Current Understanding of the Emerging Role of Prolidase in Cellular Metabolism


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
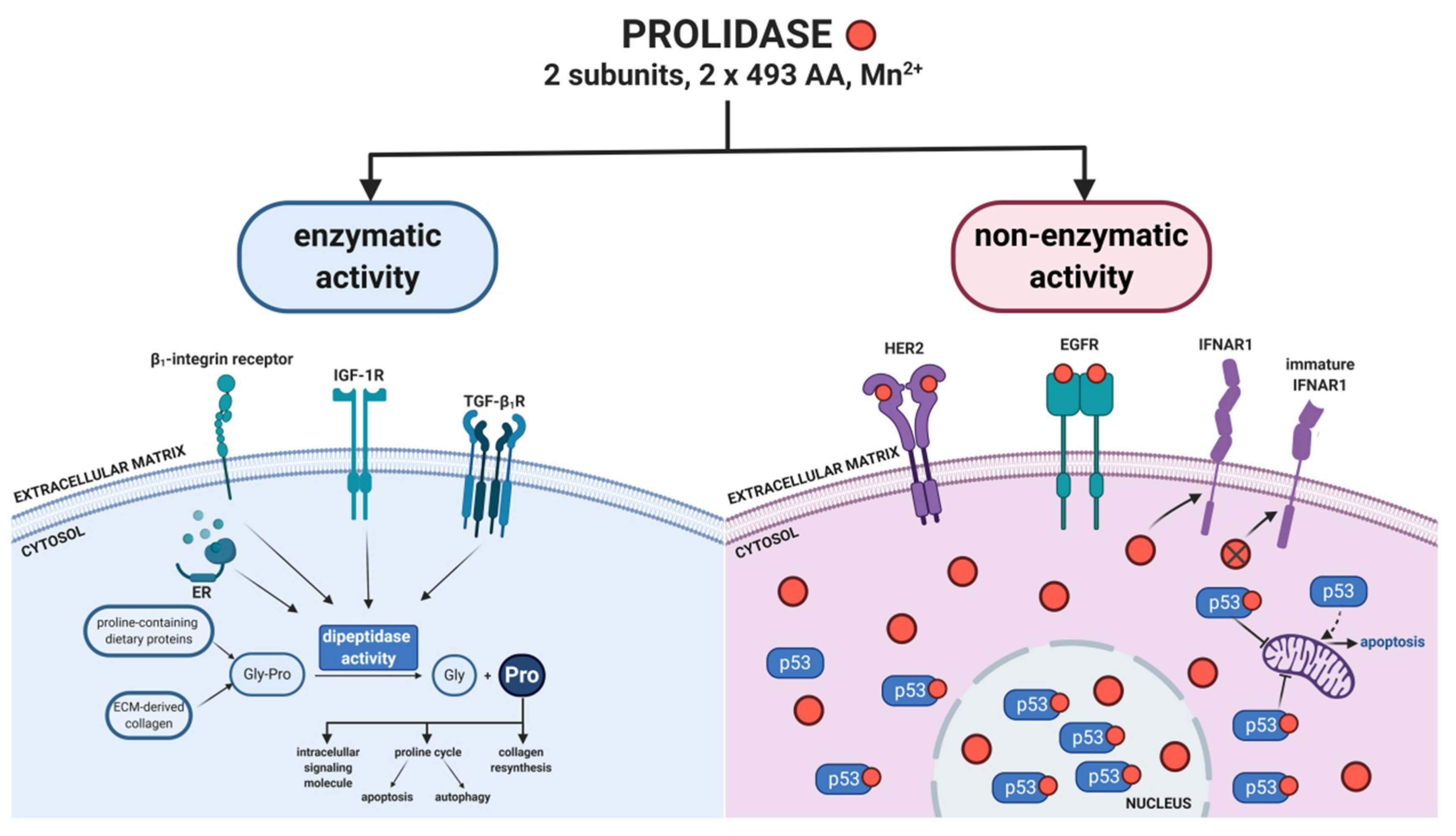
2. Regulatory Functions of Prolidase
2.1. Prolidase as an Epidermal Growth Factor Receptor (ErbB1/EGFR) Ligand
2.2. Prolidase as an ErbB2/HER2 Ligand
2.3. Prolidase as a p53 Activity Regulator
2.4. Prolidase as a Regulator of Interferon α/β Receptor
3. Enzyme-Dependent Activity of Prolidase
3.1. Prolidase as a Dipeptidase: General Structure, Physical Properties, and Substrate Specificity
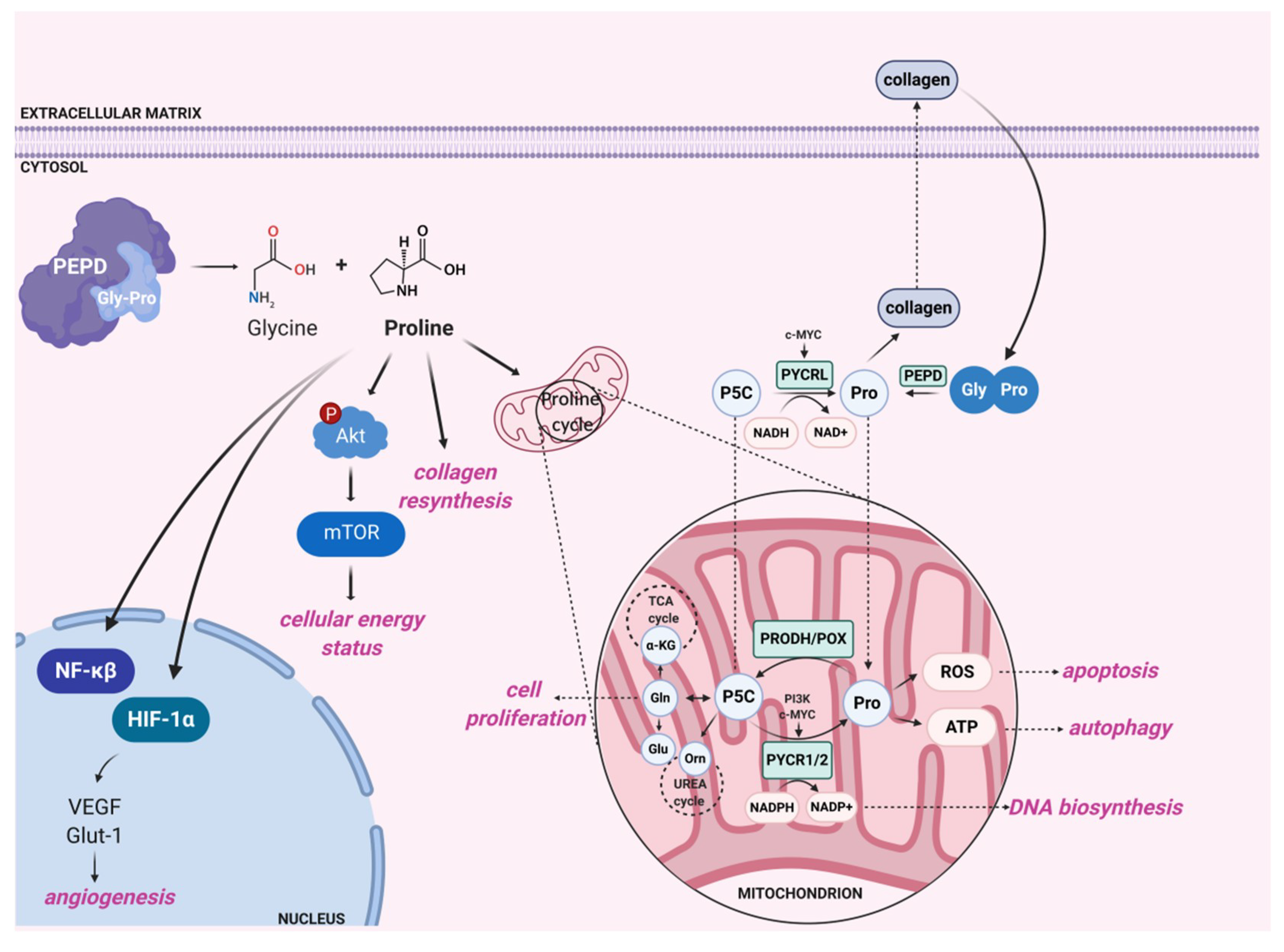
3.2. Biological Significance of Prolidase as a Dipeptidase
3.3. Clinical Significance of Prolidase as a Dipeptidase
4. Concluding Remarks and Future Perspectives
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Akt | Protein kinase B |
| Ala-Pro | Alanyl-proline |
| ATM | Mutated in ataxia telangiectasia |
| ATR | ATM and RAD3-related |
| BAX | Bcl-2-associated X |
| BCL-2 | B-cell lymphoma 2 |
| cGMP | Cyclic guanosine monophosphate |
| CHK1 | Checkpoint kinase 1 |
| CHK2 | Checkpoint kinase 2 |
| EGF | Epidermal growth factor |
| EP | Enoxaparin |
| ErbB1/EGFR | Epidermal growth factor receptor |
| ErbB2/HER2 | Epidermal growth factor receptor 2 |
| ErbB3 | Epidermal growth factor receptor 3 |
| ECM | Extracellular matrix |
| FAK | Focal adhesion kinase |
| Glut-1 | Focal adhesion kinase |
| Gly | Glycine |
| Gly-Pro | Glycyl-proline |
| Grb2 | Growth factor receptor-bound protein 2 |
| HB-EGF | Heparin-binding EGF-like growth factor |
| HIF-1α | Hypoxia-inducible factor 1 alpha |
| Hyp | Hydroxyproline |
| IFNAR1 | Interferon alpha/beta receptor 1 |
| IGF-1R | Insulin-like growth factor 1 receptor |
| iNOS | Inducible nitric oxide synthase |
| IκBα | Nuclear factor kappa alpha |
| JAK | Janus kinase |
| Kd | Dissociation constant |
| Leu-Pro | Leucyl-proline |
| MAPK/ERK | MAP kinase/Extracellular signal-regulated kinase |
| MDM2 | Murine double minute 2 |
| MDM4 | Murine double minute 4 |
| Met-Pro | Methionyl-proline |
| mTOR | Mammalian target of rapamycin |
| NADPH | Reduced nicotinamide adenine dinucleotide phosphate |
| NADP+ | Nicotinamide adenine dinucleotide phosphate |
| NADH | Reduced nicotinamide adenine dinucleotide |
| NAD+ | Nicotinamide adenine dinucleotide |
| NF-κβ | Nuclear factor kappa beta |
| NMDA | N-methyl-D-aspartate |
| NO | Nitric oxide |
| NS5 | Non-structural protein 5 |
| NSAID | Nonsteroidal anti-inflammatory drug |
| OATT | Ornithine aminotransferase |
| P5C | Pyrroline-5-carboxylic acid |
| P5CS | Pyrroline-5-carboxylic acid synthase |
| PD | Prolidase deficiency |
| PEPD | Prolidase |
| Phe-Pro | Phenylalanyl-proline |
| PI3K | Phosphoinositide 3-kinase |
| PPA | Plasma prolidase activity |
| Pro | Proline |
| PRODH/POX | Proline dehydrogenase/proline oxidase |
| PYCR1/2/L | Pyrroline-5-carboxylic acid reductase 1/2/L |
| ROS | Reactive oxygen species |
| SNP | Single nucleotide polymorphism |
| Src | Proto-oncogene tyrosine-protein kinase |
| STAT3 | Signal transducer and activator of transcription 3 |
| T2D | Type 2 diabetes |
| TGF-β | Transforming growth factor beta |
| TGF-β1R | Transforming growth factor beta 1 receptor |
| Val-Pro | Valyl-proline |
| VEGF | Vascular endothelial growth factor |
| VHL | Von Hippel–Lindau tumor suppressor |
| WIP1 | Wild-type p53-induced phosphatase |
References
