Multilineage Differentiation Potential of Human Dental Pulp Stem Cells—Impact of 3D and Hypoxic Environment on Osteogenesis In Vitro

 , ,
, ,  , ,
, , 
Abstract
:1. Introduction
2. Results
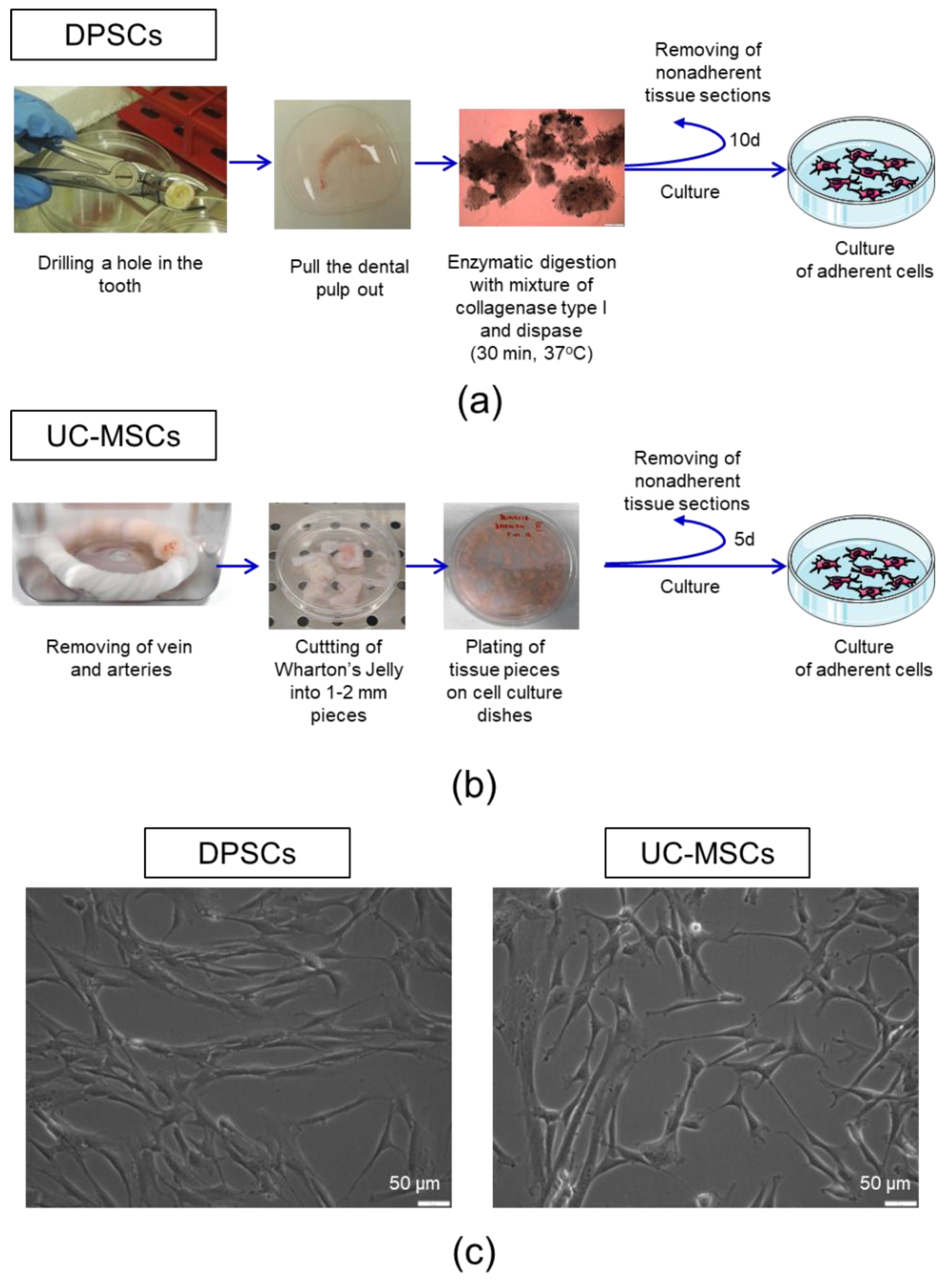
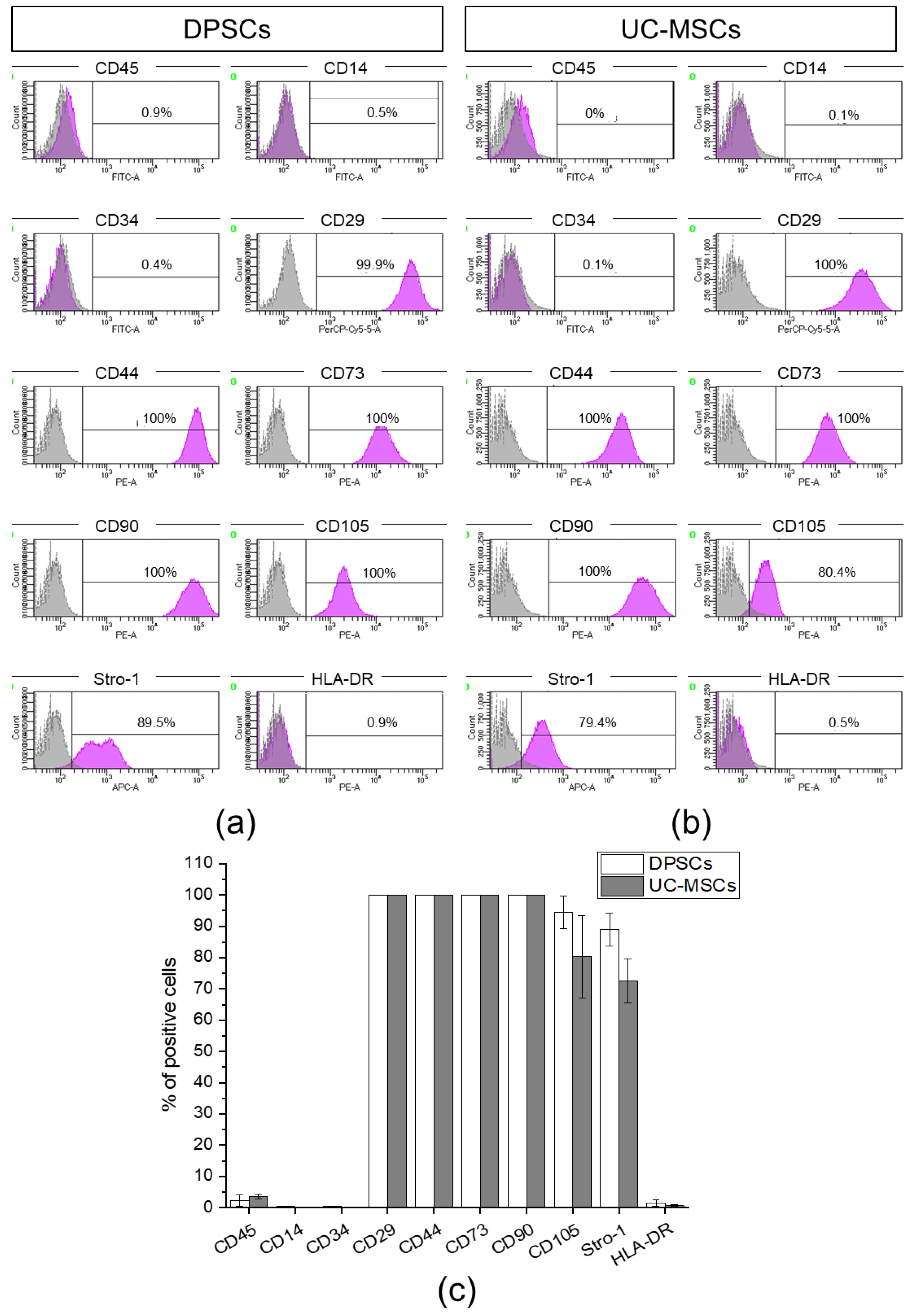
2.1. Human Dental Pulp Harbours a Population of Adherent Cells with MSC Characteristics
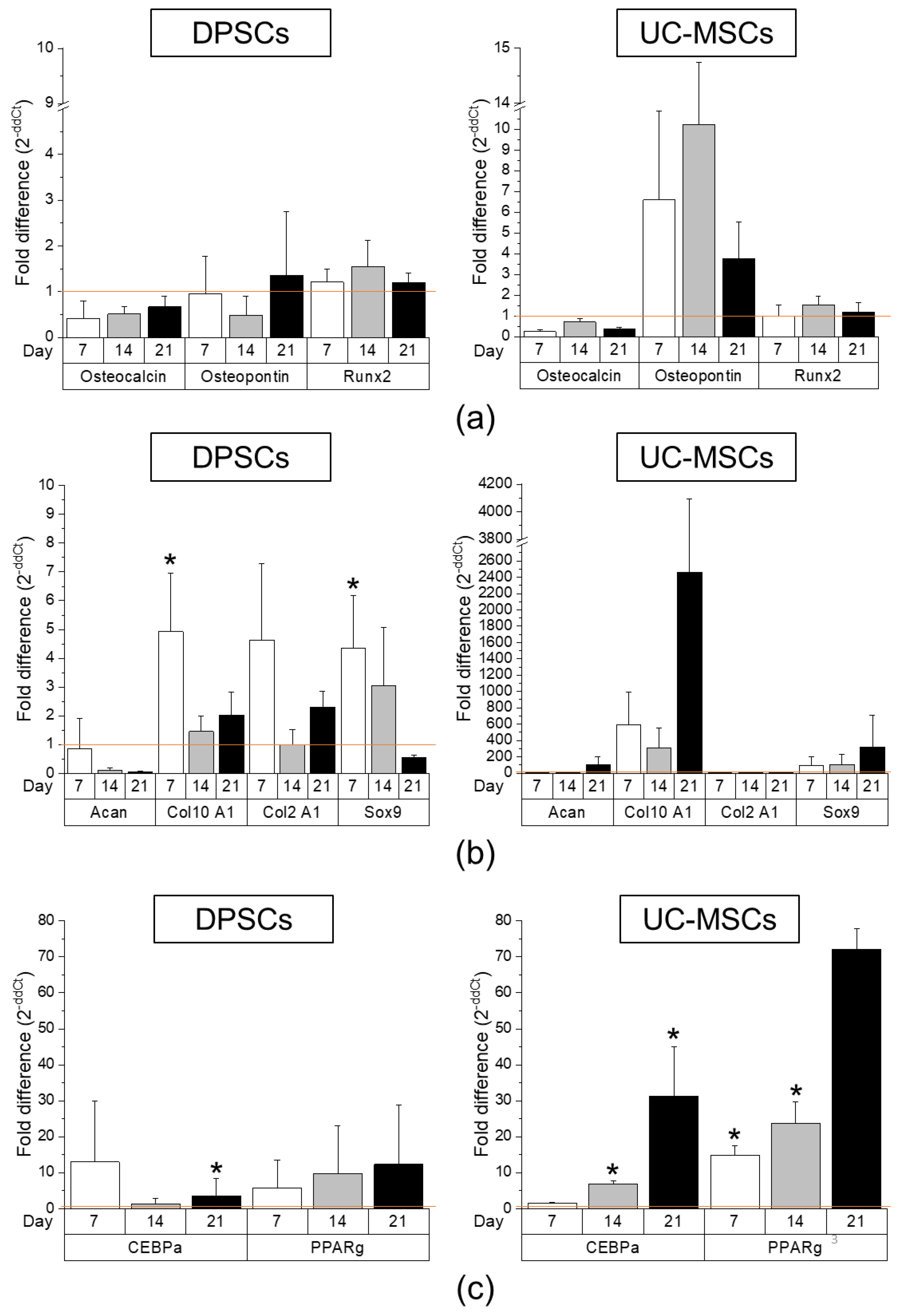
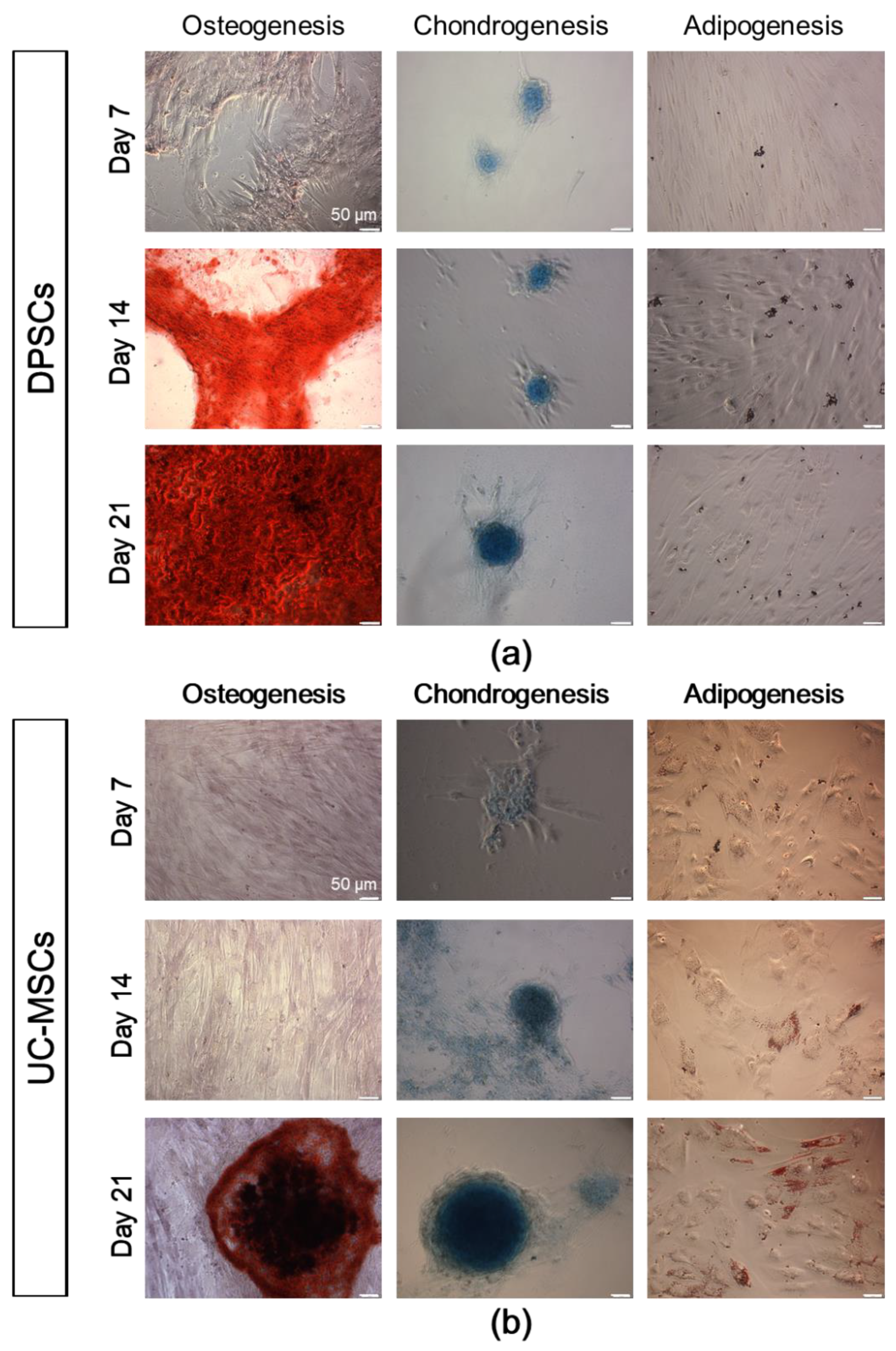
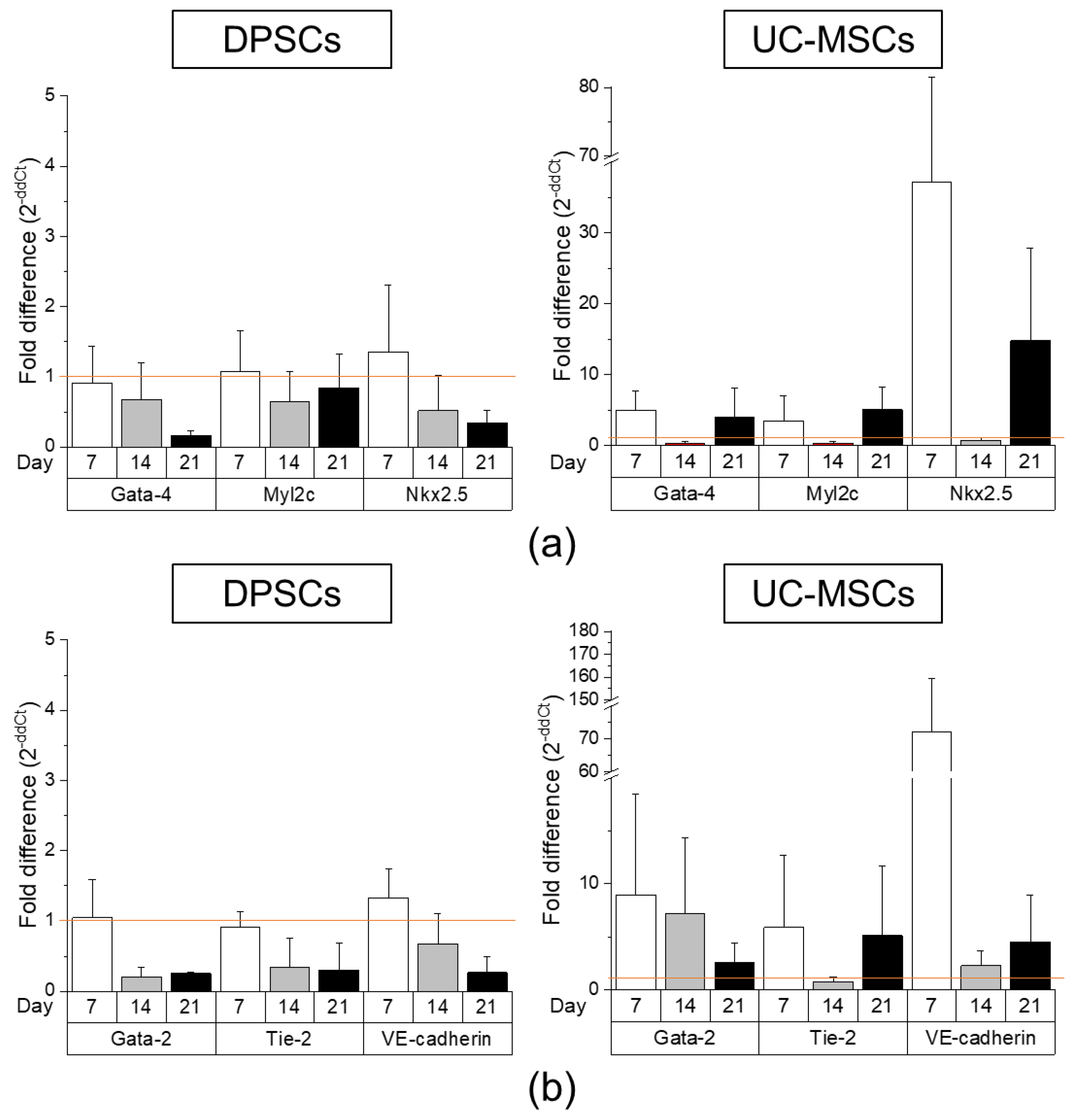
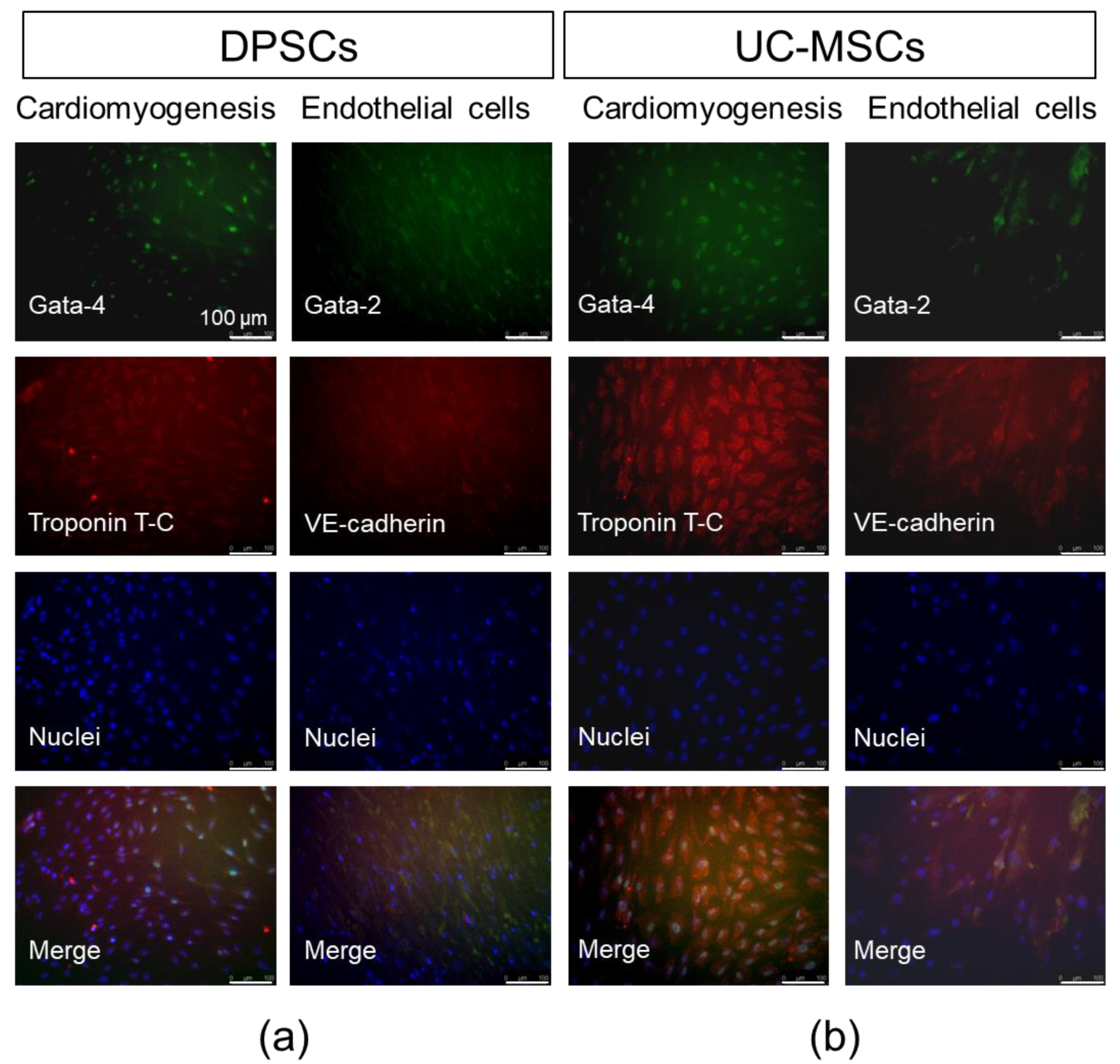
2.2. DPSCs Exhibit Wide Differentiation Potential In Vitro
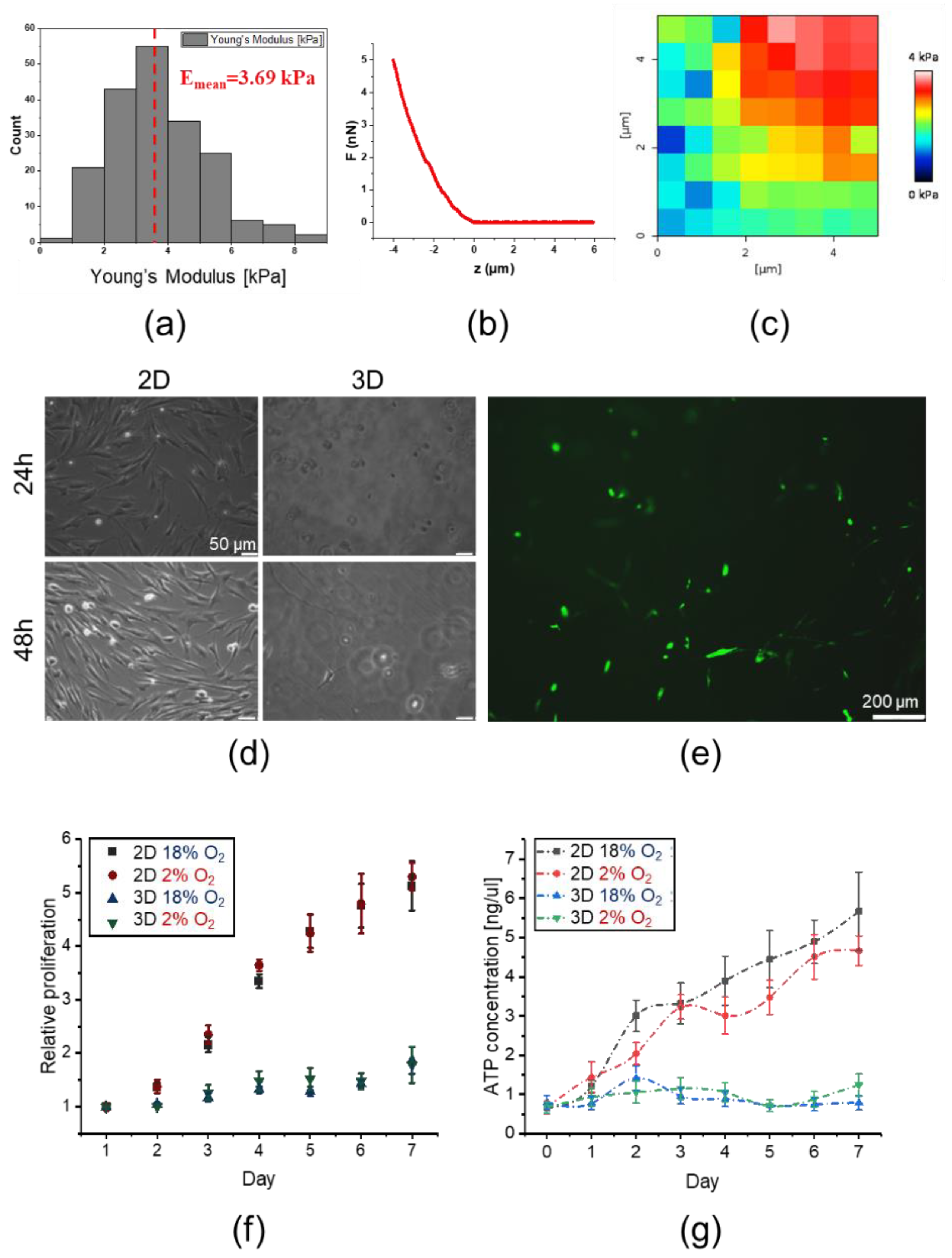
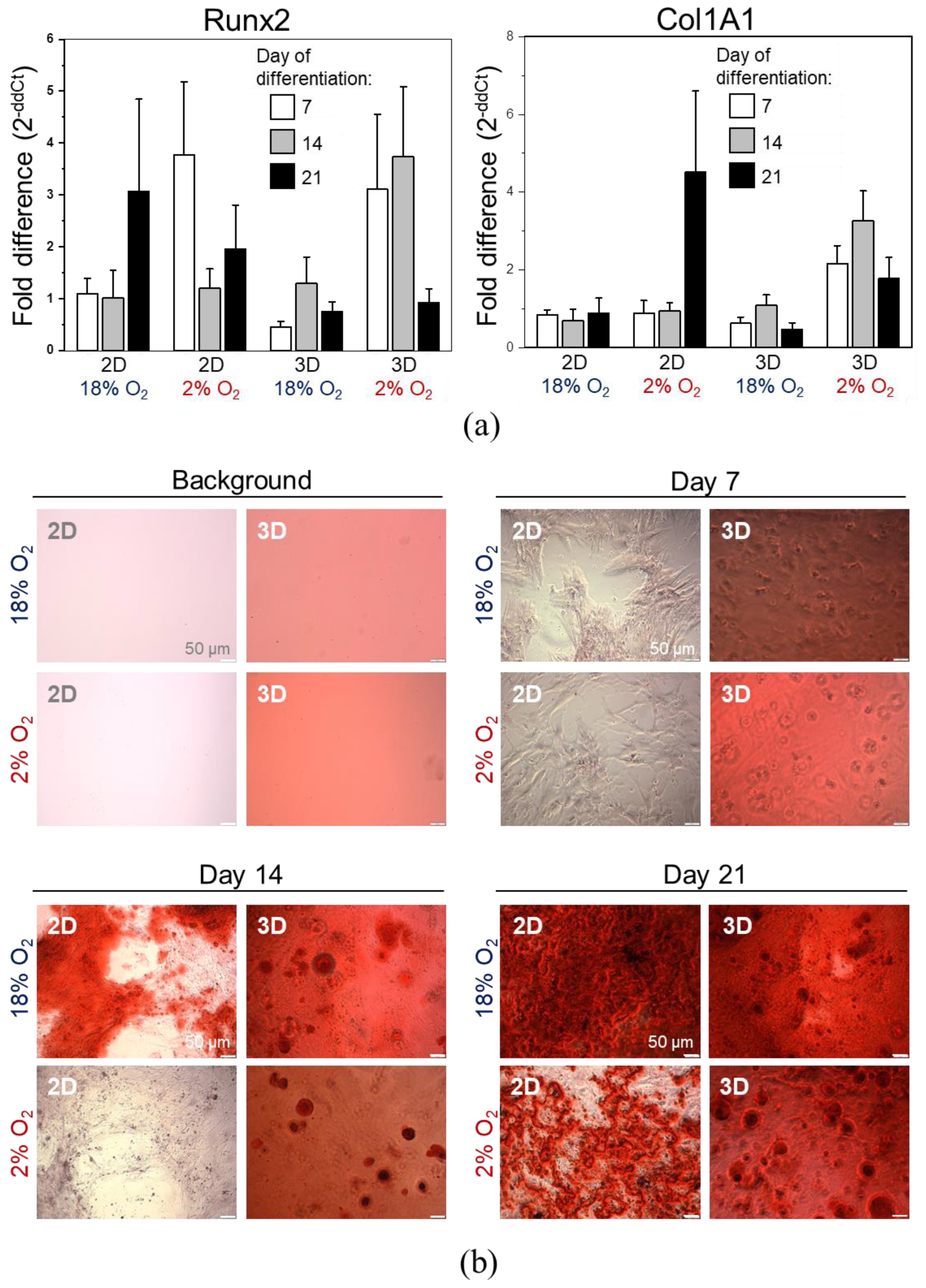
2.3. 3D Encapsulated DPSCs Exhibit Higher Differentiation Capacity into Osteoblasts in Vitro
3. Discussion
4. Materials and Methods
4.1. Isolation of Human DPSCs
4.2. Isolation of Human UC-MSCs
4.3. Cell Counting and Viability Assessment
4.4. Antigenic Phenotyping by Flow Cytometry
