The MUDENG Augmentation: A Genesis in Anti-Cancer Therapy?


 , ,
, ,
Abstract
1. Introduction
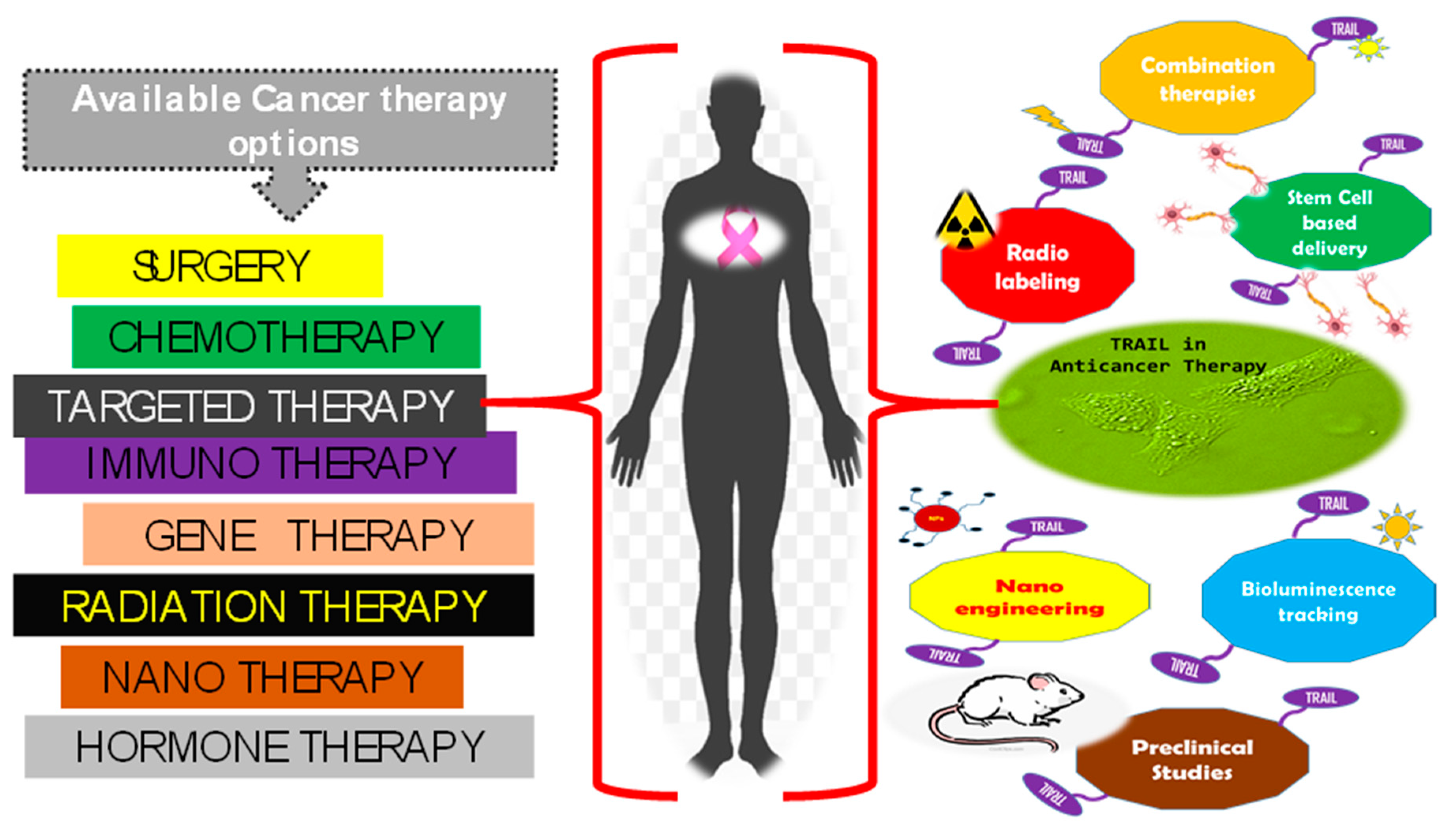
2. Trail of TRAIL in Cancer Therapeutics
3. Evolution of MUDENG
3.1. What Is MUDENG
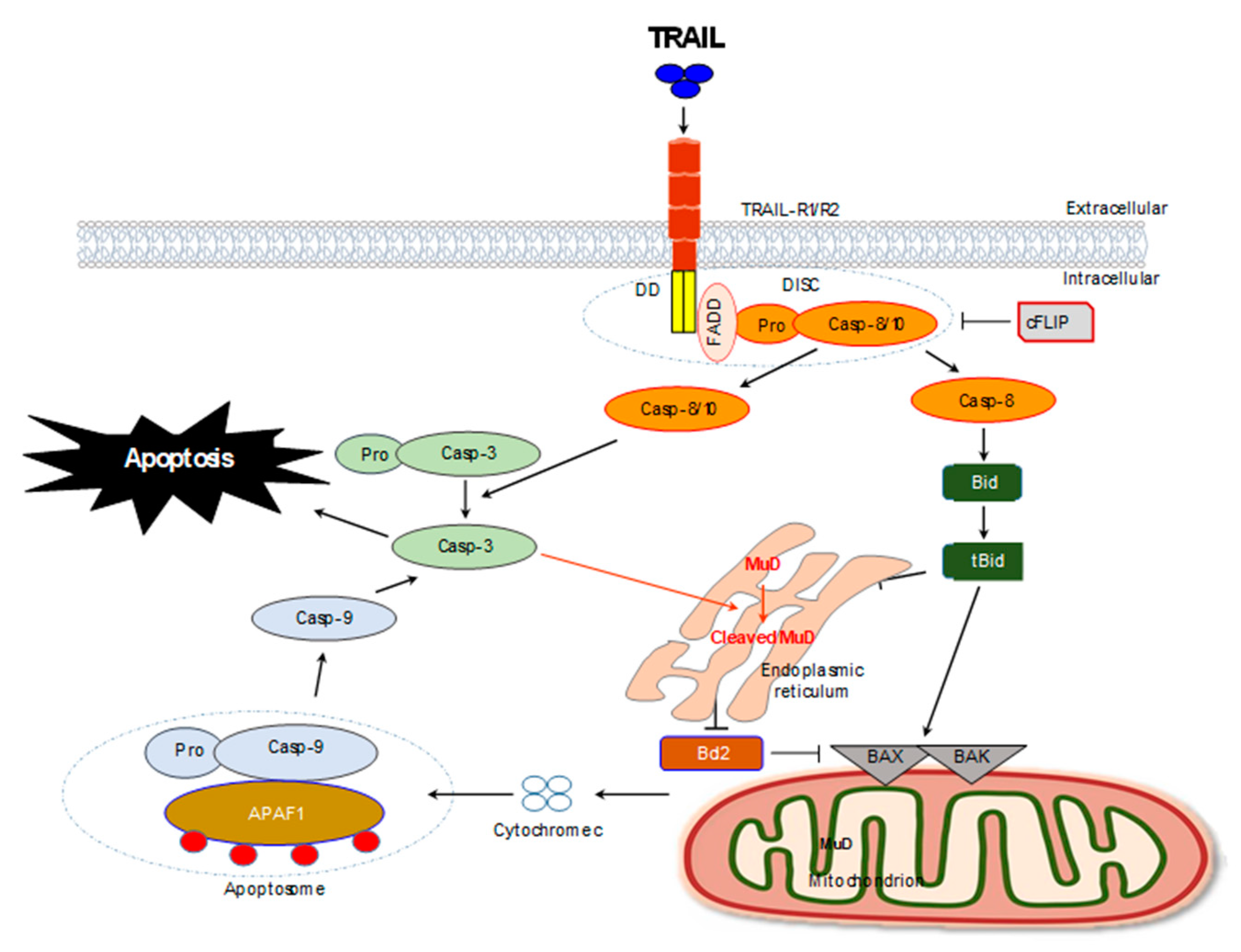
3.2. Mechanism of MuD Based Anti-Cancer Therapy
3.3. MuD Milestones in Therapeutics
4. Future of MuD: Challenges and Scope
Funding
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Vogelstein, B.; Fearon, E.R.; Hamilton, S.R.; Kern, S.E.; Preisinger, A.C.; Leppert, M.; Nakamura, Y.; White, R.; Smits, A.M.; Bos, J.L. Genetic alterations during colorectal-tumor development. N. Engl. J. Med. 1988, 319, 525–532. [Google Scholar] [CrossRef]
- Balmain, A.; Harris, C.C. Carcinogenesis in mouse and human cells: Parallels and paradoxes. Carcinogenesis 2000, 21, 371–377. [Google Scholar] [CrossRef]
- Evan, G.I.; Vousden, K.H. Proliferation, cell cycle and apoptosis in cancer. Nature 2001, 411, 342–348. [Google Scholar] [CrossRef]
- Wanjek, C. Exciting New Cancer Treatments Emerge Amid Persistent Myths. Live Science 2006. Available online: https://www.livescience.com/4211-exciting-cancer-treatments-emerge-persistent-myths.html (accessed on 1 July 2020).
- Hayden, E.C. Cutting off cancer’s supply lines. Nature 2009, 458, 686–687. [Google Scholar] [CrossRef]
- Subotic, S.; Wyler, S.; Bachmann, A. Surgical Treatment of Localised Renal Cancer. Eur. Urol. Suppl. 2012, 11, 60–65. [Google Scholar] [CrossRef]