- Cunningham, D.F.; O’Connor, B. Proline specific peptidases. Biochim. Biophys. Acta 1997, 1343, 160–186. [Google Scholar] [CrossRef]
- Namiduru, E.S. Prolidase. Bratisl. Lek. Listy. 2016, 117, 480–485. [Google Scholar] [CrossRef] [PubMed]
- Spodenkiewicz, M.; Cleary, M.; Massier, M.; Fitsialos, G.; Cottin, V.; Jouret, G.; Poirsier, C.; Doco-Fenzy, M.; Lèbre, A.S. Clinical Genetics of Prolidase Deficiency: An Updated Review. Biology 2020, 9, 108. [Google Scholar] [CrossRef] [PubMed]
- Bhatnager, R.; Nanda, S.; Dang, A.S. Plasma prolidase levels as a biomarker for polycystic ovary syndrome. Biomark. Med. 2018, 12, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Citak Kurt, A.N.; Ustundag, B.; Akarsu, S.; Kurt, A.; Yilmaz, E.; Ocal, C.; Aygun, A.D. Cord blood prolidase activity correlates with gestational age and birth weight. Neonatology 2008, 94, 110–112. [Google Scholar] [CrossRef]
- Sayın, R.; Aslan, M.; Kucukoglu, M.E.; Luleci, A.; Atmaca, M.; Esen, R.; Demir, H. Serum prolidase enzyme activity and oxidative stress levels in patients with diabetic neuropathy. Endocrine 2014, 47, 146–151. [Google Scholar] [CrossRef]
- Rabus, M.; Demirbag, R.; Yildiz, A.; Tezcan, O.; Yilmaz, R.; Ocak, A.R.; Alp, M.; Erel, O.; Aksoy, N.; Yakut, C. Association of prolidase activity, oxidative parameters, and presence of atrial fibrillation in patients with mitral stenosis. Arch. Med. Res. 2008, 39, 519–524. [Google Scholar] [CrossRef]
- Akcakoyun, M.; Pala, S.; Esen, O.; Acar, G.; Kargin, R.; Emiroglu, Y.; Tigen, K.; Ozcan, O.; Ipcioglu, O.M.; Esen, A.M. Dilatation of the ascending aorta is associated with low serum prolidase activity. Tohoku. J. Exp. Med. 2010, 220, 273–277. [Google Scholar] [CrossRef] [Green Version]
- Vural, M.; Toy, H.; Camuzcuoglu, H.; Aksoy, N. Comparison of prolidase enzyme activities of maternal serum and placental tissue in patients with early pregnancy failure. Arch. Gynecol. Obstet. 2011, 283, 953–958. [Google Scholar] [CrossRef]
- Horoz, M.; Aslan, M.; Bolukbas, F.F.; Bolukbas, C.; Nazligul, Y.; Celik, H.; Aksoy, N. Serum prolidase enzyme activity and its relation to histopathological findings in patients with non-alcoholic steatohepatitis. J. Clin. Lab. Anal. 2010, 24, 207–211. [Google Scholar] [CrossRef]
- Pehlivan, M.; Ozün Ozbay, P.; Temur, M.; Yılmaz, O.; Verit, F.F.; Aksoy, N.; Korkmazer, E.; Üstünyurt, E. Is there any role of prolidase enzyme activity in the etiology of preeclampsia? J. Matern. Fetal Neonatal Med. 2017, 30, 1108–1113. [Google Scholar] [CrossRef] [PubMed]
- Ceylan, M.F.; Tural Hesapcioglu, S.; Kasak, M.; Senat, A.; Erel, O. Increased prolidase activity and high blood monocyte counts in pediatric bipolar disorder. Psychiatry Res. 2019, 271, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Sultan, A.; Zheng, Y.; Trainor, P.J.; Siow, Y.; Amraotkar, A.R.; Hill, B.G.; DeFilippis, A.P. Circulating Prolidase Activity in Patients with Myocardial Infarction. Front. Cardiovasc. Med. 2017, 4, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, Y.S.; Chen, C.H.; Hu, C.; Long, J.; Ong, R.T.; Sim, X.; Takeuchi, F.; Wu, Y.; Go, M.J.; Yamauchi, T.; et al. Meta-analysis of genome-wide association studies identifies eight new loci for type 2 diabetes in east Asians. Nat. Genet. 2011, 44, 67–72. [Google Scholar] [CrossRef]
