HSV-1 Cytoplasmic Envelopment and Egress


Abstract
:1. Introduction
2. Emergence of Non-Enveloped Capsids from the Nucleus and Recruitment of Inner Tegument
3. Capsid Transport and Envelopment in the Cytoplasm
3.1. Reorganization of Microtubules in the HSV-1 Infected Cell
3.2. Trafficking of Non-Enveloped HSV-1 Capsids along Microtubules
3.3. Identity of the HSV-1 Cytoplasmic Envelopment Organelle
3.4. Delivery of Envelope Proteins to the Site of Envelopment: Roles for gK/UL20p and gM
3.5. Capsid Docking to the Surface of the Envelopment Organelle
3.6. Capsid Envelopment and the Roles of HSV-1 Encoded Envelope and Tegument Proteins
3.7. The Cellular ESCRT Apparatus in Envelopment and Scission
3.8. MT-Directed Transport of Enveloping Capsids Is Arrested until Envelopment Is Complete
4. Sorting of Virions in Polarized and Non-Polarized Cells
4.1. The Virally Encoded Membrane Proteins gE, gI, and US9p: An Overview
4.2. Sorting of HSV-1 in Polarized and Non-Polarized Epithelial Cells
4.3. Sorting of HSV-1 in Neurons, the Married and Separate Models
4.4. Molecular Functions for gE/gI and US9p in the Married and Separate Mechanisms
4.5. HSV-1 Transmission via Tunneling Nanotubes
5. Late Exocytic Events: Emergence from the Cell Surface
6. Conclusions
- How do HSV-1 capsids recognize and select the lipid bilayer of a specific organelle for their envelopment? How does the reorganization of MTs and the MTOC ensure efficient delivery of non-enveloped capsids to that location?
- If the capsid-bound or TGN-associated UL7p/(UL51p)2 complex triggers ESCRT-III assembly, how does it choose the location and timing of filament polymerization? How is this function coordinated with the activity of other viral structural proteins known to be important for envelopment?
- What mechanisms exist to ensure that all necessary envelope and outer tegument proteins are loaded into the nascent envelopment site before ESCRT-III-driven envelope scission, the “point of no return”, occurs? How is MT-directed motility of the envelopment intermediate suppressed during assembly?
- How do gE/gI and US9p interact with kinesin motors during egress of capsids and enveloped virions? Which motors are utilized before and after envelopment, and how are they recruited and exchanged? Does motor recruitment and modification of cell–cell junctions explain all of the features of HSV-1 particle sorting in epithelial cells, or do gE/gI serve other functions?
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| HSV-1 | Herpes simplex virus type 1 |
| PRV | Pseudorabies virus (suid alphaherpesvirus 1) |
| ULnumber | Unique long (position of gene in HSV-1 genome) |
| USnumber | Unique short (position of gene in HSV-1 genome) |
| VPnumber | Virus protein |
| gletter. | Virally encoded membrane glycoprotein |
| ER | Endoplasmic reticulum |
| ESCRT | Endosomal sorting complex required for transport |
| MT | Microtubules |
| OEV | Organelle-associated enveloped virion |
| TGN | Trans-Golgi network |
| TNT | Tunneling nanotube |
References
- Roizman, B.; Knipe, D.M.; Whitley, R.J. Herpes Simplex Viruses. In Fields Virology; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; Volume 2, pp. 1824–1898. [Google Scholar]
- Pellett, P.E.; Roizman, B. Herpesviridae. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; Volume 2, pp. 1802–1822. [Google Scholar]
- Steiner, I.; Kennedy, P.G.; Pachner, A.R. The neurotropic herpes viruses: Herpes simplex and varicella-zoster. Lancet Neurol. 2007, 6, 1015–1028. [Google Scholar] [CrossRef]
- Kramer, T.; Enquist, L.W. Directional spread of alphaherpesviruses in the nervous system. Viruses 2013, 5, 678–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diwaker, D.; Wilson, D.W. Microtubule-Dependent Trafficking of Alphaherpesviruses in the Nervous System: The Ins and Outs. Viruses 2019, 11, 1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pomeranz, L.E.; Reynolds, A.E.; Hengartner, C.J. Molecular biology of pseudorabies virus: Impact on neurovirology and veterinary medicine. Microbiol. Mol. Biol. Rev. 2005, 69, 462–500. [Google Scholar] [CrossRef] [Green Version]
- Smith, G. Herpesvirus transport to the nervous system and back again. Annu. Rev. Microbiol. 2012, 66, 153–176. [Google Scholar] [CrossRef] [Green Version]
- Denes, C.E.; Miranda-Saksena, M.; Cunningham, A.L.; Diefenbach, R.J. Cytoskeletons in the Closet-Subversion in Alphaherpesvirus Infections. Viruses 2018, 10, 79. [Google Scholar] [CrossRef] [Green Version]
- Valerio, G.S.; Lin, C.C. Ocular manifestations of herpes simplex virus. Curr. Opin. Ophthalmol. 2019, 30, 525–531. [Google Scholar] [CrossRef]
- Smith, G.A. Assembly and Egress of an Alphaherpesvirus Clockwork. Adv. Anat. Embryol. Cell Biol. 2017, 223, 171–193. [Google Scholar] [CrossRef] [Green Version]
- Owen, D.J.; Crump, C.M.; Graham, S.C. Tegument Assembly and Secondary Envelopment of Alphaherpesviruses. Viruses 2015, 7, 5084–5114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, J.; Wilson, D.W. Seeking Closure: How Do Herpesviruses Recruit the Cellular ESCRT Apparatus? J. Virol. 2019, 93, e00392-00319. [Google Scholar] [CrossRef] [Green Version]
- Crump, C. Virus Assembly and Egress of HSV. Adv. Exp. Med. Biol. 2018, 1045, 23–44. [Google Scholar] [CrossRef] [PubMed]
- Heming, J.D.; Conway, J.F.; Homa, F.L. Herpesvirus Capsid Assembly and DNA Packaging. Adv. Anat. Embryol. Cell Biol. 2017, 223, 119–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bigalke, J.M.; Heldwein, E.E. Nuclear Exodus: Herpesviruses Lead the Way. Annu. Rev. Virol. 2016, 3, 387–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bigalke, J.M.; Heldwein, E.E. Have NEC Coat, Will Travel: Structural Basis of Membrane Budding During Nuclear Egress in Herpesviruses. Adv. Virus Res. 2017, 97, 107–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mettenleiter, T.C. Vesicular Nucleo-Cytoplasmic Transport-Herpesviruses as Pioneers in Cell Biology. Viruses 2016, 8, 266. [Google Scholar] [CrossRef] [Green Version]
- Arii, J.; Watanabe, M.; Maeda, F.; Tokai-Nishizumi, N.; Chihara, T.; Miura, M.; Maruzuru, Y.; Koyanagi, N.; Kato, A.; Kawaguchi, Y. ESCRT-III mediates budding across the inner nuclear membrane and regulates its integrity. Nat. Commun. 2018, 9, 3379. [Google Scholar] [CrossRef] [Green Version]
- Banfield, B.W. Beyond the NEC: Modulation of Herpes Simplex Virus Nuclear Egress by Viral and Cellular Components. Curr. Clin. Microbiol. Rep. 2019, 6, 1–9. [Google Scholar] [CrossRef]
- Yuan, S.; Wang, J.; Zhu, D.; Wang, N.; Gao, Q.; Chen, W.; Tang, H.; Wang, J.; Zhang, X.; Liu, H.; et al. Cryo-EM structure of a herpesvirus capsid at 3.1 A. Science 2018, 360. [Google Scholar] [CrossRef] [Green Version]
- Dai, X.; Zhou, Z.H. Structure of the herpes simplex virus 1 capsid with associated tegument protein complexes. Science 2018, 360, eaao7298. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.C.; Baines, J.D. Herpesviruses remodel host membranes for virus egress. Nat. Rev. Microbiol. 2011, 9, 382–394. [Google Scholar] [CrossRef]
- Mettenleiter, T.C.; Klupp, B.G.; Granzow, H. Herpesvirus assembly: An update. Virus Res. 2009, 143, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.H.; Roberts, A.P.; McElwee, M.; Bhella, D.; Rixon, F.J.; Lauder, R. The large tegument protein pUL36 is essential for formation of the capsid vertex-specific component at the capsid-tegument interface of herpes simplex virus 1. J. Virol. 2015, 89, 1502–1511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koenigsberg, A.L.; Heldwein, E.E. The dynamic nature of the conserved tegument protein UL37 of herpesviruses. J. Biol. Chem. 2018, 293, 15827–15839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynolds, A.E.; Wills, E.G.; Roller, R.J.; Ryckman, B.J.; Baines, J.D. Ultrastructural localization of the herpes simplex virus type 1 UL31, UL34, and US3 proteins suggests specific roles in primary envelopment and egress of nucleocapsids. J. Virol. 2002, 76, 8939–8952. [Google Scholar] [CrossRef] [Green Version]
- Henaff, D.; Remillard-Labrosse, G.; Loret, S.; Lippe, R. Analysis of the early steps of herpes simplex virus 1 capsid tegumentation. J. Virol. 2013, 87, 4895–4906. [Google Scholar] [CrossRef] [Green Version]
- Kato, A.; Kawaguchi, Y. Us3 Protein Kinase Encoded by HSV: The Precise Function and Mechanism on Viral Life Cycle. Adv. Exp. Med. Biol. 2018, 1045, 45–62. [Google Scholar] [CrossRef]
- Newcomb, W.W.; Brown, J.C. Structure and capsid association of the herpesvirus large tegument protein UL36. J. Virol. 2010, 84, 9408–9414. [Google Scholar] [CrossRef] [Green Version]
- Conway, J.F.; Cockrell, S.K.; Copeland, A.M.; Newcomb, W.W.; Brown, J.C.; Homa, F.L. Labeling and localization of the herpes simplex virus capsid protein UL25 and its interaction with the two triplexes closest to the penton. J. Mol. Biol. 2010, 397, 575–586. [Google Scholar] [CrossRef] [Green Version]