- Mieog, J.S.; van der Hage, J.A.; van de Velde, C.J. Neoadjuvant chemotherapy for operable breast cancer. Br. J. Surg. 2007, 94, 1189–1200. [Google Scholar] [CrossRef]
- Jafri, S.H.; Mills, G. Neoadjuvant chemotherapy in lung cancer. Therapy 2011, 8, 23–31. [Google Scholar] [CrossRef]
- Takimoto, C.; Calvo, E. Principles of Oncologic Pharmacotherapy. In Cancer Management: A Multidisciplinary Approach, 11th ed.; Pazdur, R., Wagman, L.D., Camphausen, K.A., Hoskins, W.J., Eds.; CMP United Business Media: Manhasset, NY, USA, 2009. [Google Scholar]
- Duarte, F.J.E. Tunable Laser Applications, 2nd ed.; CRC: Boca Raton, FL, USA, 2009. [Google Scholar]
- Dolmans, D.E.; Fukumura, D.; Jain, R.K. Photodynamic therapy for cancer. Nat. Rev. Cancer 2003, 3, 380–387. [Google Scholar] [CrossRef]
- Zhang, P.; Huang, H.; Banerjee, S.; Clarkson, G.J.; Ge, C.; Imberti, C.; Sadler, P.J. Nucleus-Targeted Organoiridium-Albumin Conjugate for Photodynamic Cancer Therapy. Angew. Chem. Int. Ed. Engl. 2019, 58, 2350–2354. [Google Scholar] [CrossRef]
- Mo, S.; Carlisle, R.; Laga, R.; Myers, R.; Graham, S.; Cawood, R.; Ulbrich, K.; Seymour, L.; Coussios, C.C. Increasing the density of nanomedicines improves their ultrasound-mediated delivery to tumours. J. Control. Release 2015, 210, 10–18. [Google Scholar] [CrossRef]
- University of Warwick. Simply Shining Light on ‘Dinosaur Metal’ Compound Kills Cancer Cells; 2019. ScienceDaily, 2019. Available online: https://www.sciencedaily.com/releases/2019/02/190204085956.htm (accessed on 1 July 2020).
