P38 Regulates Kainic Acid-Induced Seizure and Neuronal Firing via Kv4.2 Phosphorylation



Abstract
:1. Introduction
2. Results
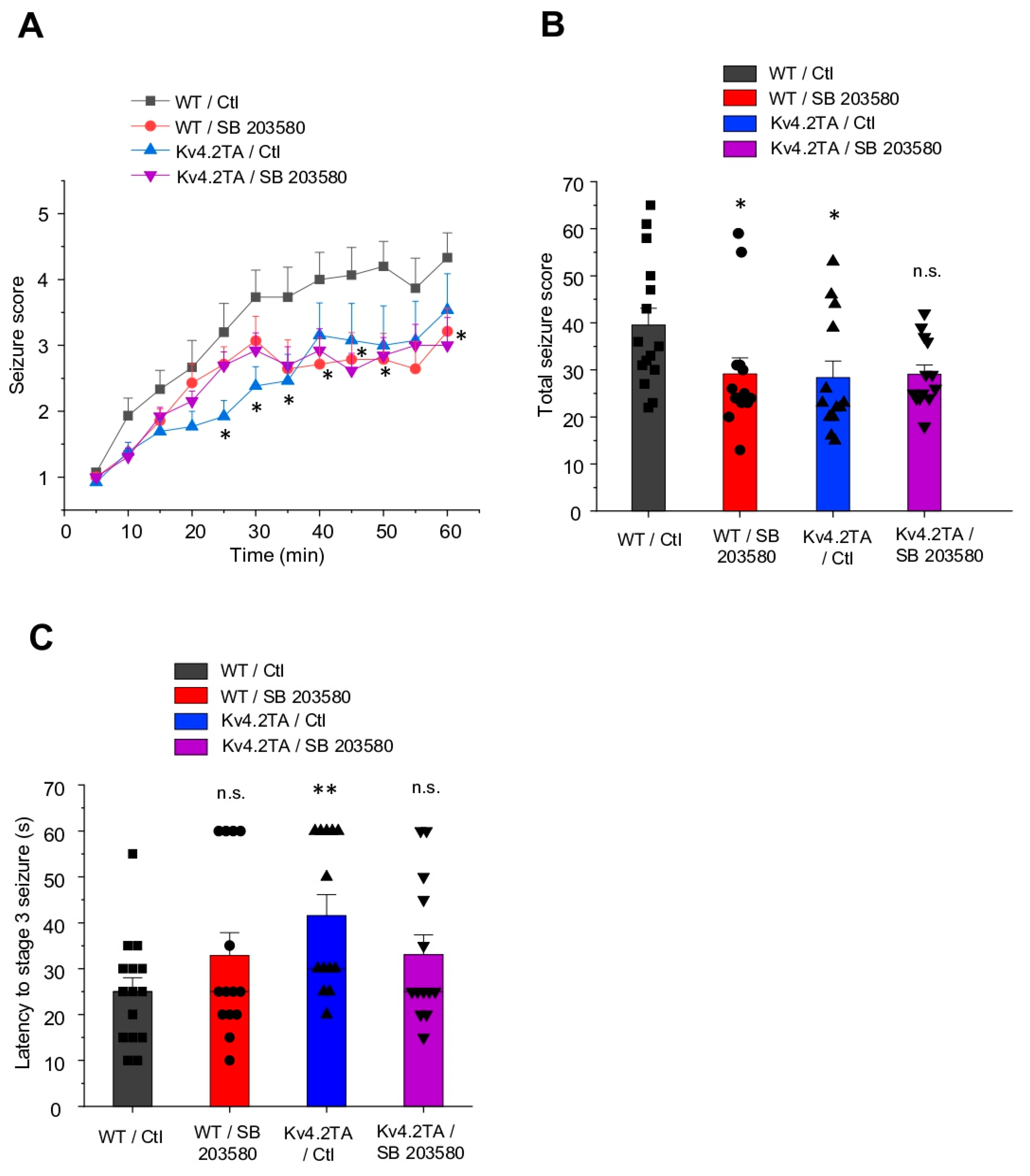
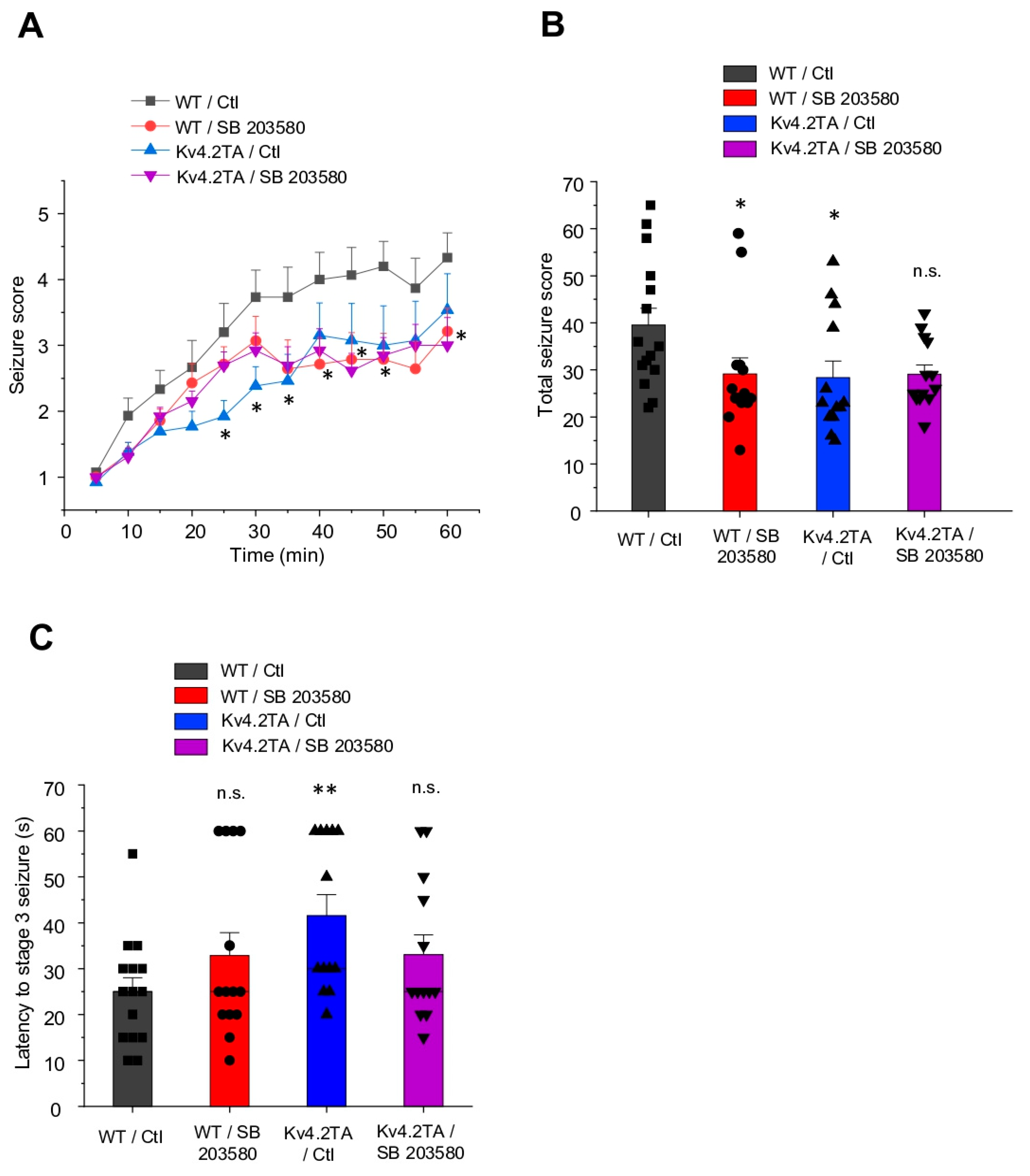
2.1. p38 MAPK Contributes to Kainic Acid-Induced Seizure in WT but Not Kv4.2TA Mice
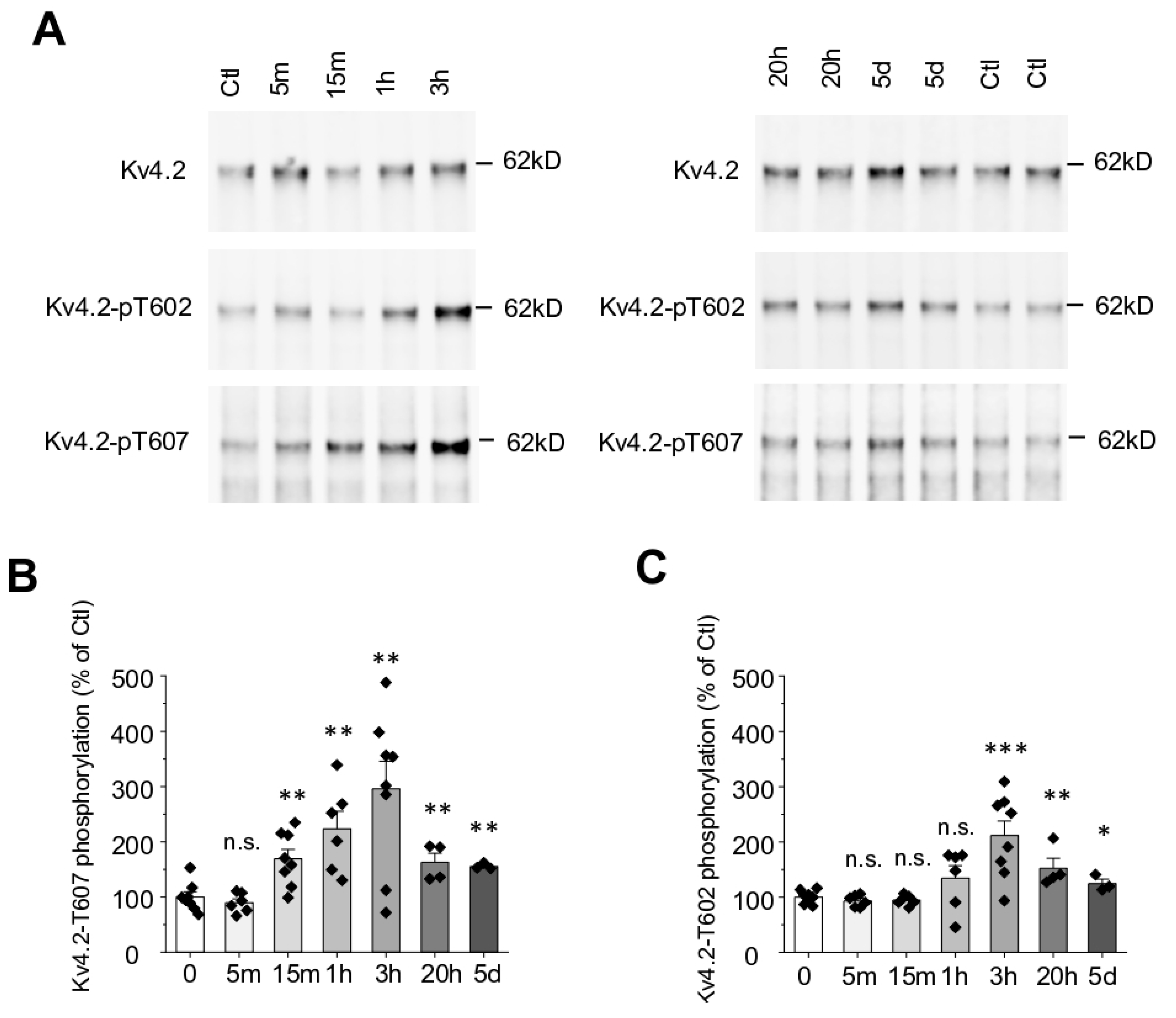
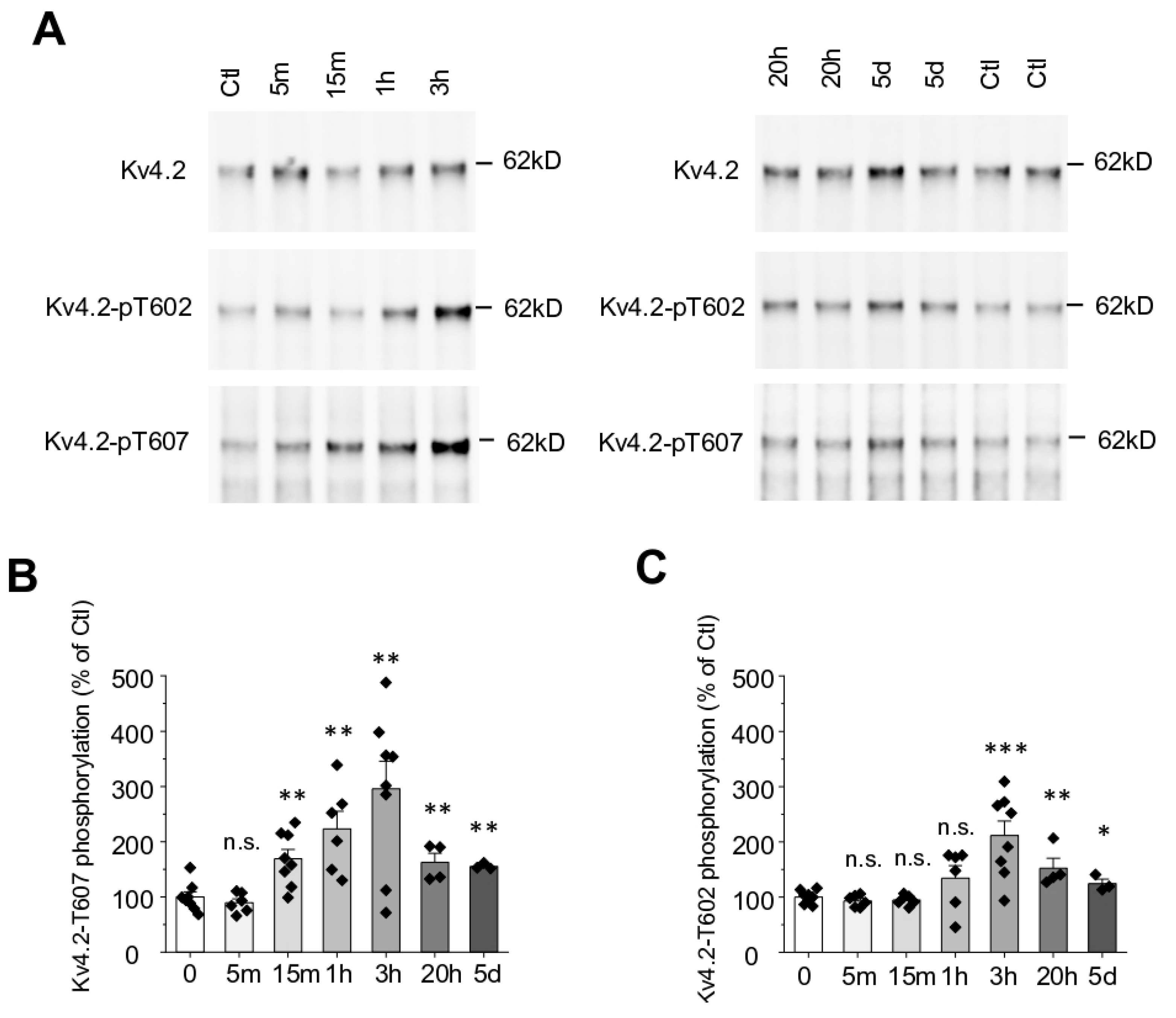
2.2. Seizure Induced by Kainic Acid Triggers Kv4.2 T607 Phosphorylation in a Time-Dependent Manner
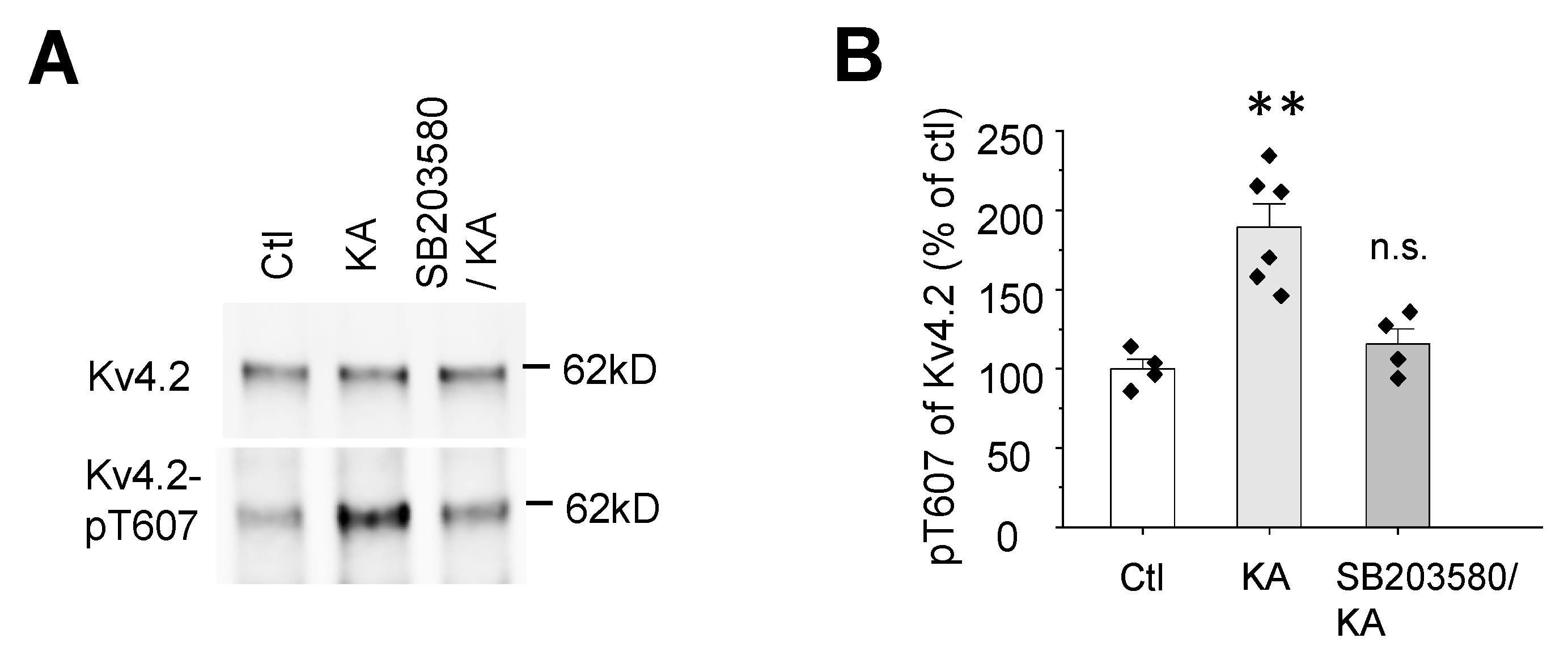
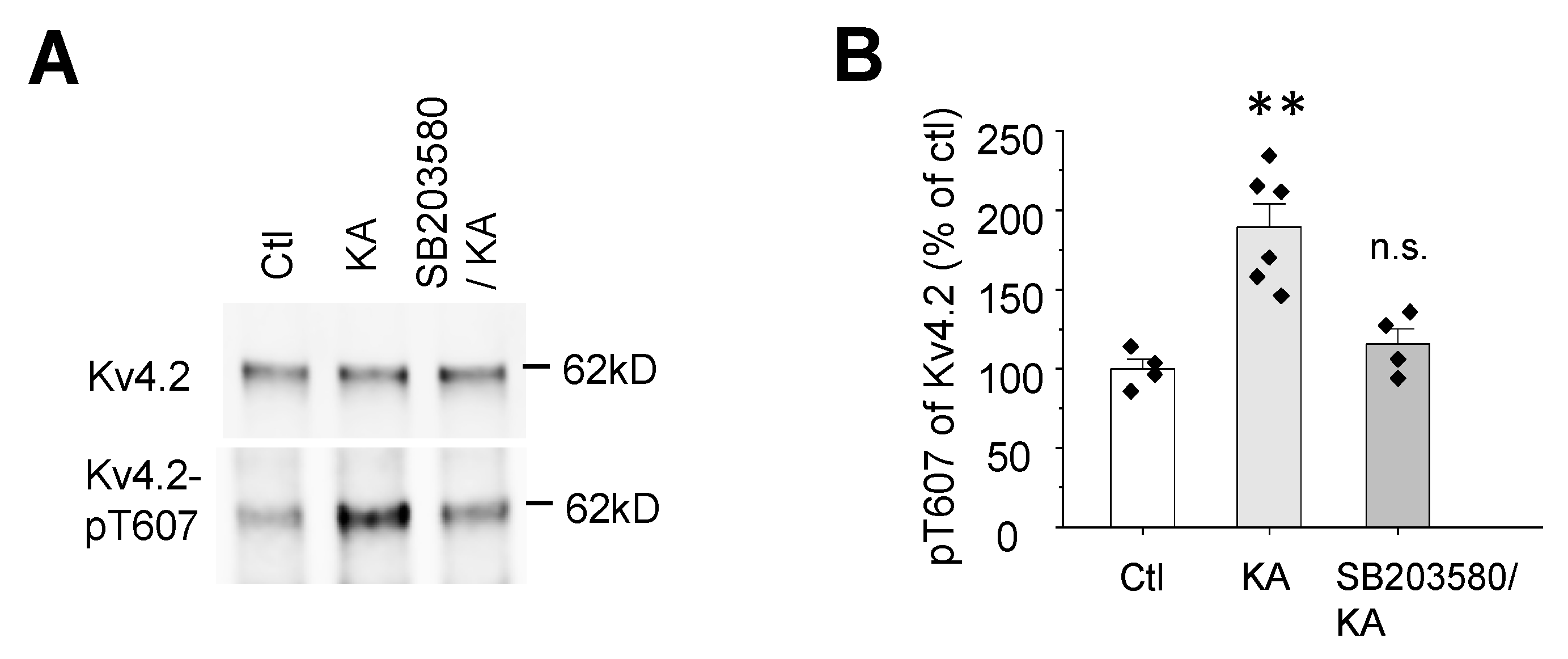
2.3. Kainic Acid-Induced Kv4.2 Phosphorylation at T607 Is Dependent on p38 MAPK
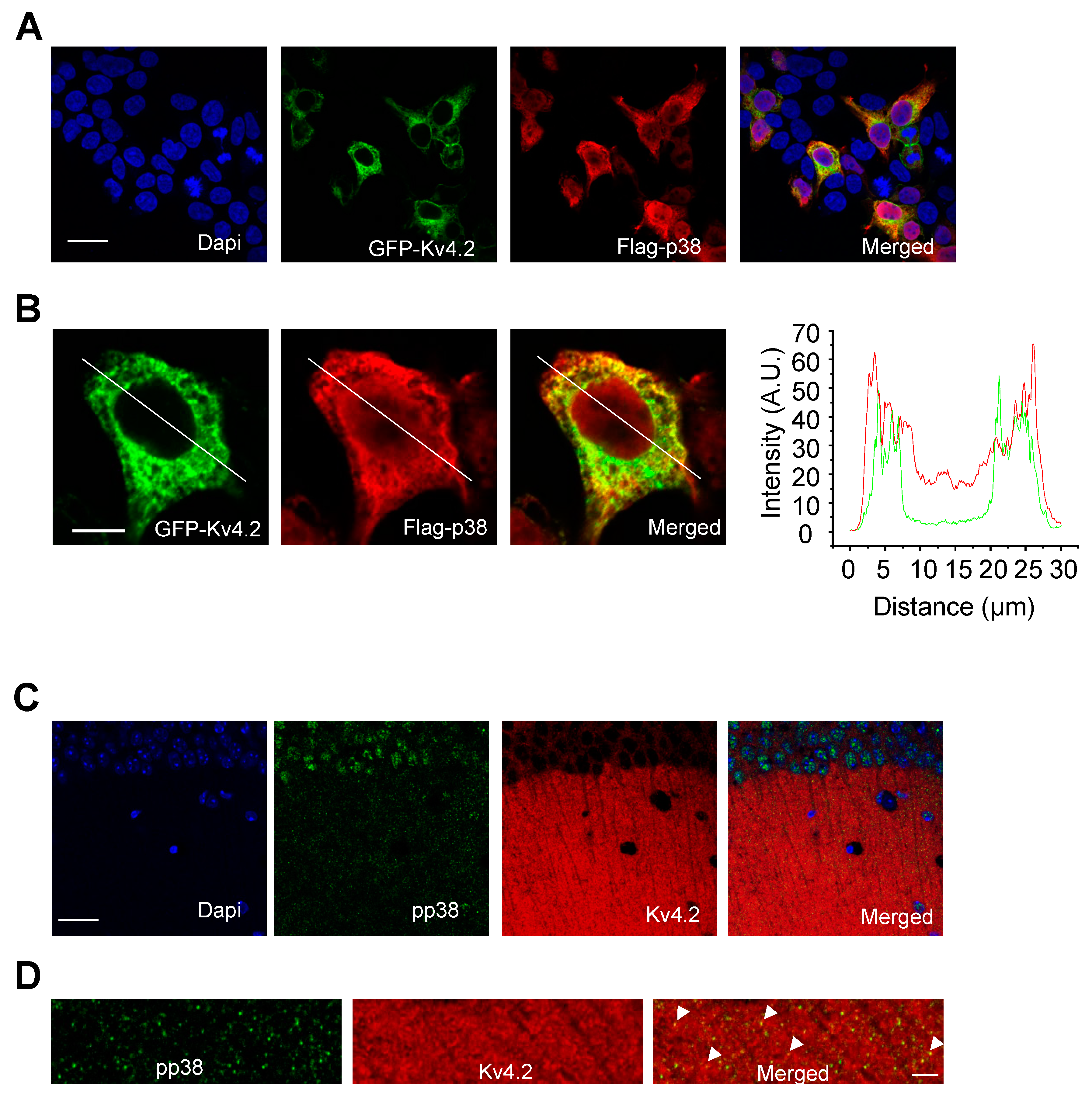
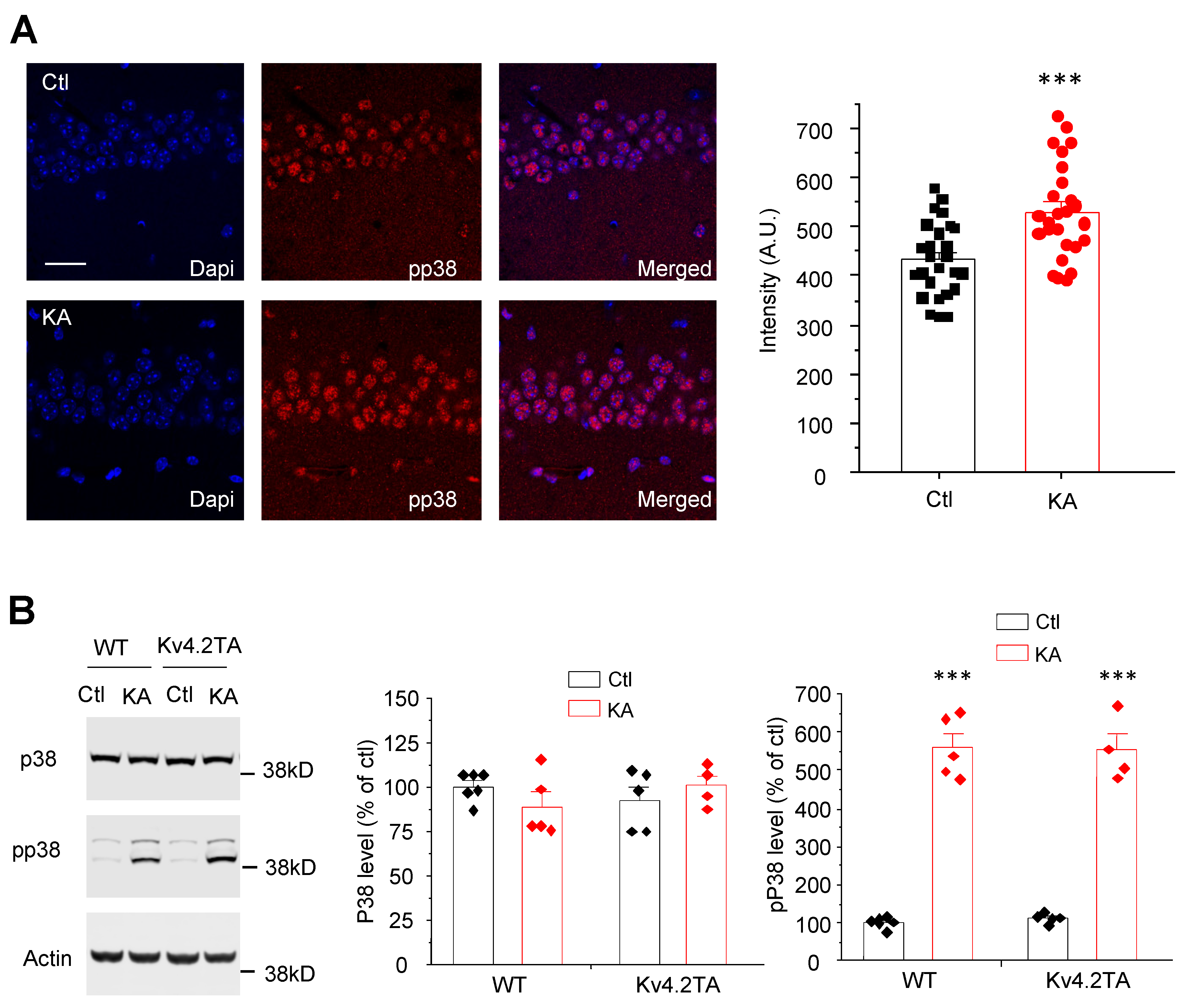
2.4. p38 MAPK Colocalizes with Kv4.2
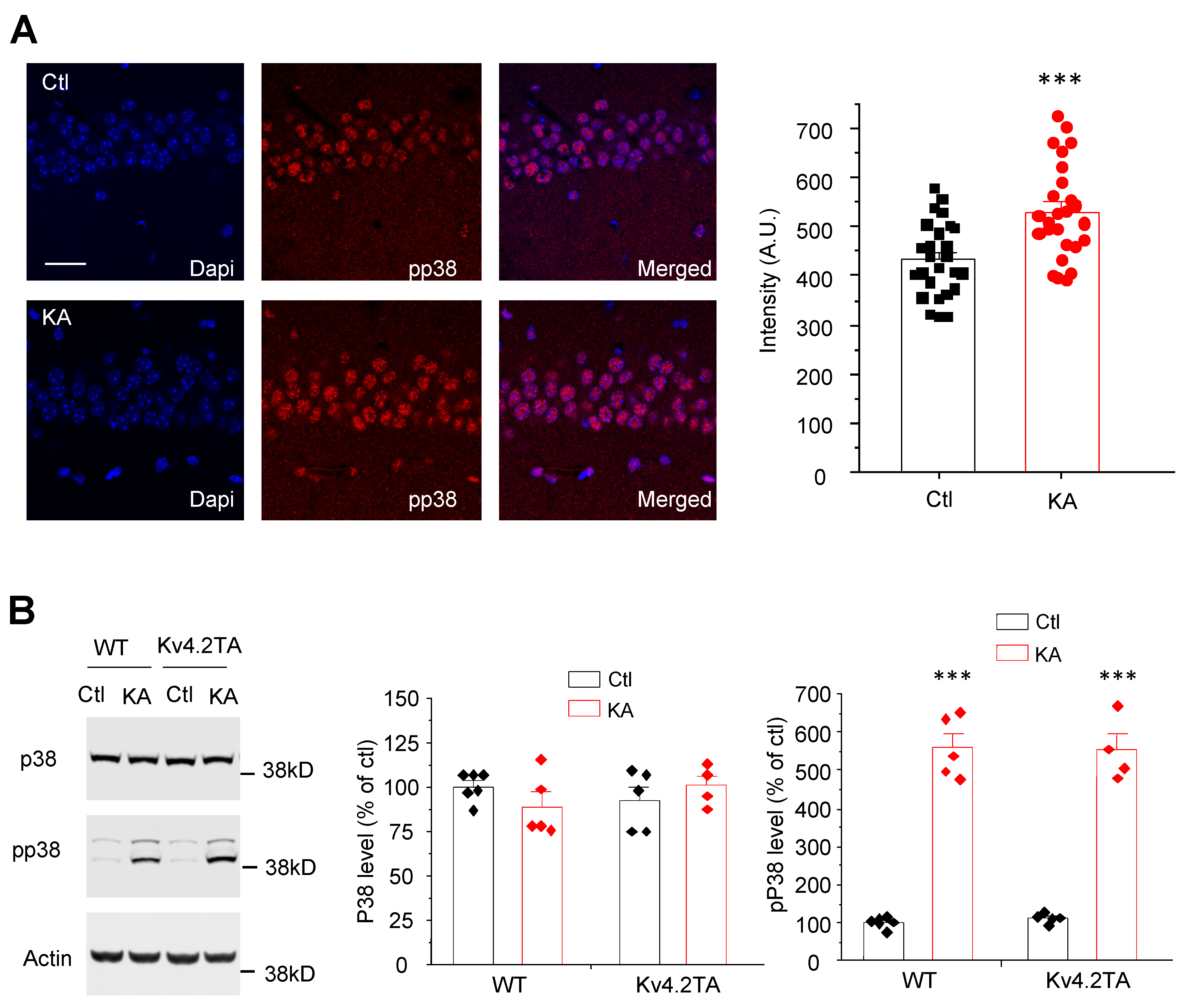
2.5. Kainic Acid Activates p38 MAPK in both WT and Kv4.2TA Mice
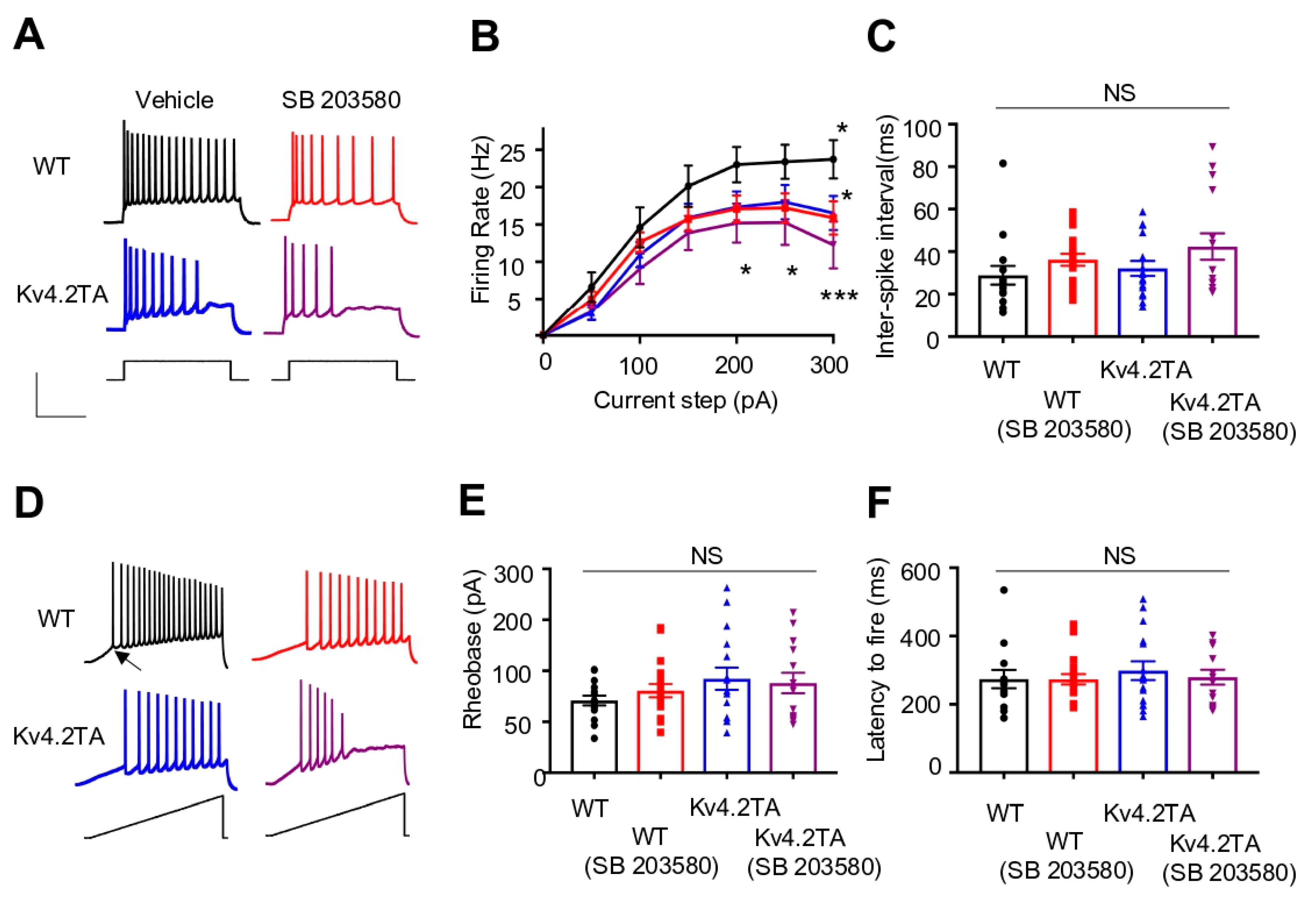
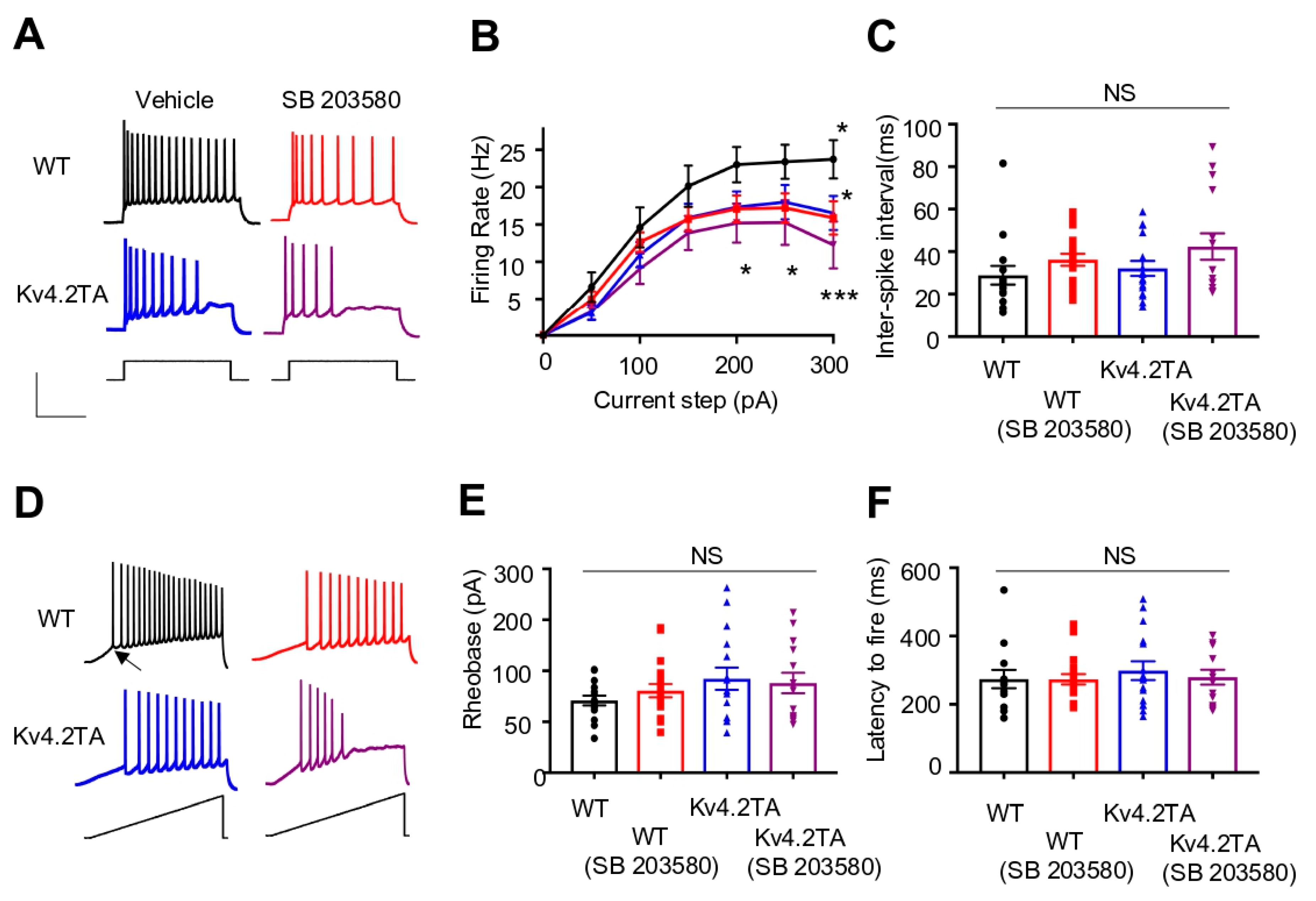
2.6. p38 MAPK Modulates Neuronal Excitability through Kv4.2
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Expression Constructs
4.3. Chemicals
4.4. Antibodies
4.5. Cell Culture and Transfection
4.6. Western Blot and Quantification
4.7. Immunostaining
4.8. Acute Hippocampal Slice Preparation
4.9. Whole-Cell Current Clamp Recordings
4.10. Seizure Behavioral Assays
4.11. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kwan, P.; Brodie, M.J. Early identification of refractory epilepsy. N. Engl. J. Med. 2000, 342, 314–319. [Google Scholar] [CrossRef]
- Bien, C.G.; Kurther, M.; Baron, K.; Lux, S.; Helmstaedter, C.; Schramm, J.; Elger, C.E. Long-term seizure outcome and antiepileptic drug treatment in surgically treated temporal lobe epilepsy patients: A controlled study. Epilepsia 2001, 42, 1416–1421. [Google Scholar] [CrossRef] [PubMed]
- Engel, J.J. Mesial temporal lobe epilepsy: What have we learned? Neuroscientist 2001, 7, 340–352. [Google Scholar] [CrossRef] [PubMed]