- Schipke, J.; Pohlmann, A.; Diestel, R.; Binz, A.; Rudolph, K.; Nagel, C.H.; Bauerfeind, R.; Sodeik, B. The C terminus of the large tegument protein pUL36 contains multiple capsid binding sites that function differently during assembly and cell entry of herpes simplex virus. J. Virol. 2012, 86, 3682–3700. [Google Scholar] [CrossRef] [Green Version]
- Cardone, G.; Newcomb, W.W.; Cheng, N.; Wingfield, P.T.; Trus, B.L.; Brown, J.C.; Steven, A.C. The UL36 tegument protein of herpes simplex virus 1 has a composite binding site at the capsid vertices. J. Virol. 2012, 86, 4058–4064. [Google Scholar] [CrossRef] [Green Version]
- Vittone, V.; Diefenbach, E.; Triffett, D.; Douglas, M.W.; Cunningham, A.L.; Diefenbach, R.J. Determination of interactions between tegument proteins of herpes simplex virus type 1. J. Virol. 2005, 79, 9566–9571. [Google Scholar] [CrossRef] [Green Version]
- Svobodova, S.; Bell, S.; Crump, C.M. Analysis of the interaction between the essential herpes simplex virus 1 tegument proteins VP16 and VP1/2. J. Virol. 2012, 86, 473–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamen, D.E.; Gross, S.T.; Girvin, M.E.; Wilson, D.W. Structural basis for the physiological temperature dependence of the association of VP16 with the cytoplasmic tail of herpes simplex virus glycoprotein H. J. Virol. 2005, 79, 6134–6141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, S.T.; Harley, C.A.; Wilson, D.W. The cytoplasmic tail of Herpes simplex virus glycoprotein H binds to the tegument protein VP16 in vitro and in vivo. Virology 2003, 317, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fossum, E.; Friedel, C.C.; Rajagopala, S.V.; Titz, B.; Baiker, A.; Schmidt, T.; Kraus, T.; Stellberger, T.; Rutenberg, C.; Suthram, S.; et al. Evolutionarily conserved herpesviral protein interaction networks. PLoS Pathog. 2009, 5, e1000570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diefenbach, R.J. Conserved tegument protein complexes: Essential components in the assembly of herpesviruses. Virus Res. 2015, 210, 308–317. [Google Scholar] [CrossRef]
- Kelly, B.J.; Mijatov, B.; Fraefel, C.; Cunningham, A.L.; Diefenbach, R.J. Identification of a single amino acid residue which is critical for the interaction between HSV-1 inner tegument proteins pUL36 and pUL37. Virology 2012, 422, 308–316. [Google Scholar] [CrossRef] [Green Version]
- Kelly, B.J.; Bauerfeind, R.; Binz, A.; Sodeik, B.; Laimbacher, A.S.; Fraefel, C.; Diefenbach, R.J. The interaction of the HSV-1 tegument proteins pUL36 and pUL37 is essential for secondary envelopment during viral egress. Virology 2014, 454–455, 67–77. [Google Scholar] [CrossRef] [Green Version]
- Mijatov, B.; Cunningham, A.L.; Diefenbach, R.J. Residues F593 and E596 of HSV-1 tegument protein pUL36 (VP1/2) mediate binding of tegument protein pUL37. Virology 2007, 368, 26–31. [Google Scholar] [CrossRef] [Green Version]
- Pitts, J.D.; Klabis, J.; Richards, A.L.; Smith, G.A.; Heldwein, E.E. Crystal structure of the herpesvirus inner tegument protein UL37 supports its essential role in control of viral trafficking. J. Virol. 2014, 88, 5462–5473. [Google Scholar] [CrossRef] [Green Version]
- Koenigsberg, A.L.; Heldwein, E.E. Crystal Structure of the N-Terminal Half of the Traffic Controller UL37 from Herpes Simplex Virus 1. J. Virol. 2017, 91, e01244-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grzesik, P.; Pryce, E.N.; Bhalala, A.; Vij, M.; Ahmed, R.; Etienne, L.; Perez, P.; McCaffery, J.M.; Desai, A.P.J. Functional Domains of the Herpes Simplex Virus Type 1 Tegument Protein pUL37: The Amino Terminus is Dispensable for Virus Replication in Tissue Culture. Viruses 2019, 11, 853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metrick, C.M.; Koenigsberg, A.L.; Heldwein, E.E. Conserved Outer Tegument Component UL11 from Herpes Simplex Virus 1 Is an Intrinsically Disordered, RNA-Binding Protein. mBio 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Leege, T.; Fuchs, W.; Granzow, H.; Kopp, M.; Klupp, B.G.; Mettenleiter, T.C. Effects of simultaneous deletion of pUL11 and glycoprotein M on virion maturation of herpes simplex virus type 1. J. Virol. 2009, 83, 896–907. [Google Scholar] [CrossRef] [Green Version]
- Loomis, J.S.; Courtney, R.J.; Wills, J.W. Binding partners for the UL11 tegument protein of herpes simplex virus type 1. J. Virol. 2003, 77, 11417–11424. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Chadha, P.; Meckes, D.G., Jr.; Baird, N.L.; Wills, J.W. Interaction and interdependent packaging of tegument protein UL11 and glycoprotein E of herpes simplex virus. J. Virol. 2011, 85, 9437–9446. [Google Scholar] [CrossRef] [Green Version]
- Yeh, P.C.; Meckes, D.G., Jr.; Wills, J.W. Analysis of the interaction between the UL11 and UL16 tegument proteins of herpes simplex virus. J. Virol. 2008, 82, 10693–10700. [Google Scholar] [CrossRef] [Green Version]
- Meckes, D.G., Jr.; Marsh, J.A.; Wills, J.W. Complex mechanisms for the packaging of the UL16 tegument protein into herpes simplex virus. Virology 2010, 398, 208–213. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Chadha, P.; Starkey, J.L.; Wills, J.W. Function of glycoprotein E of herpes simplex virus requires coordinated assembly of three tegument proteins on its cytoplasmic tail. Proc. Natl. Acad. Sci. USA 2012, 109, 19798–19803. [Google Scholar] [CrossRef] [Green Version]
- Haugo, A.C.; Szpara, M.L.; Parsons, L.; Enquist, L.W.; Roller, R.J. Herpes simplex virus 1 pUL34 plays a critical role in cell-to-cell spread of virus in addition to its role in virus replication. J. Virol. 2011, 85, 7203–7215. [Google Scholar] [CrossRef] [Green Version]
- Radtke, K.; Kieneke, D.; Wolfstein, A.; Michael, K.; Steffen, W.; Scholz, T.; Karger, A.; Sodeik, B. Plus- and minus-end directed microtubule motors bind simultaneously to herpes simplex virus capsids using different inner tegument structures. PLoS Pathog. 2010, 6, e1000991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanova, L.; Buch, A.; Dohner, K.; Pohlmann, A.; Binz, A.; Prank, U.; Sandbaumhuter, M.; Bauerfeind, R.; Sodeik, B. Conserved Tryptophan Motifs in the Large Tegument Protein pUL36 Are Required for Efficient Secondary Envelopment of Herpes Simplex Virus Capsids. J. Virol. 2016, 90, 5368–5383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, P.J. A null mutation in the UL36 gene of herpes simplex virus type 1 results in accumulation of unenveloped DNA-filled capsids in the cytoplasm of infected cells. J. Virol. 2000, 74, 11608–11618. [Google Scholar] [CrossRef] [Green Version]
- Sandbaumhuter, M.; Dohner, K.; Schipke, J.; Binz, A.; Pohlmann, A.; Sodeik, B.; Bauerfeind, R. Cytosolic herpes simplex virus capsids not only require binding inner tegument protein pUL36 but also pUL37 for active transport prior to secondary envelopment. Cell. Microbiol. 2013, 15, 248–269. [Google Scholar] [CrossRef] [PubMed]
- Shanda, S.K.; Wilson, D.W. UL36p is required for efficient transport of membrane-associated herpes simplex virus type 1 along microtubules. J. Virol. 2008, 82, 7388–7394. [Google Scholar] [CrossRef] [Green Version]
- Kharkwal, H.; Furgiuele, S.S.; Smith, C.G.; Wilson, D.W. Herpes Simplex Virus Capsid-Organelle Association in the Absence of the Large Tegument Protein UL36p. J. Virol. 2015, 89, 11372–11382. [Google Scholar] [CrossRef] [Green Version]
- Kharkwal, H.; Smith, C.G.; Wilson, D.W. HSV capsid localization to ESCRT-VPS4 complexes in the presence and absence of the large tegument protein UL36p. J. Virol. 2016, 90, 7257–7267. [Google Scholar] [CrossRef] [Green Version]
- Chouljenko, D.V.; Jambunathan, N.; Chouljenko, V.N.; Naderi, M.; Brylinski, M.; Caskey, J.R.; Kousoulas, K.G. Herpes Simplex Virus 1 UL37 Protein Tyrosine Residues Conserved among All Alphaherpesviruses Are Required for Interactions with Glycoprotein K, Cytoplasmic Virion Envelopment, and Infectious Virus Production. J. Virol. 2016, 90, 10351–10361. [Google Scholar] [CrossRef] [Green Version]
- Jambunathan, N.; Chouljenko, D.; Desai, P.; Charles, A.S.; Subramanian, R.; Chouljenko, V.N.; Kousoulas, K.G. Herpes simplex virus 1 protein UL37 interacts with viral glycoprotein gK and membrane protein UL20 and functions in cytoplasmic virion envelopment. J. Virol. 2014, 88, 5927–5935. [Google Scholar] [CrossRef] [Green Version]
- Desai, P.; Sexton, G.L.; McCaffery, J.M.; Person, S. A null mutation in the gene encoding the herpes simplex virus type 1 UL37 polypeptide abrogates virus maturation. J. Virol. 2001, 75, 10259–10271. [Google Scholar] [CrossRef] [Green Version]
