Learning from Monocyte-Macrophage Fusion and Multinucleation: Potential Therapeutic Targets for Osteoporosis and Rheumatoid Arthritis




Abstract
:
1. Introduction
2. M-FM during Normal Osteoclastogenesis: Therapeutic Perspectives for OP
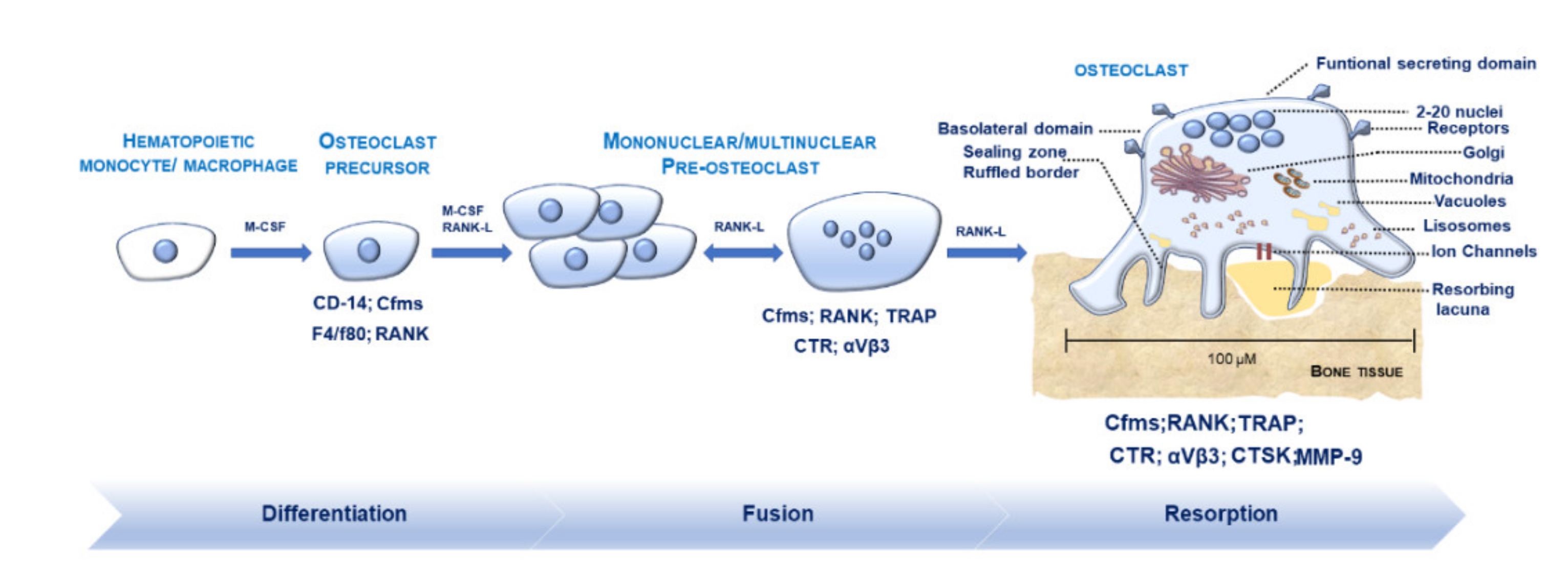
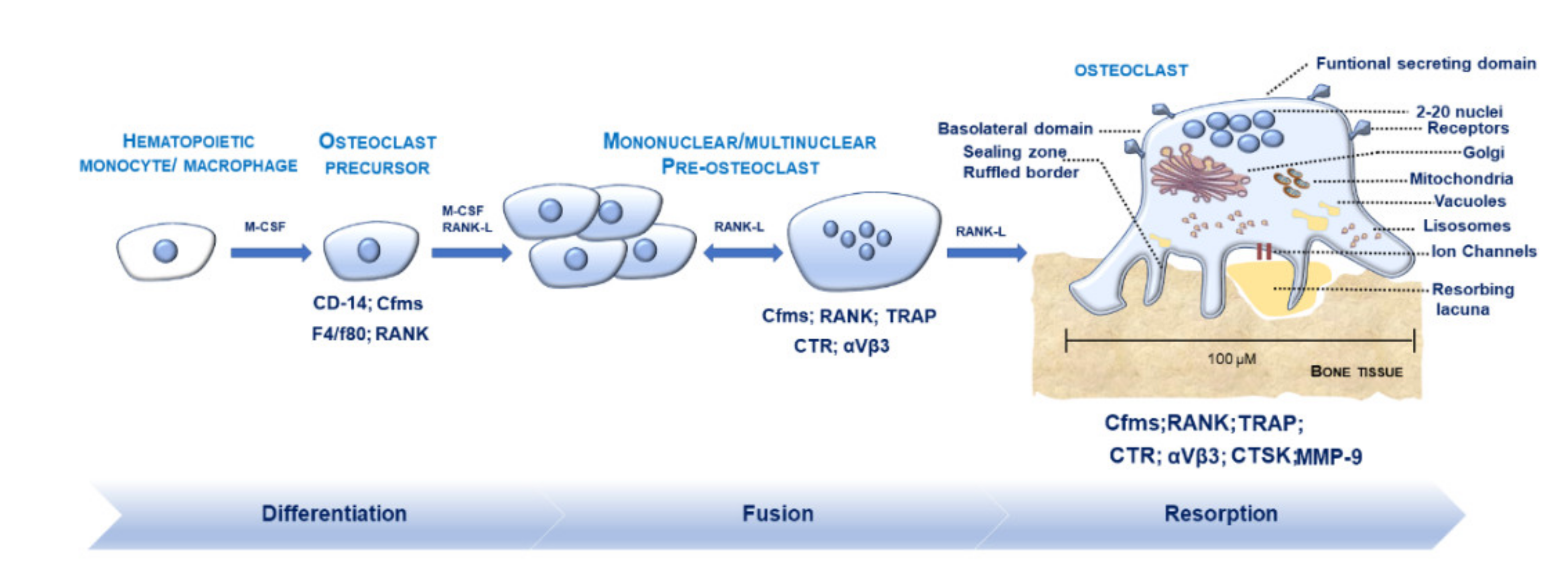
2.1. Morphological Features of OCs in Physiological Conditions
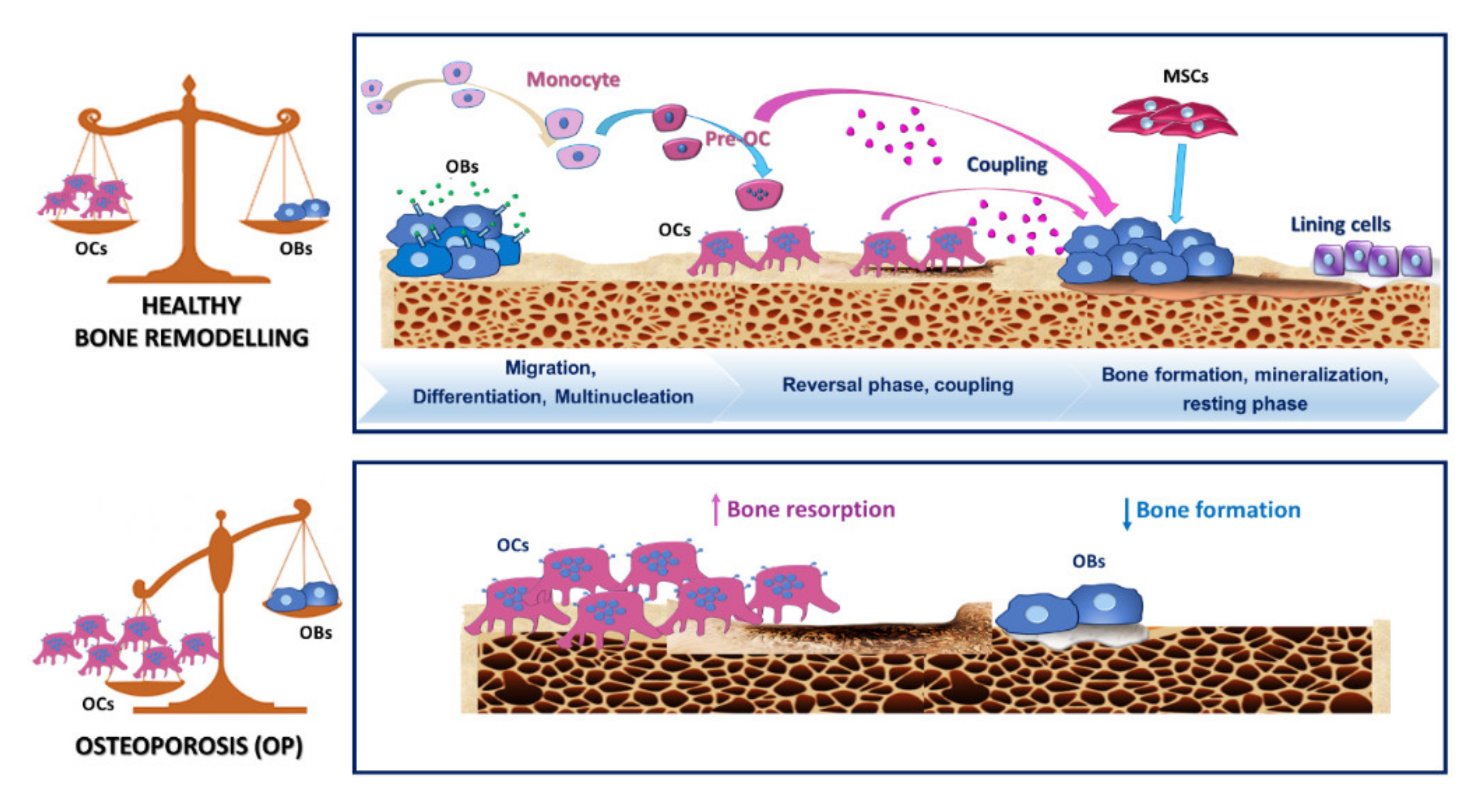
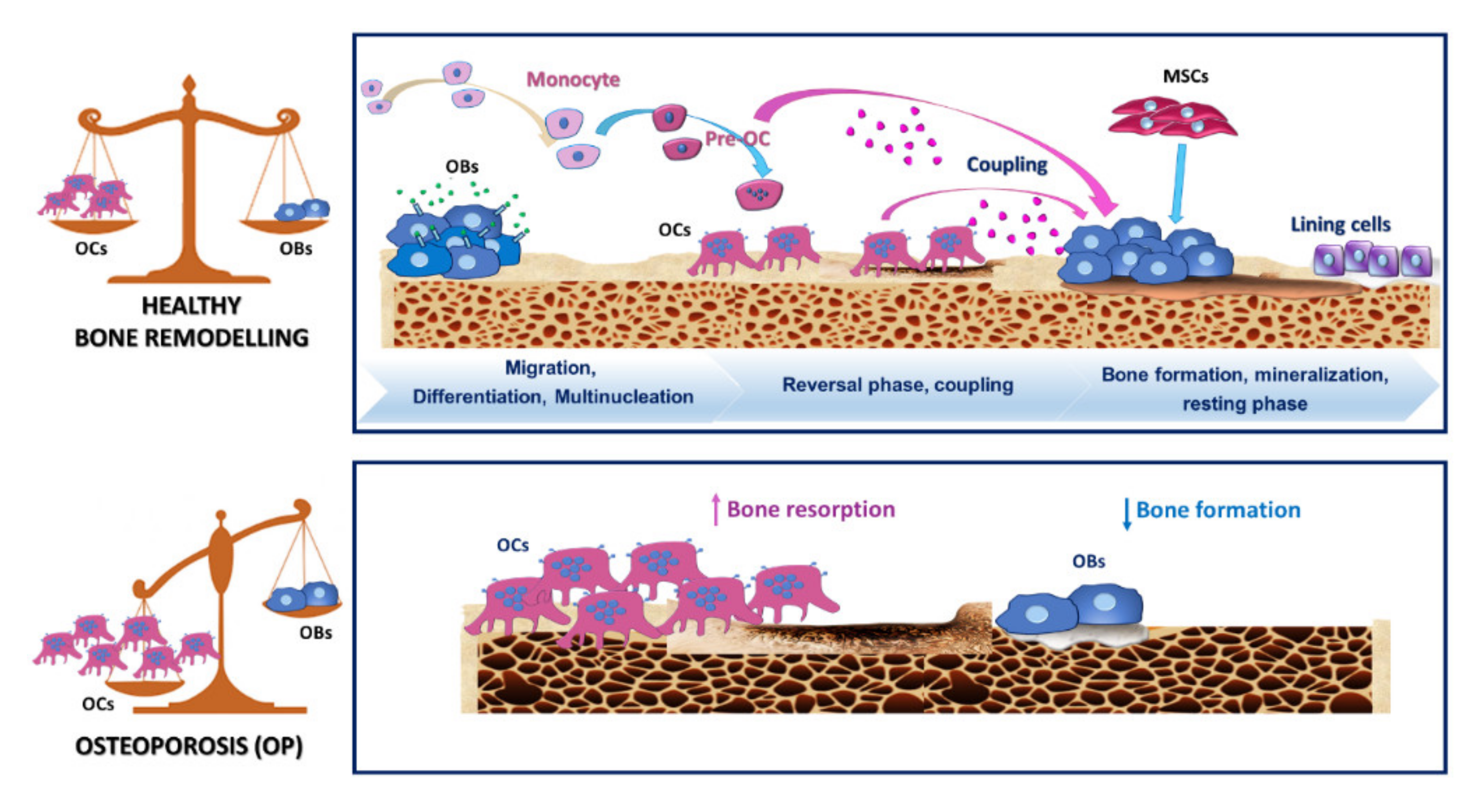
2.2. Osteoclastogenesis and Osteoclastic Bone Resorption
2.3. Regulators Modulating M-FM
2.4. OCs and Osteoclastogenesis during OP
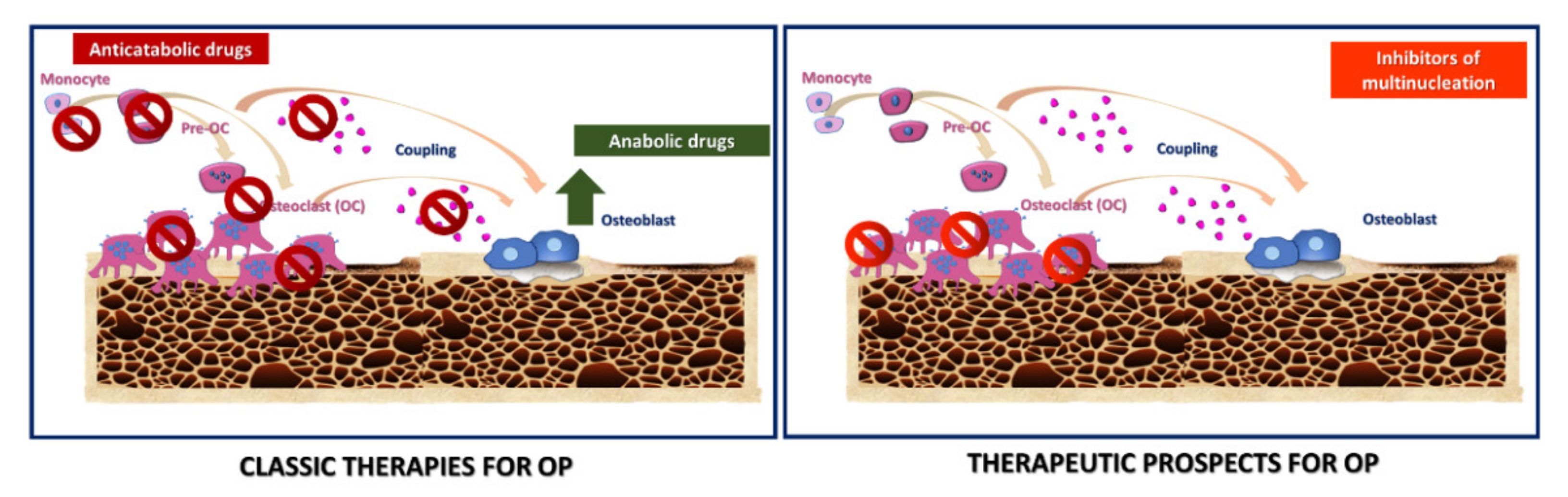
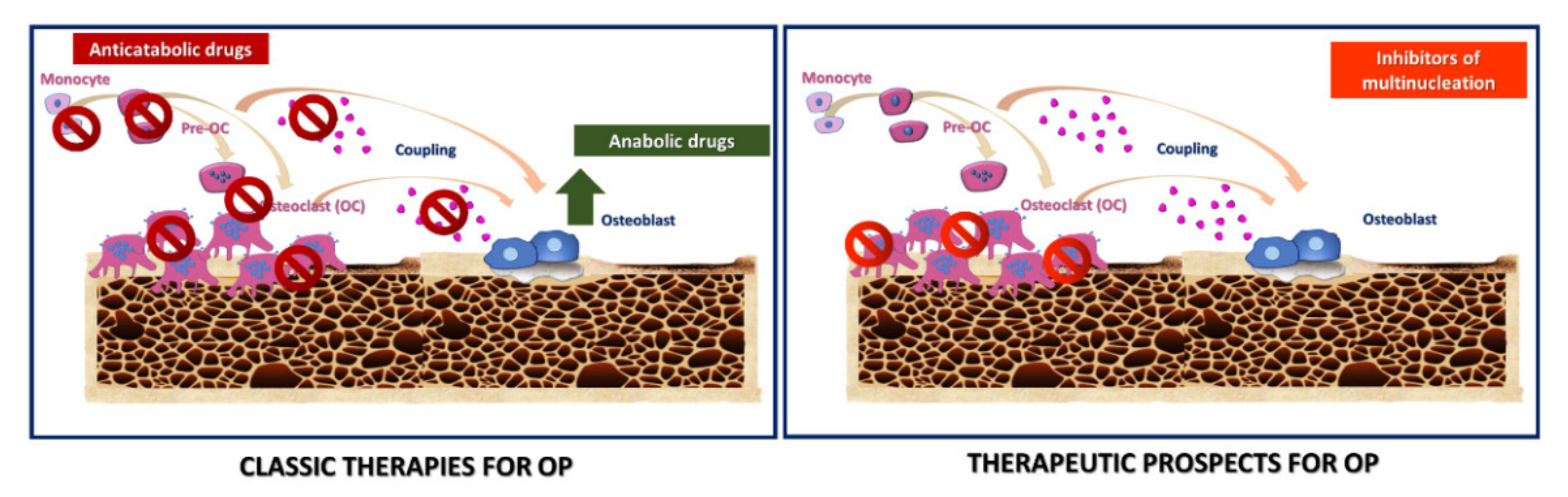
2.5. Therapeutic Strategies in OP
Novel Targets for Anti-Osteoclastogenic Therapies for OP
3. GCs and OCs in RA: Relationships with Bone Erosion and Therapeutic Alternatives
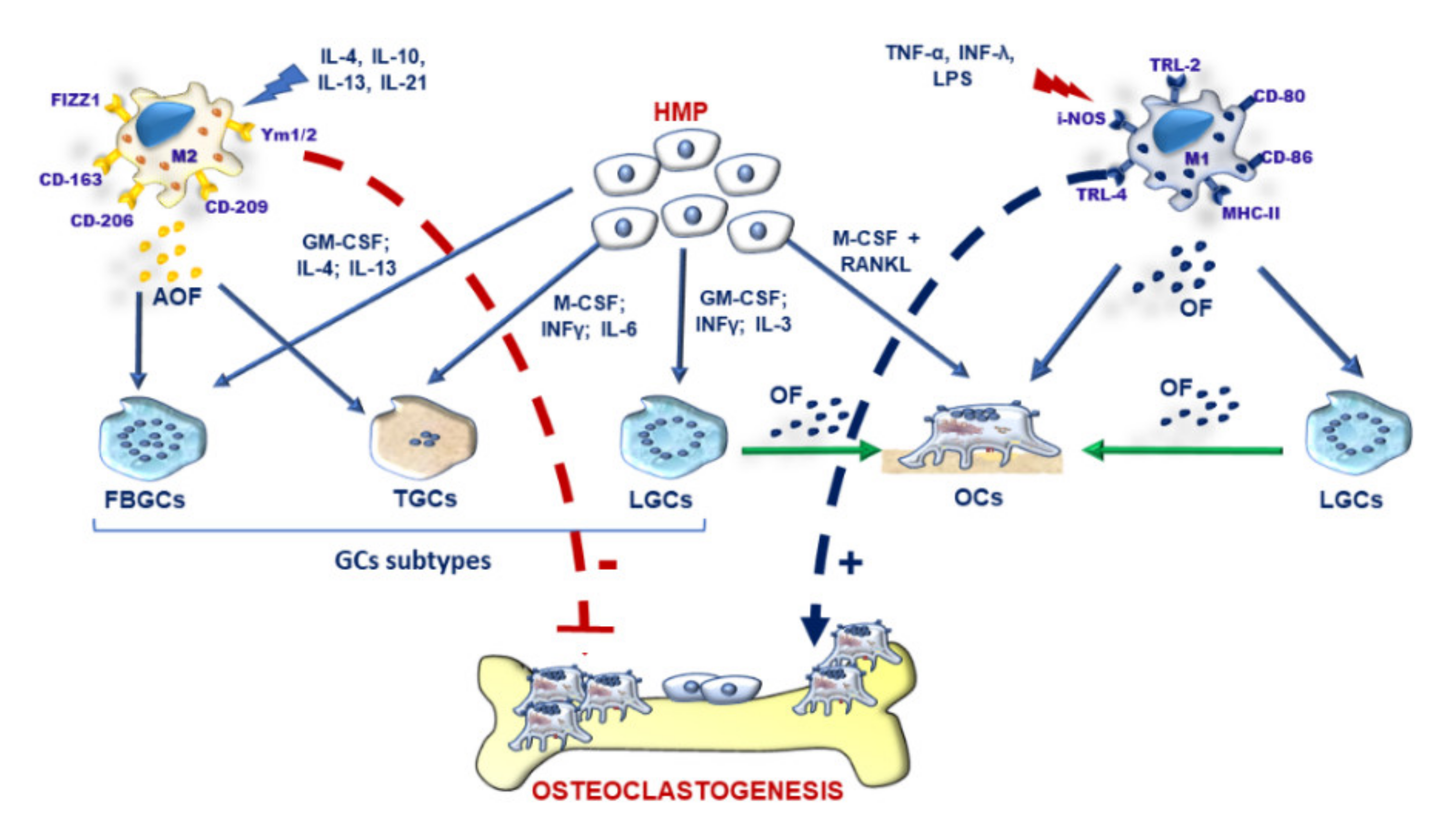
3.1. Leading Characteristics of GCs: Similarities and Differences with OCs
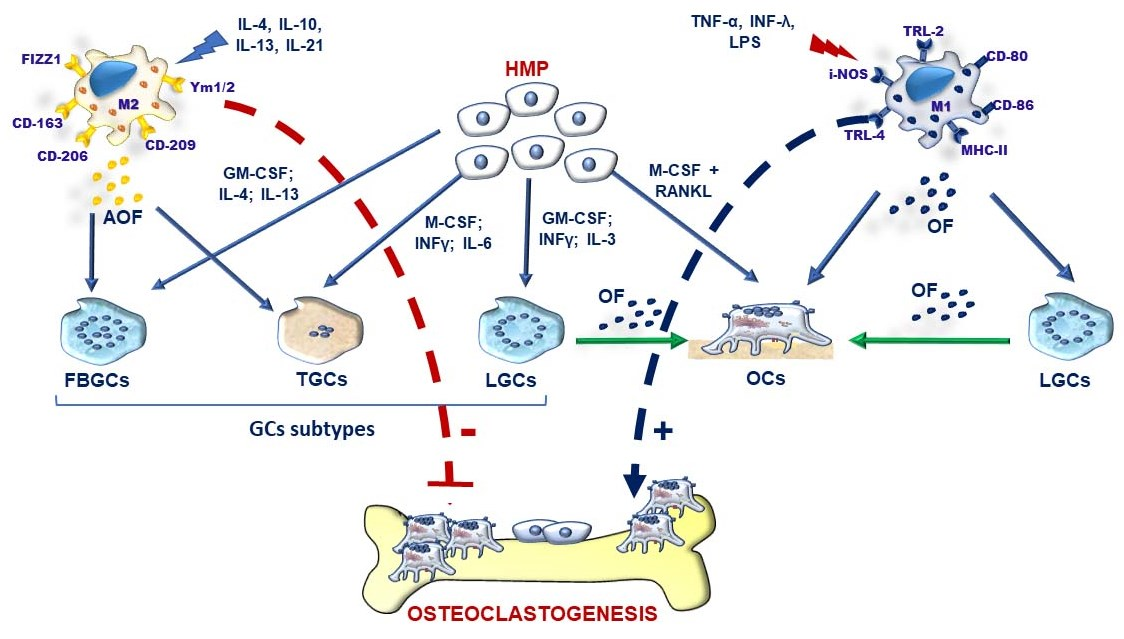
3.2. M-FM of OCs and GCs and Osteoclastogenesis during Inflammation
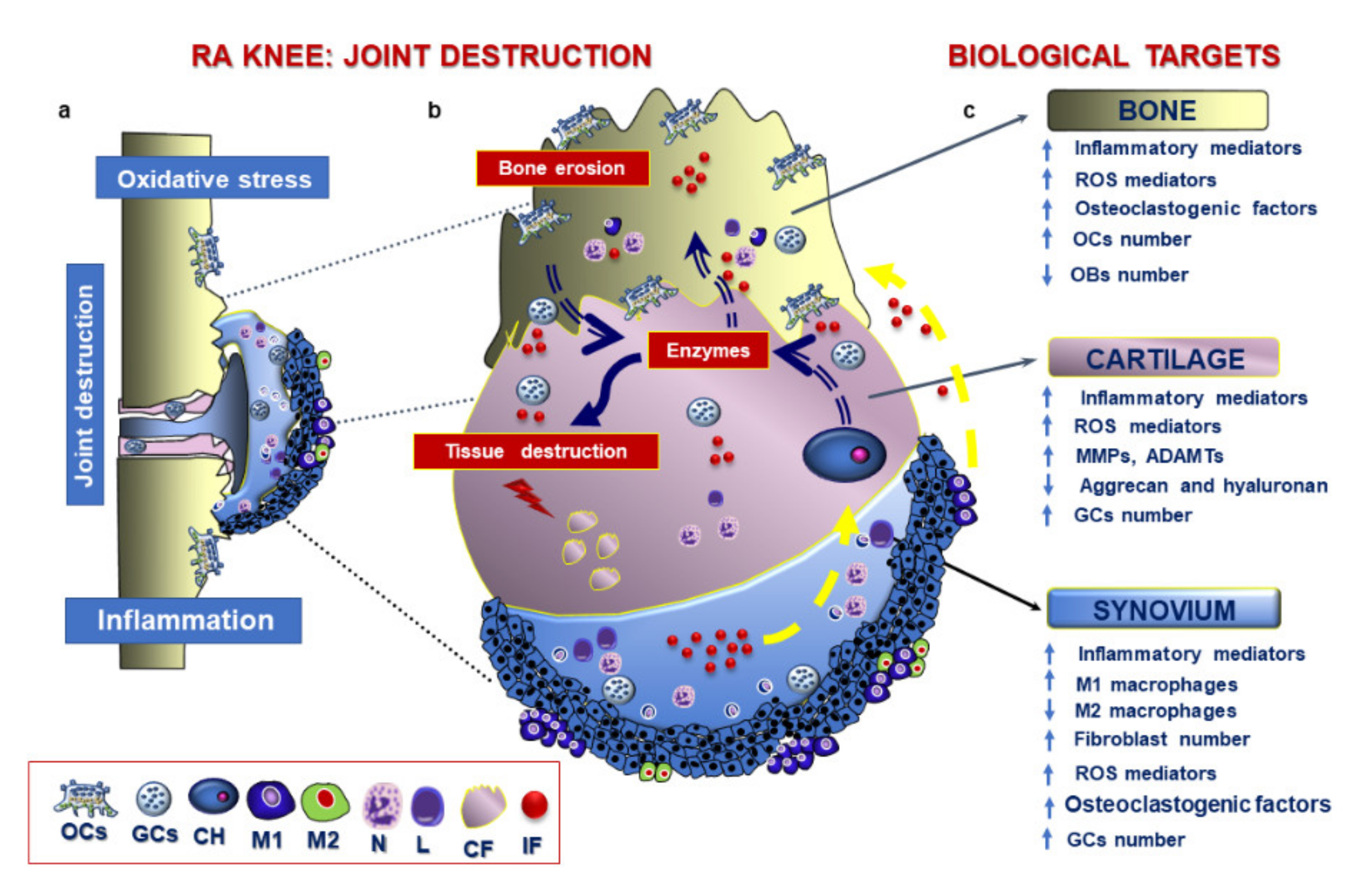
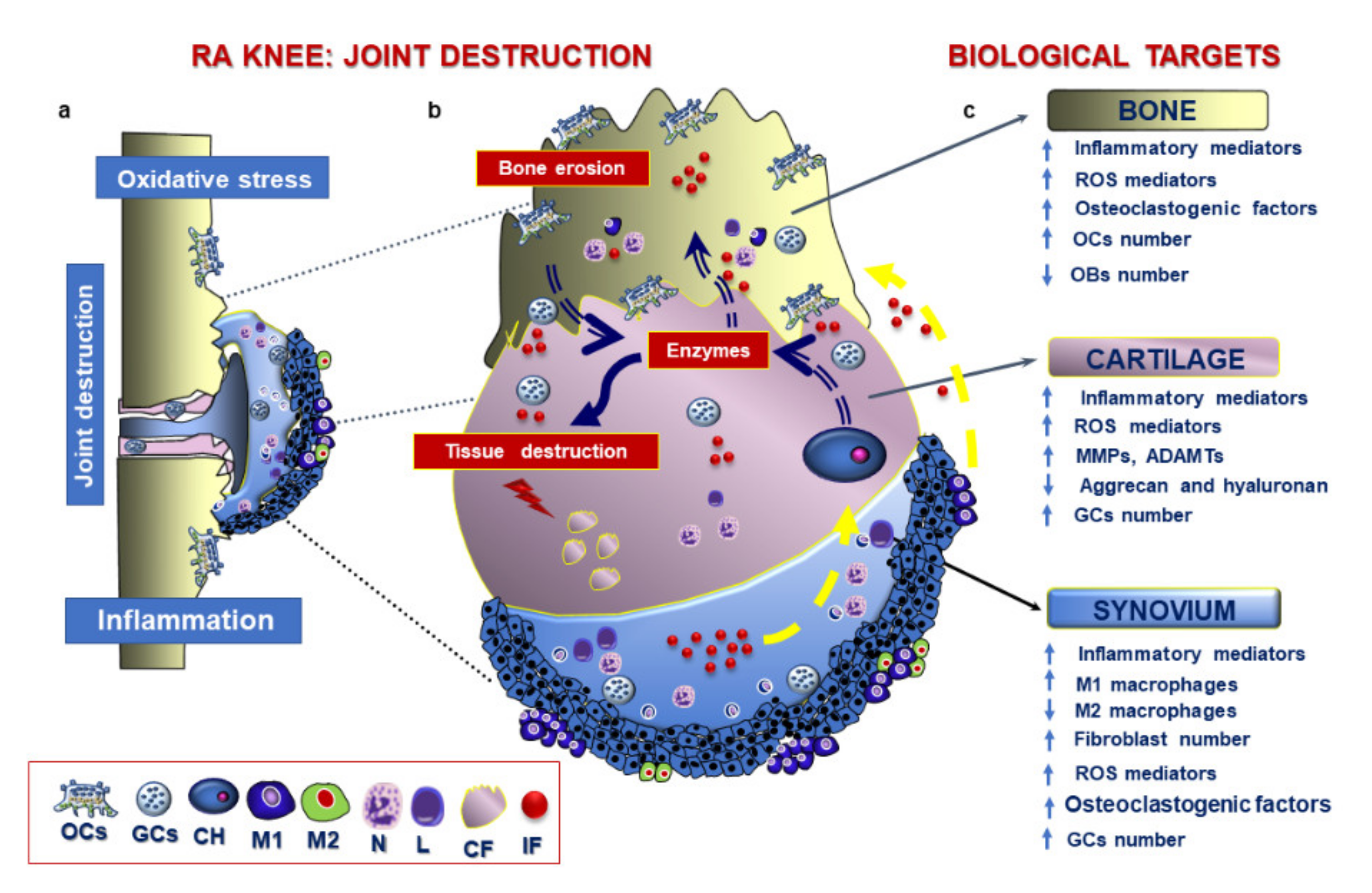
3.3. Pathological Features of RA
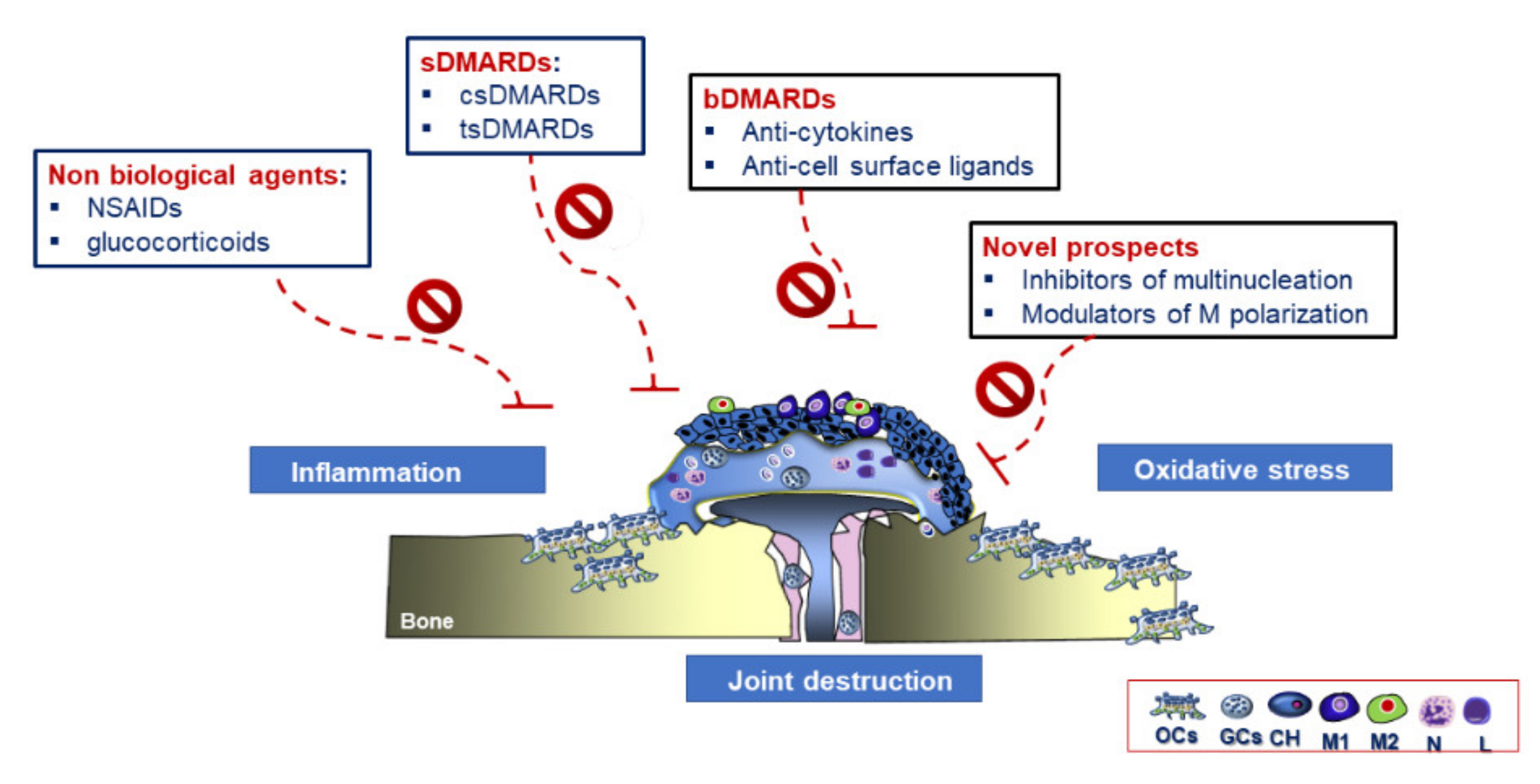
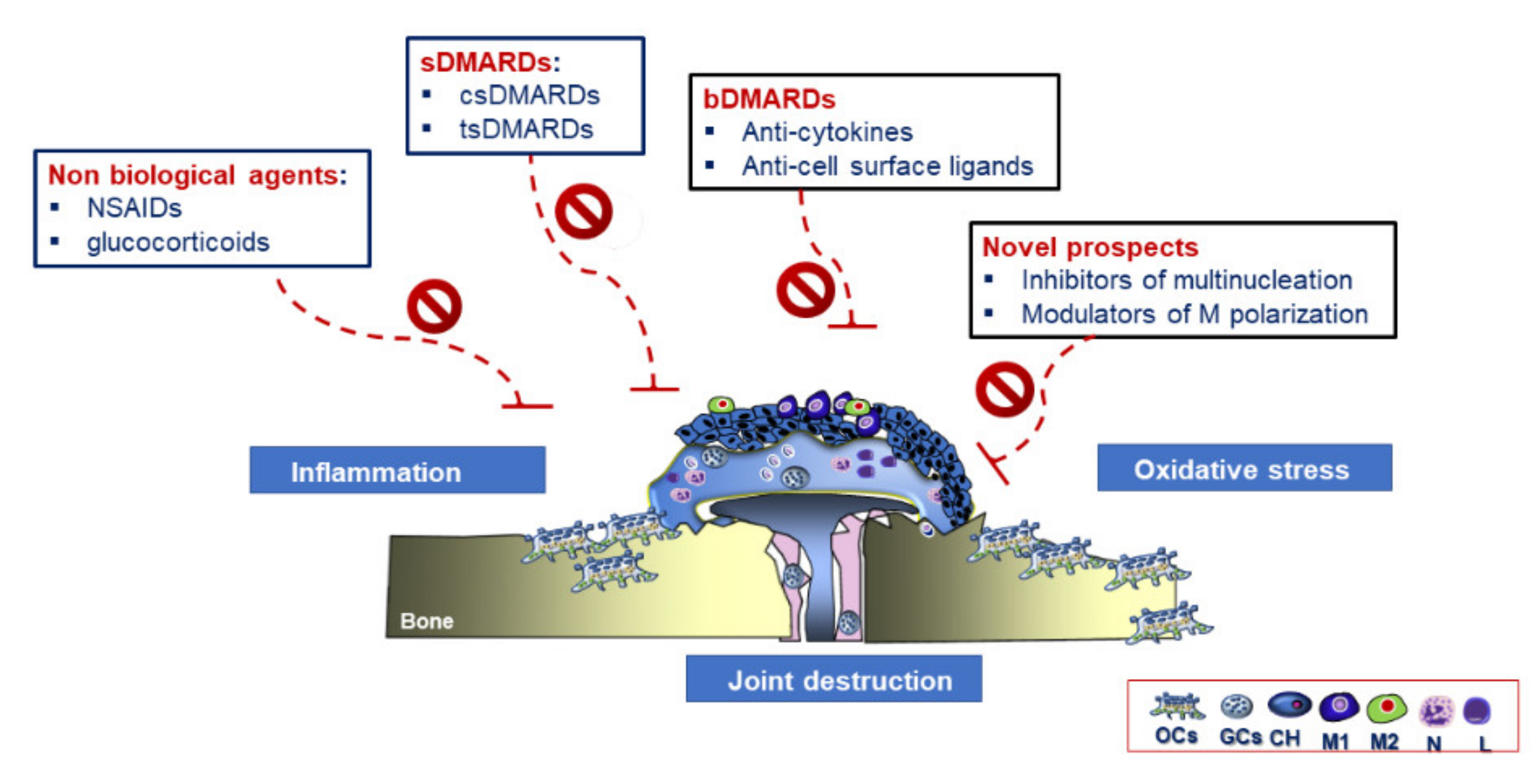
3.4. Therapeutic Strategies in RA
Future Therapeutic Perspectives in RA Treatment
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fu, S.Q.; Wang, Z.Y.; Jiang, Z.M.; Bi, Z.M.; Liu, E.H. Integration of zebrafish model and network pharmacology to explore possible action mechanisms of morinda officinalis for treating osteoporosis. Chem. Biodivers. 2020. [Google Scholar] [CrossRef]
- Bombak, A.E.; Hanson, H.M. Qualitative insights from the osteoporosis research: A narrative review of the literature. J. Osteoporos 2016, 7915041. [Google Scholar] [CrossRef] [Green Version]
- Goldring, S.R.; Gravallese, E.M. Mechanisms of bone loss in inflammatory arthritis: Diagnosis and therapeutic implications. Arthritis Res. 2000, 2, 33–37. [Google Scholar] [CrossRef] [Green Version]
- Walsh, N.C.; Reinwald, S.; Manning, C.A.; Condon, K.W.; Iwata, K.; Burr, D.B.; Gravallese, E.M. Osteoblast function is compromised at sites of focal bone erosion in inflammatory arthritis. J. Bone Miner. Res. 2009, 24, 1572–1585. [Google Scholar] [CrossRef] [Green Version]
- Karmakar, S.; Kay, J.; Gravallese, E.M. Bone damage in rheumatoid arthritis: Mechanistic insights and approaches to prevention. Rheum. Dis. Clin. N. Am. 2010, 36, 385–404. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.; Kerloc, A.; Rotival, M.; Xu, X.; Zhang, Q.; Souza, Z.D.; Kim, M.; Scholz, J.C.; Ko, J.; Srivastava, P.K.; et al. Kcnn4 is a regulator of macrophage multinucleation in bone homeostasis and inflammatory disease. Cell Rep. 2014, 8, 1210–1224. [Google Scholar] [CrossRef] [Green Version]
- Pereira, M.; Petretto, E.; Gordon, S.; Bassett, J.H.D.; Williams, G.R.; Behmoaras, J. Common signalling pathways in macrophage and osteoclast multinucleation. J. Cell Sci. 2018, 131. [Google Scholar] [CrossRef] [Green Version]
- Brodbeck, W.G.; Anderson, J.M. Giant cell formation and function. Curr. Opin. Hematol. 2009, 16, 53–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphrey, M.B.; Daws, M.R.; Spusta, S.C.; Niemi, E.C.; Torchia, J.A.; Lanier, L.L.; Seaman, W.E.; Nakamura, M.C. TREM2, a DAP12-associated receptor, regulates osteoclast differentiation and function. J. Bone Miner. Res. 2006, 21, 237–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helming, L.; Gordon, S. Molecular mediators of macrophage fusion. Trends Cell Biol. 2009, 19, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Soysa, N.S.; Alles, N. Positive and negative regulators of osteoclast apoptosis. Bone Rep. 2019, 11, 100225. [Google Scholar] [CrossRef] [PubMed]
- Enelow, R.I.; Sullivan, G.W.; Carper, H.T.; Mandell, G.L. Cytokine-induced human multinucleated giant cells have enhanced candidacidal activity and oxidative capacity compared with macrophages. J. Infect. Dis. 1992, 166, 664–668. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, T.; Nakashima, N.; Matsuda, T.; Nakamura, H.; Uchida, S.; Abe, T. Induction of multinucleated giant cells from rheumatoid arthritis (RA) synovial adherent cells by anti-DR antibody. Clin. Exp. Immunol. 1994, 98, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Quinn, M.T.; Schepetkin, I.A. Role of NADPH oxidase in formation and function of multinucleated giant cells. J. Innate Immun. 2009, 1, 509–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prieto-Potin, I.; Largo, R.; Roman-Blas, J.A.; Herrero-Beaumont, G.; Walsh, D.A. Characterization of multinucleated giant cells in synovium and subchondral bone in knee osteoarthritis and rheumatoid arthritis. BMC Musculoskelet. Disord. 2015, 16, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lampiasi, N.; Russo, R.; Zito, F. The alternative faces of macrophage generate osteoclasts. Biomed Res. Int. 2016, 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youn, M.-Y.; Takada, I.; Imai, Y.; Yasuda, H.; Kato, S. Transcriptionally active nuclei are selective in mature multinucleated osteoclasts. Genes Cells 2010, 15, 1025–1035. [Google Scholar] [CrossRef] [PubMed]
- Ying, P.; Ribet, A.M.B.; Pavlos, N.J. Membrane trafficking in osteoclasts and implications for osteoporosis. Biochem. Soc. Trans. 2019, 47, 639–650. [Google Scholar]
- Takito, J.; Inoue, S.; Nakamura, M. The sealing zone in osteoclasts: A self-organized structure on the bone. Int. J. Mol. Sci. 2018, 19, 984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenbeck, G. Formation and function of the ruffled border in osteoclasts. Semin. Cell Dev. Biol. 2002, 13, 285–292. [Google Scholar] [CrossRef]
- Itzstein, C.; Coxon, F.P.; Rogers, M.J. The regulation of osteoclast function and bone resorption by small GTPases. Small GTPases 2011, 2, 117–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takito, J.; Otsuka, H.; Yanagisawa, N.; Arai, H.; Shiga, M.; Inoue, M.; Nonaka, N.; Nakamura, M. Regulation of osteoclast multinucleation by the actin cytoskeleton signaling network. J. Cell. Physiol. 2015, 230, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Tiedemann, K.; Le Nihouannen, D.; Fong, J.E.; Hussein, O.; Barralet, J.E.; Komarova, S.V. Regulation of osteoclast growth and fusion by mTOR/raptor and mTOR/rictor/Akt. Front. Cell Dev. Biol. 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Oikawa, T.; Oyama, M.; Kozuka-Hata, H.; Uehara, S.; Udagawa, N.; Saya, H.; Matsuo, K. Tks5-dependent formation of circumferential podosomes/invadopodia mediates cell-cell fusion. J. Cell Biol. 2012, 197, 553–568. [Google Scholar] [CrossRef] [Green Version]