- Helmstaedter, C.; Kockelmann, E. Cognitive outcomes in patients with chronic temporal lobe epilepsy. Epilepsia 2006, 47, 96–98. [Google Scholar] [CrossRef] [PubMed]
- French, J.A. Refractory epilepsy: Clinical overview. Epilepsia 2007, 48, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Coan, A.C.; Cendes, F. Understanding the spectrum of temporal lobe epilepsy: Contributions for the development of individual therapies. Expert Rev. Neurother. 2013, 13, 1383–1394. [Google Scholar] [CrossRef]
- Brodie, M.J.; Dichter, M.D. Antiepileptic drugs. N. Engl. J. Med. 1996, 334, 168–175. [Google Scholar] [CrossRef]
- Ryvlin, P.; Rheims, S. Epilepsy surgery: Eligibility criteria and presurgical evaluation. Dialogues Clin. Neurosci. 2008, 10, 91–103. [Google Scholar]
- Jung, S.; Jones, T.D.; Lugo, J.N.J.; Sheerin, A.H.; Miller, J.W.; D’Ambrosio, R.; Anderson, A.E.; Poolos, N.P. Progressive dendritic HCN channelopathy during epileptogenesis in the rat pilocarpine model of epilepsy. J. Neurosci. 2007, 27, 13012–13021. [Google Scholar] [CrossRef]
- Powell, K.L.; Ng, C.; O’Brien, T.J.; Xu, S.H.; Williams, D.A.; Foote, S.J.; Reid, C.A. Decreases in HCN mRNA expression in the hippocampus after kindling and status epilepticus in adult rats. Epilepsia 2008, 49, 1686–1695. [Google Scholar] [CrossRef]
- Marcelin, B.; Chauviere, L.; Becker, A.; Migliore, M.; Esclapez, M.; Bernard, C. H channel-dependent deficit of theta-oscillation resonance and phase shift in temporal lobe epilepsy. Neurobiol. Dis. 2009, 33, 436–447. [Google Scholar] [CrossRef] [PubMed]
- Arnold, E.C.; McMurray, C.; Gray, R.; Johnston, D. Epilepsy-induced reduction in HCN channel expression contributes to an increased excitability in dorsal, but not ventral, hippocampal CA1 neurons. eNeuro 2019, 6, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foote, K.M.; Lyman, K.A.; Han, Y.; Michailidis, I.E.; Heuermann, R.J.; Mandikian, D.; Trimmer, J.S.; Swanson, G.T.; Chetkovich, D.M. Phosphorylation