A Practical Review of NMR Lineshapes for Spin-1/2 and Quadrupolar Nuclei in Disordered Materials


Abstract
1. Introduction
2. NMR Detection of Spin-1/2 Nuclei in Disordered Environments
2.1. Impacts of Disorder on 1D NMR Spectroscopy
2.1.1. NMR Lineshapes of Spin-1/2 Nuclei
- Structural disorder, chemical shielding;
- CSA (chemical shift anisotropy), chemical shielding;
- Imperfect shimming, external field;
- Bulk magnetic susceptibility (BMS), internal field;
- Anisotropy bulk magnetic susceptibility (ABMS), internal field;
- Dipolar coupling from nearby spins, dipole–dipole interaction;
- J-coupling, through-bond interaction.
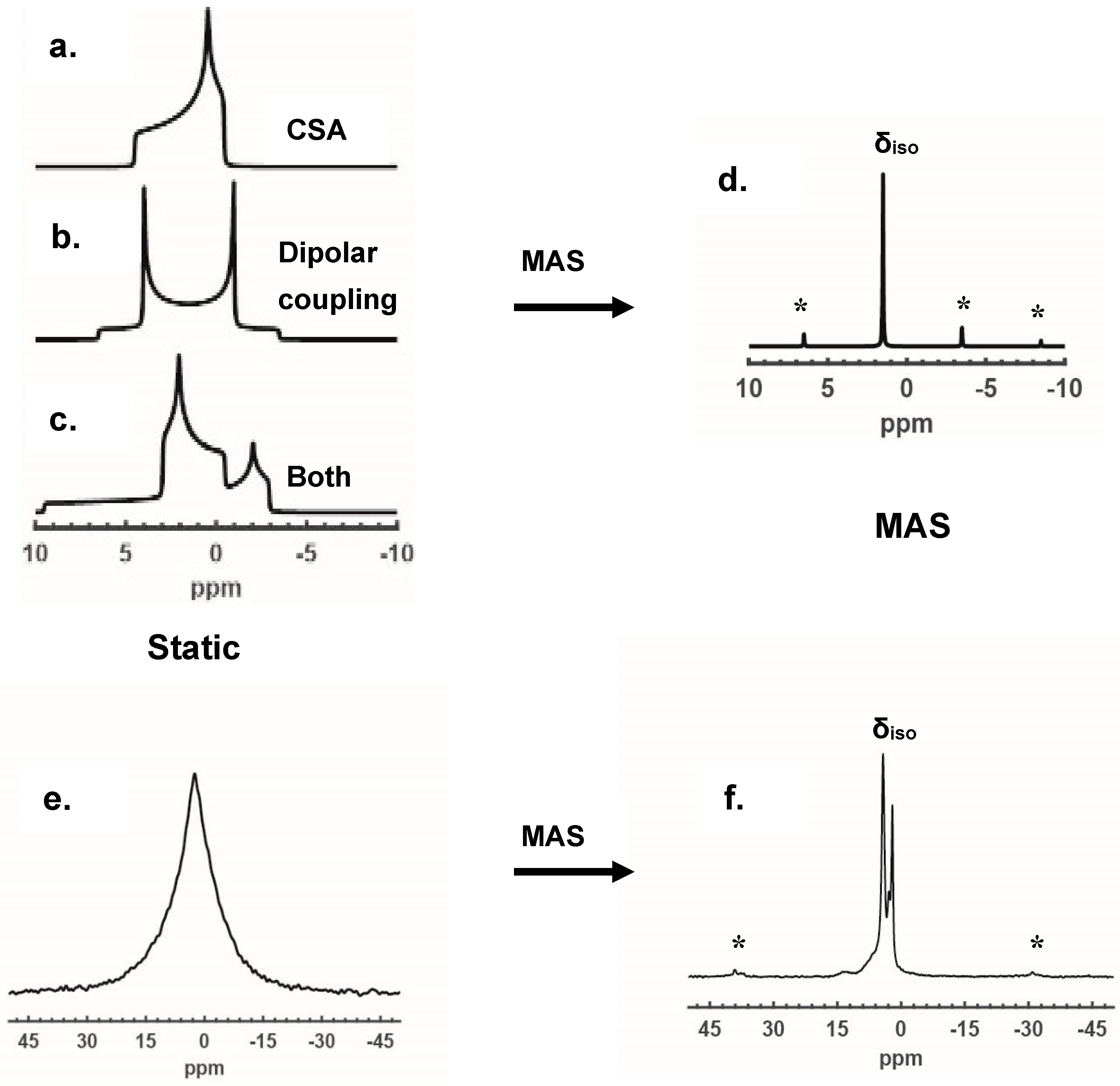
2.1.2. Removal of CSA and Dipolar Coupling by MAS
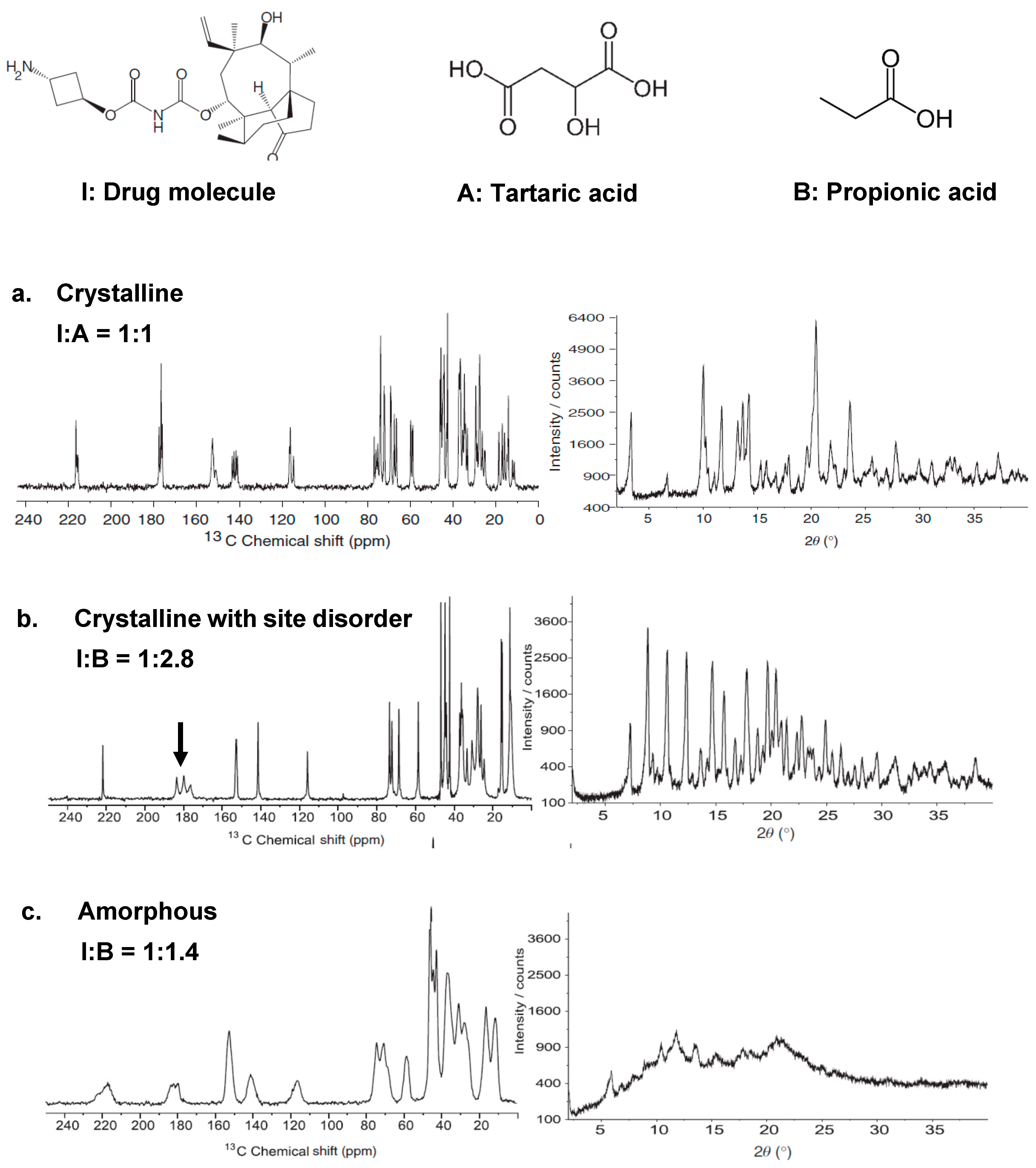
2.1.3. Effect of Disorder on 1D MAS NMR Spectroscopy
2.2. Impacts of Disorder on 2D NMR Spectroscopy
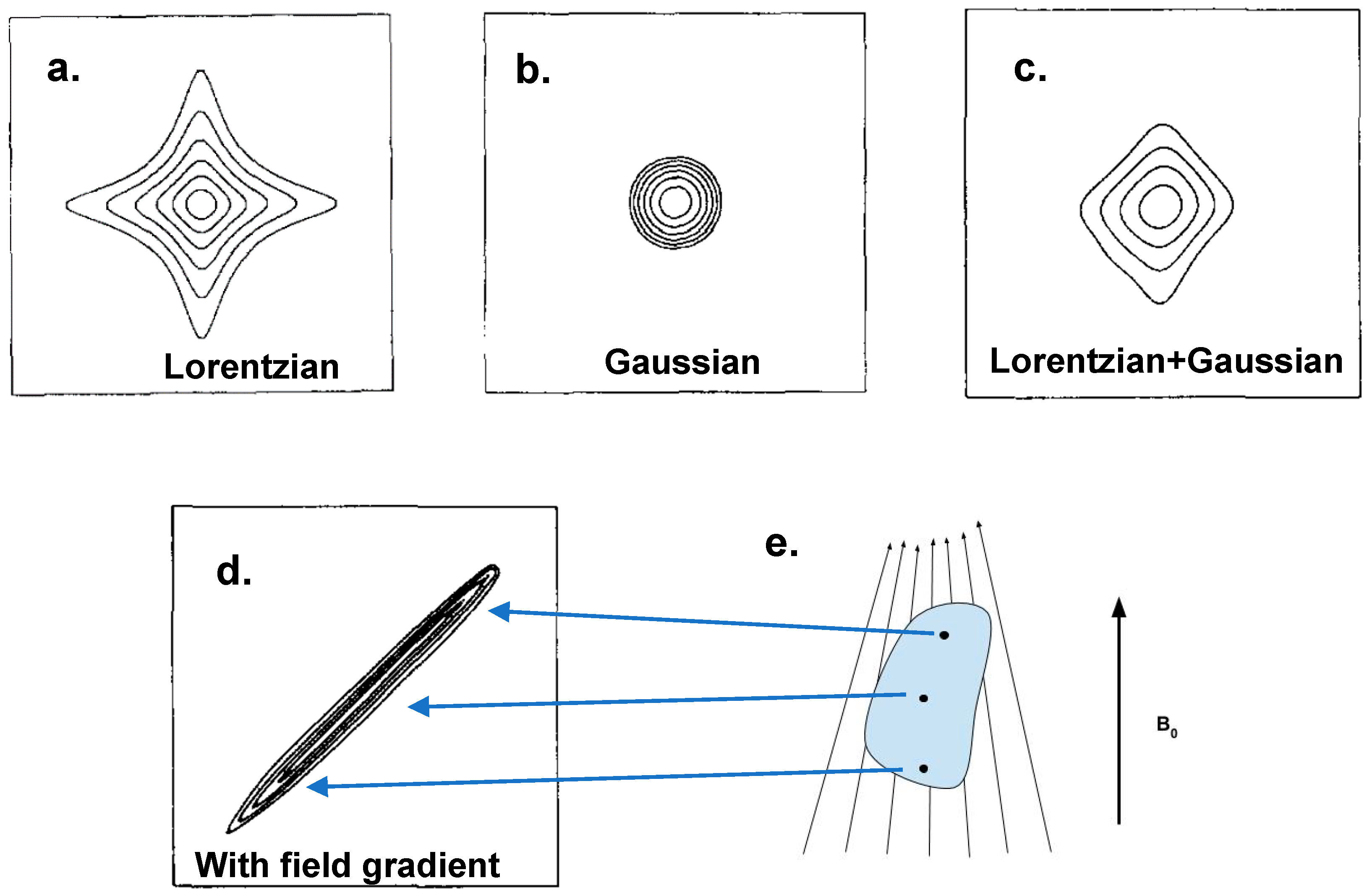
2.2.1. Lineshapes in 2D NMR
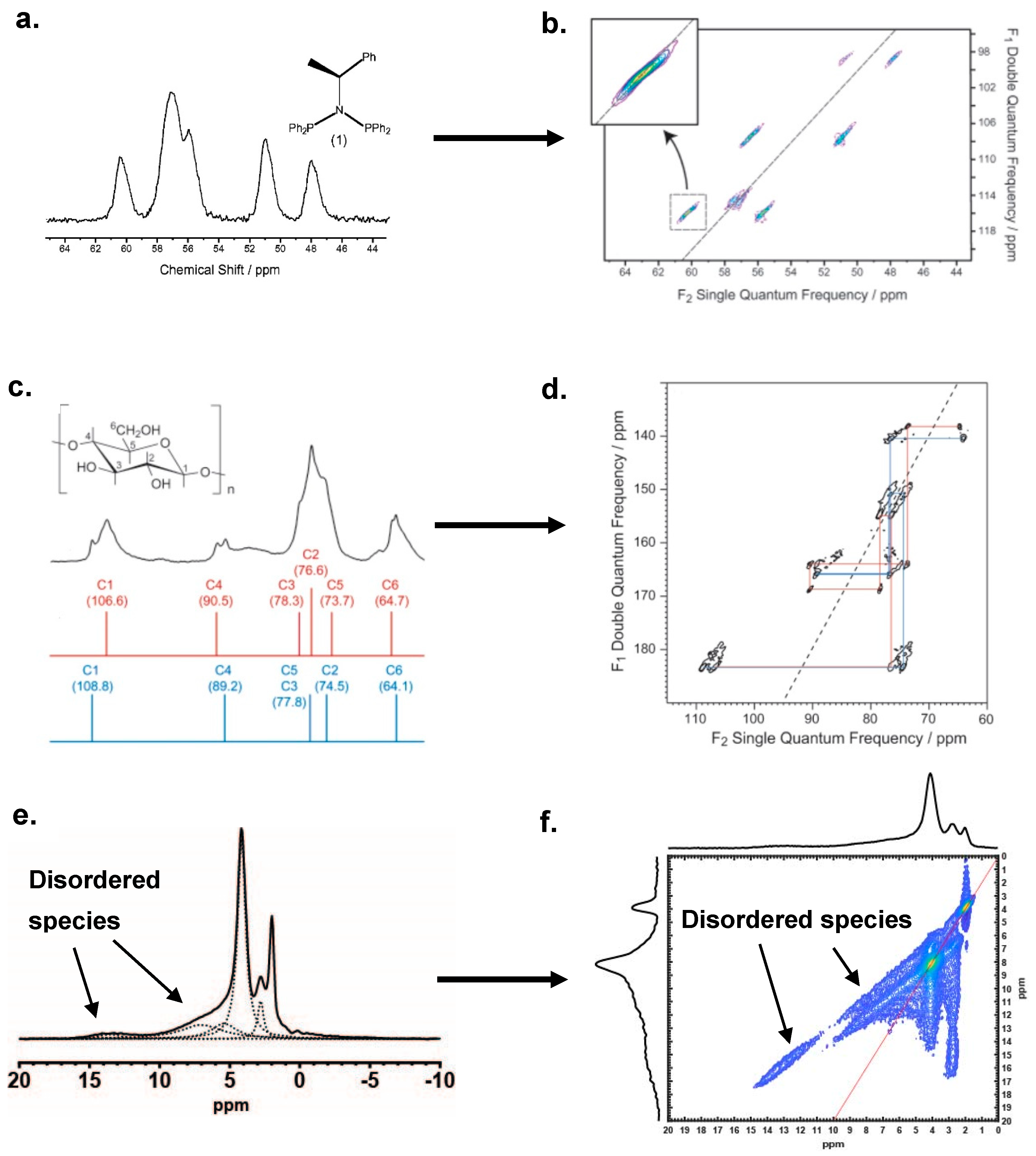
2.2.2. Effect of the Disorder on 2D MAS NMR Spectroscopy
3. NMR Detection of Quadrupolar Nuclei in Disordered Environments
3.1. NMR Spectroscopy of Quadrupolar Nuclei
3.1.1. Quadrupolar Effect
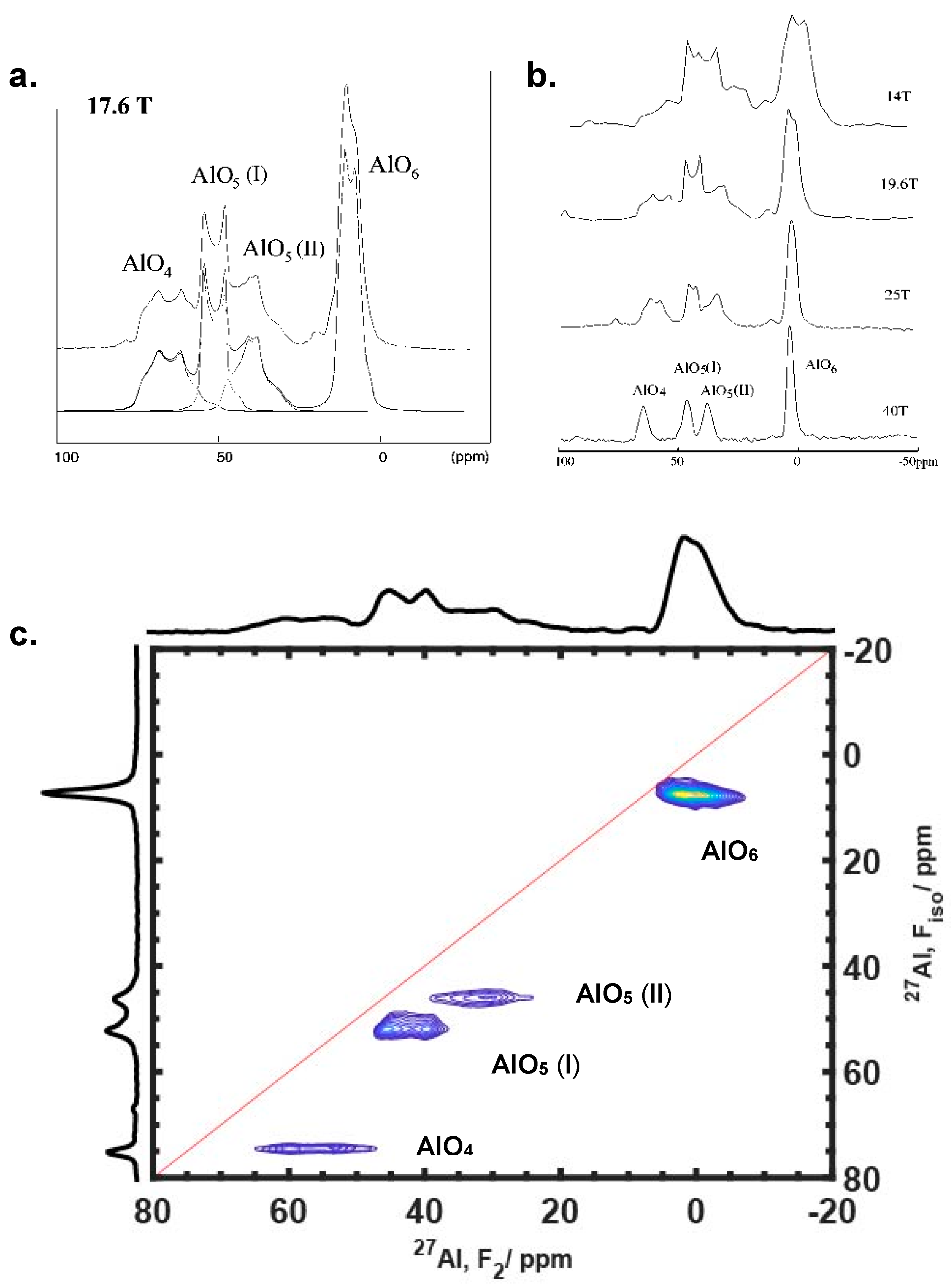
3.1.2. Resolving Quadrupolar Species with Higher Fields and/or MQMAS Method
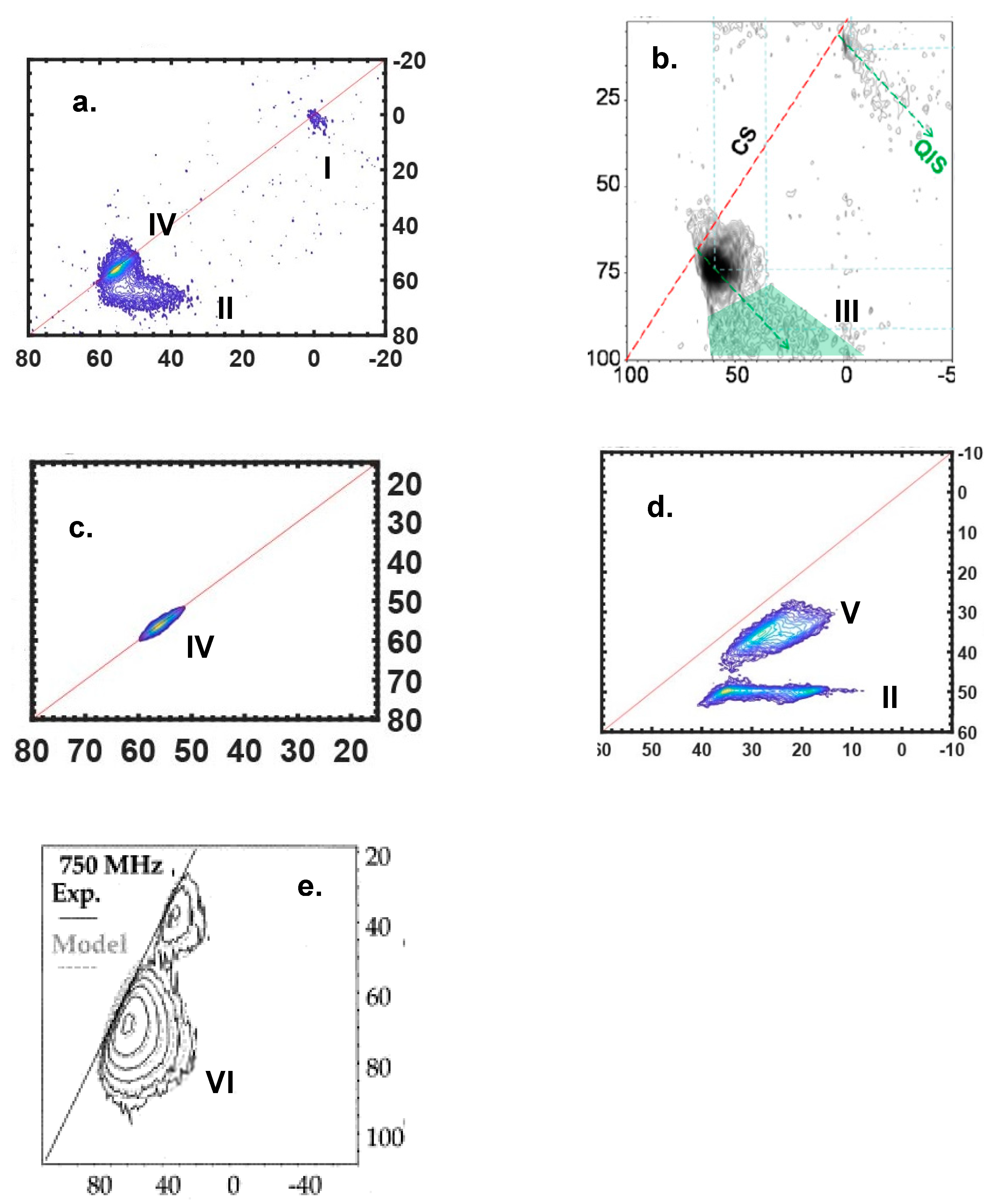
3.2. Disorder Observed by MQMAS Spectroscopy
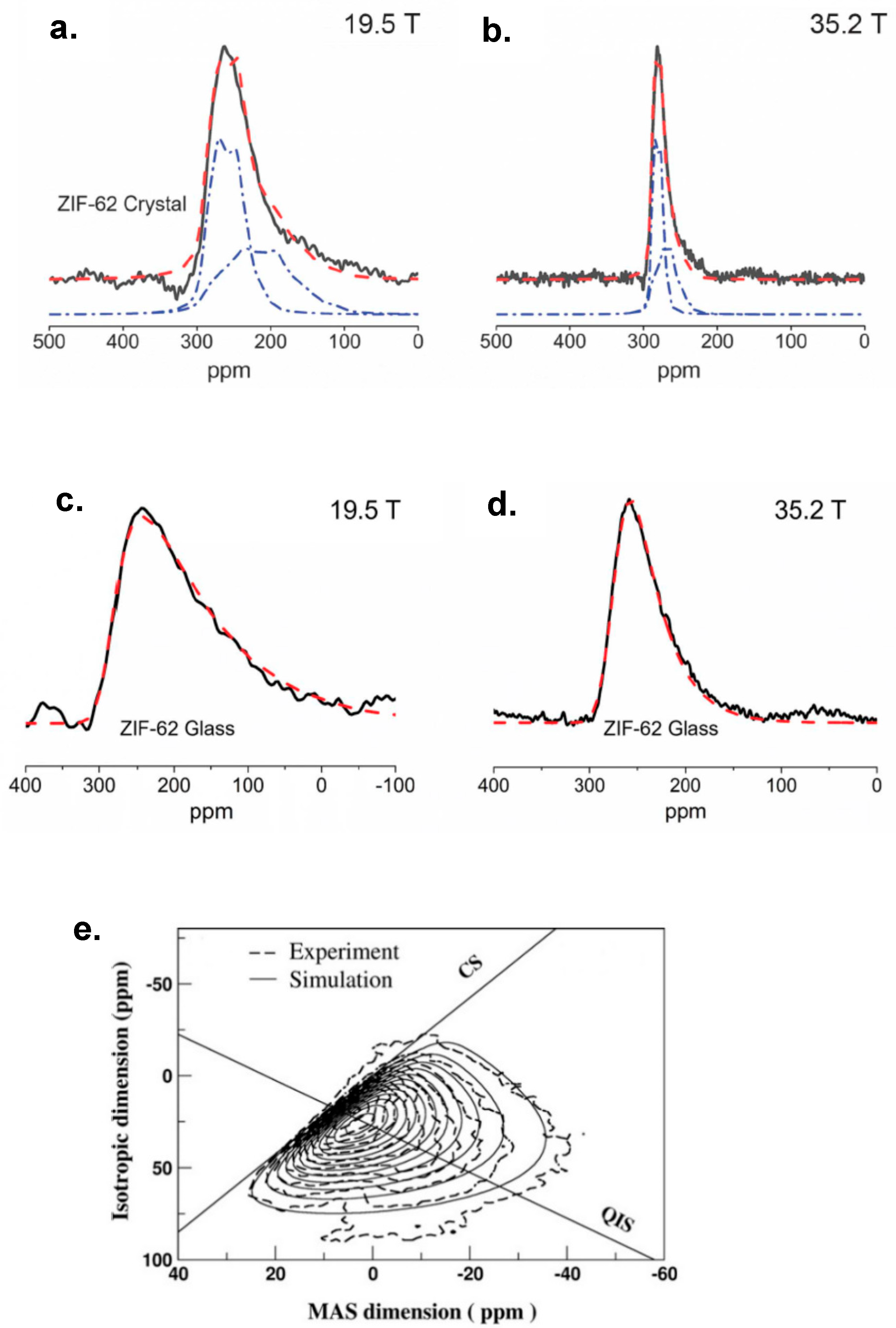
3.3. Fitting Disordered Lineshapes with the Czjzek Model
3.4. Practical Problems and the Strategy for Disorder Characterizations
3.4.1. Weak Sensitivity and Broad-Line Problems
3.4.2. General Strategy for Disorder Characterization
4. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Madsen, R.S.K.; Qiao, A.; Sen, J.; Hung, I.; Chen, K.; Gan, Z.; Sen, S.; Yue, Y. Ultrahigh-field 67Zn NMR reveals short-range disorder in zeolitic imidazolate framework glasses. Science 2020, 367, 1473–1476. [Google Scholar] [CrossRef]
- Neuville, D.R.; Cormier, L.; Massiot, D. Al environment in tectosilicate and peraluminous glasses: A 27Al MQ-MAS NMR, Raman, and XANES investigation. Geochim. Cosmochim. Acta 2004, 68, 5071–5079. [Google Scholar] [CrossRef]
- White, J.L.; Lohse, D.J. Spin diffusion analysis of miscibility in an anomalous nonpolar blend. Macromolecules 1999, 32, 958–960. [Google Scholar] [CrossRef]
- White, J.L.; Dias, A.J.; Ashbaugh, J.R. Detailed analysis of differential dynamics and morphology in polyisobutylene and poly(isobutylene-co-p-methylstyrene) by solid state NMR. Macromolecules 1998, 31, 1880–1888. [Google Scholar] [CrossRef]