- Dastani, Z.; Hivert, M.F.; Timpson, N.; Perry, J.R.; Yuan, X.; Scott, R.A.; Henneman, P.; Heid, I.M.; Kizer, J.R.; Lyytikäinen, L.P.; et al. Novel loci for adiponectin levels and their influence on type 2 diabetes and metabolic traits: A multi-ethnic meta-analysis of 45,891 individuals. PLoS Genet. 2012, 8, e1002607. [Google Scholar] [CrossRef] [Green Version]
- Willer, C.J.; Schmidt, E.M.; Sengupta, S.; Peloso, G.M.; Gustafsson, S.; Kanoni, S.; Ganna, A.; Chen, J.; Buchkovich, M.L.; Mora, S.; et al. Discovery and refinement of loci associated with lipid levels. Nat. Genet. 2013, 45, 1274–1283. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Gao, H.; Li, H.; Tabara, Y.; Nakatochi, M.; Chiu, Y.F.; Park, E.J.; Wen, W.; Adair, L.S.; Borja, J.B.; et al. A meta-analysis of genome-wide association studies for adiponectin levels in East Asians identifies a novel locus near WDR11-FGFR2. Hum. Mol. Genet. 2014, 23, 1108–1119. [Google Scholar] [CrossRef] [Green Version]
- Verma, A.K.; Chandra, S.; Singh, R.G.; Singh, T.B.; Srivastava, S.; Srivastava, R. Serum prolidase activity and oxidative stress in diabetic nephropathy and end stage renal disease: A correlative study with glucose and creatinine. Biochem. Res. Int. 2014, 2014, 291458. [Google Scholar] [CrossRef] [Green Version]
- Eren, M.A.; Torun, A.N.; Tabur, S.; Ulas, T.; Demir, M.; Sabuncu, T.; Aksoy, N. Serum prolidase activity in diabetic foot ulcers. Acta Diabetol. 2013, 50, 423–427. [Google Scholar] [CrossRef]
- Sabuncu, T.; Boduroglu, O.; Eren, M.A.; Torun, A.N.; Aksoy, N. The Value of Serum Prolidase Activity in Progression of Microalbuminuria in Patients With Type 2 Diabetes Mellitus. J. Clin. Lab. Anal. 2016, 30, 557–562. [Google Scholar] [CrossRef]
- Mittal, S.; Song, X.; Vig, B.S.; Landowski, C.P.; Kim, I.; Hilfinger, J.M.; Amidon, G.L. Prolidase, a potential enzyme target for melanoma: Design of proline-containing dipeptide-like prodrugs. Mol. Pharm. 2005, 2, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Cechowska-Pasko, M.; Pałka, J.; Wojtukiewicz, M.Z. Enhanced prolidase activity and decreased collagen content in breast cancer tissue. Int. J. Exp. Pathol. 2006, 87, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Karna, E.; Surazynski, A.; Palka, J. Collagen metabolism disturbances are accompanied by an increase in prolidase activity in lung carcinoma planoepitheliale. Int. J. Exp. Pathol. 2000, 81, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Camuzcuoglu, H.; Arioz, D.T.; Toy, H.; Kurt, S.; Celik, H.; Aksoy, N. Assessment of preoperative serum prolidase activity in epithelial ovarian cancer. Eur. J. Obstet. Gynecol. Reprod. Biol. 2009, 147, 97–100. [Google Scholar] [CrossRef]
- Arioz, D.T.; Camuzcuoglu, H.; Toy, H.; Kurt, S.; Celik, H.; Aksoy, N. Serum prolidase activity and oxidative status in patients with stage I endometrial cancer. Int. J. Gynecol. Cancer 2009, 19, 1244–1247. [Google Scholar] [CrossRef]
- Yang, L.; Li, Y.; Ding, Y.; Choi, K.S.; Kazim, A.L.; Zhang, Y. Prolidase directly binds and activates epidermal growth factor receptor and stimulates downstream signaling. J. Biol. Chem. 2013, 288, 2365–2375. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Li, Y.; Zhang, Y. Identification of prolidase as a high affinity ligand of the ErbB2 receptor and its regulation of ErbB2 signaling and cell growth. Cell Death Dis. 2014, 5, e1211. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Li, Y.; Bhattacharya, A.; Zhang, Y. Inhibition of ERBB2-overexpressing Tumors by Recombinant Human Prolidase and Its Enzymatically Inactive Mutant. EBioMedicine 2015, 2, 396–405. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Li, Y.; Bhattacharya, A.; Zhang, Y. Dual inhibition of ErbB1 and ErbB2 in cancer by recombinant human prolidase mutant hPEPD-G278D. Oncotarget 2016, 7, 42340–42352. [Google Scholar] [CrossRef]