of the HCN channel auxiliary subunit TRIP8b is altered in an animal model of temporal lobe epilepsy and modulates channel function. J. Biol. Chem. 2019, 294, 15743–15758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ketelaars, S.O.; Gorter, J.A.; van Vliet, E.A.; Lopes da Silva, F.H.; Wadman, W.J. Sodium currents in isolated rat CA1 pyramidal and dentate granule neurones in the post-status epilepticus model of spilepsy. Neuroscience 2001, 105, 109–120. [Google Scholar] [CrossRef]
- Kile, K.B.; Tian, N.; Durand, D.M. Scn2a channel mutation results in hyperexcitability in the hippocampus in vitro. Epilepsia 2008, 49, 488–499. [Google Scholar] [CrossRef] [Green Version]
- Djamshidian, A.; Grassi, R.; Seltenhammer, M.; Czech, T.; Baumgartner, C.; Schmidbauer, M.; Ulrich, W.; Zimprich, F. Altered expression of voltage-gated calcium channel alpha(1) subunits in temporal lobe epilepsy with Ammon’s horn sclerosis. Neuroscience 2002, 111, 57–69. [Google Scholar] [CrossRef]
- Su, H.; Sochivko, D.; Becker, A.; Chen, J.; Jiang, Y.; Yaari, Y.; Beck, H. Upregulation of a T-type Ca2+ channel causes long-lasting modification of neuronal firing mode after status epilepticus. J. Neurosci. 2002, 22, 3645–3655. [Google Scholar] [CrossRef] [Green Version]
- D’Adamo, M.C.; Catacuzzeno, L.; Di Giovanni, G.; Franciolini, F.; Pessia, M. K+ channelepsy: Progress in the neurobiology of potassium channels and epilepsy. Front. Cell Neurosci. 2013, 7, 134. [Google Scholar] [CrossRef] [Green Version]
- Kohling, R.; Wolfart, J. Potassium channels in Epilepsy. Cold Spring Harb. Perspect. Med. 2016, 6, a022871. [Google Scholar] [CrossRef] [Green Version]
- Singh, B.; Ogiwara, I.; Kaneda, M.; Tokonami, N.; Mazaki, E.; Baba, K.; Matsuda, K.; Inoue, Y.; Yamakawa, K. A Kv4.2 truncation mutation in a patient with temporal lobe epilepsy. Neurobiol. Dis. 2006, 24, 245–253. [Google Scholar] [CrossRef]
