Cell Bank Origin of MDCK Parental Cells Shapes Adaptation to Serum-Free Suspension Culture and Canine Adenoviral Vector Production

 ,
, 
Abstract
1. Introduction
2. Results
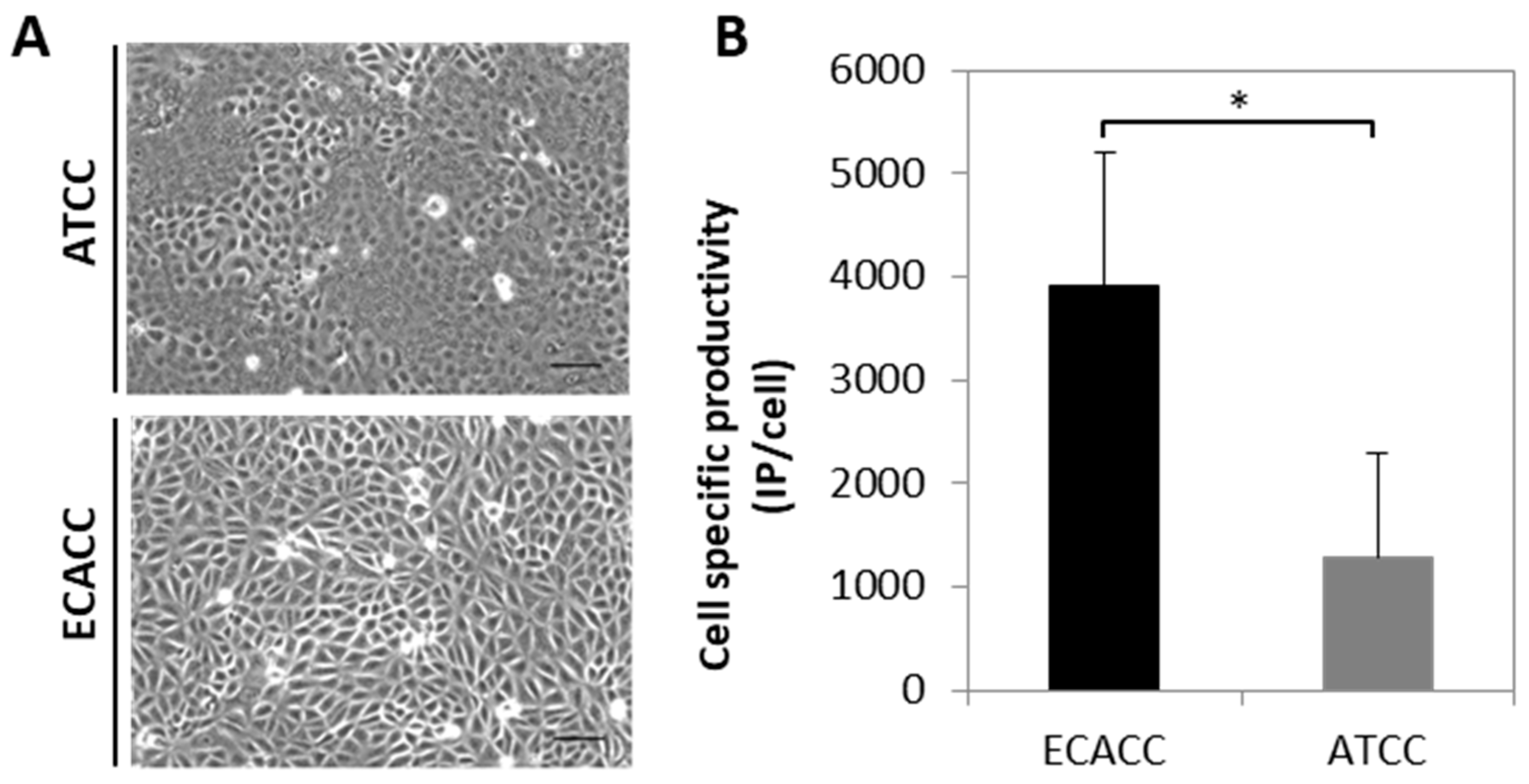
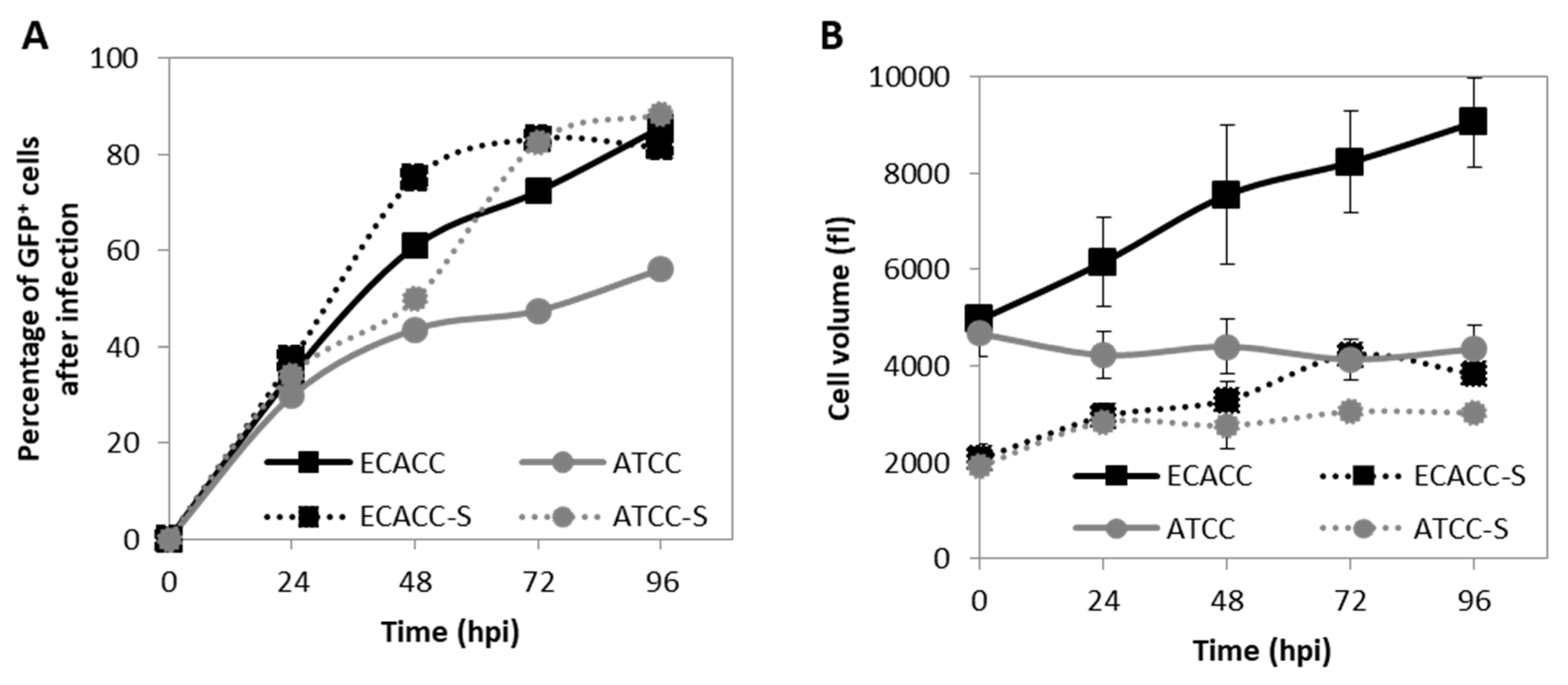
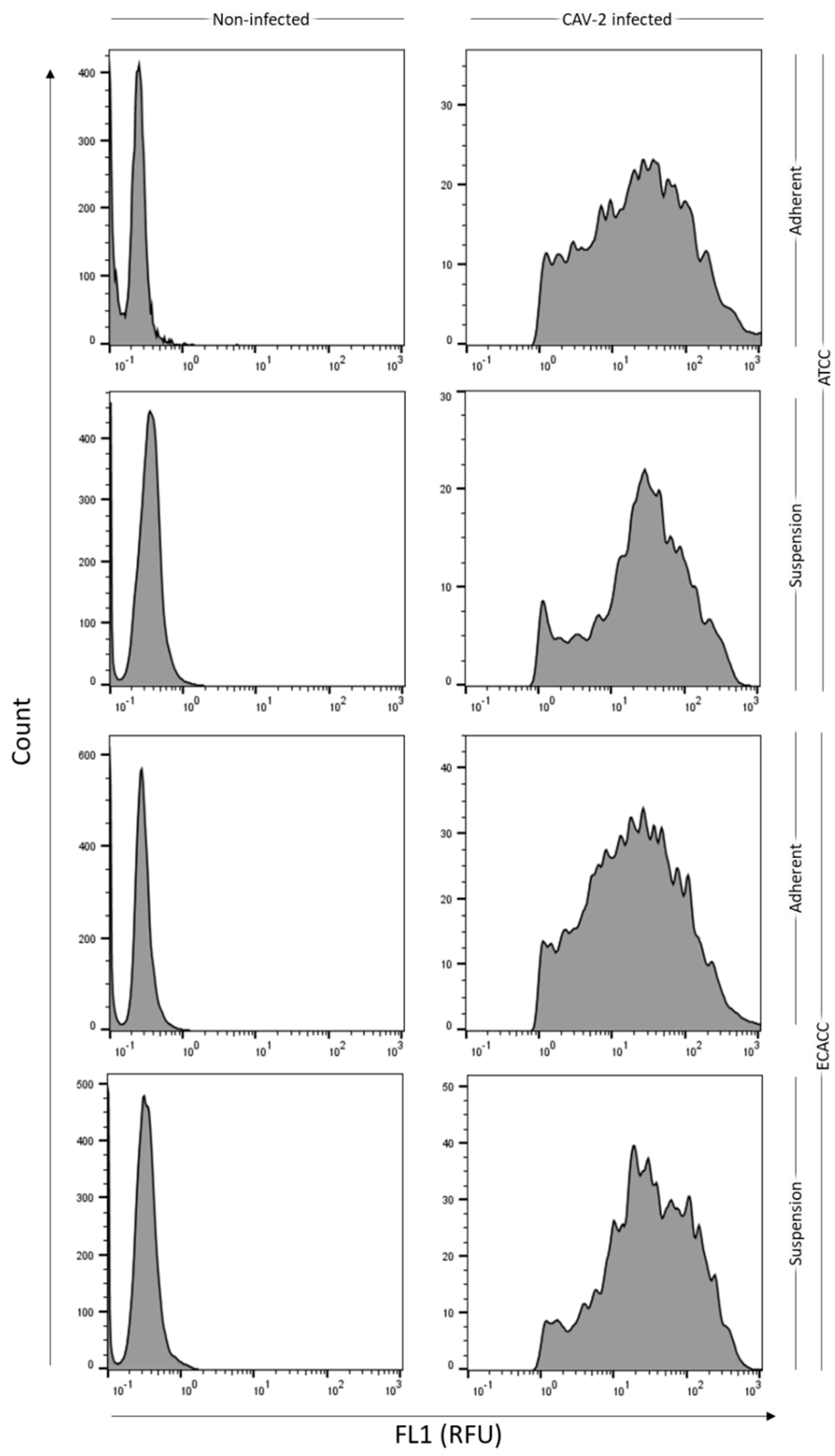
2.1. Cell Growth and Virus Production in Adherent MDCK Cultures
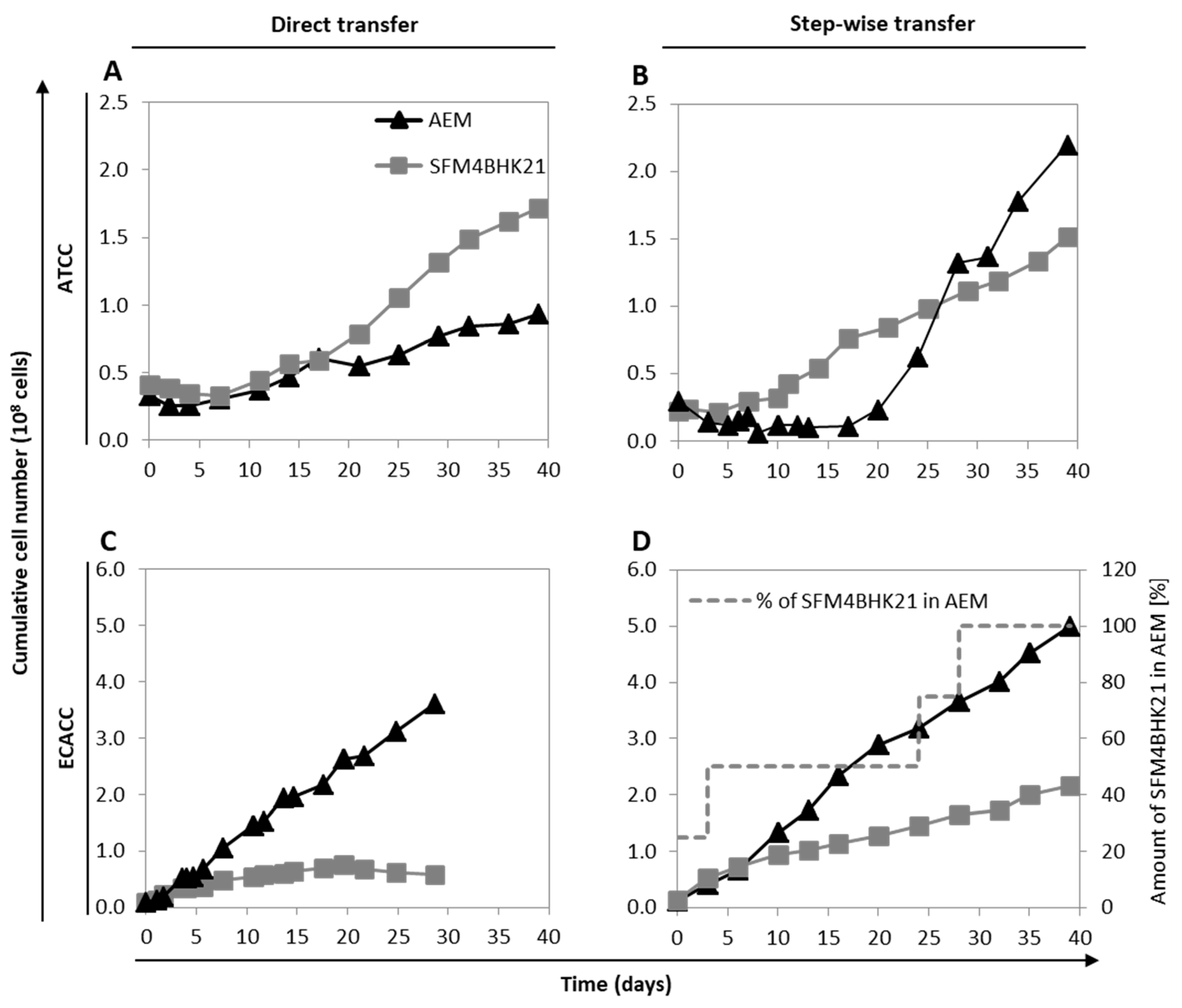
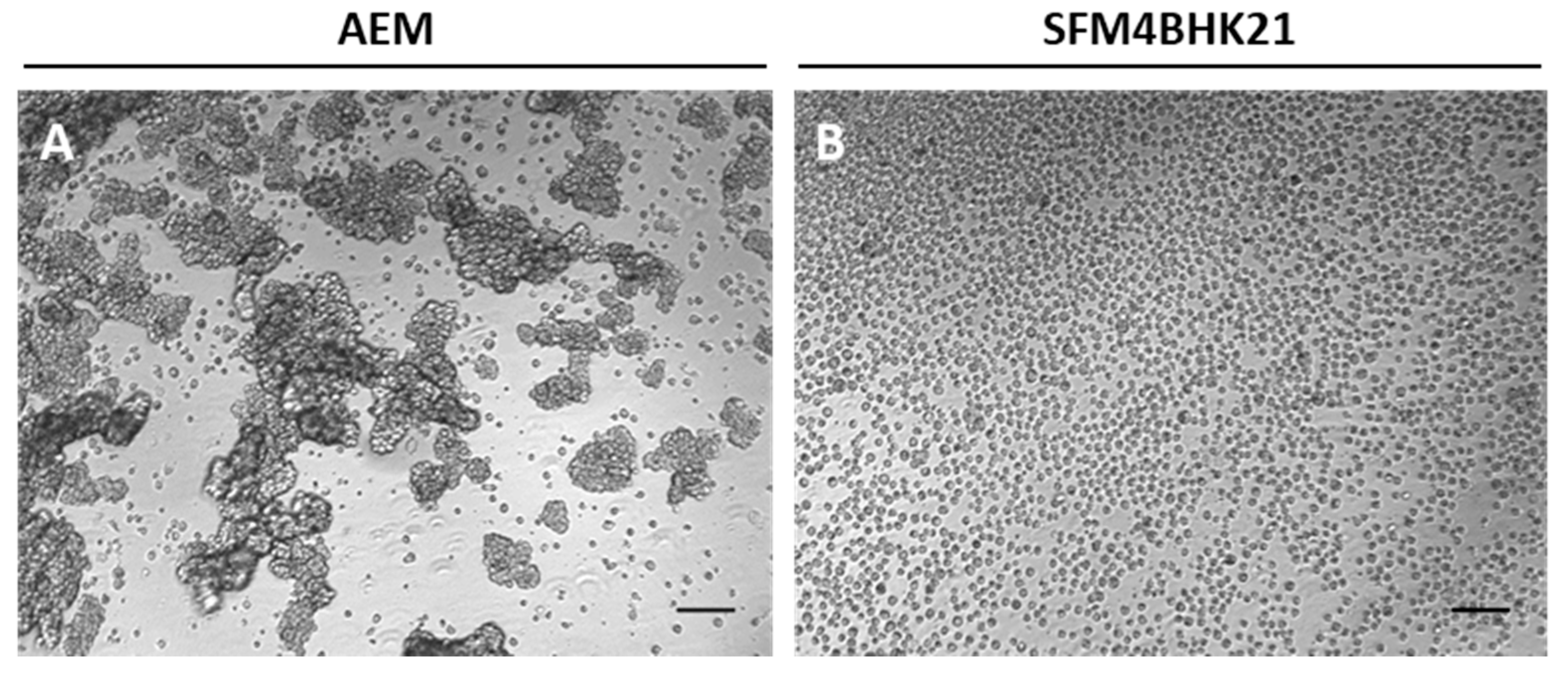
2.2. Adaptation of MDCK Cells to Serum-Free Suspension Growth
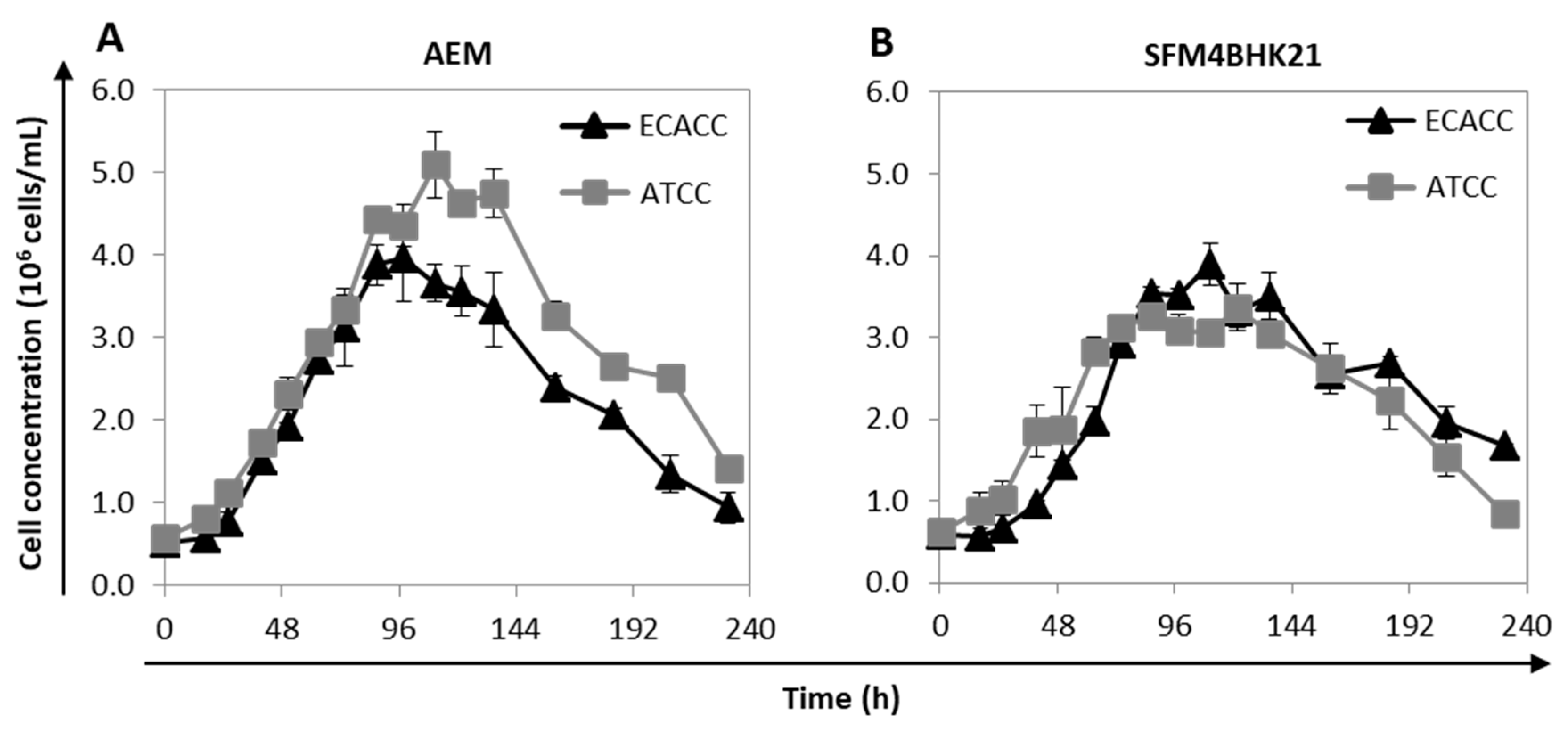
2.3. Growth of MDCK Cells in Suspension Cultures
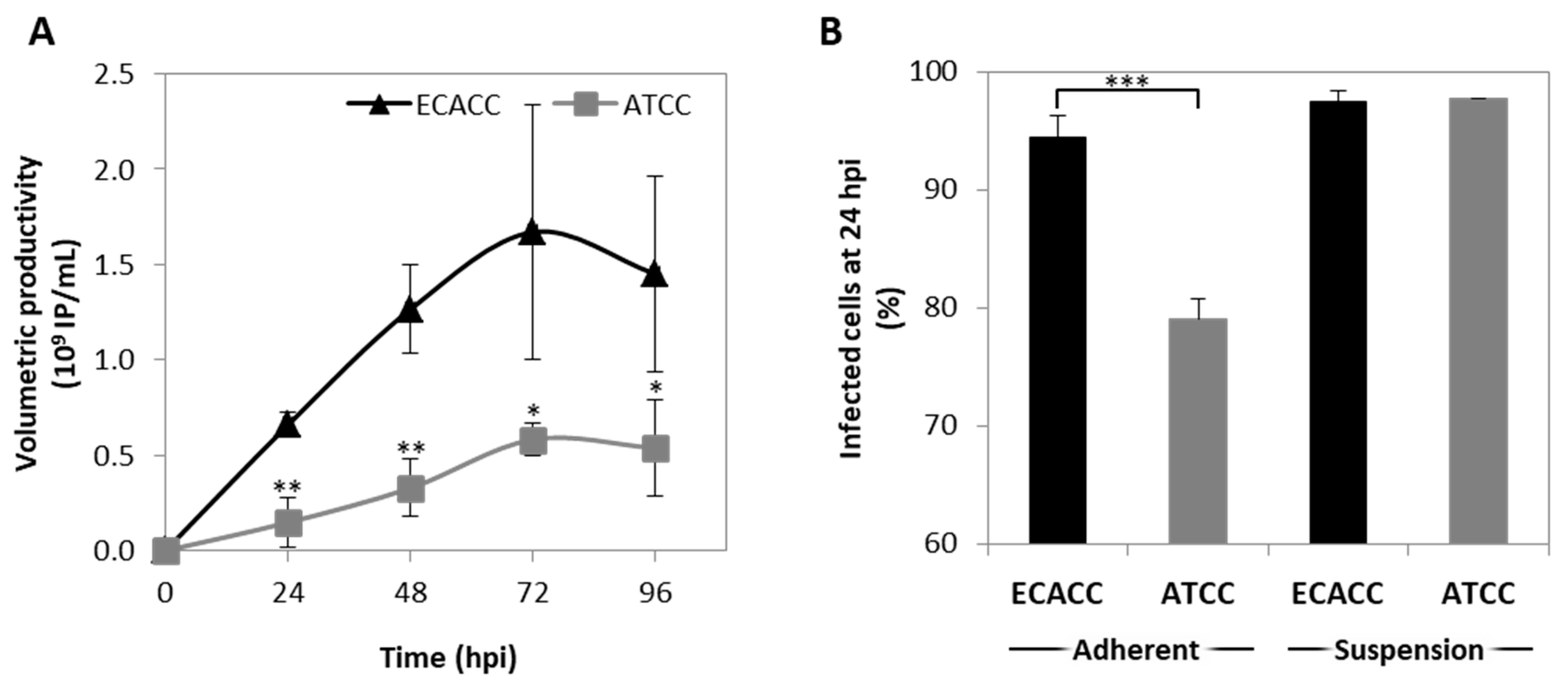
2.4. CAV-2 Prodution and Infection Progression in Suspension Cultures with AEM Medium
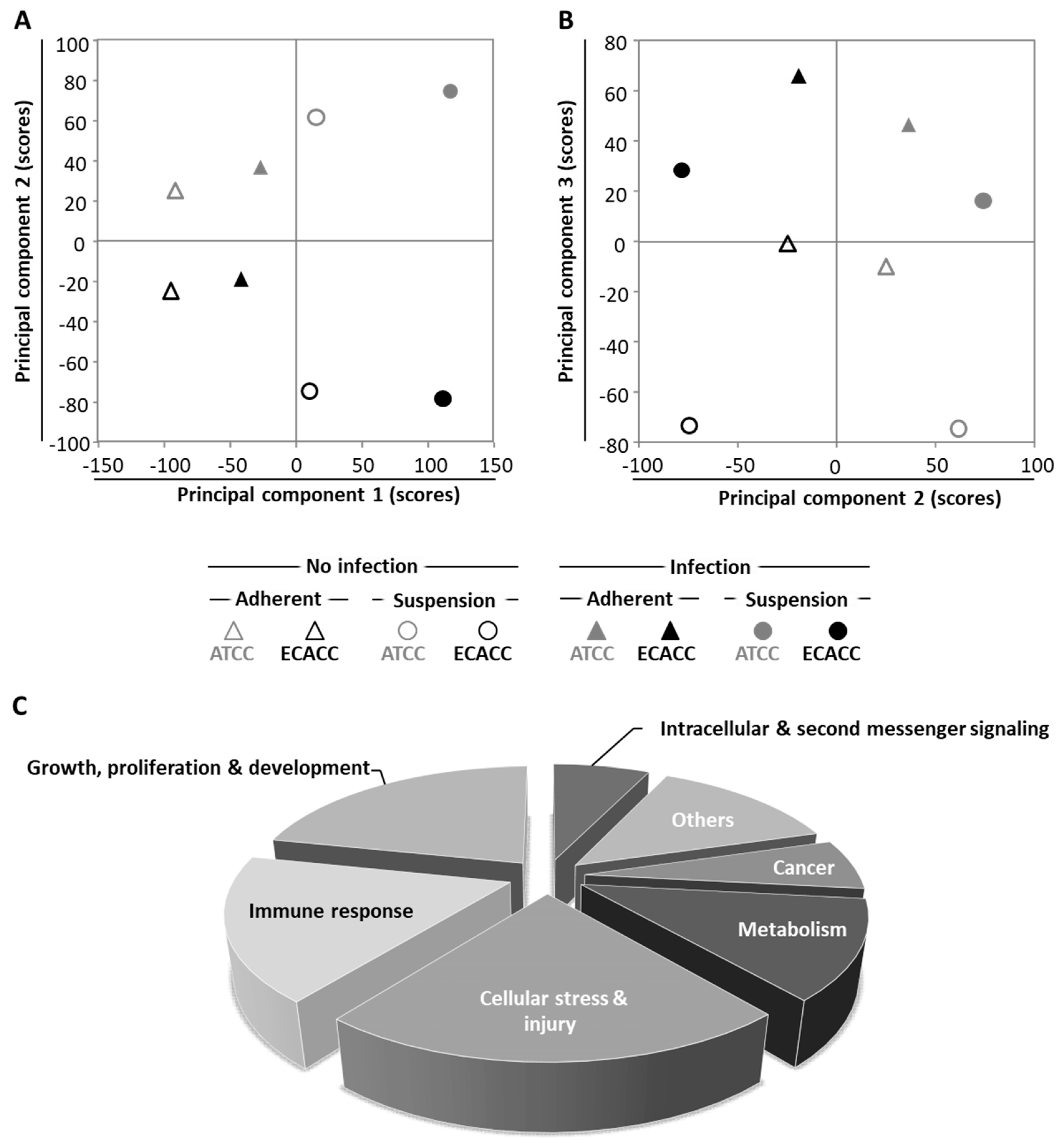
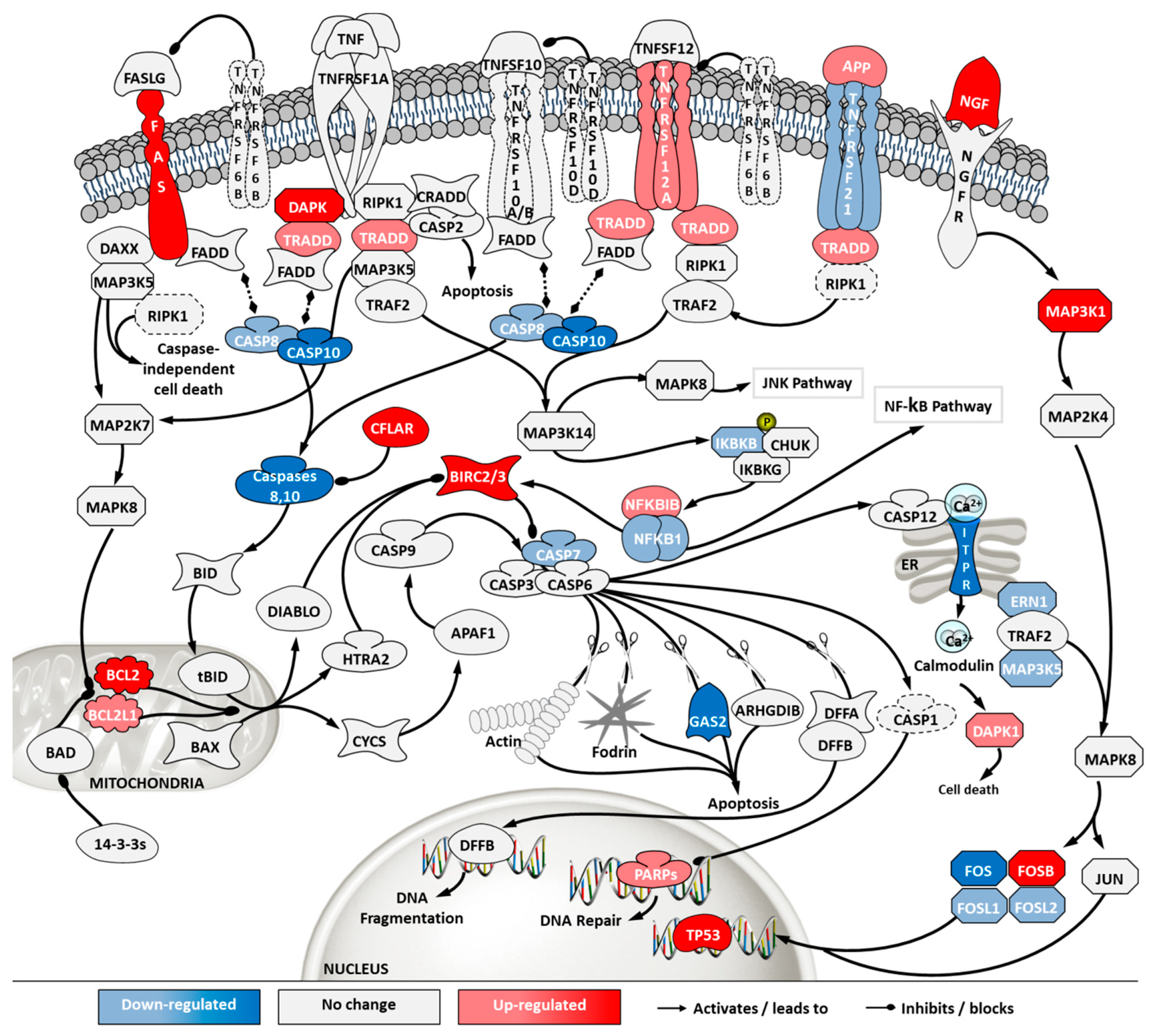
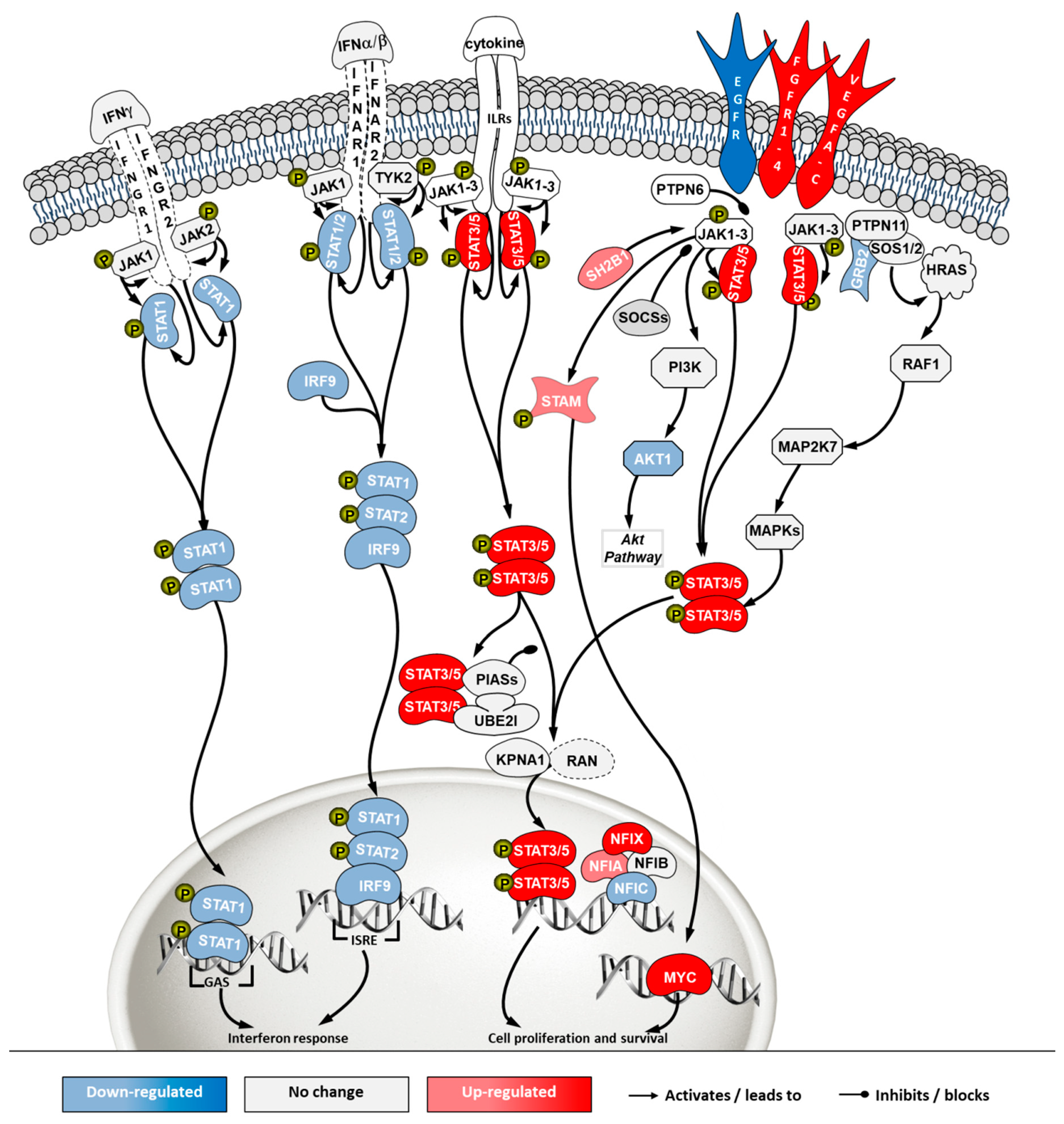
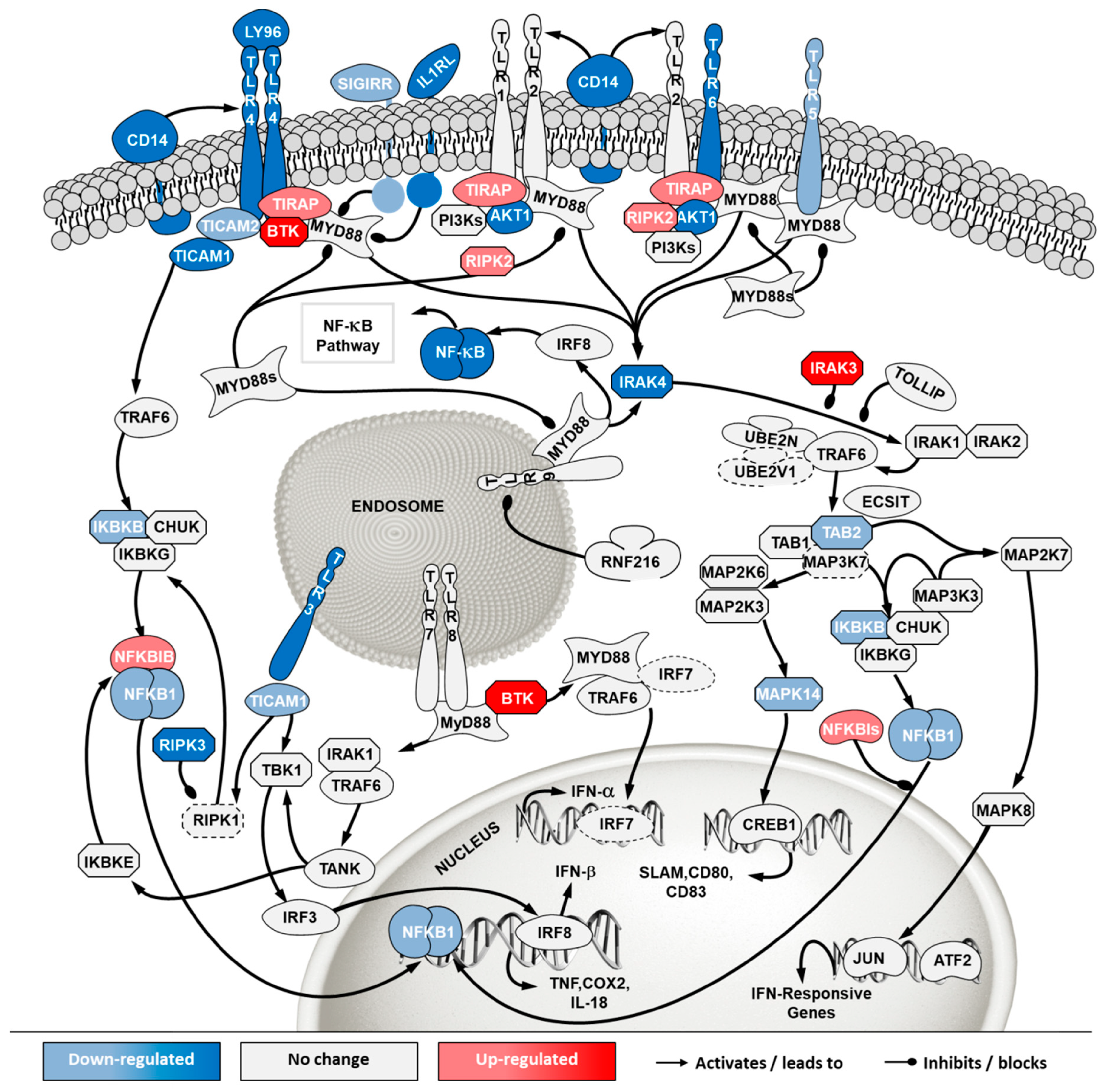
2.5. Transcriptome Analysis
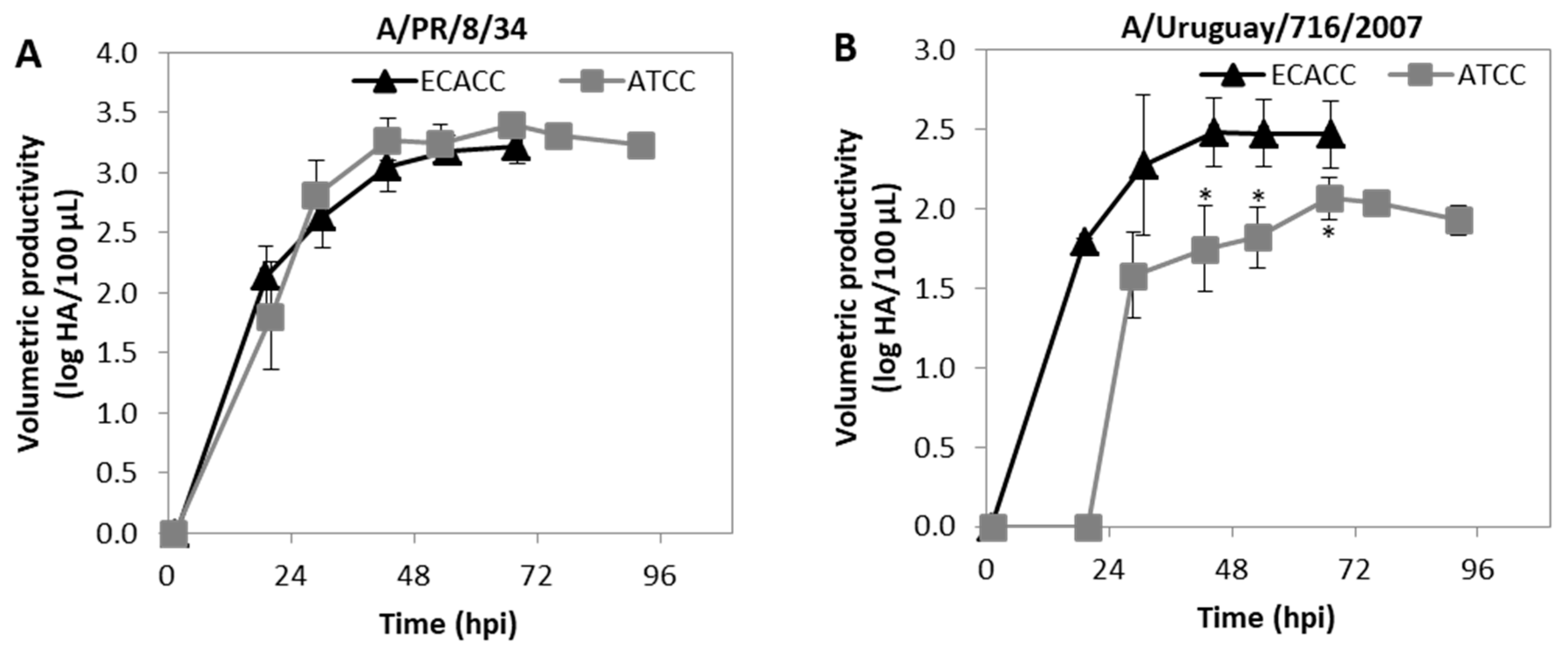
2.6. Influenza Virus Production in MDCK Cells Suspension Cultures with AEM Medium
3. Discussion
4. Materials and Methods
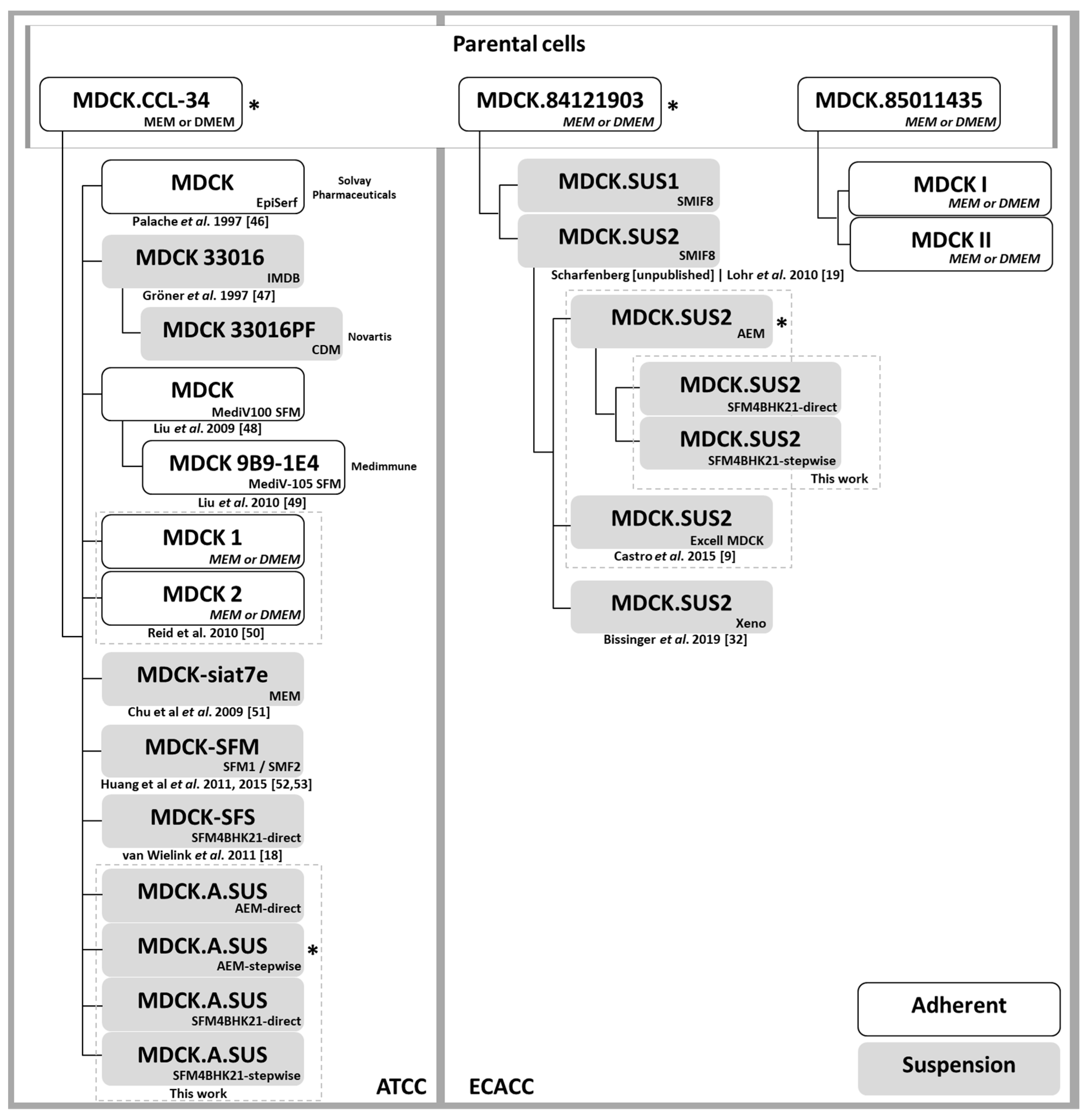
4.1. Cell Lines and Culture Media
4.2. Viruses
4.3. Adaptation to Serum-Free Media and Suspension and Growth Assays
4.4. Cell Permissiveness to CAV-2 Infection
4.5. Progression of Infection and Production of CAV-2
4.6. Production of Influenza Virus
4.7. Cell Concentration and Size
4.8. CAV-2 Quantification
4.9. Influenza Virus Titer Determinations
4.10. RNA Extraction for Transcriptome Analysis
4.11. Amplification, Labelling and Hybridization of RNA Samples for Microarray Analysis
4.12. Transcriptome Data Processing and Analysis
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AEM | Adenovirus expression medium |
| ATCC | American Type Culture Collection |
| BCL2 | BCL2 apoptosis regulator |
| BIRC | Baculoviral IAP repeat containing |
| CAV | Canine adenoviral vector |
| CD14 | CD14 molecule |
| CFLAR | CASP8 and FADD like apoptosis regulator |
| DK | Dog kidney (cells) |
| E1A | Control protein E1A |
| E1B | Control protein E1B |
| ECACC | European Collection of Authenticated Cell Cultures |
| FAS | Fas cell surface death receptor |
| GFP | Green fluorescent protein |
| GMP | Good manufacturing practices |
| HA | Hemagglutination |
