.-Y.; Studd, J.B.; Joshi, O.; Ongen, H.; Bramsen, J.B.; Yan, J.; et al. The non-coding variant rs1800734 enhances DCLK3 expression through long-range interaction and promotes colorectal cancer progression. Nat. Commun. 2017, 8, 14418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perera, S.; Mrkonjic, M.; Rawson, J.R.; Bapatet, B. Functional effects of the MLH1-93G>A polymorphism on MLH1/EPM2AIP1 promoter activity. Oncol. Rep. 2011, 25, 809–815. [Google Scholar]
- Pan, X.-M.; Yang, W.-Z.; Xu, G.-H.; Bai, P.; Qin, H.-J.; Zhang, L.-S.; Zhai, X.-D.; Tang, M.; Deng, W.; Gao, L.-B. The association between MLH1 −93 G>A polymorphism of DNA mismatch repair and cancer susceptibility: A meta-analysis. Mutagenesis 2011, 26, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Tulupova, E.; Kumar, R.; Hanova, M.; Slyskova, J.; Pardini, B.; Polakova, V.; Naccarati, A.; Vodickova, L.; Novotny, J.; Halamkova, J.; et al. Do polymorphisms and haplotypes of mismatch repair genes modulate risk of sporadic colorectal cancer. Mutat. Res. Mol. Mech. Mutagen. 2008, 648, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Pardini, B.; Corrado, A.; Paolicchi, E.; Cugliari, G.; Berndt, S.I.; Bezieau, S.; Bien, S.A.; Brenner, H.; Caan, B.J.; Campbell, P.T.; et al. DNA repair and cancer in colon and rectum: Novel players in genetic susceptibility. Int. J. Cancer 2019, 146, 363–372. [Google Scholar] [CrossRef]
- Cervena, K.; Siskova, A.; Buchler, T.; Vodicka, P.; Vymetalkova, V. Methylation-Based Therapies for Colorectal Cancer. Cells 2020, 9, 1540. [Google Scholar] [CrossRef]
- Savio, A.J.; Lemire, M.; Mrkonjic, M.; Gallinger, S.; Zanke, B.W.; Hudson, T.J.; Bapat, B. MLH1 Region Polymorphisms Show a Significant Association with CpG Island Shore Methylation in a Large Cohort of Healthy Individuals. PLoS ONE 2012, 7, e51531. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Nicolaides, N.C.; Markowitz, S.; Willson, J.K.V.; Parsons, R.; Jen, J.; Papadopolous, N.; Peltomäki, P.; De La Chapelle, A.; Hamilton, S.R.; et al. Mismatch repair gene defects in sporadic colorectal cancers with microsatellite instability. Nat. Genet. 1995, 9, 48–55. [Google Scholar] [CrossRef]
- Nejda, N.; Iglesias, D.; Azcoita, M.M.; Arana, V.M.; González-Aguilera, J.J.; Fernández-Peralta, A.M. A MLH1 polymorphism that increases cancer risk is associated with better outcome in sporadic colorectal cancer. Cancer Genet. Cytogenet. 2009, 193, 71–77. [Google Scholar] [CrossRef]
- Smith, T.R.; Levine, E.A.; Freimanis, R.I.; Akman, S.A.; Allen, G.O.; Hoang, K.N.; Liu-Mares, W.; Hu, J.J. Polygenic model of DNA repair genetic polymorphisms in human breast cancer risk. Carcinogenesis 2008, 29, 2132–2138. [Google Scholar] [CrossRef] [Green Version]
- An, Y.; Jin, G.; Wang, H.; Liu, H.; Li, R.; Wang, H.; Qian, J.; Sun, W.; Wang, Y.; Ma, H.; et al. Polymorphisms in hMLH1 and risk of early-onset lung cancer in a southeast Chinese population. Lung Cancer 2008, 59, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Langeberg, W.J.; Kwon, E.M.; Koopmeiners, J.S.; Ostrander, E.A.; Stanford, J.L. Population-based study of the association of variants in mismatch repair genes with prostate cancer risk and outcomes. Cancer Epidemiol. Biomark. Prev. 2010, 19, 258–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, D.; Rasi, S.; Di Rocco, A.; Fabbri, A.; Forconi, F.; Gloghini, A.; Bruscaggin, A.; Franceschetti, S.; Fangazio, M.; De Paoli, L.; et al. The host genetic background of DNA repair mechanisms is an independent predictor of survival in diffuse large B-cell lymphoma. Blood 2011, 117, 2405–2413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picelli, S.; Zajac, P.; Zhou, X.-L.; Edler, D.; Lenander, C.; Dalén, J.; Hjern, F.; Lundqvist, N.; Lindforss, U.; Påhlman, L.; et al. Common variants in human CRC genes as low-risk alleles. Eur. J. Cancer 2010, 46, 1041–1048. [Google Scholar] [CrossRef]
- Landi, S.; Gemignani, F.; Canzian, F.; Gaborieau, V.; Barale, R.; Landi, D.; Szeszenia-Dabrowska, N.; Zaridze, D.G.; Lissowska, J.; Rudnai, P.; et al. DNA Repair and Cell Cycle Control Genes and the Risk of Young-Onset Lung Cancer. Cancer Res. 2006, 66, 11062–11069. [Google Scholar] [CrossRef] [Green Version]
- Campbell, P.T.; Curtin, K.; Ulrich, C.M.; Samowitz, W.S.; Bigler, J.; Velicer, C.M.; Caan, B.; Potter, J.D.; Slattery, M.L. Mismatch repair polymorphisms and risk of colon cancer, tumour microsatellite instability and interactions with lifestyle factors. Gut 2008, 58, 661–667. [Google Scholar] [CrossRef] [Green Version]
- Picelli, S.; Bermejo, J.L.; Chang-Claude, J.; Hoffmeister, M.; Fernandez-Rozadilla, C.; Carracedo, A.; Castells, A.; Castellvi-Bel, S.; Naccarati, A.; Pardini, B.; et al. Meta-Analysis of Mismatch Repair Polymorphisms within the Cogent Consortium for Colorectal Cancer Susceptibility. PLoS ONE 2013, 8, e72091. [Google Scholar] [CrossRef] [Green Version]