- Takito, J.; Nakamura, M.; Yoda, M.; Tohmonda, T.; Uchikawa, S.; Horiuchi, K.; Toyama, Y.; Chiba, K. The transient appearance of zipper-like actin superstructures during the fusion of osteoclasts. J. Cell Sci. 2012, 125, 662–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, A.; Kukita, A.; Li, Y.J.; Zhang, J.Q.; Nomiyama, H.; Yamaza, T.; Ayukawa, Y.; Koyano, K.; Kukita, T. Tunneling nanotube formation is essential for the regulation of osteoclastogenesis. J. Cell. Biochem. 2013, 114, 1238–1247. [Google Scholar] [CrossRef]
- Takito, J.; Nakamura, M. Precursors linked via the zipper-like structure or the filopodium during the secondary fusion of osteoclasts. Commun. Integr. Biol. 2012, 5, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Lemma, S.; Sboarina, M.; Porporato, P.E.; Zini, N.; Sonveaux, P.; Di Pompo, G.; Baldini, N.; Avnet, S. Energy metabolism in osteoclast formation and activity. Int. J. Biochem. Cell Biol. 2016, 79, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Golan, K.; Kollet, O.; Lapidot, T. Dynamic cross talk between S1P and CXCL12 regulates hematopoietic stem cells migration, development and bone remodeling. Pharmaceuticals 2013, 6, 1145–1169. [Google Scholar] [CrossRef] [PubMed]
- Aubin, J.E.; Bonnelye, E. Osteoprotegerin and its ligand: A new paradigm for regulation of osteoclastogenesis and bone resorption. Osteoporos. Int. 2000, 11, 905–913. [Google Scholar] [CrossRef]
- Wada, T.; Nakashima, T.; Hiroshi, N.; Penninger, J.M. RANKL-RANK signaling in osteoclastogenesis and bone disease. Trends Mol. Med. 2006, 12, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Grössinger, E.M.; Kang, M.; Bouchareychas, L.; Sarin, R.; Haudenschild, D.R.; Borodinsky, L.N.; Adamopoulos, I.E. Ca2+-dependent regulation of NFATc1 via KCa3.1 in inflammatory osteoclastogenesis. J. Immunol. 2018, 200, 749–757. [Google Scholar] [CrossRef] [Green Version]
- Nishikawa, K.; Nakashima, T.; Hayashi, M.; Fukunaga, T.; Kato, S.; Kodama, T.; Takahashi, S.; Calame, K.; Takayanagi, H. Blimp1-mediated repression of negative regulators is required for osteoclast differentiation. Proc. Natl. Acad. Sci. USA 2010, 107, 3117–3122. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Zhang, C.; Guo, H.; Chen, J.; Tao, Y.; Wang, F.; Lin, X.; Liu, Q.; Su, L.; Qin, A. Pregnenolone inhibits osteoclast differentiation and protects against lipopolysaccharide-induced inflammatory bone destruction and ovariectomy-induced bone loss. Front. Pharmacol. 2020, 11, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Xiao, J.; Xiang, C.; Xijie, Y. MicroRNAs in osteoclastogenesis and function: Potential therapeutic targets for osteoporosis. Int. J. Mol. Sci. 2016, 17, 349. [Google Scholar] [CrossRef]
- Li, K.; Chen, S.; Cai, P.; Chen, K.; Li, L.; Yang, X.; Yi, J.; Luo, X.; Du, Y.; Zheng, H. MiRNA-483–5p is involved in the pathogenesis of osteoporosis by promoting osteoclast differentiation. Mol. Cell. Probes 2020, 49. [Google Scholar] [CrossRef]
- Dou, C.; Zhang, C.; Kang, F.; Yang, X.; Jiang, H.; Bai, Y.; Xiang, J.; Xu, J.; Dong, S. MiR-7b directly targets DC-STAMP causing suppression of NFATc1 and c-Fos signaling during osteoclast fusion and differentiation. Biochim. Biophys. Acta Gene Regul. Mech. 2014, 1839, 1084–1096. [Google Scholar] [CrossRef]
- Lee, N.K.; Choi, Y.G.; Baik, J.Y.; Han, S.Y.; Jeong, D.W.; Bae, Y.S.; Kim, N.; Lee, S.Y. A crucial role for reactive oxygen species in RANKL-induced osteoclast differentiation. Blood 2005, 106. [Google Scholar] [CrossRef] [Green Version]
- Irie, N.; Takada, Y.; Watanabe, Y.; Matsuzaki, Y.; Naruse, C.; Asano, M.; Iwakura, Y.; Suda, T.; Matsuo, K. Bidirectional signaling through EphrinA2-EphA2 enhances osteoclastogenesis and suppresses osteoblastogenesis. J. Biol. Chem. 2009, 284, 14637–14644. [Google Scholar] [CrossRef] [Green Version]
- Costa, A.G.; Cusano, N.E.; Silva, B.C.; Cremers, S.; Bilezikian, J.P. Cathepsin K: Its skeletal actions and role as a therapeutic target in osteoporosis. Nat. Rev. Rheumatol. 2011, 7, 447–456. [Google Scholar] [CrossRef]
- Chiu, W.S.M.; McManus, J.F.; Notini, A.J.; Cassady, A.I.; Zajac, J.D.; Davey, R.A. Transgenic mice that express Cre recombinase in osteoclasts. Genesis 2004, 39, 178–185. [Google Scholar] [CrossRef]
- Takito, J.; Otsuka, H.; Inoue, S.; Kawashima, T.; Nakamura, M. Symmetrical retrograde actin flow in the actin fusion structure is involved in osteoclast fusion. Biol. Open 2017, 6, 1104–1114. [Google Scholar] [CrossRef] [Green Version]
- Søe, K.; Hobolt-Pedersen, A.S.; Delaisse, J.M. The elementary fusion modalities of osteoclasts. Bone 2015, 73, 181–189. [Google Scholar] [CrossRef]
- Ishii, T.; Ruiz-Torruella, M.; Ikeda, A.; Shindo, S.; Movila, A.; Mawardi, H.; Albassam, A.; Kayal, R.A.; Al-Dharrab, A.A.; Egashira, K.; et al. OC-STAMP promotes osteoclast fusion for pathogenic bone resorption in periodontitis via up-regulation of permissive fusogen CD9. FASEB J. 2018, 32, 4016–4030. [Google Scholar] [CrossRef] [Green Version]
- Samanna, V.; Ma, T.; Mak, T.W.; Rogers, M.; Chellaiah, M.A. Actin polymerization modulates CD44 surface expression, MMP-9 activation, and osteoclast function. J. Cell. Physiol. 2007, 213, 710–720. [Google Scholar] [CrossRef]
- Sodek, J.; Zhu, B.; Huynh, M.-H.; Brown, T.J.; Ringuette, M. Novel functions of the matricellular proteins Osteopontin and Osteonectin/SPARC. Connect. Tissue Res. 2002, 43, 308–319. [Google Scholar] [CrossRef]
- Georgess, D.; Machuca-Gayet, I.; Blangy, A.; Jurdic, P. Podosome organization drives osteoclast-mediated bone resorption. Cell Adhes. Migr. 2014, 8, 192–204. [Google Scholar] [CrossRef] [Green Version]
- Chellaiah, M.A.; Ma, T. Membrane localization of membrane type 1 matrix metalloproteinase by CD44 regulates the activation of pro-matrix Metalloproteinase 9 in Osteoclasts. Biomed Res. Int. 2013, 2013, 13. [Google Scholar] [CrossRef] [Green Version]
- Khan, U.A.; Hashimi, S.M.; Bakr, M.M.; Forwood, M.R.; Morrison, N.A. CCL2 and CCR2 are essential for the formation of Osteoclasts and foreign body giant cells. J. Cell. Biochem. 2016, 117, 382–389. [Google Scholar] [CrossRef] [Green Version]
- Miyamoto, K.; Ninomiya, K.; Sonoda, K.H.; Miyauchi, Y.; Hoshi, H.; Iwasaki, R.; Miyamoto, H.; Yoshida, S.; Sato, Y.; Morioka, H.; et al. MCP-1 expressed by osteoclasts stimulates osteoclastogenesis in an autocrine/paracrine manner. Biochem. Biophys. Res. Commun. 2009, 383, 373–377. [Google Scholar] [CrossRef]
- Ishii, M.; Iwai, K.; Koike, M.; Ohshima, S.; Kudo-Tanaka, E.; Ishii, T.; Mima, T.; Katada, Y.; Miyatake, K.; Uchiyama, Y.; et al. RANKL-induced expression of tetraspanin CD9 in lipid raft membrane microdomain is essential for cell fusion during osteoclastogenesis. J. Bone Miner. Res. 2006, 21, 965–976. [Google Scholar] [CrossRef]
- Kim, K.; Lee, S.H.; Jung, H.K.; Choi, Y.; Kim, N. NFATc1 induces osteoclast fusion via up-regulation of Atp6v0d2 and the Dendritic Cell-Specific Transmembrane Protein (DC-STAMP). Mol. Endocrinol. 2008, 22, 176–185. [Google Scholar] [CrossRef] [Green Version]
- Mensah, K.A.; Ritchlin, C.T.; Schwarz, E.M. RANKL induces heterogeneous DC-STAMPlo and DC-STAMPhi osteoclast precursors of which the DC-STAMPlo precursors are the master fusogens. J. Cell. Physiol. 2010, 223, 76–83. [Google Scholar] [CrossRef] [Green Version]