- Lee, H.; Lin, M.C.; Kornblum, H.I.; Papazian, D.M.; Nelson, S.F. Exome sequencing identifies de novo gain of function missense mutation in KCND2 in identical twins with autism and seizures that slows potassium channel inactivation. Hum. Mol. Genet. 2014, 23, 3481–3489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, M.A.; Cannon, S.C.; Papazian, D.M. Kv4.2 autism and epilepsy mutation enhances inactivation of closed channels but impairs access to inactivated state after opening. Proc. Natl. Acad. Sci. USA 2018, 115, E3559–E3568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffman, D.A.; Magee, J.C.; Colbert, C.M.; Johnston, D. K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature 1997, 387, 869–875. [Google Scholar] [CrossRef] [PubMed]
- Menegola, M.; Trimmer, J.S. Unanticipated region-and cell-specific downregulation of individual KChIP auxiliary subunit isotypes in Kv4.2 knock-out mouse brain. J. Neurosci. 2006, 26, 12137–12142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Yuan, L.L.; Zhao, C.; Birnbaum, S.G.; Frick, A.; Jung, W.E.; Schwarz, T.L.; Sweatt, J.D.; Johnston, D. Deletion of Kv4.2 gene eliminates dendritic A-type K+ current and enhances induction of long-term potentiation in hippocampal CA1 pyramidal neurons. J. Neurosci. 2006, 26, 12143–12151. [Google Scholar] [CrossRef] [Green Version]
- Magee, J.C.; Carruth, M. Dendritic voltage-gated ion channels regulate the action potential firing mode of hippocampal CA1 pyramidal neurons. J. Neurophysiol. 1999, 82, 1895–1901. [Google Scholar] [CrossRef]
- Johnston, D.; Hoffman, D.A.; Magee, J.C.; Poolos, N.P.; Watanabe, S.; Colbert, C.M.; Migliore, M. Dendritic potassium channels in hippocampal pyramidal neurons. J. Physiol. 2000, 525, 75–81. [Google Scholar] [CrossRef]