- Pecher, O.; Carretero-González, J.; Griffith, K.J.; Grey, C.P. Materials’ methods: NMR in battery research. Chem. Mater. 2017, 29, 213–242. [Google Scholar] [CrossRef]
- Karasulu, B.; Emge, S.P.; Groh, M.F.; Grey, C.P.; Morris, A.J. Al/Ga-doped Li7La3Zr2O12 garnets as li-ion solid-state battery electrolytes: Atomistic insights into local coordination environments and their influence on 17O, 27Al, and 71Ga NMR spectra. J. Am. Chem. Soc. 2020, 142, 3132–3148. [Google Scholar] [CrossRef]
- Wang, Z.; Jiang, Y.; Lafon, O.; Trébosc, J.; Duk Kim, K.; Stampfl, C.; Baiker, A.; Amoureux, J.-P.; Huang, J. Brønsted acid sites based on penta-coordinated aluminum species. Nat. Commun. 2016, 7, 13820. [Google Scholar] [CrossRef]
- Chen, K.; Horstmeier, S.; Nguyen, V.T.; Wang, B.; Crossley, S.P.; Pham, T.; Gan, Z.; Hung, I.; White, J.L. Structure and catalytic characterization of a second framework Al(IV) site in zeolite catalysts revealed by NMR at 35.2 T. J. Am. Chem. Soc. 2020, 142, 7514–7523. [Google Scholar] [CrossRef]
- Cheetham, A.K.; Bennett, T.D.; Coudert, F.-X.; Goodwin, A.L. Defects and disorder in metal organic frameworks. Dalton Trans. 2016, 45, 4113–4126. [Google Scholar] [CrossRef]
- Lucier, B.E.G.; Chen, S.; Huang, Y. Characterization of metal–organic frameworks: Unlocking the potential of solid-state NMR. Acc. Chem. Res. 2018, 51, 319–330. [Google Scholar] [CrossRef]
- Moran, R.F.; Dawson, D.M.; Ashbrook, S.E. Exploiting NMR spectroscopy for the study of disorder in solids. Int. Rev. Phys. Chem. 2017, 36, 39–115. [Google Scholar] [CrossRef]
- Clawson, J.S.; Kennedy-Gabb, S.; Lee, A.Y.; Copley, R.C.B. One-phase crystal disorder in pharmaceutical solids and its implication for solid-state stability. J. Pharm. Sci. 2011, 100, 4302–4316. [Google Scholar] [CrossRef] [PubMed]
- Sakellariou, D.; Brown, S.P.; Lesage, A.; Hediger, S.; Bardet, M.; Meriles, C.A.; Pines, A.; Emsley, L. High-resolution NMR correlation spectra of disordered solids. J. Am. Chem. Soc. 2003, 125, 4376–4380. [Google Scholar] [CrossRef] [PubMed]
- Paudel, A.; Geppi, M.; Van den Mooter, G. Structural and dynamic properties of amorphous solid dispersions: The role of solid-state nuclear magnetic resonance spectroscopy and relaxometry. J. Pharm. Sci. 2014, 103, 2635–2662. [Google Scholar] [CrossRef]
- Samoson, A.; Lippmaa, E.; Pines, A. High resolution solid-state N.M.R. Mol. Phys. 1988, 65, 1013–1018. [Google Scholar] [CrossRef]