- Chiu, Y.H.; Mensah, K.A.; Schwarz, E.M.; Ju, Y.; Takahata, M.; Feng, C.; McMahon, L.A.; Hicks, D.G.; Panepento, B.; Keng, P.C.; et al. Regulation of human osteoclast development by dendritic cell-specific transmembrane protein (DC-STAMP). J. Bone Miner. Res. 2012, 27, 79–92. [Google Scholar] [CrossRef] [Green Version]
- Hobolt-Pedersen, A.S.; Delaissé, J.M.; Søe, K. Osteoclast fusion is based on heterogeneity between fusion partners. Calcif. Tissue Int. 2014, 95, 73–82. [Google Scholar] [CrossRef] [Green Version]
- Yi, T.G.; Kim, H.J.; Cho, J.Y.; Woo, K.M.; Ryoo, H.M.; Kim, G.S.; Baek, J.H. Tetraspanin CD9 regulates osteoclastogenesis via regulation of p44/42 MAPK activity. Biochem. Biophys. Res. Commun. 2006, 347, 178–184. [Google Scholar] [CrossRef]
- Witwicka, H.; Hwang, S.Y.; Reyes-Gutierrez, P.; Jia, H.; Odgren, P.E.; Donahue, L.R.; Birnbaum, M.J.; Odgren, P.R. Studies of OC-STAMP in osteoclast fusion: A new knockout mouse model, rescue of cell fusion, and transmembrane topology. PLoS ONE 2015, 10, e0128275. [Google Scholar] [CrossRef] [Green Version]
- Møller, A.M.J.; Delaissé, J.M.; Søe, K. Osteoclast fusion: Time-lapse reveals involvement of CD47 and Syncytin-1 at different stages of nuclearity. J. Cell. Physiol. 2017, 232, 1396–1403. [Google Scholar] [CrossRef]
- Kameda, Y.; Takahata, M.; Mikuni, S.; Shimizu, T.; Hamano, H.; Angata, T.; Hatakeyama, S.; Kinjo, M.; Iwasaki, N. Siglec-15 is a potential therapeutic target for postmenopausal osteoporosis. Bone 2015, 71, 217–226. [Google Scholar] [CrossRef]
- Hiruma, Y.; Hirai, T.; Tsuda, E. Siglec-15, a member of the sialic acid-binding lectin, is a novel regulator for osteoclast differentiation. Biochem. Biophys. Res. Commun. 2011, 409, 424–429. [Google Scholar] [CrossRef]
- Ishida-Kitagawa, N.; Tanaka, K.; Bao, X.; Kimura, T.; Miura, T.; Kitaoka, Y.; Hayashi, K.; Sato, M.; Maruoka, M.; Ogawa, T.; et al. Siglec-15 protein regulates formation of functional osteoclasts in concert with DNAX-activating protein of 12 kDa (DAP12). J. Biol. Chem. 2012, 287, 17493–17502. [Google Scholar] [CrossRef] [Green Version]
- Hiruma, Y.; Tsuda, E.; Maeda, N.; Okada, A.; Kabasawa, N.; Miyamoto, M.; Hattori, H.; Fukuda, C. Impaired osteoclast differentiation and function and mild osteopetrosis development in Siglec-15-deficient mice. Bone 2013, 53, 87–93. [Google Scholar] [CrossRef]
- Stuible, M.; Moraitis, A.; Fortin, A.; Saragosa, S.; Kalbakji, A.; Filion, M.; Tremblay, G.B. Mechanism and function of monoclonal antibodies targeting Siglec-15 for therapeutic inhibition of osteoclastic bone resorption. J. Biol. Chem. 2014, 289, 6498–6512. [Google Scholar] [CrossRef] [Green Version]
- Shimada-Sugawara, M.; Sakai, E.; Okamoto, K.; Fukuda, M.; Izumi, T.; Yoshida, N.; Tsukuba, T. Rab27A regulates transport of cell surface receptors modulating multinucleation and lysosome-related Organelles in Osteoclasts. Sci. Rep. 2015, 5, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Pata, M.; Vacher, J. Ostm1 bifunctional roles in Osteoclast maturation : Insights from a mouse model mimicking a human OSTM1 mutation. J. Bone Miner. Res. 2018, 33, 888–898. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Kim, J.H.; Kim, I.; Lee, J.; Seong, S.; Park, Y.W.; Kim, N. MicroRNA-26a regulates RANKL-induced osteoclast formation. Mol. Cells 2014, 38, 75–80. [Google Scholar] [CrossRef] [Green Version]
- Tang, L.; Yin, Y.; Liu, J.; Li, Z.; Lu, X. MIR-124 attenuates osteoclastogenic differentiation of bone marrow monocytes via targeting Rab27a. Cell. Physiol. Biochem. 2017, 43, 1663–1672. [Google Scholar] [CrossRef]
- Nishida, H.; Suzuki, H.; Madokoro, H.; Hayashi, M.; Morimoto, C.; Sakamoto, M.; Yamada, T. Blockade of CD26 signaling inhibits human osteoclast development. J. Bone Miner. Res. 2014, 29, 2439–2455. [Google Scholar] [CrossRef]
- Maile, L.A.; Demambro, V.E.; Wai, C.; Aday, A.W.; Capps, B.E.; Beamer, W.G.; Rosen, C.J.; Clemmons, D.R. An essential role for the association of CD47 to SHPS-1 in skeletal remodeling. J. Bone Miner. Res. 2011, 26, 2068–2081. [Google Scholar] [CrossRef]
- Kanemoto, S.; Kobayashi, Y.; Yamashita, T.; Miyamoto, T.; Cui, M.; Asada, R.; Cui, X.; Hino, K.; Kaneko, M.; Takai, T.; et al. Luman is involved in osteoclastogenesis through the regulation of DC-STAMP expression, stability and localization. J. Cell Sci. 2015, 128, 4353–4365. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Rho, J.; Jeong, D.; Sul, J.Y.; Kim, T.; Kim, N.; Kang, J.S.; Miyamoto, T.; Suda, T.; Lee, S.K.; et al. V-ATPase V0 subunit d2-deficient mice exhibit impaired osteoclast fusion and increased bone formation. Nat. Med. 2006, 12, 1403–1409. [Google Scholar] [CrossRef] [PubMed]
- Helming, L.; Tomasello, E.; Kyriakides, T.R.; Martinez, F.O.; Takai, T.; Gordon, S.; Vivier, E. Essential role of DAP12 signaling in macrophage programming into a fusion-competent state. Sci. Signal. 2008, 1, ra11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Søe, K.; Andersen, T.L.; Hinge, M.; Rolighed, L.; Marcussen, N.; Delaisse, J.-M. Coordination of fusion and trafficking of pre-osteoclasts at the marrow-bone interface. Calcif. Tissue Int. 2019, 105, 430–445. [Google Scholar] [CrossRef]
- Sun, H.; Kaartinen, M.T. Assessment of expression and specific activities of transglutaminases TG1, TG2, and FXIII-A during osteoclastogenesis. Anal. Biochem. 2020, 591, 113512. [Google Scholar] [CrossRef]
- Agrawal, A.; Buckley, K.A.; Bowers, K.; Furber, M.; Gallagher, J.A.; Gartland, A. The effects of P2X7 receptor antagonists on the formation and function of human osteoclasts in vitro. Purinergic Signal. 2010, 6, 307–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Walsh, M.C.; Yu, J.; Laskoski, P.; Takigawa, K.; Takegahara, N.; Choi, Y. Methylosome protein 50 associates with the purinergic receptor P2X5 and is involved in osteoclast maturation. FEBS Lett. 2020, 594, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Walsh, M.C.; Takegahara, N.; Middleton, S.A.; Kim, J.; Choi, Y. The purinergic receptor P2X5 regulates inflammasome activity and hyper-multinucleation of murine osteoclasts. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Kajitani, N.; Yamada, T.; Kawakami, K.; Matsumoto, K. Ichi TNX deficiency results in bone loss due to an increase in multinucleated osteoclasts. Biochem. Biophys. Res. Commun. 2019, 512, 659–664. [Google Scholar] [CrossRef]
- Shin, N.Y.; Choi, H.; Neff, L.; Wu, Y.; Saito, H.; Ferguson, S.M.; De Camilli, P.D.; Baron, R. Dynamin and endocytosis are required for the fusion of osteoclasts and myoblasts. J. Cell Biol. 2014, 207, 73–89. [Google Scholar] [CrossRef]
- Notomi, T.; Ezura, Y.; Noda, M. Identification of two-pore channel 2 as a novel regulator of osteoclastogenesis. J. Biol. Chem. 2012, 287, 35057–35064. [Google Scholar] [CrossRef] [Green Version]