- Dreussi, E.; Cecchin, E.; Polesel, J.; Canzonieri, V.; Agostini, M.; Boso, C.; Belluco, C.; Buonadonna, A.; Lonardi, S.; Bergamo, F.; et al. Pharmacogenetics Biomarkers and Their Specific Role in Neoadjuvant Chemoradiotherapy Treatments: An Exploratory Study on Rectal Cancer Patients. Int. J. Mol. Sci. 2016, 17, 1482. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Li, G.; Hu, F.; Bi, H.; Wu, Z.; Zhao, X.; Li, Y.; Li, S.; Li, D.; Cui, B.; et al. The prognostic significance of polymorphisms in hMLH1/hMSH2 for colorectal cancer. Med. Oncol. 2014, 31, 975. [Google Scholar] [CrossRef]
- Shinozuka, K.; Tang, H.; Jones, R.B.; Li, D.; Nieto, Y. Impact of Polymorphic Variations of Gemcitabine Metabolism, DNA Damage Repair, and Drug-Resistance Genes on the Effect of High-Dose Chemotherapy for Relapsed or Refractory Lymphoid Malignancies. Biol. Blood Marrow Transplant. 2016, 22, 843–849. [Google Scholar] [CrossRef] [Green Version]
- Sapkota, Y.; Mackey, J.R.; Lai, R.; Franco-Villalobos, C.; Lupichuk, S.; Robson, P.; Kopciuk, K.; Cass, C.E.; Yasui, Y.; Damaraju, S. Assessing SNP-SNP Interactions among DNA Repair, Modification and Metabolism Related Pathway Genes in Breast Cancer Susceptibility. PLoS ONE 2013, 8, e64896. [Google Scholar] [CrossRef] [PubMed]
- Kan, R.; Sun, X.; Kolas, N.K.; Avdievich, E.; Kneitz, B.; Edelmann, W.; Cohen, P.E. Comparative Analysis of Meiotic Progression in Female Mice Bearing Mutations in Genes of the DNA Mismatch Repair Pathway. Biol. Reprod. 2008, 78, 462–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conde, J.; Silva, S.N.; Azevedo, A.P.; Teixeira, V.; Pina, J.E.; Rueff, J.; Gaspar, J. Association of common variants in mismatch repair genes and breast cancer susceptibility: A multigene study. BMC Cancer 2009, 9, 344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santucci-Darmanin, S.; Neyton, S.; Lespinasse, F.; Saunières, A.; Gaudray, P.; Paquis-Flucklinger, V. The DNA mismatch-repair MLH3 protein interacts with MSH4 in meiotic cells, supporting a role for this MutL homolog in mammalian meiotic recombination. Hum. Mol. Genet. 2002, 11, 1697–1706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michiels, S.; Danoy, P.; Dessen, P.; Bera, A.; Boulet, T.; Bouchardy, C.; Lathrop, M.; Sarasin, A.; Benhamou, S. Polymorphism discovery in 62 DNA repair genes and haplotype associations with risks for lung and head and neck cancers. Carcinogenesis 2007, 28, 1731–1739. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, X.; Jia, J.; Tang, L.; Gao, X.; Yan, L.; Wang, L.; Yu, F.; Ma, N.; Liu, W.; et al. Correlation between polymorphisms in DNA mismatch repair genes and the risk of primary hepatocellular carcinoma for the Han population in northern China. Scand. J. Gastroenterol. 2015, 50, 1–7. [Google Scholar] [CrossRef]
- Ye, F.; Cheng, Q.; Shen, J.; Zhou, C.; Chen, H. Mismatch Repair Gene MLH3 Pro844Leu and Thr942Ile Polymorphisms and the Susceptibility to Cervical Carcinoma and HPV Infection: A Case-Control Study in a Chinese Population. PLoS ONE 2014, 9, e96224. [Google Scholar] [CrossRef]
- Li, N.; Duell, E.J.; Yu, K.; Risch, H.A.; Olson, S.H.; Kooperberg, C.; Wolpin, B.M.; Jiao, L.; Dong, X.; Wheeler, B.; et al. Pathway analysis of genome-wide association study data highlights pancreatic development genes as susceptibility factors for pancreatic cancer. Carcinogenesis 2012, 33, 1384–1390. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Chen, Z.; Gao, P.; Gao, Z.; Chen, H.; Qi, J.; Liu, F.; Ye, D.; Jiang, H.; Na, R.; et al. TEX15: A DNA repair gene associated with prostate cancer risk in Han Chinese. Prostate 2017, 77, 1271–1278. [Google Scholar] [CrossRef]
- Yang, Q.; Zhang, R.; Wang, X.W.; Linke, S.P.; Sengupta, S.; Hickson, I.D.; Pedrazzi, G.; Perrera, C.; Stagljar, I.; Littman, S.J.; et al. The mismatch DNA repair heterodimer, hMSH2/6, regulates BLM helicase. Oncogene 2004, 23, 3749–3756. [Google Scholar] [CrossRef] [Green Version]
- Seifert, M.; Scherer, S.J.; Edelmann, W.; Böhm, M.; Meineke, V.; Löbrich, M.; Tilgen, W.; Reichrath, J. The DNA-Mismatch Repair Enzyme hMSH2 Modulates UV-B-Induced Cell Cycle Arrest and Apoptosis in Melanoma Cells. J. Investig. Dermatol. 2008, 128, 203–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slováková, P.; Majerová, L.; Matakova, T.; Skerenova, M.; Kavcová, E.; Halasova, E. Mismatch Repair Gene Polymorphisms and Association with Lung Cancer Development. Retin. Degener. Dis. 2014, 833, 15–22. [Google Scholar] [CrossRef]