| hAdV | human adenoviral vectors |
| HPI | Hours post infection |
| IP | Infectious particle |
| IPA | Ingenuity Pathway Analysis |
| JAK/STAT | Janus kinase/signal transducers and activators of transcription |
| MDBK | Madin-Darby bovine kidney (cells) |
| MDCK | Madin-Darby canine kidney (cells) |
| MDOK | Madin-Darby ovine kidney (cells) |
| MOI | Multiplicity of infection |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NIBSC | National Institute for Biological Standards and Control |
| PCA | Principal component analysis |
| RFU | Relative fluorescence units |
| RKI | Robert Koch-Institut |
| SFM | Serum-free medium |
| TCID50 | Fifty-percent tissue culture infective dose |
| TLR | Toll-like receptor |
| TNFRSF12A | TNF receptor superfamily member 12A |
| TRADD | TNFRSF1A associated via death domain |
| VG | Viral genomes |
Appendix A






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Culture System Δ | Cell Bank | Infection Status ΔΔ | Scores | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| PC1 | PC2 | PC3 | PC4 | PC5 | PC6 | PC7 | PC8 | |||
| Adherent | ATCC | Non-infected | −91.6 | 24.9 | −10.0 | 39.0 | −51.5 | −7.7 | −37.8 | −3.7 × 10−14 |
| Adherent | ATCC | Infected | −26.7 | 36.4 | 46.1 | 65.6 | 36.9 | −6.0 | 31.7 | 2.6 × 10−14 |
| Adherent | ECACC | Non-infected | −95.0 | −24.8 | 1.4 | −48.6 | −35.2 | 3.2 | 42.1 | −7.0 × 10−15 |
| Adherent | ECACC | Infected | −41.4 | −19.1 | 65.7 | −45.0 | 46.3 | 6.6 | −33.0 | 1.8 × 10−13 |
| Suspension | ATCC | Non-infected | 15.2 | 61.5 | −74.4 | −10.8 | 23.7 | 49.6 | −1.3 | −1.4 × 10−13 |
| Suspension | ATCC | Infected | 117.4 | 74.3 | 16.0 | −31.4 | −21.0 | −38.4 | 1.0 | −3.8 × 10−14 |
| Suspension | ECACC | Non-infected | 10.3 | −74.8 | −73.2 | 8.0 | 30.8 | −42.1 | −2.3 | 9.9 × 10−14 |
| Suspension | ECACC | Infected | 111.8 | −78.5 | 28.4 | 23.3 | −30.0 | 34.8 | −0.5 | 1.1 × 10−14 |
| Strain | Replicate | TCID50/mL | FCECACC/ATCC | |
|---|---|---|---|---|
| ECACC | ATCC | |||
| A/PR/8/34 | #1 | 1.3 × 109 | 1.8 × 109 | 0.8 |
| #2 | 4.2 × 109 | 2.4 × 109 | 1.8 | |
| #3 | 7.5 × 109 | 5.6 × 109 | 1.3 | |
| average | 4.4 × 109 | 3.3 × 109 | 1.3 | |
| A/Uruguay/716/2007 | #1 | 1.0 × 109 | 5.6 × 107 | 17.8 |
| #2 | 4.2 × 108 | 4.2 × 107 | 10.0 | |