- Shirakawa, J.; Takegahara, N.; Kim, H.; Lee, S.H.; Sato, K.; Yamagishi, S.; Choi, Y. Flrt2 is involved in fine-tuning of osteoclast multinucleation. BMB Rep. 2019, 52, 514–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Lewis, T.L.; Robinson, L.J.; Brundage, K.M.; Schafer, R.; Martin, K.H.; Blair, H.C.; Soboloff, J.; Barnett, J.B. The role of calcium release activated calcium channels in osteoclast differentiation. J. Cell. Physiol. 2011, 226, 1082–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Go, E.M.; Oh, J.H.; Park, J.H.; Lee, S.Y.; Lee, N.K. Spi-C positively regulates RANKL-mediated osteoclast differentiation and function. Exp. Mol. Med. 2020, 52, 691–701. [Google Scholar] [CrossRef]
- Hoshino, A.; Iimura, T.; Ueha, S.; Hanada, S.; Maruoka, Y.; Mayahara, M.; Suzuki, K.; Imai, T.; Ito, M.; Manome, Y.; et al. Deficiency of chemokine receptor CCR1 causes osteopenia due to impaired functions of osteoclasts and osteoblasts. J. Biol. Chem. 2010, 285, 28826–28837. [Google Scholar] [CrossRef] [Green Version]
- Nagai, Y.; Osawa, K.; Fukushima, H.; Tamura, Y.; Aoki, K.; Ohya, K.; Yasuda, H.; Hikiji, H.; Takahashi, M.; Seta, Y.; et al. P130Cas, Crk-associated substrate, plays important roles in osteoclastic bone resorption. J. Bone Miner. Res. 2013, 28, 2449–2462. [Google Scholar] [CrossRef]
- Lizneva, D.; Yuen, T.; Sun, L.; Kim, S.M.; Atabiekov, I.; Munshi, L.B.; Epstein, S.; New, M.; Zaidi, M. Emerging concepts in the epidemiology, pathophysiology, and clinical care of osteoporosis across the menopausal transition. Matrix Biol. 2018, 71, 70–81. [Google Scholar] [CrossRef]
- Cagnetta, V.; Patella, V. The role of the immune system in the physiopathology of osteoporosis. Clin. Cases Miner. Bone Metab. 2012, 9, 85–88. [Google Scholar] [PubMed]
- Cummings, S.R.; Nevitt, M.C.; Browner, W.S.; Stone, K.; Fox, K.M.; Ensrud, K.E.; Cauley, J.; Black, D.; Vogt, T.M. Risk factors for hip fracture in white women. Study of osteoporotic fractures research group. N. Engl. J. Med. 1995, 332. [Google Scholar] [CrossRef] [PubMed]
- Raisz, L.G. Pathogenesis of osteoporosis: Concepts, conflicts, and prospects. J. Clin. Investig. 2005, 115. [Google Scholar] [CrossRef] [Green Version]
- Eriksen, E.F.; Hodgson, S.F.; Eastell, R.; Cedel, S.L.; O’ Fallon, W.M.; Riggs, B.L. Cancellous bone remodeling in type I (postmenopausal) osteoporosis: Quantitative assessment of rates of formation, resorption, and bone loss at tissue and cellular levels. J. Bone Miner. Res. 1990, 5. [Google Scholar] [CrossRef]
- Lean, J.M.; Davis, J.T.; Fuller, K.; Jagger, C.J.; Kirstein, B.; Partington, G.A.; Urry, Z.L.; Chambers, T.J. A crucial role for thiol antioxidants in estrogen-deficiency bone loss. J. Clin. Investig. 2003, 112. [Google Scholar] [CrossRef] [Green Version]
- De Martinis, M.; Sirufo, M.M.; Suppa, M. IL-33 / IL-31 axis in osteoporosis. Int. J. Mol. Sci. 2020, 21, 1239. [Google Scholar] [CrossRef] [Green Version]
- Faienza, M.F.; Ventura, A.; Marzano, F.; Cavallo, L. Postmenopausal osteoporosis: The role of immune system cells. Clin. Dev. Immunol. 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Z.H.; Huang, T.; Xiao, J.W.; Gu, R.C.; Ouyang, J.; Wu, G.; Liao, H. Estrogen signaling effects on muscle-specific immune responses through controlling the recruitment and function of macrophages and T cells. Skelet. Muscle 2019, 9, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Brunetti, G.; Storlino, G.; Oranger, A.; Colaianni, G.; Faienza, M.F.; Ingravallo, G.; Di Comite, M.; Reseland, J.E.; Celi, M.; Tarantino, U.; et al. LIGHT/TNFSF14 regulates estrogen deficiency-induced bone loss. J. Pathol. 2020, 250, 440–451. [Google Scholar] [CrossRef]
- Pacifici, R. The inmune system and bone. Arch. Biochem. Biophys. 2010, 503, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Chassaing, B.; Tyagi, A.M.; Vaccaro, C.; Luo, T.; Adams, J.; Darby, T.M.; Weitzmann, M.N.; Mulle, J.G.; Gewirtz, A.T.; et al. Sex steroid deficiency-associated bone loss is microbiota dependent and prevented by probiotics. J. Clin. Investig. 2016, 126, 2049–2063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, D.H.; Yang, M.Y. The role of macrophage in the pathogenesis of osteoporosis. Int. J. Mol. Sci. 2019, 20, 2093. [Google Scholar] [CrossRef] [Green Version]
- Dou, C.; Ding, N.; Zhao, C.; Hou, T.; Kang, F.; Cao, Z.; Liu, C.; Bai, Y.; Dai, Q.; Ma, Q.; et al. Estrogen deficiency–mediated M2 macrophage osteoclastogenesis contributes to M1/M2 ratio alteration in ovariectomized osteoporotic mice. J. Bone Miner. Res. 2018, 33, 899–908. [Google Scholar] [CrossRef] [Green Version]
- Agidigbi, T.S.; Kim, C. Reactive oxygen species in osteoclast differentiation and possible pharmaceutical targets of ROS-mediated Osteoclast diseases. Int. J. Mol. Sci. 2019, 20, 3576. [Google Scholar] [CrossRef] [Green Version]
- Manolagas, S.C. From estrogen-centric to aging and oxidative stress: A revised perspective of the pathogenesis of osteoporosis. Endocr. Rev. 2010, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eghbali-Fatourechi, G.; Khosla, S.; Sanyal, A.; Boyle, W.J.; Lacey, D.L.; Riggs, B.L. Role of RANK ligand in mediating increased bone resorption in early postmenopausal women. J. Clin. Investig. 2003, 111. [Google Scholar] [CrossRef]
- Wang, H.; Yang, G.; Xiao, Y.; Luo, G.; Li, G.; Li, Z. Friend or foe? Essential roles of osteoclast in maintaining skeletal health. Biomed. Res. Int. 2020, 2020. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.H.; Park, M.; Baek, S.H.; Kim, H.J.; Kim, S.H. Molecules and signaling pathways involved in the expression of OC-STAMP during osteoclastogenesis. Amino Acids 2011, 40, 1447–1459. [Google Scholar] [CrossRef]
- Levine, J.P. Long-term estrogen and hormone replacement therapy for the prevention and treatment of osteoporosis. Curr. Womens Health Rep. 2003, 3, 181–186. [Google Scholar]
- Levin, V.A.; Jiang, X.; Kagan, R. Estrogen therapy for osteoporosis in the modern era. Osteoporos. Int. 2018, 29, 1049–1055. [Google Scholar] [CrossRef]
- Komm, B.S.; Chines, A.A. An update on selective estrogen receptor modulators for the prevention and treatment of osteoporosis. Maturitas 2012, 71, 221–226. [Google Scholar] [CrossRef]
- Pinkerton, J.V.; Thomas, S. Use of SERMs for treatment in postmenopausal women. J. Steroid Biochem. Mol. Biol. 2014, 142, 142–154. [Google Scholar] [CrossRef]
- Gałęzowska, J. Interactions between clinically used bisphosphonates and bone mineral: From coordination chemistry to biomedical applications and beyond. ChemMedChem 2018, 13, 289–302. [Google Scholar] [CrossRef]
- Papapoulos, S.E. Bisphosphonates: How do they work? Best Pract. Res. Clin. Endocrinol. Metab. 2008, 22, 831–847. [Google Scholar] [CrossRef]