- Yang, L.; Li, Y.; Bhattacharya, A.; Zhang, Y. A recombinant human protein targeting HER2 overcomes drug resistance in HER2-positive breast cancer. Sci. Transl. Med. 2019, 11, eaav1620. [Google Scholar] [CrossRef]
- Yang, L.; Li, Y.; Bhattacharya, A.; Zhang, Y. PEPD is a pivotal regulator of p53 tumor suppressor. Nat. Commun. 2017, 8, 2052. [Google Scholar] [CrossRef] [Green Version]
- Lubick, K.J.; Robertson, S.J.; McNally, K.L.; Freedman, B.A.; Rasmussen, A.L.; Taylor, R.T.; Walts, A.D.; Tsuruda, S.; Sakai, M.; Ishizuka, M.; et al. Flavivirus Antagonism of Type I Interferon Signaling Reveals Prolidase as a Regulator of IFNAR1 Surface Expression. Cell Host Microbe 2015, 18, 61–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ADAMS, E.; SMITH, E.L. Peptidases of erythrocytes. II. Isolation and properties of prolidase. J. Biol. Chem. 1952, 198, 671–682. [Google Scholar] [PubMed]
- Hynes, N.E.; Lane, H.A. ERBB receptors and cancer: The complexity of targeted inhibitors. Nat. Rev. Cancer 2005, 5, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.T.; Akita, R.W.; Sliwkowski, M.X. Binding specificities and affinities of egf domains for ErbB receptors. FEBS Lett. 1999, 447, 227–231. [Google Scholar] [CrossRef] [Green Version]
- Arteaga, C.L.; Engelman, J.A. ERBB receptors: From oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell 2014, 25, 282–303. [Google Scholar] [CrossRef] [Green Version]
- Tong, J.; Wang, Z. Analysis of Epidermal Growth Factor Receptor-Induced Cell Motility by Wound Healing Assay. Methods Mol. Biol. 2017, 1652, 159–163. [Google Scholar] [CrossRef]
- Yang, L.; Li, Y.; Bhattacharya, A.; Zhang, Y. A plasma proteolysis pathway comprising blood coagulation proteases. Oncotarget 2016, 7, 40919–40938. [Google Scholar] [CrossRef] [Green Version]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef]
- Kruse, J.P.; Gu, W. Modes of p53 regulation. Cell 2009, 137, 609–622. [Google Scholar] [CrossRef] [Green Version]
- Hafner, A.; Bulyk, M.L.; Jambhekar, A.; Lahav, G. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol. 2019, 20, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Lupi, A.; Della Torre, S.; Campari, E.; Tenni, R.; Cetta, G.; Rossi, A.; Forlino, A. Human recombinant prolidase from eukaryotic and prokaryotic sources. Expression, purification, characterization and long-term stability studies. FEBS J. 2006, 273, 5466–5478. [Google Scholar] [CrossRef] [PubMed]
- Klimant, E.; Wright, H.; Rubin, D.; Seely, D.; Markman, M. Intravenous vitamin C in the supportive care of cancer patients: A review and rational approach. Curr. Oncol. 2018, 25, 139–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Andrea, G.M. Use of antioxidants during chemotherapy and radiotherapy should be avoided. CA Cancer J. Clin. 2005, 55, 319–321. [Google Scholar] [CrossRef] [PubMed]
- Best, S.M. The Many Faces of the Flavivirus NS5 Protein in Antagonism of Type I Interferon Signaling. J. Virol. 2017, 91, e01970-16. [Google Scholar] [CrossRef] [Green Version]
- Thurmond, S.; Wang, B.; Song, J.; Hai, R. Suppression of Type I Interferon Signaling by Flavivirus NS5. Viruses 2018, 10, 712. [Google Scholar] [CrossRef] [Green Version]