| #3 | 3.2 × 108 | 7.5 × 106 | 42.1 | |
| average | 5.8 × 108 | 3.5 × 107 | 23.3 | |
| Strain | Replicate | Cell Specific Productivity | |||||
|---|---|---|---|---|---|---|---|
| ECACC | ATCC | ||||||
| PP/Cell | IP/Cell | PP/IP Ratio | PP/Cell | IP/Cell | PP/IP Ratio | ||
| A/PR/8/34 | #1 | 4376 | 667 | 7 | 2254 | 736 | 3 |
| #2 | 4620 | 253 | 16 | 5890 | 859 | 7 | |
| #3 | 6640 | 3282 | 2 | 8404 | 2204 | 4 | |
| #4 | 5809 | 1543 | 4 | - | - | - | |
| #5 | 10648 | 2564 | 4 | - | - | - | |
| average | 6419 | 1662 | 7 | 5516 | 1266 | 5 | |
| SD | 2536 | 1268 | 6 | 3092 | 814 | 2 | |
| SD% | 39.5% | 76.3% | 84.1% | 56.1% | 64.3% | 44.6% | |
| A/Uruguay/716/2007 | #1 | 602 | 512 | 1 | 776 | 22 | 34 |
| #2 | 2320 | 182 | 13 | 661 | 16 | 40 | |
| #3 | 3029 | 117 | 26 | 738 | 3 | 261 | |
| average | 1984 | 270 | 13 | 725 | 14 | 112 | |
| SD | 1248 | 212 | 13 | 59 | 10 | 129 | |
| SD% | 62.9% | 78.3% | 93.8% | 8.1% | 71.1% | 115.8% | |
References
- Del Rio, D.; Beucher, B.; Lavigne, M.; Wehbi, A.; Dopeso-Reyes, I.G.; Saggio, I.; Kremer, E.J. CAV-2 Vector Development and Gene Transfer in the Central and Peripheral Nervous Systems. Front. Mol. Neurosci. 2019, 12, 71. [Google Scholar] [CrossRef]
- Junyent, F.; Kremer, E.J. CAV-2—Why a canine virus is a neurobiologist’s best friend. Curr. Opin. Pharmacol. 2015, 24, 86–93. [Google Scholar] [CrossRef]
- Bru, T.; Salinas, S.; Kremer, E.J. An Update on Canine Adenovirus Type 2 and Its Vectors. Viruses 2010, 2, 2134–2153. [Google Scholar] [CrossRef] [PubMed]
- Kremer, E.J.; Boutin, S.; Chillon, M.; Danos, O. Canine Adenovirus Vectors: An Alternative for Adenovirus-Mediated Gene Transfer. J. Virol. 2000, 74, 505–512. [Google Scholar] [CrossRef] [PubMed]
- Soudais, C.; Skander, N.; Kremer, E.J. Long-term in vivo transduction of neurons throughout the rat CNS using novel helper-dependent CAV-2 vectors. FASEB J. 2004, 18, 391–393. [Google Scholar] [CrossRef] [PubMed]