- Coxon, F.P.; Thompson, K.; Roelofs, A.J.; Ebetino, F.H.; Rogers, M.J. Visualizing mineral binding and uptake of bisphosphonate by osteoclasts and non-resorbing cells. Bone 2008, 42, 848–860. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.G.G. Bisphosphonates: From bench to bedside. Ann. N. Y. Acad. Sci. 2006, 1068, 367–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorentzon, M. Treating osteoporosis to prevent fractures: Current concepts and future developments. J. Intern. Med. 2019, 285, 381–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pazianas, M.; Van Der Geest, S.; Miller, P. Bisphosphonates and bone quality. Bonekey Rep. 2014, 3, 529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weivoda, M.M.; Chew, C.K.; Monroe, D.G.; Farr, J.N.; Atkinson, E.J.; Geske, J.R.; Eckhardt, B.; Thicke, B.; Ruan, M.; Tweed, A.J.; et al. Identification of osteoclast-osteoblast coupling factors in humans reveals links between bone and energy metabolism. Nat. Commun. 2020, 11. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.; Goh, E.L.; Jin, A.; Bhattacharya, R.; Boughton, O.R.; Patel, B.; Karunaratne, A.; Vo, N.T.; Atwood, R.; Cobb, J.P.; et al. Long-term effects of bisphosphonate therapy: Perforations, microcracks and mechanical properties. Sci. Rep. 2017, 7, 43399. [Google Scholar] [CrossRef] [Green Version]
- Iwasaki, R.; Ninomiya, K.; Miyamoto, K.; Suzuki, T.; Sato, Y.; Kawana, H.; Nakagawa, T.; Suda, T.; Miyamoto, T. Cell fusion in osteoclasts plays a critical role in controlling bone mass and osteoblastic activity. Biochem. Biophys. Res. Commun. 2008, 377, 899–904. [Google Scholar] [CrossRef]
- Ott, S.M. Long-term safety of bisphosphonates. J. Clin. Endocrinol. Metab. 2005, 90. [Google Scholar] [CrossRef]
- Hanley, D.A.; Adachi, J.D.; Bell, A.; Brown, V. Denosumab: Mechanism of action and clinical outcomes. Int. J. Clin. Pract. 2012, 66, 1139–1146. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Ma, X.; Wang, T.; Zhai, S. Comparative efficacy of bisphosphonates in short-term fracture prevention for primary osteoporosis: A systematic review with network meta-analyses. Osteoporos. Int. 2016, 27, 3289–3300. [Google Scholar] [CrossRef]
- Beaudoin, C.; Jean, S.; Bessette, L.; Ste-Marie, L.-G.; Moore, L.; Brown, J.P. Denosumab compared to other treatments to prevent or treat osteoporosis in individuals at risk of fracture: A systematic review and meta-analysis. Osteoporos. Int. 2016, 27, 2835–2844. [Google Scholar] [CrossRef] [PubMed]
- Bone, H.G.; Bolognese, M.A.; Yuen, C.K.; Kendler, D.L.; Miller, P.D.; Yang, Y.C.; Grazette, L.; Martin, J.S.; Gallagher, J.C. Effects of denosumab treatment and discontinuation on bone mineral density and bone turnover markers in postmenopausal women with low bone mass. J. Clin. Endocrinol. Metab. 2011, 96, 972–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, A.V.; LeBoff, M.S. Osteoanabolic agents for Osteoporosis. J. Endocr. Soc. 2018, 2, 922–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandeira, L.; Lewiecki, E.M.; Bilezikian, J.P. Romosozumab for the treatment of osteoporosis. Expert Opin. Biol. Ther. 2017, 17, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Faienza, M.F.; Chiarito, M.; D’Amato, G.; Colaianni, G.; Colucci, S.; Grano, M.; Brunetti, G. Monoclonal antibodies for treating osteoporosis. Expert Opin. Biol. Ther. 2018, 18, 149–157. [Google Scholar] [CrossRef]
- Watanabe, A.; Yoneyama, S.; Nakajima, M.; Sato, N.; Takao-Kawabata, R.; Isogai, Y.; Sakurai-Tanikawa, A.; Higuchi, K.; Shimo, A.; Yamatoya, H.; et al. Osteosarcoma in Sprague-Dawley rats after long-term treatment with teriparatide (human parathyroid hormone (1-34)). J. Toxicol. Sci. 2012, 37, 617–629. [Google Scholar] [CrossRef] [Green Version]
- Anagnostis, P.; Gkekas, N.K.; Potoupnis, M.; Kenanidis, E.; Tsiridis, E.; Goulis, D.G. New therapeutic targets for osteoporosis. Maturitas 2019, 120, 1–6. [Google Scholar] [CrossRef]
- Luo, X.; Barbieri, D.; Zhang, Y.; Yang, Y.; Bruijn, J.D.; Yuan, H. Strontium ranelate in Osteoporosis. Curr. Pharm. Des. 2005, 8, 1907–1916. [Google Scholar] [CrossRef]
- Kartner, N.; Manolson, M.F. Novel techniques in the development of osteoporosis drug therapy: The osteoclast ruffled-border vacuolar H+-ATPase as an emerging target. Expert Opin. Drug Discov. 2014, 9. [Google Scholar] [CrossRef]
- Mukherjee, K.; Chattopadhyay, N. Pharmacological inhibition of cathepsin K: A promising novel approach for postmenopausal osteoporosis therapy. Biochem. Pharmacol. 2016, 117, 10–19. [Google Scholar] [CrossRef]
- Dai, R.; Wu, Z.; Chu, H.Y.; Lu, J.; Lyu, A.; Liu, J.; Zhang, G. Cathepsin K: The action in and beyond bone. Front. Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Hao, D.; Zhang, Q.; Chen, B.; Zhang, R.; Wang, Y.; Yang, H. Natural products for treatment of bone erosive diseases: The effects and mechanisms on inhibiting osteoclastogenesis and bone resorption. Int. Immunopharmacol. 2016, 36, 118–131. [Google Scholar] [CrossRef] [PubMed]
- Rajput, R.; Wairkar, S.; Gaud, R. Nutraceuticals for better management of osteoporosis: An overview. J. Funct. Foods 2018, 47, 480–490. [Google Scholar] [CrossRef]
- Dou, C.; Ding, N.; Luo, F.; Hou, T.; Cao, Z.; Bai, Y.; Liu, C.; Xu, J.; Dong, S. Graphene-based microRNA transfection blocks preosteoclast fusion to increase bone formation and vascularization. Adv. Sci. 2018, 5. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Tang, L.; Chen, J.; Lu, X. MiR-30a attenuates osteoclastogenesis via targeting DC-STAMP-c-Fos-NFATc1 signaling. Am. J. Transl. Res. 2017, 9, 5743. [Google Scholar]
- Wisitrasameewong, W.; Kajiya, M.; Movila, A.; Rittling, S.; Ishii, T.; Suzuki, M.; Matsuda, S.; Mazda, Y.; Torruella, M.R.; Azuma, M.M.; et al. DC-STAMP is an Osteoclast fusogen engaged in periodontal bone resorption. J. Dent. Res. 2017, 96, 685–693. [Google Scholar] [CrossRef]
- Sato, D.; Takahata, M.; Ota, M.; Fukuda, C.; Hasegawa, T.; Yamamoto, T.; Amizuka, N.; Tsuda, E.; Okada, A.; Hiruma, Y.; et al. Siglec-15-targeting therapy protects against glucocorticoid-induced osteoporosis of growing skeleton in juvenile rats. Bone 2020, 135, 115331. [Google Scholar] [CrossRef]
- Sato, D.; Takahata, M.; Ota, M.; Fukuda, C.; Tsuda, E.; Shimizu, T.; Okada, A.; Hiruma, Y.; Hamano, H.; Hiratsuka, S.; et al. Siglec-15-targeting therapy increases bone mass in rats without impairing skeletal growth. Bone 2018, 116, 172–180. [Google Scholar] [CrossRef]
- Brooks, P.J.; Glogauer, M.; Mcculloch, C.A. An overview of the derivation and function of multinucleated giant cells and their role in pathologic processes. Am. J. Pathol. 2019, 189, 1145–1158. [Google Scholar] [CrossRef] [Green Version]
- Al-Maawi, S.; Orlowska, A.; Sader, R.; James Kirkpatrick, C.; Ghanaati, S. In Vivo cellular reactions to different biomaterials—Physiological and pathological aspects and their consequences. Semin. Immunol. 2017, 29, 49–61. [Google Scholar] [CrossRef]
- Firestein, G.; McIness, I. Immunopathogenesis of rheumatoid arthritis. Immunity 2017, 46, 183–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moritag, I.; Okin, T.; Uekill, T.; Sakaguchi, K.; Enornoto, S.; Murota, S.; Medical, T. Expression on outer membranes of mannose residues, which are involved in osteoclast formation via cellular fusion events. J. Biol. Chem. 1994, 169, 17572–17576. [Google Scholar]