- Hintze, J.P.; Kirby, A.; Torti, E.; Batanian, J.R. Prolidase Deficiency in a Mexican-American Patient Identified by Array CGH Reveals a Novel and the Largest PEPD Gene Deletion. Mol. Syndromol. 2016, 7, 80–86. [Google Scholar] [CrossRef]
- Ota, T.; Suzuki, Y.; Nishikawa, T.; Otsuki, T.; Sugiyama, T.; Irie, R.; Wakamatsu, A.; Hayashi, K.; Sato, H.; Nagai, K.; et al. Complete sequencing and characterization of 21,243 full-length human cDNAs. Nat. Genet. 2004, 36, 40–45. [Google Scholar] [CrossRef]
- Sjöström, H.; Norén, O. Structural properties of pig intestinal proline dipeptidase. Biochim. Biophys. Acta 1974, 359, 177–185. [Google Scholar] [CrossRef]
- Lupi, A.; Tenni, R.; Rossi, A.; Cetta, G.; Forlino, A. Human prolidase and prolidase deficiency: An overview on the characterization of the enzyme involved in proline recycling and on the effects of its mutations. Amino Acids 2008, 35, 739–752. [Google Scholar] [CrossRef]
- Surazynski, A.; Liu, Y.; Miltyk, W.; Phang, J.M. Nitric oxide regulates prolidase activity by serine/threonine phosphorylation. J. Cell Biochem. 2005, 96, 1086–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surazyński, A.; Pałka, J.; Wołczyński, S. Phosphorylation of prolidase increases the enzyme activity. Mol. Cell Biochem. 2001, 220, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Ysrayl, B.B.; Balasubramaniam, M.; Albert, I.; Villalta, F.; Pandhare, J.; Dash, C. A Novel Role of Prolidase in Cocaine-Mediated Breach in the Barrier of Brain Microvascular Endothelial Cells. Sci. Rep. 2019, 9, 2567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilk, P.; Uehlein, M.; Kalms, J.; Dobbek, H.; Mueller, U.; Weiss, M.S. Substrate specificity and reaction mechanism of human prolidase. FEBS J. 2017, 284, 2870–2885. [Google Scholar] [CrossRef] [Green Version]
- Lowther, W.T.; Matthews, B.W. Metalloaminopeptidases: Common functional themes in disparate structural surroundings. Chem. Rev. 2002, 102, 4581–4608. [Google Scholar] [CrossRef]
- Wilk, P.; Uehlein, M.; Piwowarczyk, R.; Dobbek, H.; Mueller, U.; Weiss, M.S. Structural basis for prolidase deficiency disease mechanisms. FEBS J. 2018, 285, 3422–3441. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.H.; Zhi, Q.W.; Sun, M.J. Purification and characterization of recombinant human liver prolidase expressed in Saccharomyces cerevisiae. Arch. Toxicol. 2005, 79, 253–259. [Google Scholar] [CrossRef]
- Lin, L.N.; Brandts, J.F. Evidence suggesting that some proteolytic enzymes may cleave only the trans form of the peptide bond. Biochemistry 1979, 18, 43–47. [Google Scholar] [CrossRef]
- Josefsson, L.; Sjöström, H.; Norén, O. Intracellular hydrolysis of peptides. Ciba Found. Symp. 1977, 199–207. [Google Scholar] [CrossRef]
- Fagerberg, L.; Hallström, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell Proteom. 2014, 13, 397–406. [Google Scholar] [CrossRef] [Green Version]
- Guszczyn, T.; Surażyński, A.; Zaręba, I.; Rysiak, E.; Popko, J.; Pałka, J. Differential effect of platelet-rich plasma fractions on β1-integrin signaling, collagen biosynthesis, and prolidase activity in human skin fibroblasts. Drug Des. Dev. Ther. 2017, 11, 1849–1857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitchener, R.L.; Grunden, A.M. Prolidase function in proline metabolism and its medical and biotechnological applications. J. Appl. Microbiol. 2012, 113, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Pałka, J.A. The role of prolidase as an enzyme participating in the metabolism of collagen. Rocz. Akad. Med. Bialymst. 1996, 41, 149–160. [Google Scholar] [PubMed]