- Wiley, S.R.; Schooley, K.; Smolak, P.J.; Din, W.S.; Huang, C.P.; Nicholl, J.K.; Sutherland, G.R.; Smith, T.D.; Rauch, C.; Smith, C.A.; et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity 1995, 3, 673–682. [Google Scholar] [CrossRef]
- Pitti, R.M.; Marsters, S.A.; Ruppert, S.; Donahue, C.J.; Moore, A.; Ashkenazi, A. Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J. Biol. Chem. 1996, 271, 12687–12690. [Google Scholar] [CrossRef]
- Allen, J.E.; El-Deiry, W.S. Regulation of the human TRAIL gene. Cancer Biol. Ther. 2012, 13, 1143–1151. [Google Scholar] [CrossRef]
- Walczak, H.; Miller, R.E.; Ariail, K.; Gliniak, B.; Griffith, T.S.; Kubin, M.; Chin, W.; Jones, J.; Woodward, A.; Le, T.; et al. Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat. Med. 1999, 5, 157–163. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Pai, R.C.; Fong, S.; Leung, S.; Lawrence, D.A.; Marsters, S.A.; Blackie, C.; Chang, L.; McMurtrey, A.E.; Hebert, A.; et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J. Clin. Invest. 1999, 104, 155–162. [Google Scholar] [CrossRef]
- Ross, J.S.; Schenkein, D.P.; Pietrusko, R.; Rolfe, M.; Linette, G.P.; Stec, J.; Stagliano, N.E.; Ginsburg, G.S.; Symmans, W.F.; Pusztai, L.; et al. Targeted therapies for cancer 2004. Am. J. Clin. Pathol. 2004, 122, 598–609. [Google Scholar] [CrossRef]
- Ediriwickrema, A.; Saltzman, W.M. Nanotherapy for Cancer: Targeting and Multifunctionality in the Future of Cancer Therapies. ACS Biomater. Sci. Eng. 2015, 1, 64–78. [Google Scholar] [CrossRef]
- Akhter, M.H.; Rizwanullah, M.; Ahmad, J.; Ahsan, M.J.; Mujtaba, M.A.; Amin, S. Nanocarriers in advanced drug targeting: Setting novel paradigm in cancer therapeutics. Artif. Cells Nanomed Biotechnol. 2018, 46, 873–884. [Google Scholar] [CrossRef]
- Yuan, F.; Dellian, M.; Fukumura, D.; Leunig, M.; Berk, D.A.; Torchilin, V.P.; Jain, R.K. Vascular permeability in a human tumor xenograft: Molecular size dependence and cutoff size. Cancer Res. 1995, 55, 3752–3756. [Google Scholar]
- Nakamura, Y.; Mochida, A.; Choyke, P.L.; Kobayashi, H. Nanodrug Delivery: Is the Enhanced Permeability and Retention Effect Sufficient for Curing Cancer? Bioconjug. Chem. 2016, 27, 2225–2238. [Google Scholar] [CrossRef]
- Wang, A.Z.; Langer, R.; Farokhzad, O.C. Nanoparticle delivery of cancer drugs. Annu. Rev. Med. 2012, 63, 185–198. [Google Scholar] [CrossRef]
- Zou, L.; Wang, H.; He, B.; Zeng, L.; Tan, T.; Cao, H.; He, X.; Zhang, Z.; Guo, S.; Li, Y. Current Approaches of Photothermal Therapy in Treating Cancer Metastasis with Nanotherapeutics. Theranostics 2016, 6, 762–772. [Google Scholar] [CrossRef]
- He, H.; Liu, L.; Morin, E.E.; Liu, M.; Schwendeman, A. Survey of Clinical Translation of Cancer Nanomedicines-Lessons Learned from Successes and Failures. Acc. Chem Res. 2019, 52, 2445–2461. [Google Scholar] [CrossRef]
- Hare, J.I.; Lammers, T.; Ashford, M.B.; Puri, S.; Storm, G.; Barry, S.T. Challenges and strategies in anti-cancer nanomedicine development: An industry perspective. Adv. Drug Deliv. Rev. 2017, 108, 25–38. [Google Scholar] [CrossRef]
- Youn, Y.S.; Bae, Y.H. Perspectives on the past, present, and future of cancer nanomedicine. Adv. Drug Deliv. Rev. 2018, 130, 3–11. [Google Scholar] [CrossRef]
- Wilhelm, S.; Tavares, A.J.; Dai, Q.; Ohta, S.; Audet, J.; Dvorak, H.F.; Chan, W.C.W. Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater. 2016, 1, 1–12. [Google Scholar] [CrossRef]
- Danhier, F. To exploit the tumor microenvironment: Since the EPR effect fails in the clinic, what is the future of nanomedicine? J. Control. Release 2016, 244, 108–121. [Google Scholar] [CrossRef]
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [Google Scholar] [CrossRef]
- Shi, Y.; Lammers, T. Combining Nanomedicine and Immunotherapy. Acc. Chem Res. 2019, 52, 1543–1554. [Google Scholar] [CrossRef]
- Salvioni, L.; Rizzuto, M.A.; Bertolini, J.A.; Pandolfi, L.; Colombo, M.; Prosperi, D. Thirty Years of Cancer Nanomedicine: Success, Frustration, and Hope. Cancers 2019, 11, 1855. [Google Scholar] [CrossRef] [PubMed]
- Carter, S.; Thurston, D.E. Immuno-oncology agents for cancer therapy. Pharm. J. 2020. [Google Scholar] [CrossRef]
- Merino, D.; Lalaoui, N.; Morizot, A.; Solary, E.; Micheau, O. TRAIL in cancer therapy: Present and future challenges. Expert Opin. Ther.Targets 2007, 11, 1299–1314. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, Y.; Steller, H. Programmed cell death in animal development and disease. Cell 2011, 147, 742–758. [Google Scholar] [CrossRef]
- Thorburn, A. Death receptor-induced cell killing. Cell Signal. 2004, 16, 139–144. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Kroemer, G. Mitochondria: Master regulators of danger signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 780–788. [Google Scholar] [CrossRef]
- Johnstone, R.W.; Ruefli, A.A.; Lowe, S.W. Apoptosis: A link between cancer genetics and chemotherapy. Cell 2002, 108, 153–164. [Google Scholar] [CrossRef]
- Spencer, S.L.; Sorger, P.K. Measuring and modeling apoptosis in single cells. Cell 2011, 144, 926–939. [Google Scholar] [CrossRef]
- Kantari, C.; Walczak, H. Caspase-8 and bid: Caught in the act between death receptors and mitochondria. Biochim. Biophys. Acta 2011, 1813, 558–563. [Google Scholar] [CrossRef]
- Falschlehner, C.; Emmerich, C.H.; Gerlach, B.; Walczak, H. TRAIL signalling: Decisions between life and death. Int. J. Biochem. Cell Biol. 2007, 39, 1462–1475. [Google Scholar] [CrossRef]