- Katsuyama, E.; Miyamoto, H.; Kobayashi, T.; Sato, Y.; Hao, W.; Kanagawa, H.; Fujie, A.; Tando, T.; Watanabe, R.; Morita, M.; et al. Interleukin-1 receptor-associated Kinase-4 (IRAK4) promotes inflammatory osteolysis by activating Osteoclasts and inhibiting formation of foreign body giant cells. J. Biol. Chem. 2015, 290, 716–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vignery, A. Osteoclasts and giant cells: Macrophage-macrophage fusion mechanism. Int. J. Exp. Pathol. 2000, 81, 291–304. [Google Scholar] [CrossRef]
- Han, X.; Sterling, H.; Chen, Y.; Saginario, C.; Brown, E.J.; Frazier, W.A.; Lindberg, F.P.; Vignery, A. CD47, a ligand for the macrophage fusion receptor, participates in macrophage multinucleation. J. Biol. Chem. 2000, 275, 37984–37992. [Google Scholar] [CrossRef] [Green Version]
- Yagi, M.; Ninomiya, K.; Fujita, N.; Suzuki, T.; Iwasaki, R.; Morita, K.; Hosogane, N.; Matsuo, K.; Toyama, Y.; Suda, T.; et al. Induction of DC-STAMP by alternative activation and downstream signaling mechanisms. J. Bone Miner. Res. 2007, 22, 992–1001. [Google Scholar] [CrossRef]
- Moreno, J.L.; Mikhailenko, I.; Tondravi, M.M.; Keegan, A.D. IL-4 promotes the formation of multinucleated giant cells from macrophage precursors by a STAT6-dependent, homotypic mechanism: Contribution of E-cadherin. J. Leukoc. Biol. 2007, 82, 1542–1553. [Google Scholar] [CrossRef]
- Miyamoto, T. STATs and macrophage fusion. JAK STAT 2013, 2, e24777. [Google Scholar] [CrossRef] [Green Version]
- Fiorino, C.; Harrison, R.E. E-cadherin is important for cell differentiation during osteoclastogenesis. Bone 2016, 86, 106–118. [Google Scholar] [CrossRef]
- Champion, T.C.; Partridge, L.J.; Ong, S.M.; Malleret, B.; Wong, S.C.; Monk, P.N. Monocyte subsets have distinct patterns of tetraspanin expression and different capacities to form multinucleate giant cells. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Sissons, J.R.; Peschon, J.J.; Schmitz, F.; Gilchrist, M.; Aderem, A.; Sissons, J.R.; Peschon, J.J.; Schmitz, F.; Suen, R.; Gilchrist, M.; et al. Cutting edge: microRNA regulation of macrophage fusion into multinucleated giant cells. J Immunol 2012, 189, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Chang, Y.; Wei, W. Emerging role of targeting macrophages in rheumatoid arthritis: Focus on polarization, metabolism and apoptosis. Cell Prolif. 2020, 1–12. [Google Scholar] [CrossRef]
- Degboé, Y.; Rauwel, B.; Baron, M.; Boyer, J.; Ruyssen-witrand, A.; Constantin, A.; Davignon, J.; Lucas, R. Polarization of rheumatoid macrophages by TNF targeting through an IL-10 / STAT3 mechanism. Front. Immunol. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mills, C.D. Anatomy of a discovery: M1 and M2 macrophages. Front. Immunol. 2015, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ryszer, T. Understanding the mysterious M2 macrophage through activation markers and effector mechanisms. Mediat. Inflamm. 2020, 2015, 16–18. [Google Scholar] [CrossRef] [Green Version]
- Oya, A.; Katsuyama, E.; Morita, M.; Sato, Y.; Kobayashi, T.; Miyamoto, K.; Nishiwaki, T.; Funayama, A.; Fujita, Y.; Kobayashi, T.; et al. Tumor necrosis factor receptor-associated factor 6 is required to inhibit foreign body giant cell formation and activate osteoclasts under inflammatory and infectious conditions. J. Bone Miner. Metab. 2018, 36, 679–690. [Google Scholar] [CrossRef]
- Zhu, K.; Yang, C.; Dai, H.; Li, J.; Liu, W.; Luo, Y.; Zhang, X.; Wang, Q. Crocin inhibits titanium particle-induced inflammation and promotes osteogenesis by regulating macrophage polarization. Int. Immunopharmacol. 2019, 76, 105865. [Google Scholar] [CrossRef]
- Amarasekara, D.S.; Yun, H.; Kim, S.; Lee, N.; Kim, H.; Rho, J. Regulation of osteoclast differentiation by cytokine networks. Immune Netw. 2018, 18, 1–18. [Google Scholar] [CrossRef]
- Zhao, B. TNF and bone remodeling. Curr. Osteoporos. Rep. 2019, 15, 126–134. [Google Scholar] [CrossRef]
- Adamopoulos, I.E.; Mellins, E.D. Alternative pathways of osteoclastogenesis in inflammatory arthritis. Nat. Rev. Rheumatol. 2015, 11, 189–194. [Google Scholar] [CrossRef]
- Walsh, N.C.; Gravallese, E.M. Bone remodeling in rheumatic disease: A question of balance. Immunol. Rev. 2010, 233, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Hu, W.; Ge, J. Functions of interleukin-34 and its emerging association with rheumatoid arthritis. Immunology 2016, 362–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Y.; Jansen, I.D.C.; Sprangers, S.; Stap, J.; Leenen, P.J.M.; Everts, V.; Vries, T.J. De IL-1 b differently stimulates proliferation and multinucleation of distinct mouse bone marrow osteoclast precursor subsets. J. Leukoc. Biol. 2016, 100, 513–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falzoni, S.; Chiozzi, P.; Ferrari, D.; Buell, G.; Virgilio, F. Di P2X 7 receptor and polykarion formation. Mol. Biol. Cell 2000, 11, 3169–3176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiozzi, P.; Sanz, J.M.; Ferrari, D.; Falzoni, S.; Aleotti, A.; Buell, G.N.; Collo, G.; Di Virgilio, F. Spontaneous cell fusion in macrophage cultures expressing high levels of the P2Z/P2X7 receptor. J. Cell Biol. 1997, 138, 697–706. [Google Scholar] [CrossRef] [Green Version]
- Savio, L.E.; de Andrade Mello, P.; da Silva, C.G.; Coutinho-Silva, R. The P2X7 receptor in inflammatory diseases: Angel or demon? Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Essandoh, K.; Li, Y.; Huo, J.; Fan, G.C. MiRNA-mediated macrophage polarization and its potential role in the regulation of inflammatory response. Shock 2017, 46, 122–131. [Google Scholar] [CrossRef]
- Tateiwa, D.; Yoshikawa, H.; Kaito, T. Cartilage and bone destruction in arthritis: Pathogenesis and treatment strategy: A literature review. Cells 2019, 8, 818. [Google Scholar] [CrossRef] [Green Version]
- Okada, Y.; Terao, C.; Ikari, K.; Kochi, Y.; Ohmura, K.; Suzuki, A.; Kawaguchi, T.; Stahl, E.A.; Kurreeman, F.A.; Nishida, N.; et al. Meta-analysis identifies nine new loci associated with rheumatoid arthritis in the Japanese population. Nat. Genet. 2012, 44, 511–516. [Google Scholar] [CrossRef]
- Stahl, E.A.; Raychaudhuri, S.; Remmers, E.F.; Xie, G.; Eyre, S.; Thomson, B.P.; Li, Y.; Kurreeman, F.A.; Zhernakova, A.; Hinks, A.; et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat. Genet. 2010, 42, 508–514. [Google Scholar] [CrossRef]
- Symmons, D.P.M. Epidemiology of rheumatoid arthritis: Determinants of onset, persistence and outcome. Best Pract. Res. Clin. Rheumatol. 2002, 16, 702–722. [Google Scholar] [CrossRef] [PubMed]