- Hsieh, Y.-C.; Cho, E.-C.; Tu, S.-H.; Wu, C.-H.; Hung, C.-S.; Hsieh, M.-C.; Su, C.-T.; Liu, Y.-R.; Lee, C.-H.; Ho, Y.-S.; et al. MSH2 rs2303425 Polymorphism is Associated with Early-Onset Breast Cancer in Taiwan. Ann. Surg. Oncol. 2016, 24, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Mrkonjic, M.; Raptis, S.; Green, R.C.; Monga, N.; Daftary, D.; Dicks, E.; Younghusband, H.; Parfrey, P.S.; Gallinger, S.S.; McLaughlin, J.R.; et al. MSH2 −118T>C and MSH6 −159C>T promoter polymorphisms and the risk of colorectal cancer. Carcinogenesis 2007, 28, 2575–2580. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, K.; Srivastava, A.; Kumar, A.; Mittal, B. Gallbladder Cancer Predisposition: A Multigenic Approach to DNA-Repair, Apoptotic and Inflammatory Pathway Genes. PLoS ONE 2011, 6, e16449. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, G.A.S.; Lourenço, G.J.; Oliveira, C.B.M.; Marson, F.A.L.; Lopes-Aguiar, L.; Costa, E.F.D.; Lima, T.R.P.; Liutti, V.; Leal, F.; Santos, V.C.A.; et al. Association between genetic polymorphisms in DNA mismatch repair-related genes with risk and prognosis of head and neck squamous cell carcinoma. Int. J. Cancer 2015, 137, 810–818. [Google Scholar] [CrossRef]
- Sanguansin, S.; Petmitr, S.; Punyarit, P.; Vorasubin, V.; Weerapradist, W.; Surarit, R. HMSH2 gene alterations associated with recurrence of oral squamous cell carcinoma. J. Exp. Clin. Cancer Res. 2006, 25, 251–257. [Google Scholar]
- Wang, D.; Zhou, J.; Wang, T.; Li, X.; Li, S.; Chen, S.; Ma, G.; Li, J.; Zhang, X. Polymorphisms in MSH2 gene and risk of gastric cancer, and interactions with lifestyle factors in a Chinese population. Cancer Epidemiol. 2012, 36, e171–e176. [Google Scholar] [CrossRef]
- Park, J.M.; Huang, S.; Tougeron, D.; Sinicrope, F.A. MSH3 Mismatch Repair Protein Regulates Sensitivity to Cytotoxic Drugs and a Histone Deacetylase Inhibitor in Human Colon Carcinoma Cells. PLoS ONE 2013, 8, e65369. [Google Scholar] [CrossRef] [Green Version]
- Kleczkowska, H.E.; Marra, G.; Lettieri, T.; Jiricny, J. hMSH3 and hMSH6 interact with PCNA and colocalize with it to replication foci. Genes Dev. 2001, 15, 724–736. [Google Scholar] [CrossRef] [Green Version]
- Marra, G.; Iaccarino, I.; Lettieri, T.; Roscilli, G.; Delmastro, P.; Jiricny, J. Mismatch repair deficiency associated with overexpression of the MSH3 gene. Proc. Natl. Acad. Sci. USA 1998, 95, 8568–8573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berndt, S.I.; Platz, E.A.; Fallin, M.D.; Thuita, L.W.; Hoffman, S.C.; Helzlsouer, K.J. Mismatch repair polymorphisms and the risk of colorectal cancer. Int. J. Cancer 2007, 120, 1548–1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koessler, T.; Oestergaard, M.Z.; Tyrer, J.; Perkins, B.; Dunning, A.M.; Pharoah, P.D.P.; Song, H.; Easton, D.F. Common variants in mismatch repair genes and risk of colorectal cancer. Gut 2008, 57, 1097–1101. [Google Scholar] [CrossRef] [PubMed]
- Miao, H.-K.; Chen, L.-P.; Cai, D.-P.; Kong, W.-J.; Xiao, L.; Lin, J. MSH3 rs26279 polymorphism increases cancer risk: A meta-analysis. Int. J. Clin. Exp. Pathol. 2015, 8, 11060–11067. [Google Scholar] [PubMed]
- Koessler, T.; Azzato, E.M.; Perkins, B.; Maclnnis, R.; Greenberg, D.; Easton, U.F.; Pharoah, P.D. Common germline variation in mismatch repair genes and survival after a diagnosis of colorectal cancer. Int. J. Cancer 2009, 124, 1887–1891. [Google Scholar] [CrossRef] [PubMed]
- Milne, R.L.; Herranz, J.; Michailidou, K.; Dennis, J.; Tyrer, J.P.; Zamora, M.P.; Perez, J.I.A.; González-Neira, A.; Pita, G.; Alonso, M.R.; et al. A large-scale assessment of two-way SNP interactions in breast cancer susceptibility using 46 450 cases and 42 461 controls from the breast cancer association consortium. Hum. Mol. Genet. 2013, 23, 1934–1946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.-L.; Yao, Y.-L.; Xu, W.-Z.; Feng, J.-G.; Mao, W.-M. Correlation of MSH3 polymorphisms with response and survival in advanced non-small cell lung cancer patients treated with first-line platinum-based chemotherapy. Genet. Mol. Res. 2015, 14, 3525–3533. [Google Scholar] [CrossRef]
- Schuetz, J.M.; Daley, D.; Leach, S.; Conde, L.; Berry, B.R.; Gallagher, R.P.; Connors, J.M.; Gascoyne, R.D.; Bracci, P.M.; Skibola, C.F.; et al. Non-Hodgkin Lymphoma Risk and Variants in Genes Controlling Lymphocyte Development. PLoS ONE 2013, 8, e75170. [Google Scholar] [CrossRef] [Green Version]
- Skibola, C.F.; Bracci, P.M.; Halperin, E.; Nieters, A.; Hubbard, A.; Paynter, R.A.; Skibola, D.R.; Agana, L.; Becker, N.; Tressler, P.; et al. Polymorphisms in the Estrogen Receptor 1 and Vitamin C and Matrix Metalloproteinase Gene Families Are Associated with Susceptibility to Lymphoma. PLoS ONE 2008, 3, e2816. [Google Scholar] [CrossRef] [Green Version]
- Paquis-Flucklinger, V.; Santucci-Darmanin, S.; Paul, R.; Saunières, A.; Turc-Carel, C.; Desnuelle, C. Cloning and Expression Analysis of a Meiosis-Specific MutS Homolog: The HumanMSH4Gene. Genomics 1997, 44, 188–194. [Google Scholar] [CrossRef]