- Kochanek, S. High-Capacity Adenoviral Vectors for Gene Transfer and Somatic Gene Therapy. Hum. Gene Ther. 1999, 10, 2451–2459. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, P.; Santiago, V.M.; Rodrigues, A.F.; Tomás, H.; Kremer, E.J.; Alves, P.M.; Coroadinha, A.S. Impact of E1 and Cre on Adenovirus Vector Amplification: Developing MDCK CAV-2-E1 and E1-Cre Transcomplementing Cell Lines. PLoS ONE 2013, 8, e60342. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, P.; Peixoto, C.; Santiago, V.M.; Kremer, E.J.; Coroadinha, A.S.; Alves, P.M. Bioprocess developme.nt for canine adenovirus type 2 vectors. Gene Ther. 2013, 20, 353–360. [Google Scholar] [CrossRef]
- Castro, R.; Fernandes, P.; Laske, T.; Sousa, M.F.; Genzel, Y.; Scharfenberg, K.; Alves, P.M.; Coroadinha, A.S. Production of canine adenovirus type 2 in serum-free suspension cultures of MDCK cells. Appl. Microbiol. Biotechnol. 2015, 99, 7059–7068. [Google Scholar] [CrossRef]
- World Health Organization. Cell culture as a substrate for the production of influenza vaccines: Memorandum from a WHO meeting. Bull. World Health Organ. 1995, 73, 431–435. [Google Scholar]
- Manini, I.; Domnich, A.; Amicizia, D.; Rossi, S.; Pozzi, T.; Gasparini, R.; Panatto, D.; Montomoli, E. Flucelvax (Optaflu) for seasonal influenza. Expert Rev. Vaccines 2015, 14, 789–804. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kim, Y.-H.; Lee, B.-Y.; Lee, K.S.; Lee, S.-J.; Park, M.; Park, Y.W. Mdck-Derived Cell Lines Adapted to Serum-Free Culture and Suspension Culture and Method for Preparing Vaccine Virus Using the Cells. Patent WO2012033328A2, 18 July 2013. [Google Scholar]
- Gaush, C.R.; Hard, W.L.; Smith, T.F. Characterization of an Established Line of Canine Kidney Cells (MDCK). Exp. Boil. Med. 1966, 122, 931–935. [Google Scholar] [CrossRef]
- Madin, S.H.; Darby, N.B. Established Kidney Cell Lines of Normal Adult Bovine and Ovine Origin. Exp. Boil. Med. 1958, 98, 574–576. [Google Scholar] [CrossRef] [PubMed]
- Omeir, R.L.; Teferedegne, B.; Foseh, G.S.; Beren, J.J.; Snoy, P.J.; Brinster, L.R.; Cook, J.L.; Peden, K.; Lewis, A.M. Heterogeneity of the Tumorigenic Phenotype Expressed by Madin–Darby Canine Kidney Cells. Comp. Med. 2011, 61, 243–250. [Google Scholar] [PubMed]
- Dukes, J.D.; Whitley, P.; Chalmers, A.D. The MDCK variety pack: Choosing the right strain. BMC Cell Boil. 2011, 12, 43. [Google Scholar] [CrossRef] [PubMed]
- Lugovtsev, V.Y.; Melnyk, D.; Weir, J.P. Heterogeneity of the MDCK Cell Line and Its Applicability for Influenza Virus Research. PLoS ONE 2013, 8, e75014. [Google Scholar] [CrossRef] [PubMed]