- Baum, R.; Gravallese, E.M. Bone as a target organ in rheumatic disease: Impact on Osteoclasts and Osteoblasts. Clin Rev Allergy Immunol. 2016, 51, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, M.; Lai, N.; Yu, H.; Huang, H.; Hsieh, S.; Yu, C. Anti—Citrullinated protein antibodies bind surface-expressed citrullinated Grp78 on monocyte/macrophages and stimulate tumor necrosis factor production. Arthritis Rheum. 2010, 62, 1213–1223. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, A.; Joshua, V.; Hensvold, A.H.; Jin, T.; Sun, M.; Vivar, N.; Ytterberg, A.J.; Engström, M.; Fernandes-Cerqueira, C.; Amara, K.; et al. Identi fi cation of a novel chemokine-dependent molecular mechanism underlying rheumatoid arthritis-associated autoantibody-mediated bone loss. Ann. Rheum. Dis. 2016, 75, 721–729. [Google Scholar] [CrossRef] [Green Version]
- Nogueira, E.; Gomes, A.; Preto, A.; Cavaco-Paulo, A. Update on therapeutic approaches for rheumatoid arthritis. Curr. Med. Chem. 2016, 23, 2190–2203. [Google Scholar] [CrossRef]
- Ono, T.; Hayashi, M.; Sasaki, F.; Nakashima, T. RANKL biology: Bone metabolism, the immune system, and beyond. Inflamm. Regen. 2020, 40, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Yap, H.Y.; Tee, S.; Wong, M.; Chow, S.-K.; Peh, S.-C.; Teow, S.-Y. Pathogenic role of immune cells in rheumatoid arthritis: Implications in clinical treatment and biomarker development. Cells 2018, 7, 161. [Google Scholar] [CrossRef] [Green Version]
- Mazzetti, I.; Grigolo, B.; Pulsatelli, L.; Dolzani, P.; Silvestri, T.; Roseti, L.; Meliconi, R.F.A. Differential roles of nitric oxide and oxygen radicals in chondrocytes affected by osteoarthritis and rheumatoid arthritis. Clin. Sci. 2001, 101, 593–599. [Google Scholar] [CrossRef] [Green Version]
- Quinonez-Flores, C.M.; Gonzalez-Chavez, S.A.; Del Rio Najera, D.; Pacheco-Tena, C. Oxidative stress relevance in the pathogenesis of the rheumatoid arthritis: A systematic review. Biomed Res. Int. 2016, 2016. [Google Scholar] [CrossRef] [Green Version]
- Vomero, M.; Barbati, C.; Colasanti, T.; Perricone, C.; Novelli, L.; Ceccarelli, F.; Spinelli, F.R.; Di Franco, M.; Conti, F.; Valesini, G.; et al. Autophagy and rheumatoid arthritis : Current knowledges and future perspectives. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Meijer, A.J.; Codogno, P. Signalling and autophagy regulation in health, aging and disease. Mol. Asp. Med. 2006, 27, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Coury, F.; Peyruchaud, O.; Machuca-gayet, I. Osteoimmunology of bone loss in inflammatory rheumatic diseases. Front. Immunol. 2019, 10, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smolen, J.S.; van der Heijde, D.; Machold, K.P.; Aletaha, D.L.R. Proposal for a new nomenclature of disease-modifying antirheumatic drugs. Ann. Rheum. Dis. 2014, 73, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Rein, P.; Mueller, R.B. Treatment with biologicals in rheumatoid arthritis: An overview. Rheumatol. Ther. 2017, 4, 247–261. [Google Scholar] [CrossRef] [Green Version]
- Saqib, U.; Sarkar, S.; Suk, K.; Mohammad, O.; Baig, M.S. Phytochemicals as modulators of M1-M2 macrophages in inflammation. Oncotarget 2018, 9, 17937–17950. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.; Singh, K.; Ibrahim, W.; King, B.; Damsky, W. The promise of JAK inhibitors for treatment of sarcoidosis and other inflammatory disorders with macrophage activation: A review of the literature. YALE J. Biol. Med. 2020, 93, 187–195. [Google Scholar]
- Hammarén, H.M.; Virtanen, A.T.; Raivola, J.; Silvennoinen, O. Cytokine the regulation of JAKs in cytokine signaling and its breakdown in disease. Cytokine 2019, 118, 48–63. [Google Scholar] [CrossRef]
- Labranche, T.P.; Jesson, M.I.; Radi, Z.A.; Storer, C.E.; Guzova, J.A.; Bonar, S.L.; Thompson, J.M.; Happa, F.A.; Stewart, Z.S.; Zhan, Y.; et al. JAK Inhibition with tofacitinib suppresses arthritic joint structural damage through decreased RANKL production. Arthritis Rheum. 2012, 64, 3531–3542. [Google Scholar] [CrossRef]
- Adam, S.; Simon, N.; Steffen, U.; Andes, F.T.; Scholtysek, C.; Müller, D.I.; Weidner, D.; Andreev, D.; Kleyer, A.; Culemann, S.; et al. JAK inhibition increases bone mass in steady-state conditions and ameliorates pathological bone loss by stimulating osteoblast function. Sci. Transl. Med. 2020, 12, eaay4447. [Google Scholar] [CrossRef]
- Lin, N.Y.; Beyer, C.; Gießl, A.; Kireva, T.; Scholtysek, C.; Uderhardt, S.; Munoz, L.E.; Dees, C.; Distler, A.; Wirtz, S.; et al. Autophagy regulates TNFα-mediated joint destruction in experimental arthritis. Ann. Rheum. Dis. 2013, 72, 761–768. [Google Scholar] [CrossRef]
- Lauper, K.; Mongin, D.; Alpizar-Rodriguez, D.; Codreanu, C.; Iannone, F.; Kristianslund, E.K.; Kvien, T.K.; Pavelka, K.; Pombo-Suarez, M.; Santos, M.J.; et al. Drug retention of biological DMARD in rheumatoid arthritis patients: The role of baseline characteristics and disease evolution. Rheumatology 2019, 58, 2221–2229. [Google Scholar] [CrossRef] [PubMed]
- Radner, H.; Aletaha, D. Anti-TNF in rheumatoid arthritis: An overview. Wien. Med. Wochenschr. 2015, 165, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hsu, H.C.; Mountz, J.D. Managing macrophages in rheumatoid arthritis by reform or removal. Curr. Rheumatol. Rep. 2012, 14, 445–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amantea, D.; Certo, M.; Petrelli, F.; Tassorelli, C.; Micieli, G.; Corasaniti, M.T.; Puccetti, P.; Fallarino, F.; Bagetta, G. Azithromycin protects mice against ischemic stroke injury by promoting macrophage transition towards M2 phenotype. Exp. Neurol. 2016, 275, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Dugo, L.; Belluomo, M.G.; Fanali, C.; Russo, M.; Cacciola, F.; Maccarrone, M.; Sardanelli, A.M. Effect of cocoa polyphenolic extract on macrophage polarization from proinflammatory M1 to anti-inflammatory M2 state. Oxid. Med. Cell. Longev. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Tardito, S.; Martinelli, G.; Soldano, S.; Paolino, S.; Pacini, G.; Patane, M.; Alessandri, E.; Smith, V.; Cutolo, M. Macrophage M1/M2 polarization and rheumatoid arthritis: A systematic review. Autoimmun. Rev. 2019, 18, 102397. [Google Scholar] [CrossRef] [Green Version]
- Vis, M.; Havaardsholm, E.A.; Haugeberg, G.; Uhlig, T.; Voskuyl, A.E.; Van De Stadt, R.J.; Dijkmans, B.A.C.; Woolf, A.D.; Kvien, T.K.; Lems, W.F. Evaluation of bone mineral density, bone metabolism, osteoprotegerin and receptor activator of the NFκB ligand serum levels during treatment with infliximab in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2006, 65, 1495–1499. [Google Scholar] [CrossRef] [Green Version]
- Ruscitti, P.; Masedu, F.; Alvaro, S.; Airò, P.; Battafarano, N.; Cantarini, L.; Cantatore, F.P.; Carlino, G.; Abrosca, V.; Frassi, M.; et al. Anti-interleukin-1 treatment in patients with rheumatoid arthritis and type 2 diabetes (TRACK): A multicentre, open-label, randomised controlled trial. PLoS ONE 2019, 16, e1002901. [Google Scholar] [CrossRef]