4.5. Differentiation of DPSCs and UC-MSCs
4.5.1. Osteogenic, Chondrogenic, and Adipogenic Differentiation
4.5.2. Cardiomyogenic Differentiation
4.5.3. Endothelial Differentiation
4.6. Gene Expression Analysis by Real-Time RT-PCR
4.7. Histochemical Staining
4.8. Immunocytochemistry
4.9. Mechanical Characterization of the Hydrogel Matrix
4.10. 3D Encapsulation of DPSCs within the Hydrogel Matrix
4.11. 3D and 2D Culture of DPSCs in Vitro
4.12. Assessment of DPSC Proliferation and Metabolic Activity In Vitro
4.13. Osteogenic Differentiation of DPSCs in 3D or 2D in Vitro Culture
4.14. Recovery of DPSCs from the Hydrogel Matrix
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BM | Bone marrow |
| BM-MSCS | Bone marrow-derived mesenchymal stem/stromal cells |
| DPSCs | Dental pulp stem cells |
| ISCT | International Society for Cellular Therapy |
| MSCs | Mesenchymal stem/stromal cells |
| O2 | Oxygen |
| SCs | Stem cells |
| UC-MSCs | Umbilical cord Wharton’s jelly-derived mesenchymal stem/stromal cells |
| 2D | Two-dimensional |
| 3D | Three-dimensional |
References
- Uccelli, A.; Moretta, L.; Pistoia, V. Mesenchymal stem cells in health and disease. Nat. Rev. Immunol. 2008, 8, 726–736. [Google Scholar] [CrossRef]
- Berebichez-Fridman, R.; Montero-Olvera, P.R. Sources and clinical applications of mesenchymal stem cells state-of-the-art review. Sultan Qaboos Univ. Med. J. 2018, 18, e264–e277. [Google Scholar] [CrossRef] [Green Version]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Krause, D.S.; Deans, R.J.; Keating, A.; Prockop, D.J.; Horwitz, E.M. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Labovsky, V.; Hofer, E.L.; Feldman, L.; Fernández Vallone, V.; García Rivello, H.; Bayes-Genis, A.; Hernando Insúa, A.; Levin, M.J.; Chasseing, N.A. Cardiomyogenic differentiation of human bone marrow