- Bernard, C.; Anderson, A.; Becker, A.; Poolos, N.P.; Beck, H.; Johnston, D. Acquired dendritic channelopathy in temporal lobe epilepsy. Science 2004, 305, 532–535. [Google Scholar] [CrossRef] [Green Version]
- Hall, A.M.; Throesch, B.T.; Buckingham, S.C.; Markwardt, S.J.; Peng, Y.; Wang, Q.; Hoffman, D.A.; Roberson, E.D. Tau-Dependent depletion and dendritic hyperexcitability in a mouse model of Alzheimer’s Disease. J. Neurosci. 2015, 35, 6221–6230. [Google Scholar] [CrossRef]
- Pongs, O.; Schwarz, J.R. Ancillary subunits associated with voltage-gated K+ channels. Physiol. Rev. 2010, 90, 755–796. [Google Scholar] [CrossRef] [Green Version]
- Nadal, M.S.; Ozaita, A.; Amarillo, Y.; Vega-Saenz de Miera, E.; Ma, Y.; Mo, W.; Goldberg, E.M.; Misumi, Y.; Ikehara, Y.; Neubert, T.; et al. The CD26-related dipeptidyl aminopeptidase-like protein DPPX is a critical component of neuronal A-type K+ channels. Neuron 2003, 37, 449–461. [Google Scholar] [CrossRef] [Green Version]
- Rhodes, K.J.; Carroll, K.I.; Sung, M.A.; Doliveira, L.C.; Monaghan, M.M.; Burke, S.L.; Strassle, B.W.; Buchwalder, L.; Mengola, M.; Cao, J.; et al. KChIPs anf Kv4 α subunits as integral components of A-type potassium channels in mammalian brain. J. Neurosci. 2004, 24, 7903–7915. [Google Scholar] [CrossRef] [PubMed]
- Jerng, H.H.; Pfaffinger, P.J.; Covarrubias, M. Molecular physiology and modulation of somatodendritic A-type potassium channels. Mol. Cell Neurosci. 2004, 27, 343–369. [Google Scholar] [CrossRef]
- Jerng, H.H.; Kunjilwar, K.; Pfaffinger, P.J. Multiprotein assembly of Kv4.2, KChIP3, and DPP10 produces ternary channel complexes with ISA-like properties. J. Physiol. 2005, 568, 767–788. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-G.; He, X.P.; Li, Q.; Madison, R.D.; Moore, S.D.; McNamara, J.O.; Pitt, G.S. The auxiliary subunit KChIP2 is an essential regulator of homeostatic excitability. J. Biol. Chem. 2013, 288, 13258–13268. [Google Scholar] [CrossRef] [Green Version]