- Reid, K.B. Isolation, by partial pepsin digestion, of the three collagen-like regions present in subcomponent Clq of the first component of human complement. Biochem. J. 1976, 155, 5–17. [Google Scholar] [CrossRef] [Green Version]
- Surazynski, A.; Miltyk, W.; Palka, J.; Phang, J.M. Prolidase-dependent regulation of collagen biosynthesis. Amino Acids 2008, 35, 731–738. [Google Scholar] [CrossRef]
- Kadler, K.E.; Baldock, C.; Bella, J.; Boot-Handford, R.P. Collagens at a glance. J. Cell Sci. 2007, 120, 1955–1958. [Google Scholar] [CrossRef] [Green Version]
- Surazynski, A.; Donald, S.P.; Cooper, S.K.; Whiteside, M.A.; Salnikow, K.; Liu, Y.; Phang, J.M. Extracellular matrix and HIF-1 signaling: The role of prolidase. Int. J. Cancer 2008, 122, 1435–1440. [Google Scholar] [CrossRef]
- Palka, J.A.; Phang, J.M. Prolidase activity in fibroblasts is regulated by interaction of extracellular matrix with cell surface integrin receptors. J. Cell Biochem. 1997, 67, 166–175. [Google Scholar] [CrossRef] [Green Version]
- Cui, N.; Hu, M.; Khalil, R.A. Biochemical and Biological Attributes of Matrix Metalloproteinases. Prog. Mol. Biol. Transl. Sci. 2017, 147, 1–73. [Google Scholar] [CrossRef] [Green Version]
- Phang, J.M.; Liu, W.; Zabirnyk, O. Proline metabolism and microenvironmental stress. Annu. Rev. Nutr. 2010, 30, 441–463. [Google Scholar] [CrossRef] [Green Version]
- Hui, K.S.; Lajtha, A. Activation and inhibition of cerebral prolidase. J. Neurochem. 1980, 35, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Güneş, M.; Bulut, M.; Demir, S.; İbiloğlu, A.O.; Kaya, M.C.; Atlı, A.; Kaplan, İ.; Camkurt, M.A.; Sir, A. Diagnostic performance of increased prolidase activity in schizophrenia. Neurosci. Lett. 2016, 613, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Labat-Robert, J.; Robert, L. Interaction between cells and extracellular matrix: Signaling by integrins and the elastin-laminin receptor. Prog. Mol. Subcell. Biol. 2000, 25, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Surazyński, A.; Sienkiewicz, P.; Wołczyński, S.; Pałka, J. Differential effects of echistatin and thrombin on collagen production and prolidase activity in human dermal fibroblasts and their possible implication in beta1-integrin-mediated signaling. Pharmacol. Res. 2005, 51, 217–221. [Google Scholar] [CrossRef]
- Miltyk, W.; Karna, E.; Wołczyński, S.; Pałka, J. Insulin-like growth factor I-dependent regulation of prolidase activity in cultured human skin fibroblasts. Mol. Cell Biochem. 1998, 189, 177–183. [Google Scholar] [CrossRef]
- Surazynski, A.; Miltyk, W.; Prokop, I.; Palka, J. Prolidase-dependent regulation of TGF β (corrected) and TGF β receptor expressions in human skin fibroblasts. Eur. J. Pharmacol. 2010, 649, 115–119. [Google Scholar] [CrossRef]
- Prokop, I.; Konończuk, J.; Surażyński, A.; Pałka, J. Cross-talk between integrin receptor and insulin-like growth factor receptor in regulation of collagen biosynthesis in cultured fibroblasts. Adv. Med. Sci. 2013, 58, 292–297. [Google Scholar] [CrossRef] [Green Version]