- Kelley, S.K.; Harris, L.A.; Xie, D.; Deforge, L.; Totpal, K.; Bussiere, J.; Fox, J.A. Preclinical studies to predict the disposition of Apo2L/tumor necrosis factor-related apoptosis-inducing ligand in humans: Characterization of in vivo efficacy, pharmacokinetics, and safety. J. Pharmacol. Exp. Ther. 2001, 299, 31–38. [Google Scholar] [PubMed]
- Duiker, E.W.; Dijkers, E.C.; Lambers Heerspink, H.; de Jong, S.; van der Zee, A.G.; Jager, P.L.; Kosterink, J.G.; de Vries, E.G.; Lub-de Hooge, M.N. Development of a radioiodinated apoptosis-inducing ligand, rhTRAIL, and a radiolabelled agonist TRAIL receptor antibody for clinical imaging studies. Br. J. Pharmacol. 2012, 165, 2203–2212. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A.; Holland, P.; Eckhardt, S.G. Ligand-based targeting of apoptosis in cancer: The potential of recombinant human apoptosis ligand 2/Tumor necrosis factor-related apoptosis-inducing ligand (rhApo2L/TRAIL). J. Clin. Oncol. 2008, 26, 3621–3630. [Google Scholar] [CrossRef]
- Schneider, P. Production of recombinant TRAIL and TRAIL receptor: Fc chimeric proteins. Methods Enzymol. 2000, 322, 325–345. [Google Scholar] [PubMed]
- Shah, K.; Tung, C.H.; Yang, K.; Weissleder, R.; Breakefield, X.O. Inducible release of TRAIL fusion proteins from a proapoptotic form for tumor therapy. Cancer Res. 2004, 64, 3236–3242. [Google Scholar] [CrossRef] [PubMed]
- Ganten, T.M.; Koschny, R.; Sykora, J.; Schulze-Bergkamen, H.; Buchler, P.; Haas, T.L.; Schader, M.B.; Untergasser, A.; Stremmel, W.; Walczak, H. Preclinical differentiation between apparently safe and potentially hepatotoxic applications of TRAIL either alone or in combination with chemotherapeutic drugs. Clin. Cancer Res. 2006, 12, 2640–2646. [Google Scholar] [CrossRef]
- Berg, D.; Lehne, M.; Muller, N.; Siegmund, D.; Munkel, S.; Sebald, W.; Pfizenmaier, K.; Wajant, H. Enforced covalent trimerization increases the activity of the TNF ligand family members TRAIL and CD95L. Cell Death Differ. 2007, 14, 2021–2034. [Google Scholar] [CrossRef]
- Wajant, H.; Gerspach, J.; Pfizenmaier, K. Engineering death receptor ligands for cancer therapy. Cancer Lett. 2013, 332, 163–174. [Google Scholar] [CrossRef]
- Muller, N.; Schneider, B.; Pfizenmaier, K.; Wajant, H. Superior serum half life of albumin tagged TNF ligands. Biochem. Biophys. Res. Commun. 2010, 396, 793–799. [Google Scholar] [CrossRef]
- Kim, T.H.; Jo, Y.G.; Jiang, H.H.; Lim, S.M.; Youn, Y.S.; Lee, S.; Chen, X.; Byun, Y.; Lee, K.C. PEG-transferrin conjugated TRAIL (TNF-related apoptosis-inducing ligand) for therapeutic tumor targeting. J. Control. Release 2012, 162, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Youn, Y.S.; Jiang, H.H.; Lee, S.; Chen, X.; Lee, K.C. PEGylated TNF-related apoptosis-inducing ligand (TRAIL) analogues: Pharmacokinetics and antitumor effects. Bioconjug. Chem. 2011, 22, 1631–1637. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Jeong, D.; Kang, H.E.; Lee, K.C.; Na, K. A sulfate polysaccharide/TNF-related apoptosis-inducing ligand (TRAIL) complex for the long-term delivery of TRAIL in poly(lactic-co-glycolic acid) (PLGA) microspheres. J. Pharm. Pharmacol. 2013, 65, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Stieglmaier, J.; Bremer, E.; Kellner, C.; Liebig, T.M.; ten Cate, B.; Peipp, M.; Schulze-Koops, H.; Pfeiffer, M.; Buhring, H.J.; Greil, J.; et al. Selective induction of apoptosis in leukemic B-lymphoid cells by a CD19-specific TRAIL fusion protein. Cancer Immunol. Immun. 2008, 57, 233–246. [Google Scholar] [CrossRef] [PubMed]
- Hingtgen, S.D.; Kasmieh, R.; van de Water, J.; Weissleder, R.; Shah, K. A novel molecule integrating therapeutic and diagnostic activities reveals multiple aspects of stem cell-based therapy. Stem Cells 2010, 28, 832–841. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Guo, Y.; Huang, R.; Li, J.; Huang, S.; Kuang, Y.; Han, L.; Jiang, C. Gene and doxorubicin co-delivery system for targeting therapy of glioma. Biomaterials 2012, 33, 4907–4916. [Google Scholar] [CrossRef]
- Wheatley, M.A.; Cochran, M.C.; Eisenbrey, J.R.; Oum, K.L. Cellular signal transduction can be induced by TRAIL conjugated to microcapsules. J. Biomed. Mater. Res. A 2012, 100, 2602–2611. [Google Scholar] [CrossRef]
- Perlstein, B.; Finniss, S.A.; Miller, C.; Okhrimenko, H.; Kazimirsky, G.; Cazacu, S.; Lee, H.K.; Lemke, N.; Brodie, S.; Umansky, F.; et al. TRAIL conjugated to nanoparticles exhibits increased anti-tumor activities in glioma cells and glioma stem cells in vitro and in vivo. Neuro Oncol. 2013, 15, 29–40. [Google Scholar] [CrossRef]
- Kim, S.M.; Oh, J.H.; Park, S.A.; Ryu, C.H.; Lim, J.Y.; Kim, D.S.; Chang, J.W.; Oh, W.; Jeun, S.S. Irradiation enhances the tumor tropism and therapeutic potential of tumor necrosis factor-related apoptosis-inducing ligand-secreting human umbilical cord blood-derived mesenchymal stem cells in glioma therapy. Stem Cells 2010, 28, 2217–2228. [Google Scholar] [CrossRef]
- Sasportas, L.S.; Kasmieh, R.; Wakimoto, H.; Hingtgen, S.; van de Water, J.A.; Mohapatra, G.; Figueiredo, J.L.; Martuza, R.L.; Weissleder, R.; Shah, K. Assessment of therapeutic efficacy and fate of engineered human mesenchymal stem cells for cancer therapy. Proc. Natl. Acad. Sci. USA 2009, 106, 4822–4827. [Google Scholar] [CrossRef]