- Pochart, P.; Woltering, D.; Hollingsworth, N.M. Conserved Properties between Functionally Distinct MutS Homologs in Yeast. J. Biol. Chem. 1997, 272, 30345–30349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zalevsky, J.; MacQueen, A.J.; Duffy, J.B.; Kemphues, K.J.; Villeneuve, A.M. Crossing over during Caenorhabditis elegans meiosis requires a conserved MutS-based pathway that is partially dispensable in budding yeast. Genetics 1999, 153, 1271–1283. [Google Scholar] [PubMed]
- Bocker, T.; Barusevicius, A.; Snowden, T.; Rasio, D.; Guerrette, S.; Robbins, D.; Schmidt, C.; Burczak, J.; Croce, C.M.; Copeland, T.; et al. hMSH5: A human MutS homologue that forms a novel heterodimer with hMSH4 and is expressed during spermatogenesis. Cancer Res. 1999, 59, 816–822. [Google Scholar] [PubMed]
- Santucci-Darmanin, S.; Walpita, D.; Lespinasse, F.; Desnuelle, C.; Ashley, T.; Paquis-Flucklingeret, V. MSH4 acts in conjunction with MLH1 during mammalian meiosis. FASEB J. 2000, 14, 1539–1547. [Google Scholar] [CrossRef] [PubMed]
- Kolas, N.; Cohen, P. Novel and diverse functions of the DNA mismatch repair family in mammalian meiosis and recombination. Cytogenet. Genome Res. 2004, 107, 216–231. [Google Scholar] [CrossRef] [PubMed]
- Her, C. MutS Homologues hMSH4 and hMSH5: Diverse Functional Implications in Humans. Front. Biosci. 2007, 12, 905. [Google Scholar] [CrossRef] [PubMed]
- Clark, N.; Wu, X.; Her, C. MutS Homologues hMSH4 and hMSH5: Genetic Variations, Functions, and Implications in Human Diseases. Curr. Genom. 2013, 14, 81–90. [Google Scholar] [CrossRef] [Green Version]
- Chu, Y.-L.; Wu, X.; Xu, Y.; Her, C. MutS homologue hMSH4: Interaction with eIF3f and a role in NHEJ-mediated DSB repair. Mol. Cancer 2013, 12, 51. [Google Scholar] [CrossRef] [Green Version]
- Edelmann, W.; Cohen, P.E.; Kneitz, B.; Winand, N.; Lia, M.; Heyer, J.; Kolodner, R.; Pollard, J.W.; Kucherlapati, R. Mammalian MutS homologue 5 is required for chromosome pairing in meiosis. Nat. Genet. 1999, 21, 123–127. [Google Scholar] [CrossRef]
- Kelly, K.O.; Dernburg, A.F.; Stanfield, G.M.; Villeneuve, A.M. Caenorhabditis elegans msh-5 is required for both normal and radiation-induced meiotic crossing over but not for completion of meiosis. Genetics 2000, 156, 617–630. [Google Scholar]
- Xu, Y.; Wu, X.; Her, C. hMSH5 Facilitates the Repair of Camptothecin-induced Double-strand Breaks through an Interaction with FANCJ*. J. Biol. Chem. 2015, 290, 18545–18558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bannwarth, S.; Figueroa, A.; Fragaki, K.; Destroismaisons, L.; Lacas-Gervais, S.; Lespinasse, F.; Vandenbos, F.; Pradelli, L.A.; Ricci, J.-E.; Rötig, A.; et al. The human MSH5 (MutS Homolog 5) protein localizes to mitochondria and protects the mitochondrial genome from oxidative damage. Mitochondrion 2012, 12, 654–665. [Google Scholar] [CrossRef] [PubMed]
- Doherty, J.A.; Sakoda, L.C.; Loomis, M.M.; Barnett, M.J.; Julianto, L.; Thornquist, M.D.; Neuhouser, M.L.; Weiss, N.S.; Goodman, G.E.; Chen, C. DNA repair genotype and lung cancer risk in the beta-carotene and retinol efficacy trial. Int. J. Mol. Epidemiol. Genet. 2013, 4, 11–34. [Google Scholar] [PubMed]
- Timofeeva, M.N.; Hung, R.J.; Rafnar, T.; Christiani, D.C.; Field, J.K.; Bickeböller, H.; Risch, A.; McKay, J.D.; Wang, Y.; Dai, J.; et al. Influence of common genetic variation on lung cancer risk: Meta-analysis of 14 900 cases and 29 485 controls. Hum. Mol. Genet. 2012, 21, 4980–4995. [Google Scholar] [CrossRef] [PubMed]
- Yi, W.; Wu, X.; Lee, T.-H.; Doggett, N.A.; Her, C. Two variants of MutS homolog hMSH5: Prevalence in humans and effects on protein interaction. Biochem. Biophys. Res. Commun. 2005, 332, 524–532. [Google Scholar] [CrossRef]
- Liu, J.-Y.; Qian, C.-Y.; Gao, Y.-F.; Chen, J.; Zhou, H.; Yin, J.-Y. Association between DNA mismatch repair gene polymorphisms and platinum-based chemotherapy toxicity in non-small cell lung cancer patients. Chin. J. Cancer 2017, 36, 12. [Google Scholar] [CrossRef] [Green Version]
- Kazma, R.; Babron, M.-C.; Gaborieau, V.; Génin, E.; Brennan, P.; Hung, R.J.; McLaughlin, J.R.; Krokan, H.E.; Elvestad, M.B.; Skorpen, F.; et al. Lung cancer and DNA repair genes: Multilevel association analysis from the International Lung Cancer Consortium. Carcinogenesis 2012, 33, 1059–1064. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Zhang, Y.; Breitling, L.P.; Brenner, H. Tobacco smoking and methylation of genes related to lung cancer development. Oncotarget 2016, 7, 59017–59028. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Broderick, P.; Webb, E.L.; Wu, X.; Vijayakrishnan, J.; Matakidou, A.; Qureshi, M.; Dong, Q.; Gu, X.; Chen, W.V.; et al. Common 5p15.33 and 6p21.33 variants influence lung cancer risk. Nat. Genet. 2008, 40, 1407–1409. [Google Scholar] [CrossRef]