- Nishimoto, N.; Hashimoto, J.; Miyasaka, N.; Yamamoto, K.; Kawai, S.; Takeuchi, T.; Murata, N.; van der Heijde, D.; Kishimoto, T. Study of active controlled monotherapy used for rheumatoid arthritis, an IL-6 inhibitor (SAMURAI): Evidence of clinical and radiographic benefit from an x ray reader-blinded randomised controlled trial of tocilizumab. Ann. Rheum. Dis. 2007, 66, 1162–1167. [Google Scholar] [CrossRef] [Green Version]
- Srirangan, S.; Choy, E.H. The role of Interleukin 6 in the pathophysiology of rheumatoid arthritis. Ther. Adv. Musculoskelet. Dis. 2010, 2, 247–256. [Google Scholar] [CrossRef] [Green Version]
- Brunger, J.M.; Zutshi, A.; Willard, V.P.; Gersbach, C.A. Genome engineering of stem cells for autonomously regulated, closed-loop delivery of biologic drugs. Stem Cell Rep. 2017, 8, 1202–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, H.; Kang, Y.; Zhang, H.; Zhao, D.; Xia, J.; Lu, Z.; Wang, H.; Xu, F.; Shi, L. MiR-127 modulates macrophage polarization and promotes lung inflammation and injury by activating the JNK pathway. J. Immunol. 2015, 194, 1239–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Virgilio, F. Liaisons dangereuses:P2X(7) and the inflammasome. Trends Pharmacol. Sci. 2007, 28, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Di Virgilio, F.; Vuerich, M. Purinergic signaling in the immune system. Aut. Neurosci. 2015, 191, 117–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellegatti, P.; Falzoni, S.; Donvito, G.; Lemaire, I.; Virgilio, F. Di P2X7 receptor drives osteoclast fusion by increasing the extracellular adenosine concentration. FASEB J. 2011, 25, 1264–1274. [Google Scholar] [CrossRef] [PubMed]
- Gersbach, C.A.; Guilak, F. Genome engineering for personalized arthritis therapeutics. Trends Mol. Med. 2018, 23, 917–931. [Google Scholar] [CrossRef]
- Rahul Bodkhe, B.B.; Taneja, V. The role of microbiome in rheumatoid arthritis treatment. Ther. Adv. Musculoskelet. Dis. 2019, 11, 1–16. [Google Scholar]
- Maeda, Y.; Takeda, K. Host–microbiota interactions in rheumatoid arthritis. Exp. Mol. Med. 2019, 51. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biological Target | Role in M-FM | Refs |
|---|---|---|
| CD44/Matrix metallopeptidase 9 (CD44/MMP-9) |
| [45] |
| Monocyte chemoattractant protein-1/C-C chemokine receptor type 2 (MCP-1/CCR2) |
| [49,50] |
| CD9 |
| [51] |
| Dendritic cell-specific transmembrane protein (DC-STAMP) |
| [37,53] |
| OC-stimulatory transmembrane protein (OC-STAMP) |
| [44] |
| Syncitin-1 |
| [58] |
| Sialic acid-binding immunoglobulin-type lectin 15 (Siglec-15) |
| [59,60,61,62,63] |
| Ras-related protein Rab-27a (Rab27a) |
| [64] |
| Osteoclastogenesis-associated transmembrane protein-1 (Ostm1) |
| [65] |
| miR7b |
| [37] |
| miR30a |
| [35] |
| miR-26a |
| [66] |
| CD47 |
| [58] |
| Macrophage fusion receptor (MFR) |
| [7,10] |
| E-cadherin |
| [7] |
| CD-26 |
| [68] |
| CD-47 |
| [69] |
| Src non-receptor tyrosine kinase (c-Src) |
| [22,73] |
| Human protein ‘SH3 and PX domains 2A’ (Tsk5) |
| [24] |
| C-C chemokine receptor type 1 (CCR-1) |
| [22] |
| Rapamycin-insensitive companion of TOR (RICTOR) |
| [23] |
| Tenascin x (TNX) |
| [78] |
| Dynamin |
| [79] |
| Two-pore channel 2 (TPC2) |
| [80] |
| Fibronectin leucine-rich transmembrane protein 2 (Flrt2) |
| [81] |
| Calcium release-activated channels (CRAC-C channels) |
| [82] |
| Transcription factor Spi-C (SPIC) |
| [83] |
| Crk-associated substrate (Cas) |
| [85] |
| Luman |
| [70] |
| Vacuolar ATPase (ATP6v0d2) |
| [71] |
| DAP-12 |
| [72] |
| OSCAR-FcRy |
| [73] |
| Transglutaminases |
| [74] |
| P2 × 7 |
| [75] |
| P2 × 5 |
| [76,77] |
| miR124 |
| [67] |
| Biological Targets | Therapeutic Molecules/Compounds | Effects on M-FM and OP | Refs |
|---|---|---|---|
| CD9 | Anti-CD9 antibody |
| [51] |
| DC-STAMP | Lentiviral vector pre-miR-7b |
| [37] |
| MiR-30a mimic |
| [135] | |
| anti-DC-STAMP-monoclonal antibody |
| [136] | |
| OC-STAMP | anti-OC STAMP-monoclonal antibody |
| [44] |
| CTGF/CCN2 | miR-26a mimics |
| [66] |
| Siglec-15 | anti-Siglec-15 antibody |
| [60,63,137,138] |
| Rab27a | MiR-124 mimics |
| [67] |
| Biological Target | Effects on M-FM and Inflammatory Bone Loss | Refs |
|---|---|---|
| Macrophage fusion receptor (MFR) |
| [144,145] |
| Potassium calcium-activated channel subfamily N member 4 (KCNN4) |
| [6] |
| Tetraspanins (CD-9, CD81, CD63, CD53) |
| [150] |
| Pro-inflammatory macrophages (M1) |
| [15,98,152,153] |
| Interleukin-1 beta (IL-1β) |
| [7,163] |
| Tumour necrosis factor-alpha (TNF-α) |
| [159] |
| Interleukin 6 (IL-6) |
| [152,158] |
| Signal transducer and activator of transcription-6/-1 axis (STAT-6/STAT-1 axis) |
| [7,148] |
| Tumour necrosis factor receptor-associated factor 6 (TRAF6) |
| [156,160] |
| Interleukin-1 receptor-associated kinase 4 (IRAK4) |
| [143] |
| Purinergic receptor P2X7 (P2RX7) |
| [164,165,166] |
| Purinergic receptor P2X5 (P2RX5) |
| [76] |
| Mammalian target of rapamycin (mTOR) complex 1 (mTORC1) |
| [23,101] |
| miR9 |
| [167] |
| miR127 |
| [167] |
| miR125b |
| [167] |
| miR155 |
| [167] |
| Dysbiosis |
| [97] |
| Biological Targets | Therapeutic Molecules/Compounds | Effects on M-FM and Inflammation in RA | Refs |
|---|---|---|---|
| Tetraspanins | Anti-tetraspanins antibodies |
| [150] |
| Tumour necrosis factor-alpha (TNF-α) | TNF blockers |
| [192,197] |
| Interleukin-1 (IL-1) | IL-1 inhibitors |
| [198] |
| Interleukin-6 receptor (IL-6R) | IL-6R blockers |
| [199,200] |
| JAK/STAT cascade | JAK inhibitors (JAK1, JAK2, JAK3) |
| [187,188,189] |
| P2X7 signalling | Anti-P2X7 antibody |
| [205] |
| Mir127 | Antagonist of mir127 |
| [167,202] |
| Macrophage polarization | Nutraceuticals (tripertenoids, stilbenes, flavonoids) |
| [185] |
| Dysbiosis | Disease-modifying anti-rheumatic drugs (DMARDs) |
| [207,208] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gambari, L.; Grassi, F.; Roseti, L.; Grigolo, B.; Desando, G. Learning from Monocyte-Macrophage Fusion and Multinucleation: Potential Therapeutic Targets for Osteoporosis and Rheumatoid Arthritis. Int. J. Mol. Sci. 2020, 21, 6001. https://doi.org/10.3390/ijms21176001
Gambari L, Grassi F, Roseti L, Grigolo B, Desando G. Learning from Monocyte-Macrophage Fusion and Multinucleation: Potential Therapeutic Targets for Osteoporosis and Rheumatoid Arthritis. International Journal of Molecular Sciences. 2020; 21(17):6001. https://doi.org/10.3390/ijms21176001
Chicago/Turabian StyleGambari, Laura, Francesco Grassi, Livia Roseti, Brunella Grigolo, and Giovanna Desando. 2020. "Learning from Monocyte-Macrophage Fusion and Multinucleation: Potential Therapeutic Targets for Osteoporosis and Rheumatoid Arthritis" International Journal of Molecular Sciences 21, no. 17: 6001. https://doi.org/10.3390/ijms21176001
APA StyleGambari, L., Grassi, F., Roseti, L., Grigolo, B., & Desando, G. (2020). Learning from Monocyte-Macrophage Fusion and Multinucleation: Potential Therapeutic Targets for Osteoporosis and Rheumatoid Arthritis. International Journal of Molecular Sciences, 21(17), 6001. https://doi.org/10.3390/ijms21176001