- Van Wielink, R.; Kant-Eenbergen, H.; Harmsen, M.; Martens, D.; Wijffels, R.; Coco-Martin, J. Adaptation of a Madin–Darby canine kidney cell line to suspension growth in serum-free media and comparison of its ability to produce avian influenza virus to Vero and BHK21 cell lines. J. Virol. Methods 2011, 171, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Lohr, V.; Genzel, Y.; Behrendt, I.; Scharfenberg, K.; Reichl, U. A new MDCK suspension line cultivated in a fully defined medium in stirred-tank and wave bioreactor. Vaccine 2010, 28, 6256–6264. [Google Scholar] [CrossRef]
- Nadeau, I.; Kamen, A.A. Production of adenovirus vector for gene therapy. Biotechnol. Adv. 2003, 20, 475–489. [Google Scholar] [CrossRef]
- Jolliffe, I.T.; Cadima, J. Principal component analysis: A review and recent developments. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2016, 374, 20150202. [Google Scholar] [CrossRef]
- Green, D.R.; Llambi, F. Cell Death Signaling. Cold Spring Harb. Perspect. Biol. 2015, 7, a006080. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.A. The Jak/STAT pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Kawai, T. Toll-Like Receptor Signaling Pathways. Front. Immunol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Lester, S.N.; Li, K. Toll-like receptors in antiviral innate immunity. J. Mol. Boil. 2013, 426, 1246–1264. [Google Scholar] [CrossRef]
- Cros, J.; Cagnard, N.; Woollard, K.; Patey, N.; Zhang, S.-Y.; Senechal, B.; Puel, A.; Biswas, S.K.; Moshous, D.; Picard, C.; et al. Human CD14dim Monocytes Patrol and Sense Nucleic Acids and Viruses via TLR7 and TLR8 Receptors. Immunity 2010, 33, 375–386. [Google Scholar] [CrossRef]
- Mitchell, S.; Vargas, J.; Hoffmann, A. Signaling via the NFkappaB system. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 227–241. [Google Scholar] [CrossRef]
- Hughes, P.; Marshall, D.; Reid, Y.; Parkes, H.; Gelber, C. The costs of using unauthenticated, over-passaged cell lines: How much more data do we need? Biotechniques 2007, 43, 575–586. [Google Scholar] [CrossRef]
- Kluge, S.; Benndorf, D.; Genzel, Y.; Scharfenberg, K.; Rapp, E.; Reichl, U. Monitoring changes in proteome during stepwise adaptation of a MDCK cell line from adherence to growth in suspension. Vaccine 2015, 33, 4269–4280. [Google Scholar] [CrossRef]