- Choi, S.A.; Hwang, S.K.; Wang, K.C.; Cho, B.K.; Phi, J.H.; Lee, J.Y.; Jung, H.W.; Lee, D.H.; Kim, S.K. Therapeutic efficacy and safety of TRAIL-producing human adipose tissue-derived mesenchymal stem cells against experimental brainstem glioma. Neuro Oncol. 2011, 13, 61–69. [Google Scholar] [CrossRef]
- Kim, S.W.; Kim, S.J.; Park, S.H.; Yang, H.G.; Kang, M.C.; Choi, Y.W.; Kim, S.M.; Jeun, S.S.; Sung, Y.C. Complete regression of metastatic renal cell carcinoma by multiple injections of engineered mesenchymal stem cells expressing dodecameric TRAIL and HSV-TK. Clin. Cancer Res. 2013, 19, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Quintanilla, J.; Bhere, D.; Heidari, P.; He, D.; Mahmood, U.; Shah, K. Therapeutic efficacy and fate of bimodal engineered stem cells in malignant brain tumors. Stem Cells 2013, 31, 1706–1714. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.L.; Huang, B.; Zhang, T.Y.; Miao, P.H.; Tang, G.P.; Tabata, Y.; Gao, J.Q. Mesenchymal stem cells as a novel carrier for targeted delivery of gene in cancer therapy based on nonviral transfection. Mol. Pharm. 2012, 9, 2698–2709. [Google Scholar] [CrossRef] [PubMed]
- Bagci-Onder, T.; Wakimoto, H.; Anderegg, M.; Cameron, C.; Shah, K. A dual PI3K/mTOR inhibitor, PI-103, cooperates with stem cell-delivered TRAIL in experimental glioma models. Cancer Res. 2011, 71, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Corsten, M.F.; Miranda, R.; Kasmieh, R.; Krichevsky, A.M.; Weissleder, R.; Shah, K. MicroRNA-21 knockdown disrupts glioma growth in vivo and displays synergistic cytotoxicity with neural precursor cell delivered S-TRAIL in human gliomas. Cancer Res. 2007, 67, 8994–9000. [Google Scholar] [CrossRef]
- Loebinger, M.R.; Eddaoudi, A.; Davies, D.; Janes, S.M. Mesenchymal stem cell delivery of TRAIL can eliminate metastatic cancer. Cancer Res. 2009, 69, 4134–4142. [Google Scholar] [CrossRef]
- Lee, H.J.; Yang, H.M.; Choi, Y.S.; Park, S.H.; Moon, S.H.; Lee, Y.S.; Sung, Y.C.; Kim, S.J. A therapeutic strategy for metastatic malignant fibrous histiocytoma through mesenchymal stromal cell-mediated TRAIL production. Ann. Surg. 2013, 257, 952–960. [Google Scholar] [CrossRef]
- Kauer, T.M.; Figueiredo, J.L.; Hingtgen, S.; Shah, K. Encapsulated therapeutic stem cells implanted in the tumor resection cavity induce cell death in gliomas. Nat. Neurosci. 2011, 15, 197–204. [Google Scholar] [CrossRef]
- Reagan, M.R.; Seib, F.P.; McMillin, D.W.; Sage, E.K.; Mitsiades, C.S.; Janes, S.M.; Ghobrial, I.M.; Kaplan, D.L. Stem Cell Implants for Cancer Therapy: TRAIL-Expressing Mesenchymal Stem Cells Target Cancer Cells In Situ. J. Breast Cancer 2012, 15, 273–282. [Google Scholar] [CrossRef]
- van de Water, J.A.; Bagci-Onder, T.; Agarwal, A.S.; Wakimoto, H.; Roovers, R.C.; Zhu, Y.; Kasmieh, R.; Bhere, D.; Van Bergen en Henegouwen, P.M.; Shah, K. Therapeutic stem cells expressing variants of EGFR-specific nanobodies have antitumor effects. Proc. Natl. Acad. Sci. USA 2012, 109, 16642–16647. [Google Scholar] [CrossRef]
- Maksimovic-Ivanic, D.; Stosic-Grujicic, S.; Nicoletti, F.; Mijatovic, S. Resistance to TRAIL and how to surmount it. Immunol. Res. 2012, 52, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Dimberg, L.Y.; Anderson, C.K.; Camidge, R.; Behbakht, K.; Thorburn, A.; Ford, H.L. On the TRAIL to successful cancer therapy? Predicting and counteracting resistance against TRAIL-based therapeutics. Oncogene 2013, 32, 1341–1350. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, R.W.; Frew, A.J.; Smyth, M.J. The TRAIL apoptotic pathway in cancer onset, progression and therapy. Nat. Rev. Cancer 2008, 8, 782–798. [Google Scholar] [CrossRef] [PubMed]
- Jazirehi, A.R.; Arle, D. Epigenetic regulation of the TRAIL/Apo2L apoptotic pathway by histone deacetylase inhibitors: An attractive approach to bypass melanoma immunotherapy resistance. Am. J. Clin. Exp. Immunol. 2013, 2, 55–74. [Google Scholar]
- Trivedi, R.; Mishra, D.P. Trailing TRAIL Resistance: Novel Targets for TRAIL Sensitization in Cancer Cells. Front. Oncol. 2015, 5, 69. [Google Scholar] [CrossRef] [PubMed]
- Piras, V.; Hayashi, K.; Tomita, M.; Selvarajoo, K. Enhancing apoptosis in TRAIL-resistant cancer cells using fundamental response rules. Sci. Rep. 2011, 1, 144. [Google Scholar] [CrossRef]
- Hellwig, C.T.; Rehm, M. TRAIL signaling and synergy mechanisms used in TRAIL-based combination therapies. Mol. Cancer Ther. 2012, 11, 3–13. [Google Scholar] [CrossRef]
- Elmallah, M.I.; Micheau, O. Marine Drugs Regulating Apoptosis Induced by Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand (TRAIL). Mar. Drugs 2015, 13, 6884–6909. [Google Scholar] [CrossRef]
- Klosek, M.; Mertas, A.; Krol, W.; Jaworska, D.; Szymszal, J.; Szliszka, E. Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand-Induced Apoptosis in Prostate Cancer Cells after Treatment with Xanthohumol-A Natural Compound Present in Humulus lupulus L. Int. J. Mol. Sci. 2016, 17, 837. [Google Scholar] [CrossRef]
- Woo, S.M.; Seo, S.U.; Kim, S.H.; Nam, J.O.; Kim, S.; Park, J.W.; Min, K.J.; Kwon, T.K. Hispidulin Enhances TRAIL-Mediated Apoptosis via CaMKK/AMPK/USP51 Axis-Mediated Bim Stabilization. Cancers 2019, 11, 1960. [Google Scholar] [CrossRef]