- Scarbrough, P.M.; Weber, R.P.; Iversen, E.S.; Brhane, Y.; Amos, C.I.; Kraft, P.; Hung, R.J.; Sellers, T.A.; Witte, J.S.; Pharoah, P.; et al. A Cross-Cancer Genetic Association Analysis of the DNA Repair and DNA Damage Signaling Pathways for Lung, Ovary, Prostate, Breast, and Colorectal Cancer. Cancer Epidemiol. Biomark. Prev. 2015, 25, 193–200. [Google Scholar] [CrossRef] [Green Version]
- Blackwell, L.J.; Martik, D.; Bjornson, K.P.; Bjornson, E.S.; Modrich, P. Nucleotide-promoted Release of hMutSα from Heteroduplex DNA Is Consistent with an ATP-dependent Translocation Mechanism. J. Biol. Chem. 1998, 273, 32055–32062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kansikas, M.; Kariola, R.; Nystrom-Lahti, M. Verification of the three-step model in assessing the pathogenicity of mismatch repair gene variants. Hum. Mutat. 2010, 32, 107–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.; Levine, E.A.; Franco, V.I.; Allen, G.O.; Gong, F.; Zhang, Y.; Hu, J.J. Combined Genetic and Nutritional Risk Models of Triple Negative Breast Cancer. Nutr. Cancer 2014, 66, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Zelga, P.; Przybyłowska-Sygut, K.; Zelga, M.; Dziki, A.; Majsterek, I. The 116G > A MSH6 and IVS1-1121C > T PMS2 Genes Polymorphisms Modulate the Risk of the Sporadic Colorectal Cancer Development in Polish Population. Pathol. Oncol. Res. 2017, 24, 231–235. [Google Scholar] [CrossRef] [Green Version]
- Zelga, P.; Przybylowska-Sygut, K.; Zelga, M.; Dziki, A.; Majsterek, I. Polymorphism of Gly39Glu (c.116G>A) hMSH6 is associated with sporadic colorectal cancer development in the Polish population: Preliminary results. Adv. Clin. Exp. Med. 2017, 26, 1425–1429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, L.S.; Silva, S.N.; Gil, O.M.; Ferreira, T.C.; Limbert, E.; Rueff, J. Mismatch repair single nucleotide polymorphisms and thyroid cancer susceptibility. Oncol. Lett. 2018, 15, 6715–6726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogelsang, M.; Wang, Y.; Veber, N.; Mwapagha, L.M.; Parker, M.I. The Cumulative Effects of Polymorphisms in the DNA Mismatch Repair Genes and Tobacco Smoking in Oesophageal Cancer Risk. PLoS ONE 2012, 7, e36962. [Google Scholar] [CrossRef] [Green Version]
- Curtin, K.; Samowitz, W.S.; Wolff, R.K.; Caan, B.J.; Ulrich, C.M.; Potter, J.D.; Slattery, M.L. MSH6 G39E polymorphism and CpG island methylator phenotype in colon cancer. Mol. Carcinog. 2009, 48, 989–994. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.; Li, Y.; Chang, P.; Hess, K.R.; Abbruzzese, J.L.; Li, D. DNA mismatch repair network gene polymorphism as a susceptibility factor for pancreatic cancer. Mol. Carcinog. 2011, 51, 491–499. [Google Scholar] [CrossRef] [Green Version]
- Zanusso, C.; Bortolus, R.; Dreussi, E.; Polesel, J.; Montico, M.; Cecchin, E.; Gagno, S.; Rizzolio, F.; Arcicasa, M.; Novara, G.; et al. Impact of DNA repair gene polymorphisms on the risk of biochemical recurrence after radiotherapy and overall survival in prostate cancer. Oncotarget 2017, 8, 22863–22875. [Google Scholar] [CrossRef] [Green Version]
- Cecchin, E.; D’Andrea, M.; Lonardi, S.; Zanusso, C.; Pella, N.; Errante, D.; De Mattia, E.; Polesel, J.; Innocenti, F.; Toffoli, G. A prospective validation pharmacogenomic study in the adjuvant setting of colorectal cancer patients treated with the 5-fluorouracil/leucovorin/oxaliplatin (FOLFOX4) regimen. Pharm. J. 2012, 13, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Doss, C.G.P.; Rao, S. Investigation on the role of nsSNPs in HNPCC genes—A bioinformatics approach. J. Biomed. Sci. 2009, 16, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, J.; Hu, Z.; Shu, Y.; Pan, S.; Chen, W.; Wang, Y.; Hu, L.; Jiang, Y.; Dai, J.; Ma, H.; et al. Potentially functional polymorphisms in DNA repair genes and non-small-cell lung cancer survival: A pathway-based analysis. Mol. Carcinog. 2011, 51, 546–552. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Lockman, J.C.; Frankel, W.L.; Hampel, H.; Steenblock, K.; Burgart, L.J.; Thibodeau, S.N.; De La Chapelle, A. Mismatch Repair GenePMS. Cancer Res. 2004, 64, 4721–4727. [Google Scholar] [CrossRef] [Green Version]
- Mann, A.; Høgdall, E.; Ramus, S.J.; DiCioccio, R.A.; Høgdall, C.; Quaye, L.; McGuire, V.; Whittemore, A.S.; Shah, M.; Greenberg, D.; et al. Mismatch repair gene polymorphisms and survival in invasive ovarian cancer patients. Eur. J. Cancer 2008, 44, 2259–2265. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Ramus, S.J.; Quaye, L.; DiCioccio, R.A.; Tyrer, J.; Lomas, E.; Shadforth, D.; Høgdall, E.V.S.; Høgdall, C.K.; McGuire, V.; et al. Common variants in mismatch repair genes and risk of invasive ovarian cancer. Carcinogenesis 2006, 27, 2235–2242. [Google Scholar] [CrossRef]