- Llor, A.; Virlet, J. Towards high-resolution NMR of more nuclei in solids: Sample spinning with time-dependent spinner axis angle. Chem. Phys. Lett. 1988, 152, 248–253. [Google Scholar] [CrossRef]
- Medek, A.; Harwood, J.S.; Frydman, L. Multiple-quantum magic-angle spinning NMR: A new method for the study of quadrupolar nuclei in solids. J. Am. Chem. Soc. 1995, 117, 12779–12787. [Google Scholar] [CrossRef]
- Frydman, L.; Harwood, J.S. Isotropic spectra of half-integer quadrupolar spins from bidimensional magic-angle spinning NMR. J. Am. Chem. Soc. 1995, 117, 5367–5368. [Google Scholar] [CrossRef]
- Gan, Z. Satellite transition magic-angle spinning nuclear magnetic resonance spectroscopy of half-integer quadrupolar nuclei. J. Chem. Phys. 2001, 114, 10845–10853. [Google Scholar] [CrossRef]
- Gan, Z. Isotropic NMR spectra of half-integer quadrupolar nuclei using satellite transitions and magic-angle spinning. J. Am. Chem. Soc. 2000, 122, 3242–3243. [Google Scholar] [CrossRef]
- Amoureux, J.-P.; Huguenard, C.; Engelke, F.; Taulelle, F. Unified representation of MQMAS and STMAS NMR of half-integer quadrupolar nuclei. Chem. Phys. Lett. 2002, 356, 497–504. [Google Scholar] [CrossRef]
- Gan, Z.; Gor’kov, P.; Cross, T.A.; Samoson, A.; Massiot, D. Seeking higher resolution and sensitivity for NMR of quadrupolar nuclei at ultrahigh magnetic fields. J. Am. Chem. Soc. 2002, 124, 5634–5635. [Google Scholar] [CrossRef] [PubMed]
- Gan, Z.; Hung, I.; Wang, X.; Paulino, J.; Wu, G.; Litvak, I.M.; Gor’kov, P.L.; Brey, W.W.; Lendi, P.; Schiano, J.L.; et al. NMR spectroscopy up to 35.2T using a series-connected hybrid magnet. J. Magn. Reson. 2017, 284, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Hung, I.; Gan, Z. On the practical aspects of recording wideline QCPMG NMR spectra. J. Magn. Reson. 2010, 204, 256–265. [Google Scholar] [CrossRef]
- Larsen, F.H.; Jakobsen, H.J.; Ellis, P.D.; Nielsen, N.C. Sensitivity-enhanced quadrupolar-echo nmr of half-integer quadrupolar nuclei. magnitudes and relative orientation of chemical shielding and quadrupolar coupling tensors. J. Phys. Chem. A 1997, 101, 8597–8606. [Google Scholar] [CrossRef]
- Perras, F.A.; Viger-Gravel, J.; Burgess, K.M.N.; Bryce, D.L. Signal enhancement in solid-state NMR of quadrupolar nuclei. Solid State Nucl. Magn. Reson. 2013, 51–52, 1–15. [Google Scholar] [CrossRef]