- Malm, M.; Saghaleyni, R.; Lundqvist, M.; Giudici, M.; Chotteau, V.; Field, R.; Varley, P.; Hatton, D.; Grassi, L.; Svensson, T.; et al. Evolution from adherent to suspension systems biology of HEK293 cell line development. bioRxiv 2020. [Google Scholar] [CrossRef]
- Simeone, P.; Trerotola, M.; Franck, J.; Cardon, T.; Marchisio, M.; Fournier, I.; Salzet, M.; Maffia, M.; Vergara, D.; Tristan, C. The multiverse nature of epithelial to mesenchymal transition. Semin. Cancer Boil. 2019, 58, 1–10. [Google Scholar] [CrossRef]
- Bissinger, T.; Fritsch, J.; Mihut, A.; Wu, Y.; Liu, X.; Genzel, Y.; Tan, W.-S.; Reichl, U. Semi-perfusion cultures of suspension MDCK cells enable high cell concentrations and efficient influenza A virus production. Vaccine 2019, 37, 7003–7010. [Google Scholar] [CrossRef] [PubMed]
- Gregersen, J.-P.; Schmitt, H.-J.; Trusheim, H.; Bröker, M. Safety of MDCK cell culture-based influenza vaccines. Futur. Microbiol. 2011, 6, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, A.F.; Soares, H.R.; Guerreiro, M.R.; Alves, P.M.; Coroadinha, A.S. Viral vaccines and their manufacturing cell substrates: New trends and designs in modern vaccinology. Biotechnol. J. 2015, 10, 1329–1344. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, A.F.; Carrondo, M.J.; Alves, P.M.; Coroadinha, A.S. Cellular targets for improved manufacturing of virus-based biopharmaceuticals in animal cells. Trends Biotechnol. 2014, 32, 602–607. [Google Scholar] [CrossRef]
- Zhao, H.; Dahlö, M.; Isaksson, A.; Syvänen, A.-C.; Pettersson, U. The transcriptome of the adenovirus infected cell. Virology 2012, 424, 115–128. [Google Scholar] [CrossRef]
- Rodrigues, A.F.; Formas-Oliveira, A.S.; Bandeira, V.; Alves, P.M.; Hu, W.; Coroadinha, A.S. Metabolic pathways recruited in the production of a recombinant enveloped virus: Mining targets for process and cell engineering. Metab. Eng. 2013, 20, 131–145. [Google Scholar] [CrossRef]
- White, E. Regulation of the cell cycle and apoptosis by the oncogenes of adenovirus. Oncogene 2001, 20, 7836–7846. [Google Scholar] [CrossRef]
- Hendrickx, R.; Stichling, N.; Koelen, J.; Kuryk, L.; Lipiec, A.; Greber, U.F. Innate Immunity to Adenovirus. Hum. Gene Ther. 2014, 25, 265–284. [Google Scholar] [CrossRef]