- Horii, A.; Han, H.; Sasaki, S.; Shimada, M.; Nakamura, Y. Cloning, Characterization and Chromosomal Assignment of the Human Genes Homologous to Yeast PMS1, a Member of Mismatch Repair Genes. Biochem. Biophys. Res. Commun. 1994, 204, 1257–1264. [Google Scholar] [CrossRef]
- Nicolaides, N.C.; Carter, K.C.; Shell, B.K.; Papadopoulos, N.; Vogelstein, B.; Kinzler, K.W. Genomic Organization of the HumanPMS2Gene Family. Genomics 1995, 30, 195–206. [Google Scholar] [CrossRef]
- Kondo, E.; Horii, A.; Fukushige, S. The human PMS2L proteins do not interact with hMLH1, a major DNA mismatch repair protein. J. Biochem. 1999, 125, 818–825. [Google Scholar]
- De Vos, M.; Hayward, B.E.; Picton, S.; Sheridan, E.; Bonthron, D.T. Novel PMS2 Pseudogenes Can Conceal Recessive Mutations Causing a Distinctive Childhood Cancer Syndrome. Am. J. Hum. Genet. 2004, 74, 954–964. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, R.J.; Farrington, S.M.; Dunlop, M.G.; Campbell, H. Mismatch Repair Genes hMLH1 and hMSH2 and Colorectal Cancer: A HuGE Review. Am. J. Epidemiol. 2002, 156, 885–902. [Google Scholar] [CrossRef] [PubMed]
- Kamory, E.; Kolacsek, O.; Ottó, S.; Csuka, O. hMLH1 and hMSH2 somatic inactivation mechanisms in sporadic colorectal cancer patients. Pathol. Oncol. Res. 2003, 9, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Kane, M.F.; Loda, M.; Gaida, G.M.; Lipman, J.; Mishra, R.; Goldman, H.; Jessup, J.M.; Kolodner, R. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 1997, 57, 808–811. [Google Scholar] [PubMed]
- Geisler, J.P.; Goodheart, M.J.; Sood, A.K.; Holmes, R.J.; Hatterman-Zogg, M.A.; Buller, R.E. Mismatch repair gene expression defects contribute to microsatellite instability in ovarian carcinoma. Cancer 2003, 98, 2199–2206. [Google Scholar] [CrossRef]
- Pierini, S.; Jordanov, S.H.; Mitkova, A.V.; Chalakov, I.J.; Melnicharov, M.B.; Kunev, K.V.; Mitev, V.I.; Kaneva, R.P.; Goranova, T.E. Promoter hypermethylation of CDKN2A, MGMT, MLH1, and DAPK genes in laryngeal squamous cell carcinoma and their associations with clinical profiles of the patients. Head Neck 2013, 36, 1103–1108. [Google Scholar] [CrossRef]
- Gomes, A.; Reis-Silva, M.; Alarcao, A.; Couceiro, P.; Sousa, V.; Carvalho, L. Promoter hypermethylation of DNA repair genes MLH1 and MSH2 in adenocarcinomas and squamous cell carcinomas of the lung. Rev. Por.T Pneumol. 2014, 20, 20–30. [Google Scholar] [CrossRef] [Green Version]
- Haraldsdottir, S.; Hampel, H.; Wu, C.; Weng, D.Y.; Shields, P.G.; Frankel, W.L.; Pan, X.; De La Chapelle, A.; Goldberg, R.; Bekaii-Saab, T. Patients with colorectal cancer associated with Lynch syndrome and MLH1 promoter hypermethylation have similar prognoses. Genet. Med. 2016, 18, 863–868. [Google Scholar] [CrossRef] [Green Version]
- Orimo, H.; Nakajima, E.; Yamamoto, M.; Ikejima, M.; Emi, M.; Shimada, T. Association between single nucleotide polymorphisms in the hMSH3 gene and sporadic colon cancer with microsatellite instability. J. Hum. Genet. 2000, 45, 228–230. [Google Scholar] [CrossRef]
- Gazzoli, I.; Kolodner, R.D. Regulation of the Human MSH6 Gene by the Sp1 Transcription Factor and Alteration of Promoter Activity and Expression by Polymorphisms. Mol. Cell. Biol. 2003, 23, 7992–8007. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Chen, J.; Ji, Y.; Liu, Y.; Gao, L.; Chen, G.; Shen, K.; Huang, B. Association between the hMSH2 IVS12-6 T>C polymorphism and cancer risk: A meta-analysis. Exp. Ther. Med. 2011, 2, 1193–1198. [Google Scholar] [CrossRef]
- Yuan, Z.Q.; Gottlieb, B.; Beitel, L.K.; Wong, N.; Gordon, P.H.; Wang, Q.; Puisieux, A.; Foulkes, W.D.; Trifiro, M. Polymorphisms and HNPCC: PMS2-MLH1 protein interactions diminished by single nucleotide polymorphisms. Hum. Mutat. 2002, 19, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Cortellino, S.; Turner, D.; Masciullo, V.; Schepis, F.; Albino, D.; Daniel, R.; Skalka, A.M.; Meropol, N.J.; Alberti, C.; LaRue, L.; et al. The base excision repair enzyme MED1 mediates DNA damage response to antitumor drugs and is associated with mismatch repair system integrity. Proc. Natl. Acad. Sci. USA 2003, 100, 15071–15076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murata, H.; Khattar, N.H.; Gu, L.; Li, G.-M. Roles of mismatch repair proteins hMSH2 and hMLH1 in the development of sporadic breast cancer. Cancer Lett. 2005, 223, 143–150. [Google Scholar] [CrossRef]
- Russo, A.; Corsale, S.; Cammareri, P.; Agnese, V.; Cascio, S.; Di Fede, G.; Macaluso, M.; Bazan, V. Pharmacogenomics in colorectal carcinomas: Future perspectives in personalized therapy. J. Cell. Physiol. 2005, 204, 742–749. [Google Scholar] [CrossRef]