- Siegel, R.; Nakashima, T.T.; Wasylishen, R.E. Sensitivity enhancement of NMR spectra of half-integer quadrupolar nuclei in the solid state via population transfer. Concepts Magn. Reson. Part A 2005, 26, 47–61. [Google Scholar] [CrossRef]
- Hung, I.; Gan, Z. A magic-angle turning NMR experiment for separating spinning sidebands of half-integer quadrupolar nuclei. Chem. Phys. Lett. 2010, 496, 162–166. [Google Scholar] [CrossRef]
- Massiot, D.; Montouillout, V.; Fayon, F.; Florian, P.; Bessada, C. Order-resolved sideband separation in magic angle spinning NMR of half integer quadrupolar nuclei. Chem. Phys. Lett. 1997, 272, 295–300. [Google Scholar] [CrossRef]
- Hung, I.; Edwards, T.; Sen, S.; Gan, Z. MATPASS/CPMG: A sensitivity enhanced magic-angle spinning sideband separation experiment for disordered solids. J. Magn. Reson. 2012, 221, 103–109. [Google Scholar] [CrossRef]
- Han, O.H.; Kim, C.-S.; Hong, S.B. Direct evidence for the nonrandom nature of Al substitution in zeolite ZSM-5: An investigation by 27Al MAS and MQ MAS NMR. Angew. Chem. Int. Ed. 2002, 41, 469–472. [Google Scholar] [CrossRef]
- Holzinger, J.; Beato, P.; Lundegaard, L.F.; Skibsted, J. Distribution of Aluminum over the Tetrahedral Sites in ZSM-5 Zeolites and their evolution after steam treatment. J. Phys. Chem. C 2018, 122, 15595–15613. [Google Scholar] [CrossRef]
- Huo, H.; Peng, L.; Gan, Z.; Grey, C.P. Solid-state MAS NMR studies of brønsted acid sites in zeolite H-mordenite. J. Am. Chem. Soc. 2012, 134, 9708–9720. [Google Scholar] [CrossRef] [PubMed]
- Pugh, S.M.; Wright, P.A.; Law, D.J.; Thompson, N.; Ashbrook, S.E. Facile, room-temperature 17O enrichment of zeolite frameworks revealed by solid-state NMR spectroscopy. J. Am. Chem. Soc. 2020, 142, 900–906. [Google Scholar] [CrossRef]
- Zhao, R.; Zhao, Z.; Li, S.; Zhang, W. Insights into the correlation of aluminum distribution and brönsted acidity in H-beta zeolites from solid-state NMR spectroscopy and DFT calculations. J. Phys. Chem. Lett. 2017, 8, 2323–2327. [Google Scholar] [CrossRef] [PubMed]
- Holzinger, J.; Nielsen, M.; Beato, P.; Brogaard, R.Y.; Buono, C.; Dyballa, M.; Falsig, H.; Skibsted, J.; Svelle, S. Identification of distinct framework aluminum sites in zeolite ZSM-23: A combined computational and experimental 27Al NMR study. J. Phys. Chem. C 2019, 123, 7831–7844. [Google Scholar] [CrossRef]
- Chizallet, C.; Raybaud, P. Density functional theory simulations of complex catalytic materials in reactive environments: Beyond the ideal surface at low coverage. Catal. Sci. Technol. 2014, 4, 2797–2813. [Google Scholar] [CrossRef]