- Surazynski, A.; Miltyk, W.; Prokop, I.; Palka, J. The effect of estrogen on prolidase-dependent regulation of HIF-1α expression in breast cancer cells. Mol. Cell Biochem. 2013, 379, 29–36. [Google Scholar] [CrossRef] [Green Version]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discovery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef] [Green Version]
- Park, H.S.; Kim, J.H.; Sun, B.K.; Song, S.U.; Suh, W.; Sung, J.H. Hypoxia induces glucose uptake and metabolism of adipose-derived stem cells. Mol. Med. Rep. 2016, 14, 4706–4714. [Google Scholar] [CrossRef] [Green Version]
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-β and the TGF-β Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harb. Perspect. Biol. 2016, 8, a021873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senger, D.R.; Claffey, K.P.; Benes, J.E.; Perruzzi, C.A.; Sergiou, A.P.; Detmar, M. Angiogenesis promoted by vascular endothelial growth factor: Regulation through alpha1beta1 and alpha2beta1 integrins. Proc. Natl. Acad. Sci. USA 1997, 94, 13612–13617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.S.; Cui, W. Proliferation, survival and metabolism: The role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development 2016, 143, 3050–3060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, S.; Vargas, J.; Hoffmann, A. Signaling via the NFκB system. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 227–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rippe, R.A.; Schrum, L.W.; Stefanovic, B.; Solís-Herruzo, J.A.; Brenner, D.A. NF-kappaB inhibits expression of the alpha1(I) collagen gene. DNA Cell Biol. 1999, 18, 751–761. [Google Scholar] [CrossRef] [PubMed]
- Miltyk, W.; Karna, E.; Pałka, J. Inhibition of prolidase activity by non-steroid antiinflammatory drugs in cultured human skin fibroblasts. Pol. J. Pharmacol. 1996, 48, 609–613. [Google Scholar]
- Miltyk, W.; Karna, E.; Pałka, J. Inhibition of prolidase activity by collagen-degradation products. Med. Sci. Monit. 1997, 3, 6–12. [Google Scholar]
- Phang, J.M. The regulatory functions of proline and pyrroline-5-carboxylic acid. Curr. Top. Cell Regul. 1985, 25, 91–132. [Google Scholar] [CrossRef]
- Phang, J.M. Proline Metabolism in Cell Regulation and Cancer Biology: Recent Advances and Hypotheses. Antioxid. Redox Signal. 2019, 30, 635–649. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.A.; Khalil, S.; Zhaorigetu, S.; Liu, Z.; Tyler, M.; Wan, G.; Valle, D. Human Delta1-pyrroline-5-carboxylate synthase: Function and regulation. Amino Acids 2008, 35, 665–672. [Google Scholar] [CrossRef] [Green Version]
- Fahmy, A.S.; Mohamed, S.A.; Girgis, R.B.; Abdel-Ghaffar, F.A. Enzymes of delta 1-pyrroline-5-carboxylate metabolism in the camel tick Hyalomma dromedarii during embryogenesis. Purification and characterization of delta 1-pyrroline-5-carboxylate dehydrogenases. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 1997, 118, 229–237. [Google Scholar] [CrossRef]
- Liu, W.; Phang, J.M. Proline dehydrogenase (oxidase), a mitochondrial tumor suppressor, and autophagy under the hypoxia microenvironment. Autophagy 2012, 8, 1407–1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Ingeniis, J.; Ratnikov, B.; Richardson, A.D.; Scott, D.A.; Aza-Blanc, P.; De, S.K.; Kazanov, M.; Pellecchia, M.; Ronai, Z.; Osterman, A.L.; et al. Functional specialization in proline biosynthesis of melanoma. PLoS ONE 2012, 7, e45190. [Google Scholar] [CrossRef] [Green Version]