- Naghavi, M.H.; Gundersen, G.G.; Walsh, D. Plus-end tracking proteins, CLASPs, and a viral Akt mimic regulate herpesvirus-induced stable microtubule formation and virus spread. Proc. Natl. Acad. Sci. USA 2013, 110, 18268–18273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finnen, R.L.; Roy, B.B.; Zhang, H.; Banfield, B.W. Analysis of filamentous process induction and nuclear localization properties of the HSV-2 serine/threonine kinase Us3. Virology 2010, 397, 23–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favoreel, H.W.; Van Minnebruggen, G.; Adriaensen, D.; Nauwynck, H.J. Cytoskeletal rearrangements and cell extensions induced by the US3 kinase of an alphaherpesvirus are associated with enhanced spread. Proc. Natl. Acad. Sci. USA 2005, 102, 8990–8995. [Google Scholar] [CrossRef] [Green Version]
- Roller, R.J.; Fetters, R. The herpes simplex virus 1 UL51 protein interacts with the UL7 protein and plays a role in its recruitment into the virion. J. Virol. 2015, 89, 3112–3122. [Google Scholar] [CrossRef] [Green Version]
- Albecka, A.; Owen, D.J.; Ivanova, L.; Brun, J.; Liman, R.; Davies, L.; Ahmed, M.F.; Colaco, S.; Hollinshead, M.; Graham, S.C.; et al. Dual Function of the pUL7-pUL51 Tegument Protein Complex in Herpes Simplex Virus 1 Infection. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roller, R.J.; Haugo, A.C.; Yang, K.; Baines, J.D. The herpes simplex virus 1 UL51 gene product has cell type-specific functions in cell-to-cell spread. J. Virol. 2014, 88, 4058–4068. [Google Scholar] [CrossRef] [Green Version]
- Butt, B.G.; Owen, D.J.; Jeffries, C.M.; Ivanova, L.; Hill, C.H.; Houghton, J.W.; Ahmed, M.F.; Antrobus, R.; Svergun, D.I.; Welch, J.J.; et al. Insights into herpesvirus assembly from the structure of the pUL7:pUL51 complex. Elife 2020, 9. [Google Scholar] [CrossRef]
- Scholtes, L.D.; Yang, K.; Li, L.X.; Baines, J.D. The capsid protein encoded by U(L)17 of herpes simplex virus 1 interacts with tegument protein VP13/14. J. Virol. 2010, 84, 7642–7650. [Google Scholar] [CrossRef] [Green Version]
- Mossman, K.L.; Sherburne, R.; Lavery, C.; Duncan, J.; Smiley, J.R. Evidence that herpes simplex virus VP16 is required for viral egress downstream of the initial envelopment event. J. Virol. 2000, 74, 6287–6299. [Google Scholar] [CrossRef] [Green Version]
- Weinheimer, S.P.; Boyd, B.A.; Durham, S.K.; Resnick, J.L.; O’Boyle, D.R., 2nd. Deletion of the VP16 open reading frame of herpes simplex virus type 1. J. Virol. 1992, 66, 258–269. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Q.; Courtney, R.J. Chemical crosslinking of glycoproteins on the envelope of herpes simplex virus. Virology 1988, 167, 377–384. [Google Scholar] [CrossRef]
- Chi, J.H.; Harley, C.A.; Mukhopadhyay, A.; Wilson, D.W. The cytoplasmic tail of herpes simplex virus envelope glycoprotein D binds to the tegument protein VP22 and to capsids. J. Gen. Virol. 2005, 86, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Kotsakis, A.; Pomeranz, L.E.; Blouin, A.; Blaho, J.A. Microtubule reorganization during herpes simplex virus type 1 infection facilitates the nuclear localization of VP22, a major virion tegument protein. J. Virol. 2001, 75, 8697–8711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elliott, G.; O’Hare, P. Herpes simplex virus type 1 tegument protein VP22 induces the stabilization and hyperacetylation of microtubules. J. Virol. 1998, 72, 6448–6455. [Google Scholar] [CrossRef] [Green Version]
- Farnsworth, A.; Wisner, T.W.; Johnson, D.C. Cytoplasmic residues of herpes simplex virus glycoprotein gE required for secondary envelopment and binding of tegument proteins VP22 and UL11 to gE and gD. J. Virol. 2007, 81, 319–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beitia Ortiz de Zarate, I.; Cantero-Aguilar, L.; Longo, M.; Berlioz-Torrent, C.; Rozenberg, F. Contribution of endocytic motifs in the cytoplasmic tail of herpes simplex virus type 1 glycoprotein B to virus replication and cell-cell fusion. J. Virol. 2007, 81, 13889–13903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calistri, A.; Sette, P.; Salata, C.; Cancellotti, E.; Forghieri, C.; Comin, A.; Gottlinger, H.; Campadelli-Fiume, G.; Palu, G.; Parolin, C. Intracellular trafficking and maturation of herpes simplex virus type 1 gB and virus egress require functional biogenesis of multivesicular bodies. J. Virol. 2007, 81, 11468–11478. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.C.; Wisner, T.W.; Wright, C.C. Herpes simplex virus glycoproteins gB and gD function in a redundant fashion to promote secondary envelopment. J. Virol. 2011, 85, 4910–4926. [Google Scholar] [CrossRef] [Green Version]
- Turner, A.; Bruun, B.; Minson, T.; Browne, H. Glycoproteins gB, gD, and gHgL of herpes simplex virus type 1 are necessary and sufficient to mediate membrane fusion in a Cos cell transfection system. J. Virol. 1998, 72, 873–875. [Google Scholar] [CrossRef] [Green Version]
- Beitia Ortiz de Zarate, I.; Kaelin, K.; Rozenberg, F. Effects of mutations in the cytoplasmic domain of herpes simplex virus type 1 glycoprotein B on intracellular transport and infectivity. J. Virol. 2004, 78, 1540–1551. [Google Scholar] [CrossRef] [Green Version]
- Farnsworth, A.; Goldsmith, K.; Johnson, D.C. Herpes simplex virus glycoproteins gD and gE/gI serve essential but redundant functions during acquisition of the virion envelope in the cytoplasm. J. Virol. 2003, 77, 8481–8494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DuRaine, G.; Wisner, T.W.; Howard, P.; Williams, M.; Johnson, D.C. Herpes Simplex Virus gE/gI and US9 Promote both Envelopment and Sorting of Virus Particles in the Cytoplasm of Neurons, Two Processes That Precede Anterograde Transport in Axons. J. Virol. 2017, 91, e00050-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howard, P.W.; Howard, T.L.; Johnson, D.C. Herpes simplex virus membrane proteins gE/gI and US9 act cooperatively to promote transport of capsids and glycoproteins from neuron cell bodies into initial axon segments. J. Virol. 2013, 87, 403–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, A.; Polcicova, K.; Johnson, D.C. Herpes simplex virus gE/gI and US9 proteins promote transport of both capsids and virion glycoproteins in neuronal axons. J. Virol. 2008, 82, 10613–10624. [Google Scholar] [CrossRef] [Green Version]
- Farnsworth, A.; Johnson, D.C. Herpes simplex virus gE/gI must accumulate in the trans-Golgi network at early times and then redistribute to cell junctions to promote cell-cell spread. J. Virol. 2006, 80, 3167–3179. [Google Scholar] [CrossRef] [Green Version]
- Polcicova, K.; Goldsmith, K.; Rainish, B.L.; Wisner, T.W.; Johnson, D.C. The extracellular domain of herpes simplex virus gE is indispensable for efficient cell-to-cell spread: Evidence for gE/gI receptors. J. Virol. 2005, 79, 11990–12001. [Google Scholar] [CrossRef] [Green Version]
- Alconada, A.; Bauer, U.; Sodeik, B.; Hoflack, B. Intracellular traffic of herpes simplex virus glycoprotein gE: Characterization of the sorting signals required for its trans-Golgi network localization. J. Virol. 1999, 73, 377–387. [Google Scholar] [CrossRef] [Green Version]
- Lau, S.Y.; Crump, C.M. HSV-1 gM and the gK/pUL20 complex are important for the localization of gD and gH/L to viral assembly sites. Viruses 2015, 7, 915–938. [Google Scholar] [CrossRef] [Green Version]
- Foster, T.P.; Chouljenko, V.N.; Kousoulas, K.G. Functional and physical interactions of the herpes simplex virus type 1 UL20 membrane protein with glycoprotein K. J. Virol. 2008, 82, 6310–6323. [Google Scholar] [CrossRef] [Green Version]
- Foster, T.P.; Melancon, J.M.; Olivier, T.L.; Kousoulas, K.G. Herpes simplex virus type 1 glycoprotein K and the UL20 protein are interdependent for intracellular trafficking and trans-Golgi network localization. J. Virol. 2004, 78, 13262–13277. [Google Scholar] [CrossRef] [Green Version]
- Avitabile, E.; Lombardi, G.; Gianni, T.; Capri, M.; Campadelli-Fiume, G. Coexpression of UL20p and gK inhibits cell-cell fusion mediated by herpes simplex virus glycoproteins gD, gH-gL, and wild-type gB or an endocytosis-defective gB mutant and downmodulates their cell surface expression. J. Virol. 2004, 78, 8015–8025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Kasmi, I.; Lippe, R. Herpes simplex virus 1 gN partners with gM to modulate the viral fusion machinery. J. Virol. 2015, 89, 2313–2323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, Y.; Bell, S.; Zenner, H.L.; Lau, S.K.; Crump, C.M. Glycoprotein M is important for the efficient incorporation of glycoprotein H-L into herpes simplex virus type 1 particles. J. Gen. Virol. 2012, 93, 319–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crump, C.M.; Bruun, B.; Bell, S.; Pomeranz, L.E.; Minson, T.; Browne, H.M. Alphaherpesvirus glycoprotein M causes the relocalization of plasma membrane proteins. J. Gen. Virol. 2004, 85, 3517–3527. [Google Scholar] [CrossRef]
- Brideau, A.D.; Banfield, B.W.; Enquist, L.W. The Us9 gene product of pseudorabies virus, an alphaherpesvirus, is a phosphorylated, tail-anchored type II membrane protein. J. Virol. 1998, 72, 4560–4570. [Google Scholar] [CrossRef] [Green Version]