mesenchymal cells: Role of cardiac extract from neonatal rat cardiomyocytes. Differentiation 2010, 79, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Bai, Y.; Zhang, L.; Zhang, B.; Zagidullin, N.; Carvalho, K.; Du, Z.; Cai, B. Cardiomyocyte differentiation of mesenchymal stem cells from bone marrow: New regulators and its implications. Stem Cell Res. Ther. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labedz-Maslowska, A.; Lipert, B.; Berdecka, D.; Kedracka-Krok, S.; Jankowska, U.; Kamycka, E.; Sekula, M.; Madeja, Z.; Dawn, B.; Jura, J.; et al. Monocyte chemoattractant protein-induced protein 1 (MCPIP1) enhances angiogenic and cardiomyogenic potential of murine bone marrow-derived mesenchymal stem cells. PLoS ONE 2015, 10, e0133746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janeczek Portalska, K.; Leferink, A.; Groen, N.; Fernandes, H.; Moroni, L.; van Blitterswijk, C.; de Boer, J. Endothelial Differentiation of Mesenchymal Stromal Cells. PLoS ONE 2012, 7, e46842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anjos-Afonso, F.; Siapati, E.K.; Bonnet, D. In vivo contribution of murine mesenchymal stem cells into multiple cell-types under minimal damage conditions. J. Cell Sci. 2004, 117, 5655–5664. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.Z.; Lee, J.H. Mesenchymal stem cell therapy for bone regeneration. CiOS Clin. Orthop. Surg. 2018, 10, 271–278. [Google Scholar] [CrossRef]
- Murphy, J.M.; Fink, D.J.; Hunziker, E.B.; Barry, F.P. Stem Cell Therapy in a Caprine Model of Osteoarthritis. Arthritis Rheum. 2003, 48, 3464–3474. [Google Scholar] [CrossRef]
- Alge, D.L.; Zhou, D.; Adams, L.L.; Wyss, B.K.; Shadday, M.D.; Woods, E.J.; Chu, T.M.G.; Goebel, W.S. Donor-matched comparison of dental pulp stem cells and bone marrow-derived mesenchymal stem cells in a rat model. J. Tissue Eng. Regen. Med. 2010, 4, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Chu, D.