- Seitz, C.; Frensing, T.; Höper, D.; Kochs, G.; Reichl, U. High yields of influenza A virus in Madin-Darby canine kidney cells are promoted by an insufficient interferon-induced antiviral state. J. Gen. Virol. 2010, 91, 1754–1763. [Google Scholar] [CrossRef]
- Le, L.P.; Li, J.; Ternovoi, V.V.; Siegal, G.P.; Curiel, D.T. Fluorescently tagged canine adenovirus via modification with protein IX–enhanced green fluorescent protein. J. Gen. Virol. 2005, 86, 3201–3208. [Google Scholar] [CrossRef]
- Fernandes, P.; Simão, D.; Guerreiro, M.R.; Kremer, E.J.; Coroadinha, A.S.; Alves, P.M. Impact of adenovirus life cycle progression on the generation of canine helper-dependent vectors. Gene Ther. 2014, 22, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Kalbfuss, B.; Knöchlein, A.; Kröber, T.; Reichl, U. Monitoring influenza virus content in vaccine production: Precise assays for the quantitation of hemagglutination and neuraminidase activity. Biologicals 2008, 36, 145–161. [Google Scholar] [CrossRef] [PubMed]
- Genzel, Y.; Reichl, U. Vaccine production—State of the art and future needs in upstream processing. In Animal Cell Biotechnology: Methods and Protocols; Humana Press Inc.: Totowa, NJ, USA, 2007; pp. 457–473. [Google Scholar]
- R Core Team. The R Project for Statistical Computing. Available online: http://www.r-project.org/ (accessed on 11 July 2020).
- Palache, A.M.; Brands, R.; Van Scharrenburg, G.J.M. Immunogenicity and reactogenicity of influenza subunit vaccines produced in MDCK cells or fertilized chicken eggs. J. Infect. Dis. 1997, 176, S20–S24. [Google Scholar] [CrossRef] [PubMed]
- Gröner, A.J.V. Animal Cells and Processes for the Replication of Influenza Viruses. Patent WO1997037000A1, 2 December 2003. [Google Scholar]
- Liu, J.; Shi, X.; Schwartz, R.; Kemble, G. Use of MDCK cells for production of live attenuated influenza vaccine. Vaccine 2009, 27, 6460–6463. [Google Scholar] [CrossRef]
- Liu, J.; Mani, S.; Schwartz, R.; Richman, L.; Tabor, D.E. Cloning and assessment of tumorigenicity and oncogenicity of a Madin–Darby canine kidney (MDCK) cell line for influenza vaccine production. Vaccine 2010, 28, 1285–1293. [Google Scholar] [CrossRef]