- Lyman, M.G.; Curanovic, D.; Enquist, L.W. Targeting of pseudorabies virus structural proteins to axons requires association of the viral Us9 protein with lipid rafts. PLoS Pathog. 2008, 4, e1000065. [Google Scholar] [CrossRef] [Green Version]
- Miranda-Saksena, M.; Boadle, R.A.; Diefenbach, R.J.; Cunningham, A.L. Dual Role of Herpes Simplex Virus 1 pUS9 in Virus Anterograde Axonal Transport and Final Assembly in Growth Cones in Distal Axons. J. Virol. 2015, 90, 2653–2663. [Google Scholar] [CrossRef] [Green Version]
- Diefenbach, R.J.; Davis, A.; Miranda-Saksena, M.; Fernandez, M.A.; Kelly, B.J.; Jones, C.A.; LaVail, J.H.; Xue, J.; Lai, J.; Cunningham, A.L. The Basic Domain of Herpes Simplex Virus 1 pUS9 Recruits Kinesin-1 To Facilitate Egress from Neurons. J. Virol. 2016, 90, 2102–2111. [Google Scholar] [CrossRef] [Green Version]
- McNabb, D.S.; Courtney, R.J. Characterization of the large tegument protein (ICP1/2) of herpes simplex virus type 1. Virology 1992, 190, 221–232. [Google Scholar] [CrossRef]
- Abaitua, F.; O’Hare, P. Identification of a highly conserved, functional nuclear localization signal within the N-terminal region of herpes simplex virus type 1 VP1-2 tegument protein. J. Virol. 2008, 82, 5234–5244. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, J.B.; Albright, A.G.; Kinchington, P.R.; Jenkins, F.J. The UL37 protein of herpes simplex virus type 1 is associated with the tegument of purified virions. Virology 1995, 206, 1055–1065. [Google Scholar] [CrossRef] [PubMed]
- Wolfstein, A.; Nagel, C.H.; Radtke, K.; Dohner, K.; Allan, V.J.; Sodeik, B. The inner tegument promotes herpes simplex virus capsid motility along microtubules in vitro. Traffic 2006, 7, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Bucks, M.A.; O’Regan, K.J.; Murphy, M.A.; Wills, J.W.; Courtney, R.J. Herpes simplex virus type 1 tegument proteins VP1/2 and UL37 are associated with intranuclear capsids. Virology 2007, 361, 316–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trus, B.L.; Newcomb, W.W.; Cheng, N.; Cardone, G.; Marekov, L.; Homa, F.L.; Brown, J.C.; Steven, A.C. Allosteric signaling and a nuclear exit strategy: Binding of UL25/UL17 heterodimers to DNA-Filled HSV-1 capsids. Mol. Cell 2007, 26, 479–489. [Google Scholar] [CrossRef] [Green Version]
- Boutell, C.; Everett, R.D. Regulation of alphaherpesvirus infections by the ICP0 family of proteins. J. Gen. Virol. 2013, 94, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Dremel, S.E.; DeLuca, N.A. Herpes simplex viral nucleoprotein creates a competitive transcriptional environment facilitating robust viral transcription and host shut off. Elife 2019, 8. [Google Scholar] [CrossRef]
- Maringer, K.; Elliott, G. Recruitment of herpes simplex virus type 1 immediate-early protein ICP0 to the virus particle. J. Virol. 2010, 84, 4682–4696. [Google Scholar] [CrossRef] [Green Version]
- Sedlackova, L.; Rice, S.A. Herpes simplex virus type 1 immediate-early protein ICP27 is required for efficient incorporation of ICP0 and ICP4 into virions. J. Virol. 2008, 82, 268–277. [Google Scholar] [CrossRef] [Green Version]
- Klupp, B.G.; Fuchs, W.; Granzow, H.; Nixdorf, R.; Mettenleiter, T.C. Pseudorabies virus UL36 tegument protein physically interacts with the UL37 protein. J. Virol. 2002, 76, 3065–3071. [Google Scholar] [CrossRef] [Green Version]
- Granzow, H.; Klupp, B.G.; Mettenleiter, T.C. The pseudorabies virus US3 protein is a component of primary and of mature virions. J. Virol. 2004, 78, 1314–1323. [Google Scholar] [CrossRef] [Green Version]
- Mohl, B.S.; Bottcher, S.; Granzow, H.; Kuhn, J.; Klupp, B.G.; Mettenleiter, T.C. Intracellular localization of the pseudorabies virus large tegument protein pUL36. J. Virol. 2009, 83, 9641–9651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luxton, G.W.; Haverlock, S.; Coller, K.E.; Antinone, S.E.; Pincetic, A.; Smith, G.A. Targeting of herpesvirus capsid transport in axons is coupled to association with specific sets of tegument proteins. Proc. Natl. Acad. Sci. USA 2005, 102, 5832–5837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leelawong, M.; Lee, J.I.; Smith, G.A. Nuclear egress of pseudorabies virus capsids is enhanced by a subspecies of the large tegument protein that is lost upon cytoplasmic maturation. J. Virol. 2012, 86, 6303–6314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasdeloup, D.; Labetoulle, M.; Rixon, F.J. Differing effects of herpes simplex virus 1 and pseudorabies virus infections on centrosomal function. J. Virol. 2013, 87, 7102–7112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wloga, D.; Joachimiak, E.; Fabczak, H. Tubulin Post-Translational Modifications and Microtubule Dynamics. Int. J. Mol. Sci. 2017, 18, 2207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, N.A.; Cai, D.; Blasius, T.L.; Jih, G.T.; Meyhofer, E.; Gaertig, J.; Verhey, K.J. Microtubule acetylation promotes kinesin-1 binding and transport. Curr. Biol. 2006, 16, 2166–2172. [Google Scholar] [CrossRef] [Green Version]
- Austefjord, M.W.; Gerdes, H.H.; Wang, X. Tunneling nanotubes: Diversity in morphology and structure. Commun. Integr. Biol. 2014, 7, e27934. [Google Scholar] [CrossRef]
- Rustom, A.; Saffrich, R.; Markovic, I.; Walther, P.; Gerdes, H.H. Nanotubular highways for intercellular organelle transport. Science 2004, 303, 1007–1010. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.E.; Murray, J.W.; Wolkoff, A.W.; Wilson, D.W. Reconstitution of herpes simplex virus microtubule-dependent trafficking in vitro. J. Virol. 2006, 80, 4264–4275. [Google Scholar] [CrossRef] [Green Version]
- Buch, A.; Muller, O.; Ivanova, L.; Dohner, K.; Bialy, D.; Bosse, J.B.; Pohlmann, A.; Binz, A.; Hegemann, M.; Nagel, C.H.; et al. Inner tegument proteins of Herpes Simplex Virus are sufficient for intracellular capsid motility in neurons but not for axonal targeting. PLoS Pathog. 2017, 13, e1006813. [Google Scholar] [CrossRef]
- Pasdeloup, D.; Beilstein, F.; Roberts, A.P.; McElwee, M.; McNab, D.; Rixon, F.J. Inner tegument protein pUL37 of herpes simplex virus type 1 is involved in directing capsids to the trans-Golgi network for envelopment. J. Gen. Virol. 2010, 91, 2145–2151. [Google Scholar] [CrossRef] [PubMed]
- Dodding, M.P.; Way, M. Coupling viruses to dynein and kinesin-1. EMBO J. 2011, 30, 3527–3539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodding, M.P.; Mitter, R.; Humphries, A.C.; Way, M. A kinesin-1 binding motif in vaccinia virus that is widespread throughout the human genome. EMBO J. 2011, 30, 4523–4538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pernigo, S.; Lamprecht, A.; Steiner, R.A.; Dodding, M.P. Structural basis for kinesin-1:cargo recognition. Science 2013, 340, 356–359. [Google Scholar] [CrossRef] [Green Version]
- Ferrier, A.; Boyer, J.G.; Kothary, R. Cellular and molecular biology of neuronal dystonin. Int. Rev. Cell Mol. Biol. 2013, 300, 85–120. [Google Scholar] [CrossRef]
- Pasdeloup, D.; McElwee, M.; Beilstein, F.; Labetoulle, M.; Rixon, F.J. Herpesvirus tegument protein pUL37 interacts with dystonin/BPAG1 to promote capsid transport on microtubules during egress. J. Virol. 2013, 87, 2857–2867. [Google Scholar] [CrossRef] [Green Version]
- Ryan, S.D.; Bhanot, K.; Ferrier, A.; De Repentigny, Y.; Chu, A.; Blais, A.; Kothary, R. Microtubule stability, Golgi organization, and transport flux require dystonin-a2-MAP1B interaction. J. Cell Biol. 2012, 196, 727–742. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.J.; Ding, J.; Kowal, A.S.; Nardine, T.; Allen, E.; Delcroix, J.D.; Wu, C.; Mobley, W.; Fuchs, E.; Yang, Y. BPAG1n4 is essential for retrograde axonal transport in sensory neurons. J. Cell Biol. 2003, 163, 223–229. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.J. Regulation of dynein-dynactin-driven vesicular transport. Traffic 2017, 18, 336–347. [Google Scholar] [CrossRef]
- Liu, J.J.; Ding, J.; Wu, C.; Bhagavatula, P.; Cui, B.; Chu, S.; Mobley, W.C.; Yang, Y. Retrolinkin, a membrane protein, plays an important role in retrograde axonal transport. Proc. Natl. Acad. Sci. USA 2007, 104, 2223–2228. [Google Scholar] [CrossRef] [Green Version]
- Karasneh, G.A.; Shukla, D. Herpes simplex virus infects most cell types in vitro: Clues to its success. Virol. J. 2011, 8, 481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilterbrand, A.T.; Heldwein, E.E. Go go gadget glycoprotein!: HSV-1 draws on its sizeable glycoprotein tool kit to customize its diverse entry routes. PLoS Pathog. 2019, 15, e1007660. [Google Scholar] [CrossRef] [PubMed]
- Heldwein, E.E.; Krummenacher, C. Entry of herpesviruses into mammalian cells. Cell. Mol. Life Sci. 2008, 65, 1653–1668. [Google Scholar] [CrossRef] [PubMed]
- Henaff, D.; Radtke, K.; Lippe, R. Herpesviruses exploit several host compartments for envelopment. Traffic 2012, 13, 1443–1449. [Google Scholar] [CrossRef]