- Schrader, L.A.; Birnbaum, S.G.; Nadin, B.; Bui, D.; Anderson, A.E.; Sweatt, J.D. ERK/MAPK regulates the Kv4.2 potassium channel by direct phosphorylation of the pore-forming subunit. Am. J. Physiol. 2006, 290, 852–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammond, R.S.; Lin, L.; Sidorov, M.S.; Wikenheiser, A.M.; Hoffman, D.A. Protein Kinase A mediates activity-dependent Kv4.2 channel trafficking. J. Neurosci. 2008, 28, 7513–7519. [Google Scholar] [CrossRef]
- Schrader, L.A.; Ren, Y.-J.; Cheng, F.; Bui, D.; Sweatt, J.D.; Anderson, A.E. Kv4.2 is a locus for PKC and ERK/MAPK cross-talk. Biochem. J. 2009, 41, 705–715. [Google Scholar] [CrossRef]
- Kim, J.; Jung, S.-C.; Clemens, A.M.; Petralia, R.S.; Hoffman, D.A. Regulation of dendritic excitability by activity-dependent trafficking of the A-type K+ channel subunit Kv4.2 in hippocampal neurons. Neuron 2007, 54, 933–947. [Google Scholar] [CrossRef] [Green Version]
- Jung, S.-C.; Hoffman, D.A. Biphasic somatic A-type K channel downregulation mediates intrinsic plasticity in hippocampal CA1 pyramidal neurons. PLoS ONE 2009, 4, e6549. [Google Scholar] [CrossRef] [Green Version]
- Rosenkranz, J.A.; Frick, A.; Johnston, D. Kinase-dependent modification of dendritic excitability after long-term potentiation. J. Physiol. 2009, 587, 115–125. [Google Scholar] [CrossRef]
- Hu, J.H.; Malloy, C.; Tabor, G.T.; Gutzmann, J.J.; Liu, Y.; Abebe, D.; Karlsson, R.M.; Durrell, S.; Cameron, H.A.; Hoffman, D.A. Activity-dependent isomerization of Kv4.2 by Pin1 regulates cognitive flexibility. Nat. Commun. 2020, 11, 1567. [Google Scholar] [CrossRef] [Green Version]
- Racine, R.J. Modification of seizure activity by electrical stimulation: II. Motor seizure. Electroencephalogr. Clin. Neurophysiol. 1972, 32, 281–294. [Google Scholar] [CrossRef]