- Iyer, R.R.; Pluciennik, A.; Burdett, V.; Modrich, P. DNA Mismatch Repair: Functions and Mechanisms. Chem. Rev. 2006, 37, 302–323. [Google Scholar] [CrossRef] [PubMed]
- Worrillow, L.J.; Travis, L.B.; Smith, A.G.; Rollinson, S.; Smith, A.J.; Wild, C.P.; Holowaty, E.J.; Kohler, B.A.; Wiklund, T.; Pukkala, E.; et al. An intron splice acceptor polymorphism in hMSH2 and risk of leukemia after treatment with chemotherapeutic alkylating agents. Clin. Cancer Res. 2003, 9, 3012–3020. [Google Scholar] [PubMed]
- Park, J.H.; Kim, N.S.; Park, J.Y.; Chae, Y.S.; Kim, J.G.; Sohn, S.K.; Moon, J.-H.; Kang, B.W.; Ryoo, H.M.; Bae, S.H.; et al. MGMT −535G>T polymorphism is associated with prognosis for patients with metastatic colorectal cancer treated with oxaliplatin-based chemotherapy. J. Cancer Res. Clin. Oncol. 2010, 136, 1135–1142. [Google Scholar] [CrossRef]
- Kim, J.G.; Chae, Y.S.; Sohn, S.K.; Moon, J.H.; Kang, B.W.; Park, J.Y.; Jeon, S.W.; Lee, M.-H.; Lim, K.-H.; Choi, G.S.; et al. IVS10+12A>G polymorphism in hMSH2 gene associated with prognosis for patients with colorectal cancer. Ann. Oncol. 2010, 21, 525–529. [Google Scholar] [CrossRef]
- Jung, C.Y.; Choi, J.E.; Park, J.M.; Chae, M.H.; Kang, H.-G.; Kim, K.M.; Lee, S.J.; Lee, W.-K.; Kam, S.; Cha, S.I.; et al. Polymorphisms in the hMSH2 Gene and the Risk of Primary Lung Cancer. Cancer Epidemiol. Biomark. Prev. 2006, 15, 762–768. [Google Scholar] [CrossRef] [Green Version]
- Boeckmann, L.; Thoms, K.-M.; Gutzmer, R.; Has, C.; Kunz, M.; Kuschal, C.; Laspe, P.; Struever, D.; Emmert, S. Modulation of the efficacy of temozolomide and dacarbazine melanoma treatment by DNA-repair factors in vivo and in vitro. Int. J. Clin. Pharmacol. Ther. 2009, 47, 33–35. [Google Scholar] [CrossRef]
- Vymetalkova, V.; Slyskova, J.; Korenková, V.; Bielik, L.; Langerová, L.; Procházka, P.; Rejhova, A.; Schwarzová, L.; Pardini, B.; Naccarati, A.; et al. Molecular characteristics of mismatch repair genes in sporadic colorectal tumors in Czech patients. BMC Med. Genet. 2014, 15, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomasova, K.; Cumova, A.; Seborova, K.; Horak, J.; Koucka, K.; Vodickova, L.; Vaclavikova, R.; Vodicka, P. DNA Repair and Ovarian Carcinogenesis: Impact on Risk, Prognosis and Therapy Outcome. Cancers 2020, 12, 1713. [Google Scholar] [CrossRef] [PubMed]


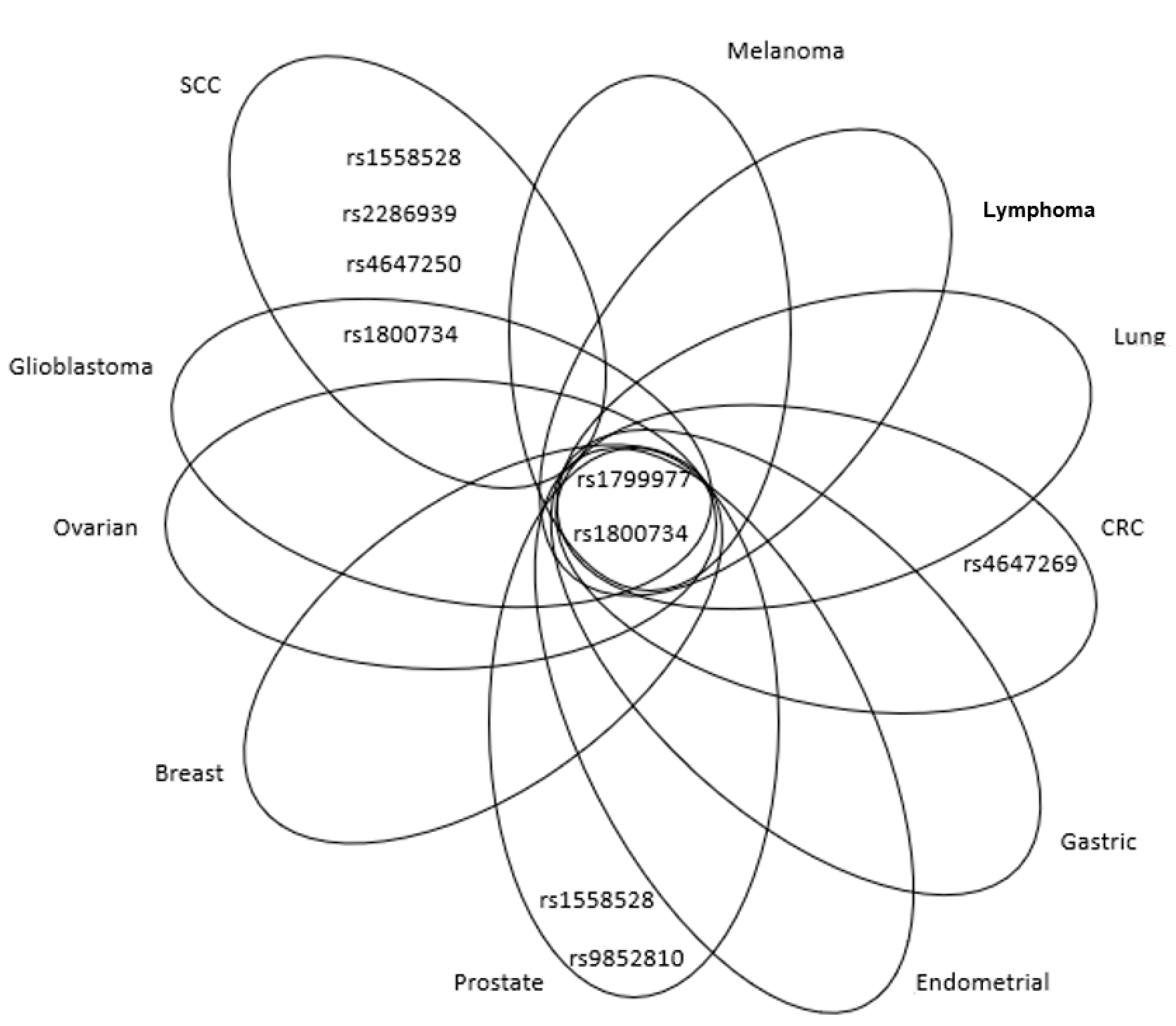
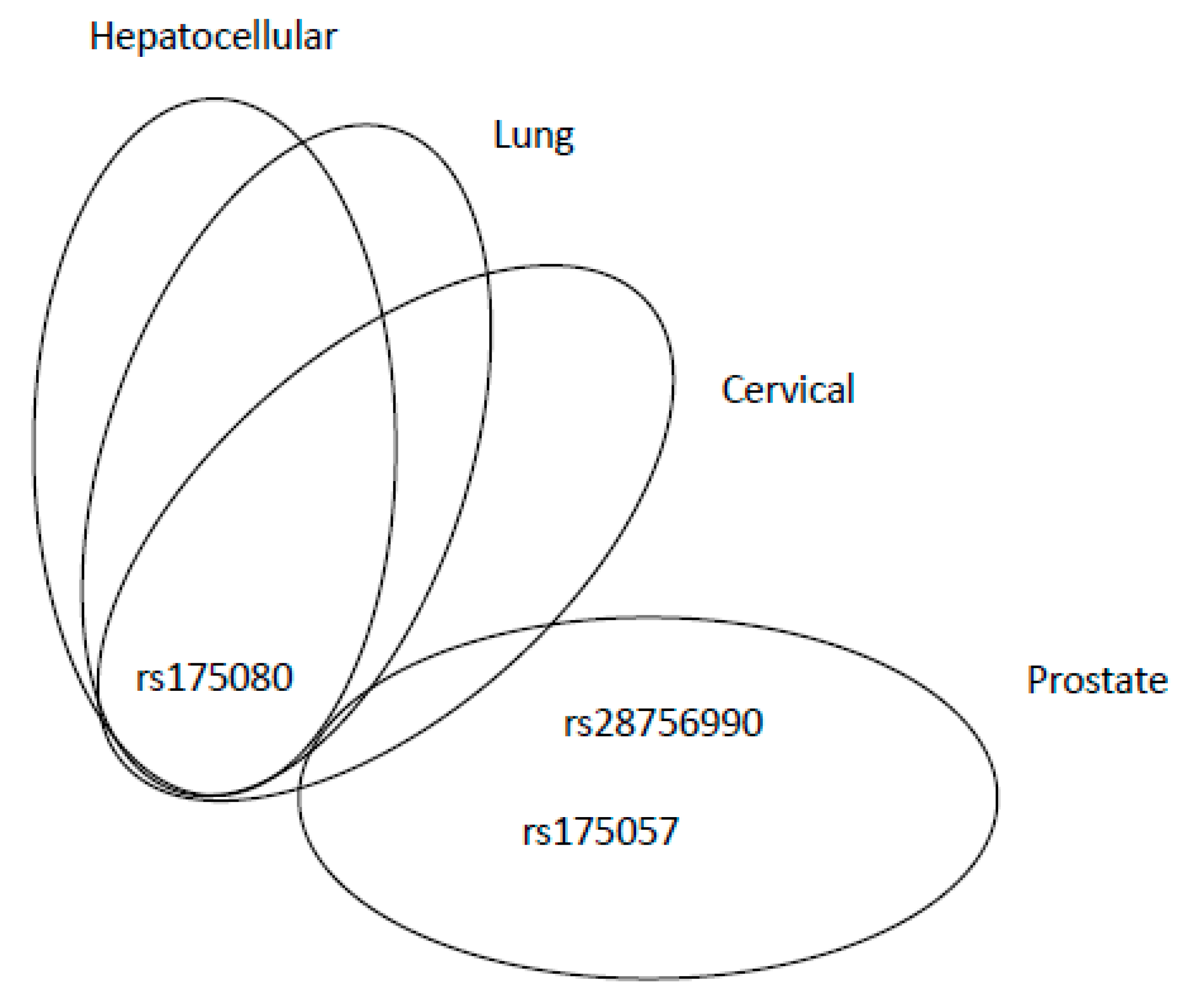
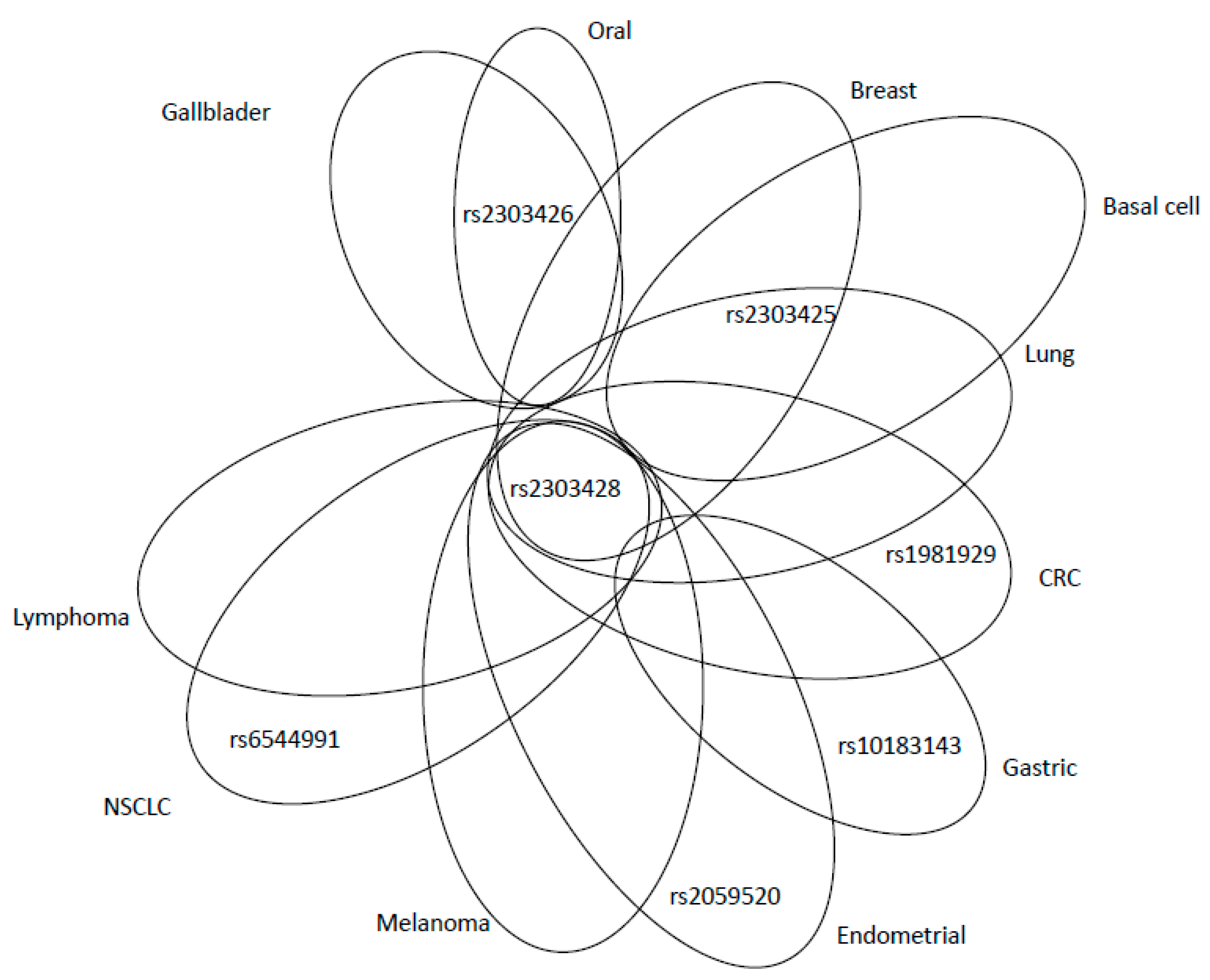
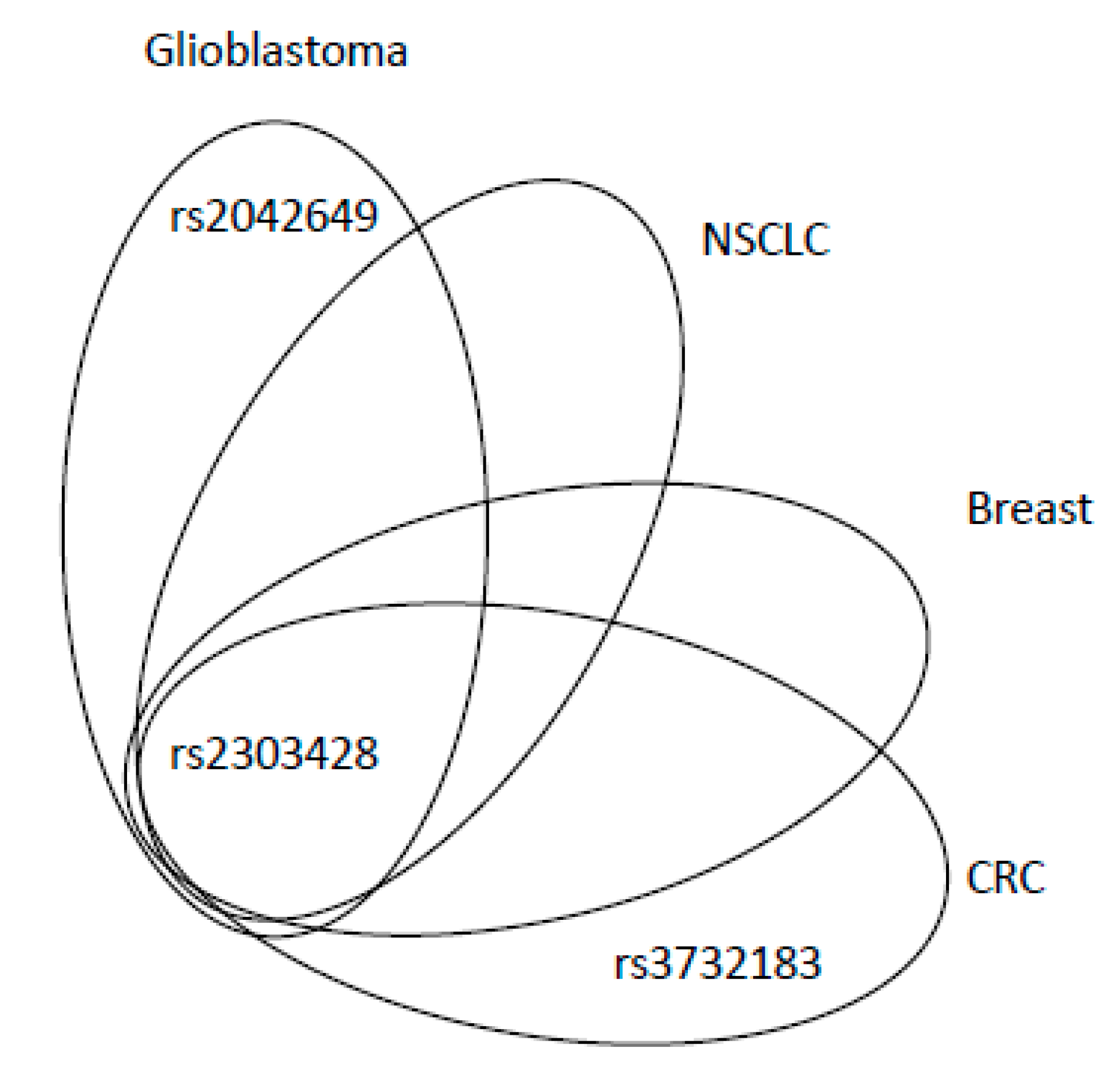


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mechanism | Error Frequency |
|---|---|
| Base pairing | 10−1 to 10−2 |
| DNA polymerases (base selection, proofreading) | 10−4 to 10−5 |
| Accessory proteins | 10−7 |
| Mismatch repair | 10−10 |
| Gene | SNP ID | AA Change | WT-MT | Ancestral Alelle | Gene Region | Gene Position | Phenotype | F in EUR Pop. |
|---|---|---|---|---|---|---|---|---|
| MLH1 | rs1540354 | - | A-T | T | intron | c.307-1403A>T | B | 16% |
| rs1558528 | - | C-A | A | intron | c.67+955C>A | P | 56% | |
| rs2286939 | - | T-C | C | intron | c.1038+86T>C | D | 56% | |
| rs4647250 | - | T-C | T | intron | c.453+544T>C | D | 44% | |
| rs1799977 | Ile219Val | A-G | A | Exon 8 | c.655A>G | A | 33% | |
| rs1800734 | - | A-G | G | 5′ prime | c.-93G>A | A | 27% | |
| rs2286940 | - | C-T | C | intron | c.1410-169C>T | B | 44% | |
| rs9852810 | - | G-A | G | intron | c.1410-1306G>A | P | 44% | |
| rs4647269 | - | C-T | C | intron | c.791-1406C>T | A | 44% | |
| MLH3 | rs108621 | - | A-G | T | UTR-3 | c.*3148A>G | B | 45% |
| rs175080 | Leu844Pro | C-T | G | Exon 2 | c.2531C>T | A | 46% | |
| rs28756990 | Val741Ile | G-A | G | Exon 2 | c.2221G>A | P | 5% | |
| rs175057 | - | G-A | T | intron | c.4012-37G>A | P | 54% | |
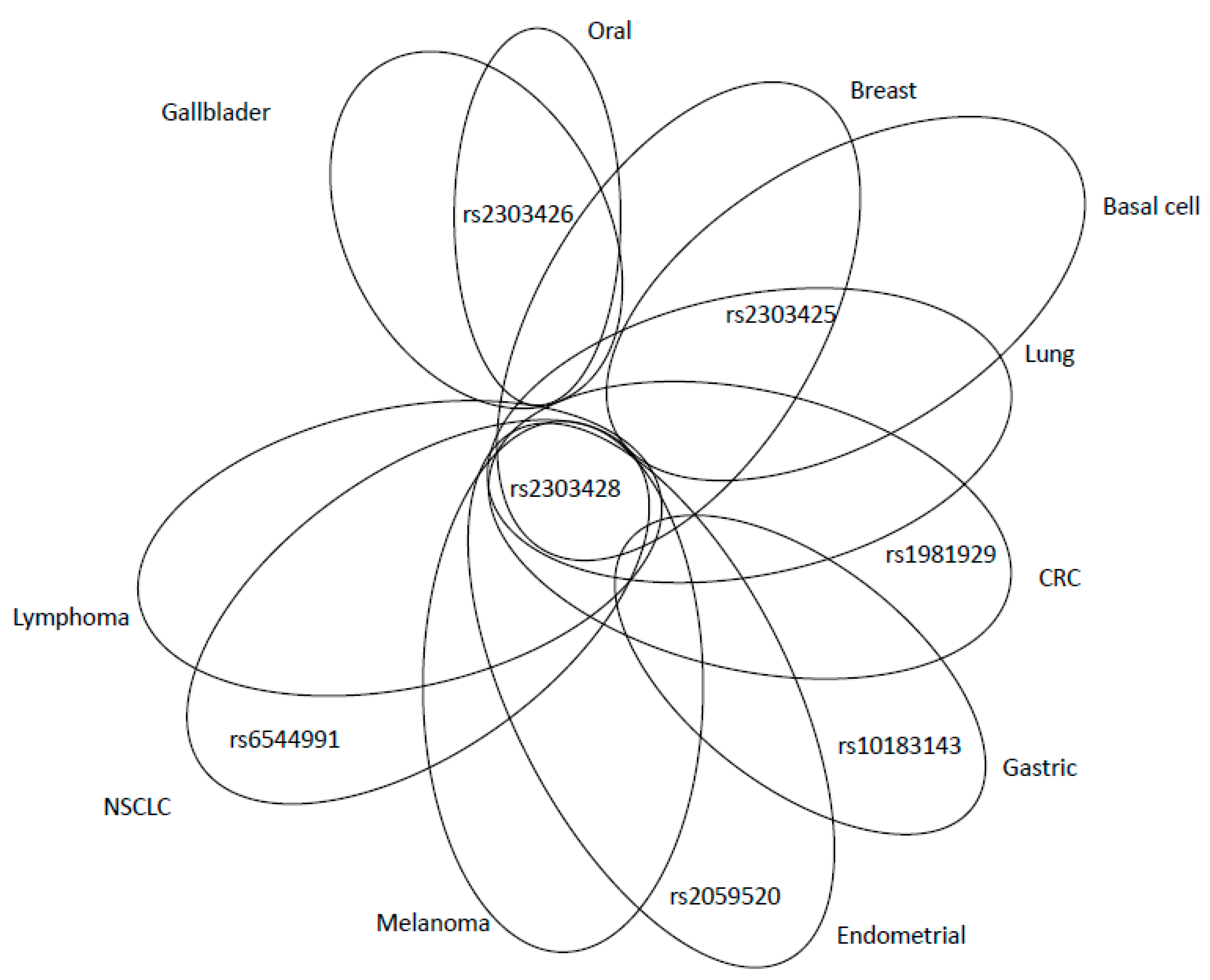
| MSH2 | rs1981929 | - | G-A | A | intron | c.1277-118G>A | P | 39% |
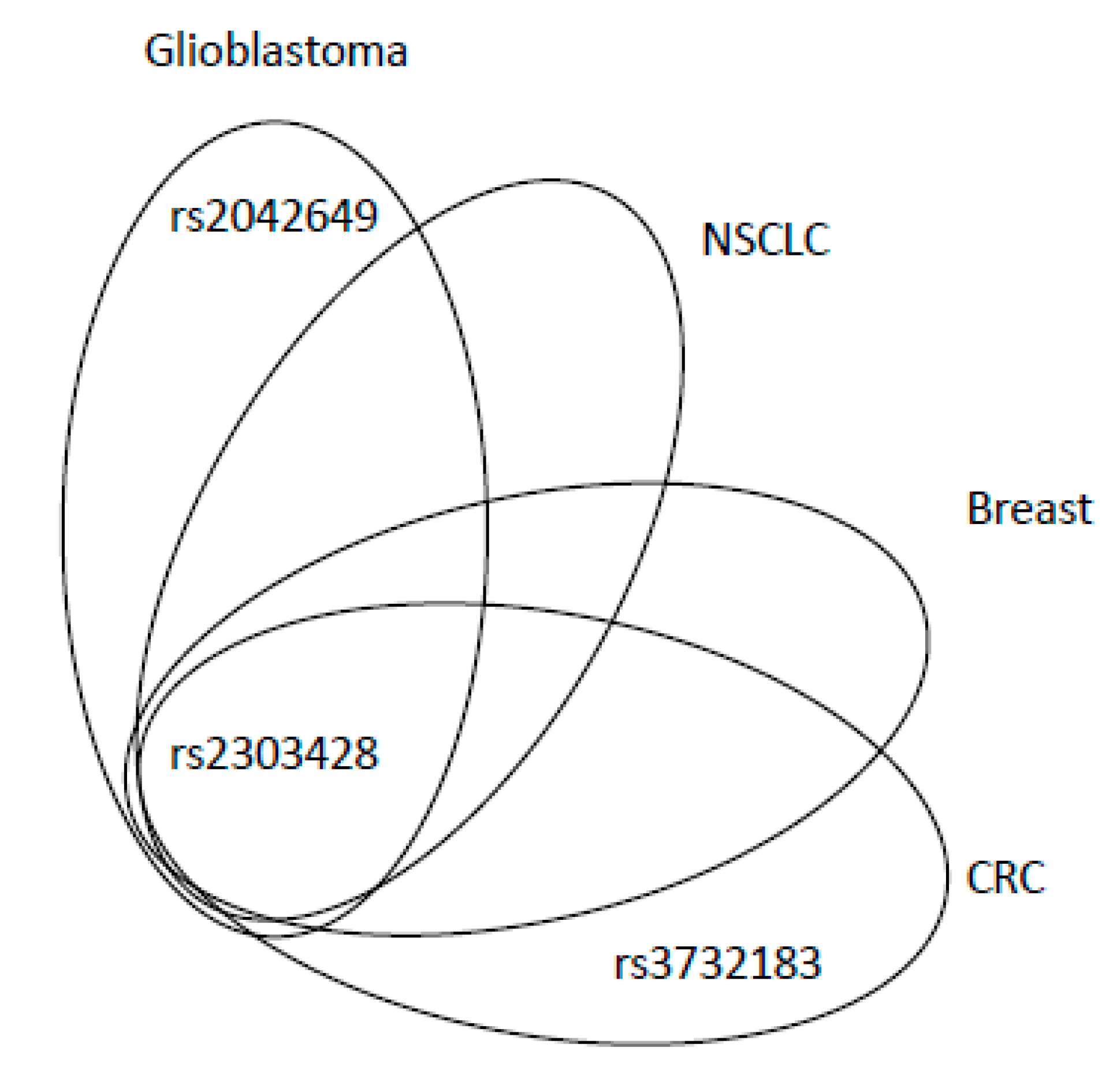
| rs2042649 | - | T-C | T | intron | c.2635-214T>C | P | 9% | |
| rs2059520 | - | A-G | A | intron | c.2006-265A>G | P | 36% | |
| rs2303425 | - | T-C | T | 5′ prime | c.-68-50T>C | P | 15% | |
| rs2303426 | - | C-G | G | intron | c.211+9C>G | P | 57% | |
| rs2303428 | - | T-A | T | intron | c.2006-6T>A | A | 9% | |
| rs3732183 | - | G-A | G | intron | c.1661+12G>A | B | 30% | |
| rs6544991 | - | A-C | A | intron | c.251+2441A>C | B | 22% | |
| rs10183143 | - | T-C | T | intron | c.1661+90T>C | D | 6% | |
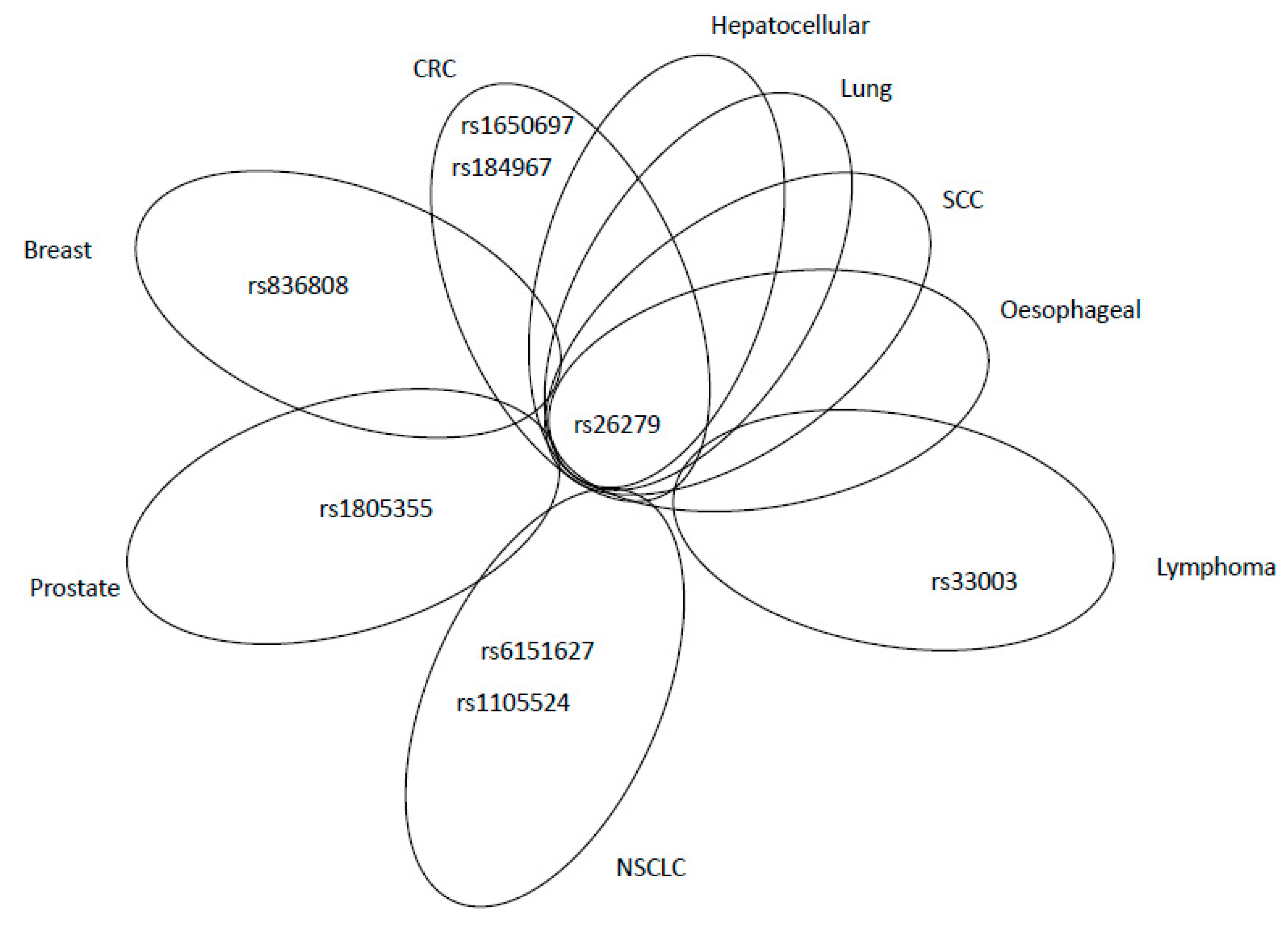
| MSH3 | rs1650697 | Ile79Val | A-G | C | Exon 1 | c.235A>G | D | 24% |
| rs26279 | Ala1045Thr | G-A | G | Exon 23 | c.3133G>A | A | 72% | |
| rs33003 | - | A-G | A | intron | c.3303-436A>G | D | 71% | |
| rs184967 | Gln949Arg | A-G | G | Exon 21 | c.2846A>G | D | 14% | |