- Reid, Y.A.; Pyla, Y.; Cedrone, E. Isolation and Characterization of MDCK Clones. ATCC Connect 2010, 30, 10. [Google Scholar]
- Chu, C.; Lugovtsev, V.; Golding, H.; Betenbaugh, M.; Shiloach, J. Conversion of MDCK cell line to suspension culture by transfecting with human siat7e gene and its application for influenza virus production. Proc. Natl. Acad. Sci. USA 2009, 106, 14802–14807. [Google Scholar] [CrossRef]
- Huang, D.; Zhao, L.; Tan, W. Adherent and single-cell suspension culture of Madin-Darby canine kidney cells in serum-free medium. Sheng Wu Gong Cheng Xue Bao 2011, 27, 645–652. [Google Scholar]
- Huang, D.; Peng, W.-J.; Ye, Q.; Liu, X.-P.; Zhao, L.; Fan, L.; Xia-Hou, K.; Jia, H.-J.; Luo, J.; Zhou, L.-T.; et al. Serum-Free Suspension Culture of MDCK Cells for Production of Influenza H1N1 Vaccines. PLoS ONE 2015, 10, e0141686. [Google Scholar] [CrossRef]











© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodrigues, A.F.; Fernandes, P.; Laske, T.; Castro, R.; Alves, P.M.; Genzel, Y.; Coroadinha, A.S. Cell Bank Origin of MDCK Parental Cells Shapes Adaptation to Serum-Free Suspension Culture and Canine Adenoviral Vector Production. Int. J. Mol. Sci. 2020, 21, 6111. https://doi.org/10.3390/ijms21176111
Rodrigues AF, Fernandes P, Laske T, Castro R, Alves PM, Genzel Y, Coroadinha AS. Cell Bank Origin of MDCK Parental Cells Shapes Adaptation to Serum-Free Suspension Culture and Canine Adenoviral Vector Production. International Journal of Molecular Sciences. 2020; 21(17):6111. https://doi.org/10.3390/ijms21176111
Chicago/Turabian StyleRodrigues, Ana Filipa, Paulo Fernandes, Tanja Laske, Rute Castro, Paula Marques Alves, Yvonne Genzel, and Ana Sofia Coroadinha. 2020. "Cell Bank Origin of MDCK Parental Cells Shapes Adaptation to Serum-Free Suspension Culture and Canine Adenoviral Vector Production" International Journal of Molecular Sciences 21, no. 17: 6111. https://doi.org/10.3390/ijms21176111
APA StyleRodrigues, A. F., Fernandes, P., Laske, T., Castro, R., Alves, P. M., Genzel, Y., & Coroadinha, A. S. (2020). Cell Bank Origin of MDCK Parental Cells Shapes Adaptation to Serum-Free Suspension Culture and Canine Adenoviral Vector Production. International Journal of Molecular Sciences, 21(17), 6111. https://doi.org/10.3390/ijms21176111