- Levitt, M.H. Spin Dynamics: Basics of Nuclear Magnetic Resonance; Wiley: Hoboken, NJ, USA, 2008; ISBN 978-0-470-51117-6. [Google Scholar]
- Petrakis, L. Spectral line shapes: Gaussian and Lorentzian functions in magnetic resonance. J. Chem. Educ. 1967, 44, 432. [Google Scholar] [CrossRef]
- Maricq, M.M.; Waugh, J.S. NMR in rotating solids. J. Chem. Phys. 1979, 70, 3300–3316. [Google Scholar] [CrossRef]
- Samoson, A.; Tuherm, T.; Gan, Z. High-field high-speed MAS resolution enhancement in 1H NMR spectroscopy of solids. Solid State Nucl. Magn. Reson. 2001, 20, 130–136. [Google Scholar] [CrossRef]
- Laws, D.D.; Bitter, H.-M.L.; Jerschow, A. Solid-state NMR spectroscopic methods in chemistry. Angew. Chem. Int. Ed. 2002, 41, 3096–3129. [Google Scholar] [CrossRef]
- Hodgkinson, P. Heteronuclear decoupling in the NMR of solids. Prog. Nucl. Magn. Reson. Spectrosc. 2005, 46, 197–222. [Google Scholar] [CrossRef]
- Ashbrook, S.E.; Griffin, J.M.; Johnston, K.E. Recent advances in solid-state nuclear magnetic resonance spectroscopy. Annu. Rev. Anal. Chem. 2018, 11, 485–508. [Google Scholar] [CrossRef]
- Madhu, P.K. High-resolution solid-state NMR spectroscopy of protons with homonuclear dipolar decoupling schemes under magic-angle spinning. Solid State Nucl. Magn. Reson. 2009, 35, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Vanderhart, D.L.; Earl, W.L.; Garroway, A.N. Resolution in 13C NMR of organic solids using high-power proton decoupling and magic-angle sample spinning. J. Magn. Reson. (1969) 1981, 44, 361–401. [Google Scholar] [CrossRef]
- Baum, J.; Munowitz, M.; Garroway, A.N.; Pines, A. Multiple-quantum dynamics in solid state NMR. J. Chem. Phys. 1985, 83, 2015–2025. [Google Scholar] [CrossRef]
- Hanrahan, M.P.; Venkatesh, A.; Carnahan, S.L.; Calahan, J.L.; Lubach, J.W.; Munson, E.J.; Rossini, A.J. Enhancing the resolution of 1H and 13C solid-state NMR spectra by reduction of anisotropic bulk magnetic susceptibility broadening. Phys. Chem. Chem. Phys. 2017, 19, 28153–28162. [Google Scholar] [CrossRef]
- Cadars, S.; Mifsud, N.; Lesage, A.; Epping, J.D.; Hedin, N.; Chmelka, B.F.; Emsley, L. Dynamics and disorder in surfactant-templated silicate layers studied by solid-state NMR dephasing times and correlated line shapes. J. Phys. Chem. C 2008, 112, 9145–9154. [Google Scholar] [CrossRef]
- Brown, S.P.; Wimperis, S. Inhomogeneous broadening of two-dimensional NMR lineshapes. Chem. Phys. Lett. 1995, 237, 509–515. [Google Scholar] [CrossRef]