| rs836808 | - | G-A | C | intron | c.1174-176G>A | P | 25% | |
| rs863221 | - | T-G | T | intron | c.1763+1841T>G | B | 41% | |
| rs1805355 | Pro231Pro | G-A | G | Exon 4 | c.693G>A | P | 5% | |
| rs6151627 | - | A-G | A | intron | c.580-380A>G | B | 27% | |
| rs1105524 | - | A-G | G | intron | c.-35A>G | P | 32% | |
| rs6151670 | - | C-G | C | intron | c.1340+8303C>G | A | 27% | |
| rs7709909 | - | C-T | C | intron | c.1341-20102C>T | A | 40% | |
| MSH4 | rs5745325 | Ala97Thr | G-A | G | Exon 2 | c.289G>A | P | 31% |
| MSH5 | rs707938 | Gln716Gln | A-G | C | Exon 22 | c.2148A>G | P | 68% |
| rs2075789 | Pro29Ser | C-T | G | Exon 2 | c.85C>T | D | 10% | |
| rs707939 | - | C-A | G | intron | c.1326+36C>A | B | 37% | |
| rs3131379 | - | G-A | C | intron | c.813-45G>A | P | 7% | |
| rs707939 | - | C-A | G | intron | c.1326+36C>A | A | 37% | |
| MSH6 | rs1800937 | Tyr214* | C-A | C | Exon 4 | c.642C>A | P | 10% |
| rs1042821 | Gly39Ala | G-A | C | Exon 1 | c.116G>A | A | 18% | |
| rs1800935 | Asp180Asp | T-C | T | Exon 2 | c.540T>C | B | 29% | |
| rs3136228 | - | T-G | G | intron | c.-152-405T>G | P | 65% | |
| rs1800932 | Pro92Pro | A-G | A | Exon 1 | c.276A>G | P | 18% | |
| PMS1 | rs5742933 | - | G-C | G | intron | c.-24G>C | D | 20% |
| rs5742938 | - | G-A | G | intron | c.-21+639G>A | P | 73% | |
| PMS2 | rs2228006 | - | G-A | G | intron | c.1621G>A | P | 12% |
| rs7797466 | - | C-T | G | intron | c.24-1121C>T | P | 18% |
| Gene | SNP ID | PubMed ID | Reference | Cancer | Cases | Controls | Phenotype |
|---|---|---|---|---|---|---|---|
| MLH1 | rs1799977 | 19203531 | [160] | Melanoma | 51 | 0 | - |
| rs1799977 | 21156845 | [63] | B-cell lymph. | 308 | 0 | ↓ effect of Dox treatment in folicullar lymphoma | |
| rs1799977 | 21156845 | [63] | B-cell lymph. | 308 | 0 | ↓ effect of Pt-based second line treatment | |
| rs1799977 | 27608007 | [68] | Rectal | 280 | 0 | protective factor in rectal c. | |
| rs1800734 | 25047469 | SCC | 185 | 0 | GA genotype ↓ OS in oral squamous cell c. | ||
| rs1800734 | 25047469 | SCC | 185 | 0 | AA genotype ↓ OS in oral squamous cell c. | ||
| rs2286940 | 26743341 | [70] | Lymphoid | 153 | 0 | sign. predictor of progression-free surv. | |
| MLH3 | rs108621 | 24755277 | [15] | CRC | 1095 | 1469 | CC genotype ↑ survival |
| MSH2 | rs2042649 | 22017238 | Glioblastoma | 121 | 0 | ↑ OS of glioblastoma patients | |
| rs2303428 | 22017238 | Glioblastoma | 121 | 0 | ↑ OS of glioblastoma patients | ||
| rs2303428 | 19741564 | Melanoma | 51 | 0 | ↑ hematologic side effects in melanoma tr. | ||
| rs2303428 | 20458443 | NSCLC | 96 | 0 | CC genotype ↑ response to Pt-based therapy | ||
| rs2303428 | 20708344 | Breast | 87 | 0 | ↑ radiosensitivity in breast c. patients | ||
| rs3732183 | 20091185 | [157] | CRC | 94 | 0 | AG/GG genotype ↑ response to therapy | |
| rs6544991 | 28093084 | [116] | NSCLC | 220 | 0 | ↑ GIT toxicity of Pt-based chemo | |
| MSH3 | rs863221 | 19115210 | [95] | CRC | 2060 | 0 | ↑ OS in patients with CRC |
| rs26279 | 25966119 | [97] | NSCLC | 180 | 0 | ↑ sensitivity to Pt-based therapy in NSCLC | |
| rs6151627 | 28093084 | [116] | NSCLC | 220 | 0 | ↑ hematological toxicity in NSCLC patients | |
| rs1105524 | 25966119 | [97] | NSCLC | 180 | 0 | G/A and A/A genotype ↓ OS in NSCLC | |
| MSH5 | rs707939 | 28093084 | [116] | NSCLC | 220 | 0 | ↑ hematological toxicity in NSCLC patients |
| MSH6 | rs3136228 | 22868256 | [131] | CRC | 144 | 0 | grade 3–4 neutropenia |
| rs3136228 | 27608007 | [68] | Rectal | 280 | 0 | ↓ response to F-pyrimidine therapy | |
| rs3136228 | 28206966 | [130] | Prostate | 542 | 0 | ↓ OS in prostate c. patients | |
| rs1800935 | 24755277 | [15] | CRC | 1095 | 1469 | CC carriers had ↓ recurrence of CRC |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caja, F.; Vodickova, L.; Kral, J.; Vymetalkova, V.; Naccarati, A.; Vodicka, P. DNA Mismatch Repair Gene Variants in Sporadic Solid Cancers. Int. J. Mol. Sci. 2020, 21, 5561. https://doi.org/10.3390/ijms21155561
Caja F, Vodickova L, Kral J, Vymetalkova V, Naccarati A, Vodicka P. DNA Mismatch Repair Gene Variants in Sporadic Solid Cancers. International Journal of Molecular Sciences. 2020; 21(15):5561. https://doi.org/10.3390/ijms21155561
Chicago/Turabian StyleCaja, Fabian, Ludmila Vodickova, Jan Kral, Veronika Vymetalkova, Alessio Naccarati, and Pavel Vodicka. 2020. "DNA Mismatch Repair Gene Variants in Sporadic Solid Cancers" International Journal of Molecular Sciences 21, no. 15: 5561. https://doi.org/10.3390/ijms21155561
APA StyleCaja, F., Vodickova, L., Kral, J., Vymetalkova, V., Naccarati, A., & Vodicka, P. (2020). DNA Mismatch Repair Gene Variants in Sporadic Solid Cancers. International Journal of Molecular Sciences, 21(15), 5561. https://doi.org/10.3390/ijms21155561