- Komuro, M.; Tajima, M.; Kato, K. Transformation of Golgi membrane into the envelope of herpes simplex virus in rat anterior pituitary cells. Eur. J. Cell Biol. 1989, 50, 398–406. [Google Scholar]
- Granzow, H.; Klupp, B.G.; Fuchs, W.; Veits, J.; Osterrieder, N.; Mettenleiter, T.C. Egress of alphaherpesviruses: Comparative ultrastructural study. J. Virol. 2001, 75, 3675–3684. [Google Scholar] [CrossRef] [Green Version]
- van Genderen, I.L.; Brandimarti, R.; Torrisi, M.R.; Campadelli, G.; van Meer, G. The phospholipid composition of extracellular herpes simplex virions differs from that of host cell nuclei. Virology 1994, 200, 831–836. [Google Scholar] [CrossRef]
- Church, G.A.; Wilson, D.W. Study of herpes simplex virus maturation during a synchronous wave of assembly. J. Virol. 1997, 71, 3603–3612. [Google Scholar] [CrossRef] [Green Version]
- Harley, C.A.; Dasgupta, A.; Wilson, D.W. Characterization of herpes simplex virus-containing organelles by subcellular fractionation: Role for organelle acidification in assembly of infectious particles. J. Virol. 2001, 75, 1236–1251. [Google Scholar] [CrossRef] [Green Version]
- Hutagalung, A.H.; Novick, P.J. Role of Rab GTPases in membrane traffic and cell physiology. Physiol. Rev. 2011, 91, 119–149. [Google Scholar] [CrossRef] [Green Version]
- Desai, P.; Sexton, G.L.; Huang, E.; Person, S. Localization of herpes simplex virus type 1 UL37 in the Golgi complex requires UL36 but not capsid structures. J. Virol. 2008, 82, 11354–11361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turcotte, S.; Letellier, J.; Lippe, R. Herpes simplex virus type 1 capsids transit by the trans-Golgi network, where viral glycoproteins accumulate independently of capsid egress. J. Virol. 2005, 79, 8847–8860. [Google Scholar] [CrossRef] [Green Version]
- Matlin, K.S.; Simons, K. Reduced temperature prevents transfer of a membrane glycoprotein to the cell surface but does not prevent terminal glycosylation. Cell 1983, 34, 233–243. [Google Scholar] [CrossRef]
- Griffiths, G.; Pfeiffer, S.; Simons, K.; Matlin, K. Exit of newly synthesized membrane proteins from the trans cisterna of the Golgi complex to the plasma membrane. J. Cell Biol. 1985, 101, 949–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liljedahl, M.; Maeda, Y.; Colanzi, A.; Ayala, I.; Van Lint, J.; Malhotra, V. Protein kinase D regulates the fission of cell surface destined transport carriers from the trans-Golgi network. Cell 2001, 104, 409–420. [Google Scholar] [CrossRef] [Green Version]
- Bossard, C.; Bresson, D.; Polishchuk, R.S.; Malhotra, V. Dimeric PKD regulates membrane fission to form transport carriers at the TGN. J. Cell Biol. 2007, 179, 1123–1131. [Google Scholar] [CrossRef] [Green Version]
- Remillard-Labrosse, G.; Mihai, C.; Duron, J.; Guay, G.; Lippe, R. Protein kinase D-dependent trafficking of the large Herpes simplex virus type 1 capsids from the TGN to plasma membrane. Traffic 2009, 10, 1074–1083. [Google Scholar] [CrossRef]
- Dingwell, K.S.; Brunetti, C.R.; Hendricks, R.L.; Tang, Q.; Tang, M.; Rainbow, A.J.; Johnson, D.C. Herpes simplex virus glycoproteins E and I facilitate cell-to-cell spread in vivo and across junctions of cultured cells. J. Virol. 1994, 68, 834–845. [Google Scholar] [CrossRef] [Green Version]
- Wisner, T.W.; Johnson, D.C. Redistribution of cellular and herpes simplex virus proteins from the trans-golgi network to cell junctions without enveloped capsids. J. Virol. 2004, 78, 11519–11535. [Google Scholar] [CrossRef] [Green Version]
- Hollinshead, M.; Johns, H.L.; Sayers, C.L.; Gonzalez-Lopez, C.; Smith, G.L.; Elliott, G. Endocytic tubules regulated by Rab GTPases 5 and 11 are used for envelopment of herpes simplex virus. EMBO J. 2012, 31, 4204–4220. [Google Scholar] [CrossRef] [Green Version]
- Johns, H.L.; Gonzalez-Lopez, C.; Sayers, C.L.; Hollinshead, M.; Elliott, G. Rab6 dependent post-Golgi trafficking of HSV1 envelope proteins to sites of virus envelopment. Traffic 2014, 15, 157–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albecka, A.; Laine, R.F.; Janssen, A.F.; Kaminski, C.F.; Crump, C.M. HSV-1 Glycoproteins Are Delivered to Virus Assembly Sites Through Dynamin-Dependent Endocytosis. Traffic 2016, 17, 21–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zenner, H.L.; Yoshimura, S.; Barr, F.A.; Crump, C.M. Analysis of Rab GTPase-activating proteins indicates that Rab1a/b and Rab43 are important for herpes simplex virus 1 secondary envelopment. J. Virol. 2011, 85, 8012–8021. [Google Scholar] [CrossRef] [Green Version]
- Brideau, A.D.; Enquist, L.W.; Tirabassi, R.S. The role of virion membrane protein endocytosis in the herpesvirus life cycle. J. Clin. Virol. Off. Publ. Pan Am. Soc. Clin. Virol. 2000, 17, 69–82. [Google Scholar] [CrossRef]
- Tirabassi, R.S.; Enquist, L.W. Mutation of the YXXL endocytosis motif in the cytoplasmic tail of pseudorabies virus gE. J. Virol. 1999, 73, 2717–2728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tirabassi, R.S.; Enquist, L.W. Role of envelope protein gE endocytosis in the pseudorabies virus life cycle. J. Virol. 1998, 72, 4571–4579. [Google Scholar] [CrossRef] [Green Version]
- Brideau, A.D.; del Rio, T.; Wolffe, E.J.; Enquist, L.W. Intracellular trafficking and localization of the pseudorabies virus Us9 type II envelope protein to host and viral membranes. J. Virol. 1999, 73, 4372–4384. [Google Scholar] [CrossRef] [Green Version]
- Brack, A.R.; Klupp, B.G.; Granzow, H.; Tirabassi, R.; Enquist, L.W.; Mettenleiter, T.C. Role of the cytoplasmic tail of pseudorabies virus glycoprotein E in virion formation. J. Virol. 2000, 74, 4004–4016. [Google Scholar] [CrossRef] [Green Version]
- Tirabassi, R.S.; Townley, R.A.; Eldridge, M.G.; Enquist, L.W. Characterization of pseudorabies virus mutants expressing carboxy-terminal truncations of gE: Evidence for envelope incorporation, virulence, and neurotropism domains. J. Virol. 1997, 71, 6455–6464. [Google Scholar] [CrossRef] [Green Version]
- Brideau, A.D.; Eldridge, M.G.; Enquist, L.W. Directional transneuronal infection by pseudorabies virus is dependent on an acidic internalization motif in the Us9 cytoplasmic tail. J. Virol. 2000, 74, 4549–4561. [Google Scholar] [CrossRef]
- McMillan, T.N.; Johnson, D.C. Cytoplasmic domain of herpes simplex virus gE causes accumulation in the trans-Golgi network, a site of virus envelopment and sorting of virions to cell junctions. J. Virol. 2001, 75, 1928–1940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, D.C.; Webb, M.; Wisner, T.W.; Brunetti, C. Herpes simplex virus gE/gI sorts nascent virions to epithelial cell junctions, promoting virus spread. J. Virol. 2001, 75, 821–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chouljenko, V.N.; Iyer, A.V.; Chowdhury, S.; Kim, J.; Kousoulas, K.G. The herpes simplex virus type 1 UL20 protein and the amino terminus of glycoprotein K (gK) physically interact with gB. J. Virol. 2010, 84, 8596–8606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, I.J.; Chouljenko, V.N.; Walker, J.D.; Kousoulas, K.G. Herpes simplex virus 1 glycoprotein M and the membrane-associated protein UL11 are required for virus-induced cell fusion and efficient virus entry. J. Virol. 2013, 87, 8029–8037. [Google Scholar] [CrossRef] [Green Version]
- Harper, A.L.; Meckes, D.G., Jr.; Marsh, J.A.; Ward, M.D.; Yeh, P.C.; Baird, N.L.; Wilson, C.B.; Semmes, O.J.; Wills, J.W. Interaction domains of the UL16 and UL21 tegument proteins of herpes simplex virus. J. Virol. 2010, 84, 2963–2971. [Google Scholar] [CrossRef] [Green Version]
- Meckes, D.G., Jr.; Wills, J.W. Dynamic interactions of the UL16 tegument protein with the capsid of herpes simplex virus. J. Virol. 2007, 81, 13028–13036. [Google Scholar] [CrossRef] [Green Version]
- Loomis, J.S.; Bowzard, J.B.; Courtney, R.J.; Wills, J.W. Intracellular trafficking of the UL11 tegument protein of herpes simplex virus type 1. J. Virol. 2001, 75, 12209–12219. [Google Scholar] [CrossRef] [Green Version]
- Starkey, J.L.; Han, J.; Chadha, P.; Marsh, J.A.; Wills, J.W. Elucidation of the block to herpes simplex virus egress in the absence of tegument protein UL16 reveals a novel interaction with VP22. J. Virol. 2014, 88, 110–119. [Google Scholar] [CrossRef] [Green Version]
- McElwee, M.; Beilstein, F.; Labetoulle, M.; Rixon, F.J.; Pasdeloup, D. Dystonin/BPAG1 promotes plus-end-directed transport of herpes simplex virus 1 capsids on microtubules during entry. J. Virol. 2013, 87, 11008–11018. [Google Scholar] [CrossRef] [Green Version]
- Browne, H.; Bell, S.; Minson, T. Analysis of the requirement for glycoprotein m in herpes simplex virus type 1 morphogenesis. J. Virol. 2004, 78, 1039–1041. [Google Scholar] [CrossRef] [Green Version]