- Cadars, S.; Lesage, A.; Emsley, L. Chemical shift correlations in disordered solids. J. Am. Chem. Soc. 2005, 127, 4466–4476. [Google Scholar] [CrossRef]
- Kervern, G.; Pintacuda, G.; Zhang, Y.; Oldfield, E.; Roukoss, C.; Kuntz, E.; Herdtweck, E.; Basset, J.-M.; Cadars, S.; Lesage, A.; et al. Solid-state NMR of a paramagnetic DIAD-FeII catalyst: Sensitivity, resolution enhancement, and structure-based assignments. J. Am. Chem. Soc. 2006, 128, 13545–13552. [Google Scholar] [CrossRef] [PubMed]
- Wasylishen, R.E.; Ashbrook, S.E.; Wimperis, S. NMR of Quadrupolar Nuclei in Solid Materials; Wiley: Hoboken, NJ, USA, 2012; ISBN 978-0-470-97398-1. [Google Scholar]
- O’Dell, L.A.; Schurko, R.W. QCPMG using adiabatic pulses for faster acquisition of ultra-wideline NMR spectra. Chem. Phys. Lett. 2008, 464, 97–102. [Google Scholar] [CrossRef]
- Massiot, D.; Fayon, F.; Capron, M.; King, I.; Le Calvé, S.; Alonso, B.; Durand, J.-O.; Bujoli, B.; Gan, Z.; Hoatson, G. Modelling one- and two-dimensional solid-state NMR spectra. Magn. Reson. Chem. 2002, 40, 70–76. [Google Scholar] [CrossRef]
- Ashbrook, S.E.; Sneddon, S. New methods and applications in solid-state NMR spectroscopy of quadrupolar nuclei. J. Am. Chem. Soc. 2014, 136, 15440–15456. [Google Scholar] [CrossRef]
- Hung, I.; Trébosc, J.; Hoatson, G.L.; Vold, R.L.; Amoureux, J.-P.; Gan, Z. Q-shear transformation for MQMAS and STMAS NMR spectra. J. Magn. Reson. 2009, 201, 81–86. [Google Scholar] [CrossRef]
- Brown, S.P.; Heyes, S.J.; Wimperis, S. Two-dimensional MAS multiple-quantum NMR of quadrupolar nuclei. removal of inhomogeneous second-order broadening. J. Magn. Reson. Ser. A 1996, 119, 280–284. [Google Scholar] [CrossRef]
- Malicki, N.; Mali, G.; Quoineaud, A.-A.; Bourges, P.; Simon, L.J.; Thibault-Starzyk, F.; Fernandez, C. Aluminium triplets in dealuminated zeolites detected by 27Al NMR correlation spectroscopy. Microporous Mesoporous Mater. 2010, 129, 100–105. [Google Scholar] [CrossRef]
- Amoureux, J.-P.; Fernandez, C.; Steuernagel, S. Z-Filtering in MQMAS NMR. J. Magn. Reson. Ser. A 1996, 123, 116–118. [Google Scholar] [CrossRef]
- Amoureux, J.; Pruski, M. MQMAS NMR: Experimental strategies and applications. In EMagRes; John Wiley & Sons Ltd., Ed.; Wiley: Hoboken, NJ, USA, 2008. [Google Scholar] [CrossRef]
- Czjzek, G.; Fink, J.; Götz, F.; Schmidt, H.; Coey, J.M.D.; Rebouillat, J.P.; Liénard, A. Atomic coordination and the distribution of electric field gradients in amorphous solids. Phys. Rev. B 1981, 23, 2513–2530. [Google Scholar] [CrossRef]