- Han, L.; Dai, S.; Li, Z.; Zhang, C.; Wei, S.; Zhao, R.; Zhang, H.; Zhao, L.; Shan, B. Combination of the natural compound Periplocin and TRAIL induce esophageal squamous cell carcinoma apoptosis in vitro and in vivo: Implication in anticancer therapy. J. Exp. Clin. Cancer Res. 2019, 38, 501. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, H.; Yamada, Y.; Komiyama, K.; Hayashi, M.; Ishibashi, M.; Sunazuka, T.; Izuhara, T.; Sugahara, K.; Tsuruda, K.; Masuda, M.; et al. A novel natural compound, a cycloanthranilylproline derivative (Fuligocandin B), sensitizes leukemia cells to apoptosis induced by tumor necrosis factor related apoptosis-inducing ligand (TRAIL) through 15-deoxy-Delta 12, 14 prostaglandin J2 production. Blood 2007, 110, 1664–1674. [Google Scholar] [CrossRef] [PubMed]
- Menke, C.; Bin, L.; Thorburn, J.; Behbakht, K.; Ford, H.L.; Thorburn, A. Distinct TRAIL resistance mechanisms can be overcome by proteasome inhibition but not generally by synergizing agents. Cancer Res. 2011, 71, 1883–1892. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yang, X.; Xu, T.; Kong, Q.; Zhang, Y.; Shen, Y.; Wei, Y.; Wang, G.; Chang, K.J. Overcoming resistance to TRAIL-induced apoptosis in solid tumor cells by simultaneously targeting death receptors, c-FLIP and IAPs. Int. J. Oncol. 2016, 49, 153–163. [Google Scholar] [CrossRef]
- Lemke, J.; von Karstedt, S.; Zinngrebe, J.; Walczak, H. Getting TRAIL back on track for cancer therapy. Cell Death Differ. 2014, 21, 1350–1364. [Google Scholar] [CrossRef]
- Thorburn, A.; Behbakht, K.; Ford, H. TRAIL receptor-targeted therapeutics: Resistance mechanisms and strategies to avoid them. Drug Resist. Updat. 2008, 11, 17–24. [Google Scholar] [CrossRef]
- Zhang, L.; Fang, B. Mechanisms of resistance to TRAIL-induced apoptosis in cancer. Cancer Gene Ther. 2005, 12, 228–237. [Google Scholar] [CrossRef]
- Ngai, S.C.; Wong, H.M.S. Trailing TRAIL Resistance for Targeted Cancer Therapy. Biomed. J. Sci. Tech. Res. 2018, 4, 3668–3671. [Google Scholar]
- Kawasaki, H.; Taira, K. A functional gene discovery in the Fas-mediated pathway to apoptosis by analysis of transiently expressed randomized hybrid-ribozyme libraries. Nucleic Acids Res. 2002, 30, 3609–3614. [Google Scholar] [CrossRef][Green Version]
- Lee, M.R.; Shin, J.N.; Moon, A.R.; Park, S.Y.; Hong, G.; Lee, M.J.; Yun, C.W.; Seol, D.W.; Piya, S.; Bae, J.; et al. A novel protein, MUDENG, induces cell death in cytotoxic T cells. Biochem. Biophys. Res. Commun. 2008, 370, 504–508. [Google Scholar] [CrossRef]
- Hirst, J.; Barlow, L.D.; Francisco, G.C.; Sahlender, D.A.; Seaman, M.N.; Dacks, J.B.; Robinson, M.S. The fifth adaptor protein complex. PLoS Biol. 2011, 9, e1001170. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Lim, J.B.; Wickramanayake, D.D.; Wagley, Y.; Kim, J.; Lee, H.C.; Seo, H.G.; Kim, T.H.; Oh, J.W. Characterization of MUDENG, a novel anti-apoptotic protein. Oncogenesis 2016, 5, e221. [Google Scholar] [CrossRef] [PubMed]
- Stuckey, D.W.; Shah, K. TRAIL on trial: Preclinical advances in cancer therapy. Trends Mol. Med. 2013, 19, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Wagley, Y.; Choi, J.H.; Wickramanayake, D.D.; Choi, G.Y.; Kim, C.K.; Kim, T.H.; Oh, J.W. A monoclonal antibody against human MUDENG protein. Monoclon. Antib. Immunodiagn. Immunother. 2013, 32, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.N.; Park, S.Y.; Cha, J.H.; Park, J.Y.; Lee, B.R.; Jung, S.A.; Lee, S.T.; Yun, C.W.; Seol, D.W.; Kim, T.H. Generation of a novel proform of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) protein that can be reactivated by matrix metalloproteinases. Exp. Cell Res. 2006, 312, 3892–3898. [Google Scholar] [CrossRef]
- Cullen, S.P.; Martin, S.J. Fas and TRAIL ‘death receptors’ as initiators of inflammation: Implications for cancer. Semin. Cell Dev. Biol. 2015, 39, 26–34. [Google Scholar] [CrossRef]
- Shin, J.N.; Han, J.H.; Kim, J.Y.; Moon, A.R.; Kim, J.E.; Chang, J.H.; Bae, J.; Oh, J.W.; Kim, T.H. MUDENG is cleaved by caspase-3 during TRAIL-induced cell death. Biochem. Biophys. Res. Commun. 2013, 435, 234–238. [Google Scholar] [CrossRef]
- Won, M.; Luo, Y.; Lee, D.H.; Shin, E.; Suh, D.S.; Kim, T.H.; Jin, H.; Bae, J. BAX is an essential key mediator of AP5M1-induced apoptosis in cervical carcinoma cells. Biochem. Biophys. Res. Commun. 2019, 518, 368–373. [Google Scholar] [CrossRef]
- Kyrtsonis, M.C.; Dedoussis, G.; Zervas, C.; Perifanis, V.; Baxevanis, C.; Stamatelou, M.; Maniatis, A. Soluble interleukin-6 receptor (sIL-6R), a new prognostic factor in multiple myeloma. Br. J. Haematol. 1996, 93, 398–400. [Google Scholar] [CrossRef]
- Oltvai, Z.N.; Milliman, C.L.; Korsmeyer, S.J. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell 1993, 74, 609–619. [Google Scholar] [CrossRef]
- Choi, J.H.; Min, W.K.; Gopal, J.; Lee, Y.M.; Muthu, M.; Chun, S.; Oh, J.W. Silver nanoparticle-induced hormesis of astroglioma cells: A Mu-2-related death-inducing protein-orchestrated modus operandi. Int. J. Biol. Macromol. 2018, 117, 1147–1156. [Google Scholar] [CrossRef] [PubMed]
- Gopal, J.; Lee, Y.M.; Shin, J.; Muthu, M.; Jung, S.; Jeong, S.; Oh, J.; Oh, J.W. The graviola impact on human astroglioma cells: Functional significance of MUDENG. RSC Adv. 2019, 9, 8935–8942. [Google Scholar] [CrossRef]
- Zeng, L.; Wu, F.-E.; Gu, Z.-M.; McLaughlin, J.L. Murihexocins A and B, two novel mono-THF acetogenins with six hydroxyls, from Annona muricata (Annonaceae). Tetrahedron Lett. 1995, 36, 5291–5294. [Google Scholar] [CrossRef]