- Brack, A.R.; Dijkstra, J.M.; Granzow, H.; Klupp, B.G.; Mettenleiter, T.C. Inhibition of virion maturation by simultaneous deletion of glycoproteins E, I, and M of pseudorabies virus. J. Virol. 1999, 73, 5364–5372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, T.P.; Melancon, J.M.; Baines, J.D.; Kousoulas, K.G. The herpes simplex virus type 1 UL20 protein modulates membrane fusion events during cytoplasmic virion morphogenesis and virus-induced cell fusion. J. Virol. 2004, 78, 5347–5357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charles, A.S.; Chouljenko, V.N.; Jambunathan, N.; Subramanian, R.; Mottram, P.; Kousoulas, K.G. Phenylalanine residues at the carboxyl terminus of the herpes simplex virus 1 UL20 membrane protein regulate cytoplasmic virion envelopment and infectious virus production. J. Virol. 2014, 88, 7618–7627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chouljenko, D.V.; Kim, I.J.; Chouljenko, V.N.; Subramanian, R.; Walker, J.D.; Kousoulas, K.G. Functional hierarchy of herpes simplex virus 1 viral glycoproteins in cytoplasmic virion envelopment and egress. J. Virol. 2012, 86, 4262–4270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayachandra, S.; Baghian, A.; Kousoulas, K.G. Herpes simplex virus type 1 glycoprotein K is not essential for infectious virus production in actively replicating cells but is required for efficient envelopment and translocation of infectious virions from the cytoplasm to the extracellular space. J. Virol. 1997, 71, 5012–5024. [Google Scholar] [CrossRef] [Green Version]
- Melancon, J.M.; Luna, R.E.; Foster, T.P.; Kousoulas, K.G. Herpes simplex virus type 1 gK is required for gB-mediated virus-induced cell fusion, while neither gB and gK nor gB and UL20p function redundantly in virion de-envelopment. J. Virol. 2005, 79, 299–313. [Google Scholar] [CrossRef] [Green Version]
- Baines, J.D.; Roizman, B. The open reading frames UL3, UL4, UL10, and UL16 are dispensable for the replication of herpes simplex virus 1 in cell culture. J. Virol. 1991, 65, 938–944. [Google Scholar] [CrossRef] [Green Version]
- Diwaker, D.; Murray, J.W.; Barnes, J.; Wolkoff, A.W.; Wilson, D.W. Deletion of the Pseudorabies Virus gE/gI-US9p complex disrupts kinesin KIF1A and KIF5C recruitment during egress, and alters the properties of microtubule-dependent transport in vitro. PLoS Pathog. 2020, 16, e1008597. [Google Scholar] [CrossRef]
- O’Regan, K.J.; Bucks, M.A.; Murphy, M.A.; Wills, J.W.; Courtney, R.J. A conserved region of the herpes simplex virus type 1 tegument protein VP22 facilitates interaction with the cytoplasmic tail of glycoprotein E (gE). Virology 2007, 358, 192–200. [Google Scholar] [CrossRef]
- Nozawa, N.; Daikoku, T.; Yamauchi, Y.; Takakuwa, H.; Goshima, F.; Yoshikawa, T.; Nishiyama, Y. Identification and characterization of the UL7 gene product of herpes simplex virus type 2. Virus Genes 2002, 24, 257–266. [Google Scholar] [CrossRef]
- Nozawa, N.; Daikoku, T.; Koshizuka, T.; Yamauchi, Y.; Yoshikawa, T.; Nishiyama, Y. Subcellular localization of herpes simplex virus type 1 UL51 protein and role of palmitoylation in Golgi apparatus targeting. J. Virol. 2003, 77, 3204–3216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nozawa, N.; Kawaguchi, Y.; Tanaka, M.; Kato, A.; Kato, A.; Kimura, H.; Nishiyama, Y. Herpes simplex virus type 1 UL51 protein is involved in maturation and egress of virus particles. J. Virol. 2005, 79, 6947–6956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klupp, B.G.; Granzow, H.; Klopfleisch, R.; Fuchs, W.; Kopp, M.; Lenk, M.; Mettenleiter, T.C. Functional analysis of the pseudorabies virus UL51 protein. J. Virol. 2005, 79, 3831–3840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uversky, V.N. Protein intrinsic disorder-based liquid-liquid phase transitions in biological systems: Complex coacervates and membrane-less organelles. Adv. Colloid Interface Sci. 2017, 239, 97–114. [Google Scholar] [CrossRef] [PubMed]
- del Rio, T.; Ch’ng, T.H.; Flood, E.A.; Gross, S.P.; Enquist, L.W. Heterogeneity of a fluorescent tegument component in single pseudorabies virus virions and enveloped axonal assemblies. J. Virol. 2005, 79, 3903–3919. [Google Scholar] [CrossRef] [Green Version]
- del Rio, T.; DeCoste, C.J.; Enquist, L.W. Actin is a component of the compensation mechanism in pseudorabies virus virions lacking the major tegument protein VP22. J. Virol. 2005, 79, 8614–8619. [Google Scholar] [CrossRef] [Green Version]
- McCullough, J.; Frost, A.; Sundquist, W.I. Structures, Functions, and Dynamics of ESCRT-III/Vps4 Membrane Remodeling and Fission Complexes. Annu. Rev. Cell Dev. Biol. 2018, 34, 85–109. [Google Scholar] [CrossRef]
- Votteler, J.; Sundquist, W.I. Virus budding and the ESCRT pathway. Cell Host Microbe 2013, 14, 232–241. [Google Scholar] [CrossRef] [Green Version]
- Crump, C.M.; Yates, C.; Minson, T. Herpes simplex virus type 1 cytoplasmic envelopment requires functional Vps4. J. Virol. 2007, 81, 7380–7387. [Google Scholar] [CrossRef] [Green Version]
- Scourfield, E.J.; Martin-Serrano, J. Growing functions of the ESCRT machinery in cell biology and viral replication. Biochem. Soc. Trans. 2017, 45, 613–634. [Google Scholar] [CrossRef]
- McCullough, J.; Clippinger, A.K.; Talledge, N.; Skowyra, M.L.; Saunders, M.G.; Naismith, T.V.; Colf, L.A.; Afonine, P.; Arthur, C.; Sundquist, W.I.; et al. Structure and membrane remodeling activity of ESCRT-III helical polymers. Science 2015, 350, 1548–1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christ, L.; Raiborg, C.; Wenzel, E.M.; Campsteijn, C.; Stenmark, H. Cellular Functions and Molecular Mechanisms of the ESCRT Membrane-Scission Machinery. Trends. Biochem. Sci. 2017, 42, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Alonso, Y.A.M.; Migliano, S.M.; Teis, D. ESCRT-III and Vps4: A dynamic multipurpose tool for membrane budding and scission. FEBS J. 2016, 283, 3288–3302. [Google Scholar] [CrossRef] [PubMed]
- Zhai, Q.; Landesman, M.B.; Robinson, H.; Sundquist, W.I.; Hill, C.P. Structure of the Bro1 domain protein BROX and functional analyses of the ALIX Bro1 domain in HIV-1 budding. PLoS ONE 2011, 6, e27466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kharkwal, H.; Smith, C.G.; Wilson, D.W. Blocking ESCRT-mediated envelopment inhibits microtubule-dependent trafficking of alphaherpesviruses in vitro. J. Virol. 2014, 88, 14467–14478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawliczek, T.; Crump, C.M. Herpes simplex virus type 1 production requires a functional ESCRT-III complex but is independent of TSG101 and ALIX expression. J. Virol. 2009, 83, 11254–11264. [Google Scholar] [CrossRef] [Green Version]
- Barnes, J.; Wilson, D.W. The ESCRT-II Subunit EAP20/VPS25 and the Bro1 Domain Proteins HD-PTP and BROX Are Individually Dispensable for Herpes Simplex Virus 1 Replication. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Calistri, A.; Munegato, D.; Toffoletto, M.; Celestino, M.; Franchin, E.; Comin, A.; Sartori, E.; Salata, C.; Parolin, C.; Palu, G. Functional Interaction Between the ESCRT-I Component TSG101 and the HSV-1 Tegument Ubiquitin Specific Protease. J. Cell. Physiol. 2015, 230, 1794–1806. [Google Scholar] [CrossRef]
- Ko, D.H.; Cunningham, A.L.; Diefenbach, R.J. The major determinant for addition of tegument protein pUL48 (VP16) to capsids in herpes simplex virus type 1 is the presence of the major tegument protein pUL36 (VP1/2). J. Virol. 2010, 84, 1397–1405. [Google Scholar] [CrossRef] [Green Version]
- Szilagyi, J.F.; Cunningham, C. Identification and characterization of a novel non-infectious herpes simplex virus-related particle. J. Gen. Virol. 1991, 72 Pt 3, 661–668. [Google Scholar] [CrossRef]
- Heilingloh, C.S.; Krawczyk, A. Role of L-Particles during Herpes Simplex Virus Infection. Front. Microbiol. 2017, 8, 2565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rixon, F.J.; Addison, C.; McLauchlan, J. Assembly of enveloped tegument structures (L particles) can occur independently of virion maturation in herpes simplex virus type 1-infected cells. J. Gen. Virol. 1992, 73 Pt 2, 277–284. [Google Scholar] [CrossRef]
- McLauchlan, J.; Rixon, F.J. Characterization of enveloped tegument structures (L particles) produced by alphaherpesviruses: Integrity of the tegument does not depend on the presence of capsid or envelope. J. Gen. Virol. 1992, 73 Pt 2, 269–276. [Google Scholar] [CrossRef]
- Russell, T.; Bleasdale, B.; Hollinshead, M.; Elliott, G. Qualitative Differences in Capsidless L-Particles Released as a By-Product of Bovine Herpesvirus 1 and Herpes Simplex Virus 1 Infections. J. Virol. 2018, 92, e01259-18. [Google Scholar] [CrossRef] [Green Version]
- Ibiricu, I.; Huiskonen, J.T.; Dohner, K.; Bradke, F.; Sodeik, B.; Grunewald, K. Cryo electron tomography of herpes simplex virus during axonal transport and secondary envelopment in primary neurons. PLoS Pathog. 2011, 7, e1002406. [Google Scholar] [CrossRef] [PubMed]