- Caër, G.L.; Brand, R.A. General models for the distributions of electric field gradients in disordered solids. J. Phys. Condens. Matter 1998, 10, 10715–10774. [Google Scholar] [CrossRef]
- D’Espinose de Lacaillerie, J.-B.; Fretigny, C.; Massiot, D. MAS NMR spectra of quadrupolar nuclei in disordered solids: The Czjzek model. J. Magn. Reson. 2008, 192, 244–251. [Google Scholar]
- Van Meerten, S.G.J.; Franssen, W.M.J.; Kentgens, A.P.M. ssNake: A cross-platform open-source NMR data processing and fitting application. J. Magn. Reson. 2019, 301, 56–66. [Google Scholar] [CrossRef]
- Angeli, F.; Villain, O.; Schuller, S.; Ispas, S.; Charpentier, T. Insight into sodium silicate glass structural organization by multinuclear NMR combined with first-principles calculations. Geochim. Cosmochim. Acta 2011, 75, 2453–2469. [Google Scholar] [CrossRef]
- Colaux, H.; Dawson, D.M.; Ashbrook, S.E. Efficient amplitude-modulated pulses for triple- to single-quantum coherence conversion in MQMAS NMR. J. Phys. Chem. A 2014, 118, 6018–6025. [Google Scholar] [CrossRef] [PubMed]
- Massiot, D. Sensitivity and lineshape improvements of MQ-MAS by rotor-synchronized data acquisition. J. Magn. Reson. Ser. A 1996, 122, 240–244. [Google Scholar] [CrossRef]
- Gan, Z.; Gor’kov, P.L.; Brey, W.W.; Sideris, P.J.; Grey, C.P. Enhancing MQMAS of low-γ nuclei by using a high B1 field balanced probe circuit. J. Magn. Reson. 2009, 200, 2–5. [Google Scholar] [CrossRef] [PubMed]
- Sefzik, T.H.; Houseknecht, J.B.; Clark, T.M.; Prasad, S.; Lowary, T.L.; Gan, Z.; Grandinetti, P.J. Solid-state 17O NMR in carbohydrates. Chem. Phys. Lett. 2007, 434, 312–315. [Google Scholar] [CrossRef]
- Dey, K.K.; Prasad, S.; Ash, J.T.; Deschamps, M.; Grandinetti, P.J. Spectral editing in solid-state MAS NMR of quadrupolar nuclei using selective satellite inversion. J. Magn. Reson. 2007, 185, 326–330. [Google Scholar] [CrossRef]
- Kentgens, A.P.M.; Verhagen, R. Advantages of double frequency sweeps in static, MAS and MQMAS NMR of spin I = 3/2 nuclei. Chem. Phys. Lett. 1999, 300, 435–443. [Google Scholar] [CrossRef]
- Meiboom, S.; Gill, D. Modified spin-echo method for measuring nuclear relaxation times. Rev. Sci. Instrum. 1958, 29, 688–691. [Google Scholar] [CrossRef]
- Carr, H.Y.; Purcell, E.M. Effects of Diffusion on free precession in nuclear magnetic resonance experiments. Phys. Rev. 1954, 94, 630–638. [Google Scholar] [CrossRef]