- Namiki, K.; Nakamura, A.; Furuya, M.; Mizuhashi, S.; Matsuo, Y.; Tokuhara, N.; Sudo, T.; Hama, H.; Kuwaki, T.; Yano, S.; et al. Involvement of p38alpha in kainite-induced seizure and neuronal cell damage. J. Recept. Signal Tranduct. 2007, 27, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Bertram, E. The relevance of kindling for human epilepsy. Epilepsia 2007, 48 (Suppl. 2), 65–74. [Google Scholar] [CrossRef]
- Adams, J.P.; Sweatt, J.D. Molecular psychology: Roles for ERK MAP kinase cascade in memory. Annu. Rev. Pharmacol. Toxicol. 2002, 42, 135–163. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.H.; Kim, Y.S.; Bae, C.D.; Park, J.B. Activation of JNK and p38 in rat hippocampus after kainic acid induced seizure. Exp. Mol. Med. 2000, 32, 227–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Wei, D.-S.; Hoffman, D.A. Kv4 potassium channel subunits control action potential repolarization and frequency-dependent broadening in rat hippocampal CA1 pyramidal neurons. J. Physiol. 2005, 569 Pt 1, 41–57. [Google Scholar] [CrossRef] [PubMed]
- Carrasquillo, Y.; Burkhalter, A.; Nerbonne, J.M. A-type K+ channels encoded by Kv4.2, Kv4.3 and Kv1.4 differentially regulate intrinsic excitability of cortical pyramidal neurons. J. Physiol. 2012, 590, 3877–3890. [Google Scholar] [CrossRef] [Green Version]
- Menegola, M.; Misonou, H.; Vacher, H.; Trimmer, J.S. Dendritic A-type potassium channel subunit expression in CA1 hippocampal interneurons. Neuroscience 2008, 154, 953–964. [Google Scholar] [CrossRef] [Green Version]
- Trimmer, J.S. Subcellular localization of K+ channels in mammalian brain neurons: Remarkable precision in the midst of extraordinary complexity. Neuron 2015, 85, 238–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfaro-Ruíz, R.; Aguado, C.; Martín-Belmonte, A.; Moreno-Martínez, A.E.; Luján, R. Expression, cellular and subcellular localization of Kv4.2 and Kv4.3 channels in the rodent hippocampus. Int. J. Mol. Sci. 2019, 20, 