- Kim, G.S.; Zeng, L.; Alali, F.; Rogers, L.L.; Wu, F.E.; McLaughlin, J.L.; Sastrodihardjo, S. Two new mono-tetrahydrofuran ring acetogenins, annomuricin E and muricapentocin, from the leaves of Annona muricata. J. Nat. Prod. 1998, 61, 432–436. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.E.; Gu, Z.M.; Zeng, L.; Zhao, G.X.; Zhang, Y.; McLaughlin, J.L.; Sastrodihardjo, S. Two new cytotoxic monotetrahydrofuran Annonaceous acetogenins, annomuricins A and B, from the leaves of Annona muricata. J. Nat. Prod. 1995, 58, 830–836. [Google Scholar] [CrossRef]
- Wu, F.E.; Zeng, L.; Gu, Z.M.; Zhao, G.X.; Zhang, Y.; Schwedler, J.T.; McLaughlin, J.L.; Sastrodihardjo, S. New bioactive monotetrahydrofuran Annonaceous acetogenins, annomuricin C and muricatocin C, from the leaves of Annona muricata. J. Nat. Prod. 1995, 58, 909–915. [Google Scholar] [CrossRef]
- Ezirim, A.U.; Okochi, V.I.; James, A.B.; Adebeshi, O.A.; Ogunnowo, S.; Odeghe, O.B. Induction of Apoptosis in Myelogenous Leukemic k562 Cells by Ethanolic Leaf Extract of Annona muricata L. Glob. J. Res. Med. Plants Indigen. Med. 2013, 2, 142–151. [Google Scholar]
- Zorofchian Moghadamtousi, S.; Karimian, H.; Rouhollahi, E.; Paydar, M.; Fadaeinasab, M.; Abdul Kadir, H. Annona muricata leaves induce G(1) cell cycle arrest and apoptosis through mitochondria-mediated pathway in human HCT-116 and HT-29 colon cancer cells. J. Ethnopharmacol. 2014, 156, 277–289. [Google Scholar] [CrossRef]
- Pieme, C.A.; Kumar, S.G.; Dongmo, M.S.; Moukette, B.M.; Boyoum, F.F.; Ngogang, J.Y.; Saxena, A.K. Antiproliferative activity and induction of apoptosis by Annona muricata (Annonaceae) extract on human cancer cells. BMC Complement. Altern. Med. 2014, 14, 516. [Google Scholar] [CrossRef]
- Torres, M.P.; Rachagani, S.; Purohit, V.; Pandey, P.; Joshi, S.; Moore, E.D.; Johansson, S.L.; Singh, P.K.; Ganti, A.K.; Batra, S.K. Graviola: A novel promising natural-derived drug that inhibits tumorigenicity and metastasis of pancreatic cancer cells in vitro and in vivo through altering cell metabolism. Cancer Lett. 2012, 323, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Liu, J.; Kadouh, H.; Sun, X.; Zhou, K. Three new anti-proliferative Annonaceous acetogenins with mono-tetrahydrofuran ring from graviola fruit (Annona muricata). Bioorg Med. Chem. Lett. 2014, 24, 2773–2776. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Liu, J.; Zhou, N.; Zhu, W.; Dou, Q.P.; Zhou, K. Isolation of three new annonaceous acetogenins from Graviola fruit (Annona muricata) and their anti-proliferation on human prostate cancer cell PC-3. Bioorg Med. Chem. Lett. 2016, 26, 4382–4385. [Google Scholar] [CrossRef] [PubMed]
- Moghadamtousi, S.Z.; Kadir, H.A.; Paydar, M.; Rouhollahi, E.; Karimian, H. Annona muricata leaves induced apoptosis in A549 cells through mitochondrial-mediated pathway and involvement of NF-kappaB. BMC Complement. Altern Med. 2014, 14, 299. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Wu, F.E.; Oberlies, N.H.; McLaughlin, J.L.; Sastrodihadjo, S. Five new monotetrahydrofuran ring acetogenins from the leaves of Annona muricata. J. Nat. Prod. 1996, 59, 1035–1042. [Google Scholar] [CrossRef]
- Valencia, L.; Munoz, D.L.; Robledo, S.M.; Echeverri, F.; Arango, G.J.; Velez, I.D.; Triana, O. Trypanocidal and cytotoxic activity of extracts of Colombian plants. Biomedica 2011, 31, 552–559. [Google Scholar] [CrossRef]
- Zeng, L.; Wu, F.-E.; McLaughlin, J.L. Annohexocin, a novel mono-THF acetogenin with six hydroxyls, from Annona muricata (Annonaceae). Bioorg. Med. Chem. Lett. 1995, 5, 1865–1868. [Google Scholar] [CrossRef]
- Myint, S.H.; Cortes, D.; Laurens, A.; Hocquemiller, R.; Lebȩuf, M.; Cavé, A.; Cotteb, J.; Quéro, A.-M. Solamin, a cytotoxic mono-tetrahydrofuranic γ-lactone acetogenin from Annona muricata seeds. Phytochemistry 1991, 30, 3335–3338. [Google Scholar] [CrossRef]
- Menan, H.; Banzouzi, J.T.; Hocquette, A.; Pelissier, Y.; Blache, Y.; Kone, M.; Mallie, M.; Assi, L.A.; Valentin, A. Antiplasmodial activity and cytotoxicity of plants used in West African traditional medicine for the treatment of malaria. J. Ethnopharmacol. 2006, 105, 131–136. [Google Scholar] [CrossRef]
- Astirin, O.P.; Artanti, A.N.; Fitria, M.S.; Perwitasari, E.A.; Prayitno, A. Annonaa muricata linn leaf induce apoptosis in cancer cause virus. J. Cancer Ther. 2013, 4, 1244–1250. [Google Scholar] [CrossRef]
- Ge, H.L.; Zhang, D.-W.; Li, L.; Xie, D.; Zou, J.-H.; Si, Y.-K.; Dai, J. Two new terpenoids from endophytic fungus Periconia sp. F-31. Chem. Pharm. Bull. 2011, 59, 1541–1544. [Google Scholar] [CrossRef][Green Version]
- Adewole, S.O.; Ojewole, J.A. Protective effects of Annona muricata Linn. (Annonaceae) leaf aqueous extract on serum lipid profiles and oxidative stress in hepatocytes of streptozotocin-treated diabetic rats. Afr. J. Tradit. Complement. Altern. Med. 2008, 6, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Lin, S.Y.; Hu, F.F.; Ye, Z.; Zhang, Q.; Wang, Y.; Guo, A.Y. Landscape of cancer diagnostic biomarkers from specifically expressed genes. Brief. Bioinform. 2019. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.F.; Branon, T.C.; Rajeev, S.; Svinkina, T.; Udeshi, N.D.; Thoudam, T.; Kwak, C.; Rhee, H.W.; Lee, I.K.; Carr, S.A.; et al. Split-TurboID enables contact-dependent proximity labeling in cells. Proc. Natl. Acad. Sci. USA 2020, 117, 12143–12154. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| TRAIL Application | Modification | Deliverables | References |
|---|---|---|---|
| Addition of a leucine zipper | LZ-TRAIL | Facilitates TRAIL stability of trimeric formation in vitro and in vivo leading to enhanced anti-cancer efficiency | [19] |