- Elia, I.; Broekaert, D.; Christen, S.; Boon, R.; Radaelli, E.; Orth, M.F.; Verfaillie, C.; Grünewald, T.G.P.; Fendt, S.M. Proline metabolism supports metastasis formation and could be inhibited to selectively target metastasizing cancer cells. Nat. Commun. 2017, 8, 15267. [Google Scholar] [CrossRef] [PubMed]
- Phang, J.M.; Pandhare, J.; Liu, Y. The metabolism of proline as microenvironmental stress substrate. J. Nutr. 2008, 138, 2008S–2015S. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Le, A.; Hancock, C.; Lane, A.N.; Dang, C.V.; Fan, T.W.; Phang, J.M. Reprogramming of proline and glutamine metabolism contributes to the proliferative and metabolic responses regulated by oncogenic transcription factor c-MYC. Proc. Natl. Acad. Sci. USA 2012, 109, 8983–8988. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Hancock, C.N.; Fischer, J.W.; Harman, M.; Phang, J.M. Proline biosynthesis augments tumor cell growth and aerobic glycolysis: Involvement of pyridine nucleotides. Sci. Rep. 2015, 5, 17206. [Google Scholar] [CrossRef] [Green Version]
- Tanner, J.J.; Fendt, S.M.; Becker, D.F. The Proline Cycle As a Potential Cancer Therapy Target. Biochemistry 2018, 57, 3433–3444. [Google Scholar] [CrossRef]
- Erbağci, A.B.; Araz, M.; Erbağci, A.; Tarakçioğlu, M.; Namiduru, E.S. Serum prolidase activity as a marker of osteoporosis in type 2 diabetes mellitus. Clin. Biochem. 2002, 35, 263–268. [Google Scholar] [CrossRef]
- Monnier, V.M.; Glomb, M.; Elgawish, A.; Sell, D.R. The mechanism of collagen cross-linking in diabetes: A puzzle nearing resolution. Diabetes 1996, 45 (Suppl. 3), S67–S72. [Google Scholar] [CrossRef]
- Mittal, S.; Song, X.; Vig, B.S.; Amidon, G.L. Proline prodrug of melphalan targeted to prolidase, a prodrug activating enzyme overexpressed in melanoma. Pharm. Res. 2007, 24, 1290–1298. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; Tsume, Y.; Landowski, C.P.; Lee, K.D.; Hilfinger, J.M.; Amidon, G.L. Proline prodrug of melphalan, prophalan-L, demonstrates high therapeutic index in a murine melanoma model. Eur. J. Pharm. Biopharm. 2007, 67, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Bielawska, A.; Bielawski, K.; Chrzanowski, K.; Wołczyński, S. Prolidase-activated prodrug for cancer chemotherapy cytotoxic activity of proline analogue of chlorambucil in breast cancer MCF-7 cells. Farmaco 2000, 55, 736–741. [Google Scholar] [CrossRef]
- Bielawski, K.; Bielawska, A.; Słodownik, T.; Bołkun-Skórnicka, U.; Muszyńska, A. Proline-linked nitrosoureas as prolidase-convertible prodrugs in human breast cancer cells. Pharmacol. Rep. 2008, 60, 171–182. [Google Scholar]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Misiura, M.; Miltyk, W. Current Understanding of the Emerging Role of Prolidase in Cellular Metabolism. Int. J. Mol. Sci. 2020, 21, 5906. https://doi.org/10.3390/ijms21165906
Misiura M, Miltyk W. Current Understanding of the Emerging Role of Prolidase in Cellular Metabolism. International Journal of Molecular Sciences. 2020; 21(16):5906. https://doi.org/10.3390/ijms21165906
Chicago/Turabian StyleMisiura, Magdalena, and Wojciech Miltyk. 2020. "Current Understanding of the Emerging Role of Prolidase in Cellular Metabolism" International Journal of Molecular Sciences 21, no. 16: 5906. https://doi.org/10.3390/ijms21165906
APA StyleMisiura, M., & Miltyk, W. (2020). Current Understanding of the Emerging Role of Prolidase in Cellular Metabolism. International Journal of Molecular Sciences, 21(16), 5906. https://doi.org/10.3390/ijms21165906