246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, W.; Maffie, J.K.; Lin, L.; Petralia, R.S.; Rudy, B.; Hoffman, D.A. DPP6 establishes the A-type K+ current gradient critical for the regulation of dendritic excitability in CA1 hippocampal neurons. Neuron 2011, 71, 1102–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | WT | WT (SB 203580) | Kv4.2TA | Kv4.2TA (SB 203580) |
|---|---|---|---|---|
| RMP (mv) | −60.5 ± 0.86 | −60.1 ± 0.72 | −58.8 ± 0.62 | −58.9 ± 0.60 |
| Whole-cell capacitance (pF) | 16.2 ± 1.0 | 22.4 ± 1.6 | 17.1 ± 0.77 | 17.8 ± 1.3 |
| Rinput (MΩ) | 228.8 ± 18.2 | 273.4 ± 19.7 | 231.2 ± 13.4 | 228.9 ± 16.0 |
| Time to AP Peak (ms) | 0.84 ± 0.1 | 1.07 ± 0.1 | 0.9 ± 0.1 | 0.89 ± 0.1 |
| AP amplitude (mV) | 81.0 ± 3.3 | 79.6 ± 3.7 | 76.7 ± 2.8 | 75.1 ± 3.3 |
| AP half-width (ms) | 1.5 ± 0.1 | 1.7 ± 0.2 | 1.6 ± 0.1 | 1.6 ± 0.1 |
| AP threshold (mV) | −40.1 ± 1.1 | −35.9 ± 1.5 | −34.9 ± 1.4 a | −37.7 ± 1.4 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, J.-h.; Malloy, C.; Hoffman, D.A. P38 Regulates Kainic Acid-Induced Seizure and Neuronal Firing via Kv4.2 Phosphorylation. Int. J. Mol. Sci. 2020, 21, 5921. https://doi.org/10.3390/ijms21165921
Hu J-h, Malloy C, Hoffman DA. P38 Regulates Kainic Acid-Induced Seizure and Neuronal Firing via Kv4.2 Phosphorylation. International Journal of Molecular Sciences. 2020; 21(16):5921. https://doi.org/10.3390/ijms21165921
Chicago/Turabian StyleHu, Jia-hua, Cole Malloy, and Dax A. Hoffman. 2020. "P38 Regulates Kainic Acid-Induced Seizure and Neuronal Firing via Kv4.2 Phosphorylation" International Journal of Molecular Sciences 21, no. 16: 5921. https://doi.org/10.3390/ijms21165921
APA StyleHu, J.-h., Malloy, C., & Hoffman, D. A. (2020). P38 Regulates Kainic Acid-Induced Seizure and Neuronal Firing via Kv4.2 Phosphorylation. International Journal of Molecular Sciences, 21(16), 5921. https://doi.org/10.3390/ijms21165921