- Schimert, K.I.; Budaitis, B.G.; Reinemann, D.N.; Lang, M.J.; Verhey, K.J. Intracellular cargo transport by single-headed kinesin motors. Proc. Natl. Acad. Sci. USA 2019, 116, 6152–6161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirokawa, N.; Noda, Y.; Tanaka, Y.; Niwa, S. Kinesin superfamily motor proteins and intracellular transport. Nat. Rev. Mol. Cell Biol. 2009, 10, 682–696. [Google Scholar] [CrossRef] [PubMed]
- Hirokawa, N.; Noda, Y. Intracellular transport and kinesin superfamily proteins, KIFs: Structure, function, and dynamics. Physiol. Rev. 2008, 88, 1089–1118. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Li, L.; Yang, F.; Wang, X.; Fan, F.; Yang, M.; Chen, C.; Li, X.; Wang, H.W.; Sui, S.F. Cryo-EM structures of the ATP-bound Vps4(E233Q) hexamer and its complex with Vta1 at near-atomic resolution. Nat. Commun. 2017, 8, 16064. [Google Scholar] [CrossRef]
- Smith, G.A.; Pomeranz, L.; Gross, S.P.; Enquist, L.W. Local modulation of plus-end transport targets herpesvirus entry and egress in sensory axons. Proc. Natl. Acad. Sci. USA 2004, 101, 16034–16039. [Google Scholar] [CrossRef] [Green Version]
- Enquist, L.W.; Husak, P.J.; Banfield, B.W.; Smith, G.A. Infection and spread of alphaherpesviruses in the nervous system. Adv. Virus Res. 1998, 51, 237–347. [Google Scholar] [CrossRef] [PubMed]
- Howard, P.W.; Wright, C.C.; Howard, T.; Johnson, D.C. Herpes simplex virus gE/gI extracellular domains promote axonal transport and spread from neurons to epithelial cells. J. Virol. 2014, 88, 11178–11186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polcicova, K.; Biswas, P.S.; Banerjee, K.; Wisner, T.W.; Rouse, B.T.; Johnson, D.C. Herpes keratitis in the absence of anterograde transport of virus from sensory ganglia to the cornea. Proc. Natl. Acad. Sci. USA 2005, 102, 11462–11467. [Google Scholar] [CrossRef] [Green Version]
- Corey, L.; Spear, P.G. Infections with herpes simplex viruses (1). N. Engl. J. Med. 1986, 314, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.C.; Huber, M.T. Directed egress of animal viruses promotes cell-to-cell spread. J. Virol. 2002, 76, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Dingwell, K.S.; Johnson, D.C. The herpes simplex virus gE-gI complex facilitates cell-to-cell spread and binds to components of cell junctions. J. Virol. 1998, 72, 8933–8942. [Google Scholar] [CrossRef] [Green Version]
- Wisner, T.; Brunetti, C.; Dingwell, K.; Johnson, D.C. The extracellular domain of herpes simplex virus gE is sufficient for accumulation at cell junctions but not for cell-to-cell spread. J. Virol. 2000, 74, 2278–2287. [Google Scholar] [CrossRef] [Green Version]
- Mingo, R.M.; Han, J.; Newcomb, W.W.; Brown, J.C. Replication of herpes simplex virus: Egress of progeny virus at specialized cell membrane sites. J. Virol. 2012, 86, 7084–7097. [Google Scholar] [CrossRef] [Green Version]
- Krautwald, M.; Fuchs, W.; Klupp, B.G.; Mettenleiter, T.C. Translocation of incoming pseudorabies virus capsids to the cell nucleus is delayed in the absence of tegument protein pUL37. J. Virol. 2009, 83, 3389–3396. [Google Scholar] [CrossRef] [Green Version]
- Dingwell, K.S.; Doering, L.C.; Johnson, D.C. Glycoproteins E and I facilitate neuron-to-neuron spread of herpes simplex virus. J. Virol. 1995, 69, 7087–7098. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Zumbrun, E.E.; Huang, J.; Si, H.; Makaroun, L.; Friedman, H.M. Herpes simplex virus type 2 glycoprotein E is required for efficient virus spread from epithelial cells to neurons and for targeting viral proteins from the neuron cell body into axons. Virology 2010, 405, 269–279. [Google Scholar] [CrossRef] [Green Version]
- McGraw, H.M.; Awasthi, S.; Wojcechowskyj, J.A.; Friedman, H.M. Anterograde spread of herpes simplex virus type 1 requires glycoprotein E and glycoprotein I but not Us9. J. Virol. 2009, 83, 8315–8326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaVail, J.H.; Tauscher, A.N.; Sucher, A.; Harrabi, O.; Brandimarti, R. Viral regulation of the long distance axonal transport of herpes simplex virus nucleocapsid. Neuroscience 2007, 146, 974–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kratchmarov, R.; Taylor, M.P.; Enquist, L.W. Making the case: Married versus separate models of alphaherpes virus anterograde transport in axons. Rev. Med Virol. 2012, 22, 378–391. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, A.; Miranda-Saksena, M.; Diefenbach, R.; Johnson, D. Letter in response to: Making the case: Married versus Separate models of alphaherpes virus anterograde transport in axons. Rev. Med. Virol. 2013, 23, 414–418. [Google Scholar] [CrossRef] [PubMed]
- Diefenbach, R.J.; Miranda-Saksena, M.; Douglas, M.W.; Cunningham, A.L. Transport and egress of herpes simplex virus in neurons. Rev. Med Virol. 2008, 18, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Lazear, H.M.; Friedman, H.M. Completely assembled virus particles detected by transmission electron microscopy in proximal and mid-axons of neurons infected with herpes simplex virus type 1, herpes simplex virus type 2 and pseudorabies virus. Virology 2011, 409, 12–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wisner, T.W.; Sugimoto, K.; Howard, P.W.; Kawaguchi, Y.; Johnson, D.C. Anterograde transport of herpes simplex virus capsids in neurons by both separate and married mechanisms. J. Virol. 2011, 85, 5919–5928. [Google Scholar] [CrossRef] [Green Version]
- Miranda-Saksena, M.; Wakisaka, H.; Tijono, B.; Boadle, R.A.; Rixon, F.; Takahashi, H.; Cunningham, A.L. Herpes simplex virus type 1 accumulation, envelopment, and exit in growth cones and varicosities in mid-distal regions of axons. J. Virol. 2006, 80, 3592–3606. [Google Scholar] [CrossRef] [Green Version]
- Miranda-Saksena, M.; Boadle, R.A.; Aggarwal, A.; Tijono, B.; Rixon, F.J.; Diefenbach, R.J.; Cunningham, A.L. Herpes simplex virus utilizes the large secretory vesicle pathway for anterograde transport of tegument and envelope proteins and for viral exocytosis from growth cones of human fetal axons. J. Virol. 2009, 83, 3187–3199. [Google Scholar] [CrossRef] [Green Version]
- Penfold, M.E.; Armati, P.; Cunningham, A.L. Axonal transport of herpes simplex virions to epidermal cells: Evidence for a specialized mode of virus transport and assembly. Proc. Natl. Acad. Sci. USA 1994, 91, 6529–6533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, A.; Wisner, T.W.; Johnson, D.C. Herpes simplex virus capsids are transported in neuronal axons without an envelope containing the viral glycoproteins. J. Virol. 2006, 80, 11165–11177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, A.; Bruun, B.; Browne, H.M.; Johnson, D.C. A herpes simplex virus gD-YFP fusion glycoprotein is transported separately from viral capsids in neuronal axons. J. Virol. 2007, 81, 8337–8340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antinone, S.E.; Zaichick, S.V.; Smith, G.A. Resolving the assembly state of herpes simplex virus during axon transport by live-cell imaging. J. Virol. 2010, 84, 13019–13030. [Google Scholar] [CrossRef] [Green Version]
- Antinone, S.E.; Smith, G.A. Retrograde axon transport of herpes simplex virus and pseudorabies virus: A live-cell comparative analysis. J. Virol. 2010, 84, 1504–1512. [Google Scholar] [CrossRef] [Green Version]
- Qi, Y.; Wang, J.K.; McMillian, M.; Chikaraishi, D.M. Characterization of a CNS cell line, CAD, in which morphological differentiation is initiated by serum deprivation. J. Neurosci. 1997, 17, 1217–1225. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Hou, L.X.; Aktiv, A.; Dahlstrom, A. Studies of the central nervous system-derived CAD cell line, a suitable model for intraneuronal transport studies? J. Neurosci. Res. 2007, 85, 2601–2609. [Google Scholar] [CrossRef]
- Negatsch, A.; Granzow, H.; Maresch, C.; Klupp, B.G.; Fuchs, W.; Teifke, J.P.; Mettenleiter, T.C. Ultrastructural analysis of virion formation and intraaxonal transport of herpes simplex virus type 1 in primary rat neurons. J. Virol. 2010, 84, 13031–13035. [Google Scholar] [CrossRef] [Green Version]
- Leterrier, C. The Axon Initial Segment, 50Years Later: A Nexus for Neuronal Organization and Function. Curr. Top. Membr. 2016, 77, 185–233. [Google Scholar] [CrossRef]
- Leterrier, C. The Axon Initial Segment: An Updated Viewpoint. J. Neurosci. 2018, 38, 2135–2145. [Google Scholar] [CrossRef] [Green Version]