- Vosegaard, T.; Larsen, F.H.; Jakobsen, H.J.; Ellis, P.D.; Nielsen, N.C. Sensitivity-enhanced multiple-quantum MAS NMR of half-integer quadrupolar nuclei. J. Am. Chem. Soc. 1997, 119, 9055–9056. [Google Scholar] [CrossRef]
- Gan, Z. Perspectives on high-field and solid-state NMR methods of quadrupole nuclei. J. Magn. Reson. 2019, 306, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Li, S.; Wang, Q.; Zheng, A.; Jun, X.; Chen, L.; Deng, F. Brønsted/lewis acid synergy in H–ZSM-5 and H–MOR zeolites studied by 1H and 27Al dq-mas solid-state NMR spectroscopy. J. Phys. Chem. C 2011, 115, 22320–22327. [Google Scholar] [CrossRef]
- Chen, Y.; Smock, S.R.; Flintgruber, A.H.; Perras, F.A.; Brutchey, R.L.; Rossini, A.J. Surface termination of CsPbBr3 perovskite quantum dots determined by solid-state NMR spectroscopy. J. Am. Chem. Soc. 2020, 142, 6117–6127. [Google Scholar] [CrossRef] [PubMed]
- Love, A.M.; Thomas, B.; Specht, S.E.; Hanrahan, M.P.; Venegas, J.M.; Burt, S.P.; Grant, J.T. Cen transformation of boron nitride catalysts under oxidative dehydrogenation conditions. J. Am. Chem. Soc. 2019, 141, 182–190. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Scenarios | δiso | CQ | Features of Lineshape | |
|---|---|---|---|---|
| Distribution | Magnitude | Distribution | ||
| I | N (Non-prominent) | N | N | Round |
| II | N | P | N | Horizontal ridge |
| III | N | P | P | Triangle |
| IV | P (Prominent) | P | N | Ridge along diagonal |
| V | P | P | N | Wide ridge parallel to diagonal |
| VI | P | P | P | Need fitting for analysis |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, K. A Practical Review of NMR Lineshapes for Spin-1/2 and Quadrupolar Nuclei in Disordered Materials. Int. J. Mol. Sci. 2020, 21, 5666. https://doi.org/10.3390/ijms21165666
Chen K. A Practical Review of NMR Lineshapes for Spin-1/2 and Quadrupolar Nuclei in Disordered Materials. International Journal of Molecular Sciences. 2020; 21(16):5666. https://doi.org/10.3390/ijms21165666
Chicago/Turabian StyleChen, Kuizhi. 2020. "A Practical Review of NMR Lineshapes for Spin-1/2 and Quadrupolar Nuclei in Disordered Materials" International Journal of Molecular Sciences 21, no. 16: 5666. https://doi.org/10.3390/ijms21165666
APA StyleChen, K. (2020). A Practical Review of NMR Lineshapes for Spin-1/2 and Quadrupolar Nuclei in Disordered Materials. International Journal of Molecular Sciences, 21(16), 5666. https://doi.org/10.3390/ijms21165666