| Radioiodinated recombinant human TRAIL | (125I) rhTRAIL | Detection of in vivo biodistribution and clearance of cancer cells | [46] |
| Addition of FLAG tag to TRAIL | FLAG-TRAIL | Enhances antitumor efficiency via FLAG tag that allows crosslinking of TRAIL by using an anti-FLAG antibody | [50] |
| Fusion of the extracellular domain of Flt3L and an isoleucine zipper to the N terminus of TRAIL. GFP fused to C terminus | S-TRAIL-GFP | Enhanced apoptosis via bystander effect and stabilized oligomerization of TRAIL. GFP expression allows visualization of TRAIL expressing cells-enhanced antitumor activity and detection | [49] |
| Addition of an isoleucine zipper | iz-TRAIL | Facilitated oligomerization resulting in improved cytotoxicity-enhanced antitumor activity | [50] |
| scFv–sTRAIL bifunctional fusion proteins | scFv-sTRAIL | Enhanced antitumor activity by increasing specificity and strength of TRAIL response. Permits paracrine signaling | [52,57] |
| Human serum albumin [23] genetically fused to N-terminus of secretable TRAIL | HSA-Flag-TNC-TRAIL | Efficacy increased through increased serum half-life to improve bioavailability | [53] |
| PEGylated TRAIL attached to transferrin | Transferrin-PEG-TRAIL (Tf-PEG10K-TRAIL) | Combined enhancement in tumor targeting/killing properties | [54] |
| PEGylation of TRAIL with PEGa of different molecular weights | PEG-TRAIL | Increased serum half-life and protracted activity in vivo leading to enhanced efficacy and longevity | [55] |
| Nanocomplex system | TRAIL-loaded PLGAb microspheres | Efficacy and longevity increased via sustained TRAIL release and tumor killing properties in vivo | [56] |
| Luciferase fused to the N terminus of sTRAIL | SRL0L2TR | Direct extracellular visualization and monitoring of levels, time of delivery, and localization of stem cell-delivered proteins by bioluminescent imaging enabling detection of tumor cells | [58] |
| Multifunctional nanoparticle comprising doxorubicin (DOX) and pORF–hTRAIL | pORF-hTRAIL | Anti-glioma efficacy in vivo increased | [59] |
| Ultrasound contrast agents chemically conjugated to TRAIL | TRAIL-UCA | Detection enhanced for ultrasound imaging | [60] |
| TRAIL conjugated to ferric oxide nanoparticles | Nanoparticle-TRAIL | Enhances antitumor activity in glioma and glioma stem cells in vitro and in vivo | [61] |
| Adenoviral infection of secretable trimeric TRAIL | Human UCB-MSC | Irradiation enhances U87-MG tumor tropism and therapeutic potential of SCs | [62] |
| Lentiviral infection of secretable TRAIL | Human BM-MSCs | Use of real-time imaging to follow migration and therapeutic effect of MSCs on primary and established human GBM tumor | [63] |
| Non-viral nucleofection of TRAIL | Human A-MSCs | Reduction of tumor volume and significant survival benefit in vivo in rat glioma models | [64] |
| Adenoviral transduction of dodecameric TRAIL | Rat BM-MSCs | Complete elimination of established renal cell carcinoma (RCC) in vivo | [65] |
| Secretable TRAIL | Human MSC/mouse MSC | Stem cells are eliminated after therapeutic effect by addition of the prodrug gancyclovir established GBMs | [66] |
| Secretable TRAIL introduced using nonviral PEI(600)-Cyd | MSCs | Reduction in lung metastasis | [67] |
| Lentiviral infection of secretable TRAIL | Mouse NSCs | PI-103 augments in vivo response of GBM6/8/12 in vitro, Gli36-EGFRVIII in vivo gliomas to TRAIL | [68] |
| Lentiviral infection of secretable TRAIL | Mouse NSCs | Synergism with TRAIL resulting in eradication of Human U87-MG established glioma model tumor in vivo locked nucleic acid (LNA) anti-miR-21 oligonucleotides | [69] |
| LV-TRAIL under tet promoter | Human MSCs | Cleared metastatic disease in lung through conditional expression of TRAIL using DOX | [70] |
| Secretable TRAIL | Human A-MSCs | Decrease in malignant fibrous histiocytoma metastasis | [71] |
| Secretable TRAIL | Mouse NSC/human MSC | Stem cells encapsulated in sECM. Increased retention of stem cells within established and primary GBMs | [72] |
| Inducible TRAIL | Human MSC | Halts breast cancer growth and decreases degree of bone and lung metastasis via stem cells encapsulated in silk scaffold | [73] |
| LV-EGFR-nanobody TRAIL (ENb2-TRAIL) | Mouse NSC | Targets EGFR and TRAIL signaling pathways simultaneously on GBMs | [74] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muthu, M.; Chun, S.; Gopal, J.; Park, G.-S.; Nile, A.; Shin, J.; Shin, J.; Kim, T.-H.; Oh, J.-W. The MUDENG Augmentation: A Genesis in Anti-Cancer Therapy? Int. J. Mol. Sci. 2020, 21, 5583. https://doi.org/10.3390/ijms21155583
Muthu M, Chun S, Gopal J, Park G-S, Nile A, Shin J, Shin J, Kim T-H, Oh J-W. The MUDENG Augmentation: A Genesis in Anti-Cancer Therapy? International Journal of Molecular Sciences. 2020; 21(15):5583. https://doi.org/10.3390/ijms21155583
Chicago/Turabian StyleMuthu, Manikandan, Sechul Chun, Judy Gopal, Gyun-Seok Park, Arti Nile, Jisoo Shin, Juhyun Shin, Tae-Hyoung Kim, and Jae-Wook Oh. 2020. "The MUDENG Augmentation: A Genesis in Anti-Cancer Therapy?" International Journal of Molecular Sciences 21, no. 15: 5583. https://doi.org/10.3390/ijms21155583
APA StyleMuthu, M., Chun, S., Gopal, J., Park, G.-S., Nile, A., Shin, J., Shin, J., Kim, T.-H., & Oh, J.-W. (2020). The MUDENG Augmentation: A Genesis in Anti-Cancer Therapy? International Journal of Molecular Sciences, 21(15), 5583. https://doi.org/10.3390/ijms21155583