- Gumy, L.F.; Katrukha, E.A.; Grigoriev, I.; Jaarsma, D.; Kapitein, L.C.; Akhmanova, A.; Hoogenraad, C.C. MAP2 Defines a Pre-axonal Filtering Zone to Regulate KIF1- versus KIF5-Dependent Cargo Transport in Sensory Neurons. Neuron 2017, 94, 347–362.e347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherer, J.; Yaffe, Z.A.; Vershinin, M.; Enquist, L.W. Dual-Color Herpesvirus Capsids Discriminate Inoculum from Progeny and Reveal Axonal Transport Dynamics. J. Virol. 2016, 90, 9997–10006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramer, T.; Greco, T.M.; Taylor, M.P.; Ambrosini, A.E.; Cristea, I.M.; Enquist, L.W. Kinesin-3 mediates axonal sorting and directional transport of alphaherpesvirus particles in neurons. Cell Host Microbe 2012, 12, 806–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kratchmarov, R.; Kramer, T.; Greco, T.M.; Taylor, M.P.; Ch’Ng, T.H.; Cristea, I.M.; Enquist, L.W. Glycoproteins gE and gI are required for efficient KIF1A-dependent anterograde axonal transport of alphaherpesvirus particles in neurons. J. Virol. 2013, 87, 9431–9440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Koyuncu, O.O.; Enquist, L.W. Pseudorabies Virus Infection Accelerates Degradation of the Kinesin-3 Motor KIF1A. J. Virol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Scherer, J.; Hogue, I.B.; Yaffe, Z.A.; Tanneti, N.S.; Winer, B.Y.; Vershinin, M.; Enquist, L.W. A kinesin-3 recruitment complex facilitates axonal sorting of enveloped alpha herpesvirus capsids. PLoS Pathog. 2020, 16, e1007985. [Google Scholar] [CrossRef]
- DuRaine, G.; Wisner, T.W.; Howard, P.; Johnson, D.C. Kinesin-1 Proteins KIF5A, -5B, and -5C Promote Anterograde Transport of Herpes Simplex Virus Enveloped Virions in Axons. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Jansens, R.J.J.; Tishchenko, A.; Favoreel, H.W. Bridging the Gap: Virus Long-Distance Spread via Tunneling Nanotubes. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Panasiuk, M.; Rychlowski, M.; Derewonko, N.; Bienkowska-Szewczyk, K. Tunneling Nanotubes as a Novel Route of Cell-to-Cell Spread of Herpesviruses. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- van Leeuwen, H.; Elliott, G.; O’Hare, P. Evidence of a role for nonmuscle myosin II in herpes simplex virus type 1 egress. J. Virol. 2002, 76, 3471–3481. [Google Scholar] [CrossRef] [Green Version]
- La Boissiere, S.; Izeta, A.; Malcomber, S.; O’Hare, P. Compartmentalization of VP16 in cells infected with recombinant herpes simplex virus expressing VP16-green fluorescent protein fusion proteins. J. Virol. 2004, 78, 8002–8014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixit, R.; Tiwari, V.; Shukla, D. Herpes simplex virus type 1 induces filopodia in differentiated P19 neural cells to facilitate viral spread. Neurosci. Lett. 2008, 440, 113–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brzozowska, A.; Rychlowski, M.; Lipinska, A.D.; Bienkowska-Szewczyk, K. Point mutations in BHV-1 Us3 gene abolish its ability to induce cytoskeletal changes in various cell types. Vet. Microbiol. 2010, 143, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Ladelfa, M.F.; Kotsias, F.; Del Medico Zajac, M.P.; Van den Broeke, C.; Favoreel, H.; Romera, S.A.; Calamante, G. Effect of the US3 protein of bovine herpesvirus 5 on the actin cytoskeleton and apoptosis. Vet. Microbiol. 2011, 153, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Roberts, K.L.; Baines, J.D. Myosin Va enhances secretion of herpes simplex virus 1 virions and cell surface expression of viral glycoproteins. J. Virol. 2010, 84, 9889–9896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansens, R.J.J.; Van den Broeck, W.; De Pelsmaeker, S.; Lamote, J.A.S.; Van Waesberghe, C.; Couck, L.; Favoreel, H.W. Pseudorabies Virus US3-Induced Tunneling Nanotubes Contain Stabilized Microtubules, Interact with Neighboring Cells via Cadherins, and Allow Intercellular Molecular Communication. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Miranda-Saksena, M.; Denes, C.E.; Diefenbach, R.J.; Cunningham, A.L. Infection and Transport of Herpes Simplex Virus Type 1 in Neurons: Role of the Cytoskeleton. Viruses 2018, 10, 92. [Google Scholar] [CrossRef] [Green Version]
- Hogue, I.B.; Scherer, J.; Enquist, L.W. Exocytosis of Alphaherpesvirus Virions, Light Particles, and Glycoproteins Uses Constitutive Secretory Mechanisms. mBio 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Hogue, I.B.; Bosse, J.B.; Hu, J.R.; Thiberge, S.Y.; Enquist, L.W. Cellular mechanisms of alpha herpesvirus egress: Live cell fluorescence microscopy of pseudorabies virus exocytosis. PLoS Pathog. 2014, 10, e1004535. [Google Scholar] [CrossRef]
- Lansbergen, G.; Grigoriev, I.; Mimori-Kiyosue, Y.; Ohtsuka, T.; Higa, S.; Kitajima, I.; Demmers, J.; Galjart, N.; Houtsmuller, A.B.; Grosveld, F.; et al. CLASPs attach microtubule plus ends to the cell cortex through a complex with LL5beta. Dev. Cell 2006, 11, 21–32. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.P.; Kobiler, O.; Enquist, L.W. Alphaherpesvirus axon-to-cell spread involves limited virion transmission. Proc. Natl. Acad. Sci. USA 2012, 109, 17046–17051. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
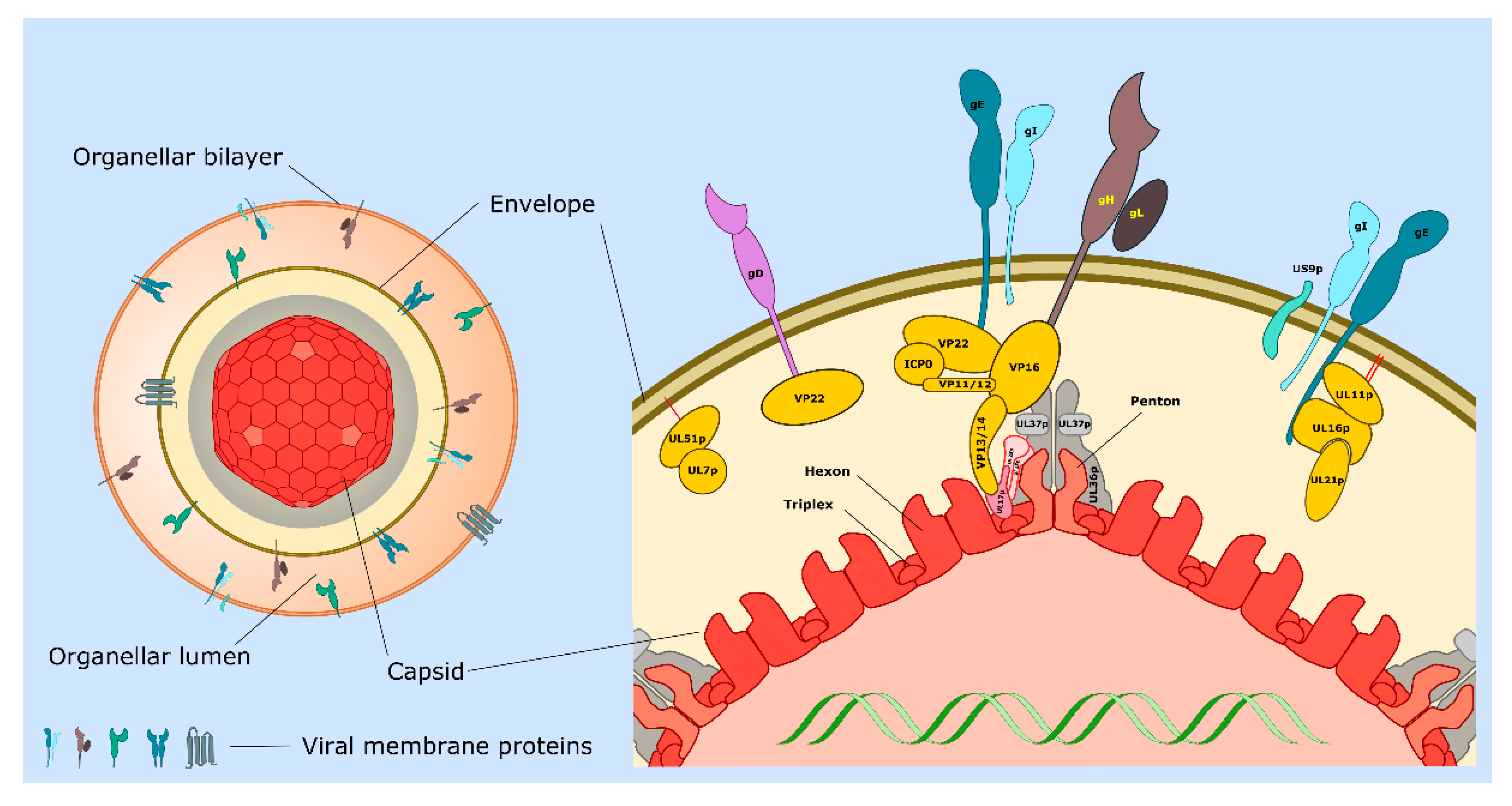
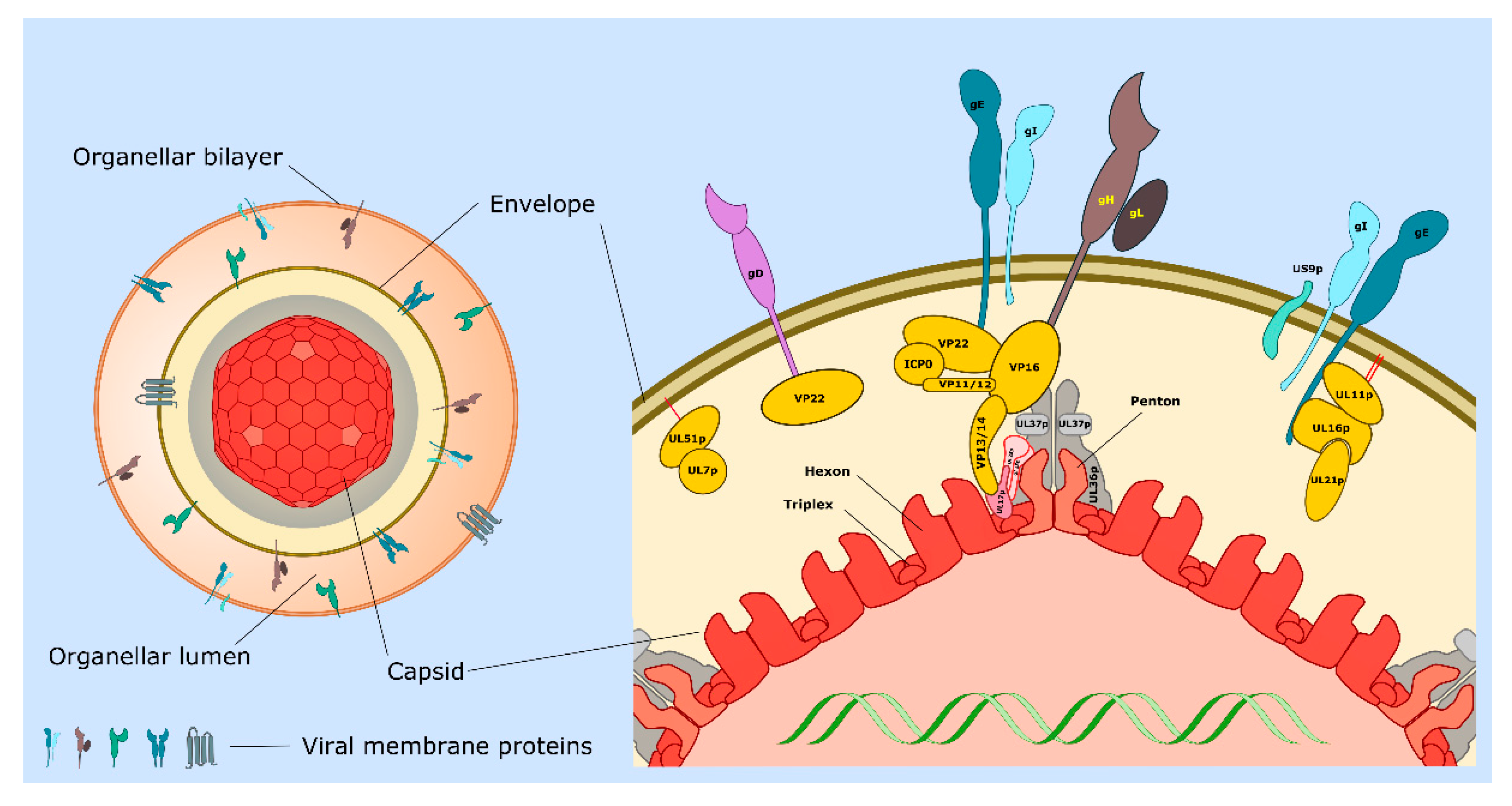
| Protein or Complex (Alternate Names) | Description and Location of Protein(s) | Functions 1 | Section 2 (Key References) |
|---|---|---|---|
| UL11p/UL16p/UL21p | Complex of tegument proteins localized to TGN. | UL11p is palmitoylated and myristoylated. Complex binds to cytoplasmic tail of gE. Envelopment. | 3.5, 3.6, 4.2 [45,46,47,48,49,50,51] |
| UL17p/UL25p | Capsid penton-associated complex. Forms part of the CVSC. | Anchors UL36p to the capsid as a UL17p/(UL25p)2 complex. Interacts with VP13/14. | 2 [21,24,31] |
| UL18p/UL38p (VP23/VP19C) | Capsid proteins. | Form VP19c/(VP23)2 triplexes that connect VP5 capsomeres. | 2 [11,14,20,21] |
| UL19p (VP5) | Major capsid protein. | Forms penton and hexon capsomeres. | 2 [14,21] |
| UL34p | Type II membrane protein. | Expression influences cell–cell spread. Component of the UL31p/UL34p nuclear export complex. | 4.2 [12,16,52] |
| UL36p (VP1/2) | Inner tegument protein. | Foundation for recruitment of outer tegument via VP16. Binds UL37p. Cooperates with UL37p to recruit kinesin-1 and kinesin-2 to capsid. Envelopment. | 2, 3.2, 3.3, 3.5, 3.6, 3.7 [53,54,55,56,57,58,59] |
| UL37p | Inner tegument protein. | Binds UL36p, dystonin and gK/UL20p. Membrane-tethering (possible mimic of cellular MTCs). Cooperates with UL36p to recruit kinesin-1 and kinesin-2 to the capsid. Envelopment. | 2, 3.2, 3.3, 3.5, 4.2 [42,43,44,56,60,61,62] |
| US3p | Serine/threonine kinase. Inner tegument protein. | MT stabilization and acetylation. Assembly of TNTs. | 2, 3.1, 4.5 [26,28,63,64,65] |
| UL7p/UL51p | Outer tegument protein complex. | May mimic or trigger assembly of cellular ESCRT-III complex. Envelopment. Cell–cell spread. Localizes to focal adhesions. | 3.6, 3.7, 4.2 [66,67,68,69] |
| UL46p (VP11/12) | Outer tegument protein. | Binds UL48p (VP16). | 3.6 [11] |
| UL47p (VP13/14) | Outer tegument protein. | Binds UL48p (VP16). Binds CVSC component UL17p. Envelopment. | 3.6 [11,70] |
| UL48p (VP16) | Outer tegument protein. | Connects UL36p to outer tegument. Binds gH carboxy-terminal tail. Envelopment. | 2, 3.6 [11,34,35,36,71,72,73] |
| UL49p (VP22) | Outer tegument protein. | MT acetylation, bundling and stabilization. Binds UL48p (VP16), gD, gE, gM. Envelopment. | 2, 3.1, 3.6 [11,74,75,76,77] |
| gB | Type I membrane protein. | Loss of gB and gD reduces envelopment. Required for fusion 3. | 3.3, 3.4, 3.6 [78,79,80,81,82] |
| gD | Type I membrane protein. | Loss of gD and gB or gE/gI reduces envelopment. Binds VP22. Required for fusion 3. | 3.4, 3.6 [80,81,83] |
| gE/gI | Heterodimer of type I membrane proteins. | Loss of gE/gI and gD disrupts envelopment. Sorting of virions to epithelial junctions and into or along axons. Loss of gE/gI and US9p reduces envelopment in neurons. gE binds VP22 and the UL11p/UL16p/UL21p complex. | 3.6, 4 [77,83,84,85,86,87,88,89] |
| gH/gL | Heterodimer of type I membrane protein (gH) with lumenal/extracellular soluble subunit (gL). | Required for fusion 3. Binds UL48p (VP16). | 3.4, 3.6 [35,36,81,90] |
| gK/UL20p | Heterodimer of multi membrane- spanning proteins. | Regulation of gB/gD/gH/gL-mediated fusion 3. Binds UL37p. Sorting of gD and gH/gL to envelopment site. Envelopment. | 3.4, 3.5, 3.6 [61,90,91,92,93] |
| gM gM/gN | Multi membrane- spanning protein (gM). Can form complex with type 1 membrane protein gN | Sorting of gD and gH/gL to envelopment site. Envelopment. | 3.4, 3.6 [90,94,95,96] |
| US9p | Type II membrane protein. | Loss of US9p and gE/gI reduces envelopment in neurons. Sorting of virions into or along axons. | 4 [97,98,99,100] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad, I.; Wilson, D.W. HSV-1 Cytoplasmic Envelopment and Egress. Int. J. Mol. Sci. 2020, 21, 5969. https://doi.org/10.3390/ijms21175969
Ahmad I, Wilson DW. HSV-1 Cytoplasmic Envelopment and Egress. International Journal of Molecular Sciences. 2020; 21(17):5969. https://doi.org/10.3390/ijms21175969
Chicago/Turabian StyleAhmad, Imran, and Duncan W. Wilson. 2020. "HSV-1 Cytoplasmic Envelopment and Egress" International Journal of Molecular Sciences 21, no. 17: 5969. https://doi.org/10.3390/ijms21175969
APA StyleAhmad, I., & Wilson, D. W. (2020). HSV-1 Cytoplasmic Envelopment and Egress. International Journal of Molecular Sciences, 21(17), 5969. https://doi.org/10.3390/ijms21175969