-T.; Phuong, T.N.T.; Tien, N.L.B.; Tran, D.K.; Van Thanh, V.; Quang, T.L.; Truong, D.T.; Pham, V.H.; Ngoc, V.T.N.; Chu-Dinh, T.; et al. An Update on the Progress of Isolation, Culture, Storage, and Clinical Application of Human Bone Marrow Mesenchymal Stem/Stromal Cells. Int. J. Mol. Sci. 2020, 21, 708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, S.M.; Glowacki, J. Age-related decline in the osteogenic potential of human bone marrow cells cultured in three-dimensional collagen sponges. J. Cell. Biochem. 2001, 82, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Cianfarani, F.; Toietta, G.; Di Rocco, G.; Cesareo, E.; Zambruno, G.; Odorisio, T. Diabetes impairs adipose tissue-derived stem cell function and efficiency in promoting wound healing. Wound Repair Regen. 2013, 21, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Nie, Y.; Lau, C.S.; Lie, A.K.W.; Chan, G.C.F.; Mok, M.Y. Defective phenotype of mesenchymal stem cells in patients with systemic lupus erythematosus. Lupus 2010, 19, 850–859. [Google Scholar] [CrossRef]
- Marmotti, A.; Mattia, S.; Castoldi, F.; Barbero, A.; Mangiavini, L.; Bonasia, D.E.; Bruzzone, M.; Dettoni, F.; Scurati, R.; Peretti, G.M. Allogeneic Umbilical Cord-Derived Mesenchymal Stem Cells as a Potential Source for Cartilage and Bone Regeneration: An in Vitro Study. Stem Cells Int. 2017, 2017, 1732094. [Google Scholar] [CrossRef] [Green Version]
- El Omar, R.; Beroud, J.; Stoltz, J.F.; Menu, P.; Velot, E.; Decot, V. Umbilical cord mesenchymal stem cells: The new gold standard for mesenchymal stem cell-based therapies? Tissue Eng. Part B Rev. 2014, 20, 523–544. [Google Scholar] [CrossRef]
- Gronthos, S.; Mankani, M.; Brahim, J.; Robey, P.G.; Shi, S. Postnatal human dental pulp stem cells (DPSCs) in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2000, 97, 13625–13630. [Google Scholar] [CrossRef] [Green Version]
- Dupin, E.; Sommer, L. Neural crest progenitors and stem cells: From early development to adulthood. Dev. Biol. 2012, 366, 83–95. [Google Scholar] [CrossRef] [Green Version]
- Kaukua, N.; Shahidi, M.K.; Konstantinidou, C.; Dyachuk, V.; Kaucka, M.; Furlan, A.; An, Z.; Wang, L.; Hultman, I.; Ährlund-Richter, L.; et al. Glial origin of mesenchymal stem cells in a tooth model system. Nature 2014, 513, 551–554. [Google Scholar] [CrossRef] [Green Version]
- D’aquino, R.; de Rosa, A.; Laino, G.; Caruso, F.; Guida, L.; Rullo, R.; Checchi, V.; Laino, L.; Tirino, V.; Papaccio, G. Human dental pulp stem cells: From biology to clinical applications. J. Exp. Zool. Part B Mol. Dev. Evol. 2009, 312, 408–415. [Google Scholar] [CrossRef]
- Arthur, A.; Rychkov, G.; Shi, S.; Koblar, S.A.; Gronthos, S. Adult Human Dental Pulp Stem Cells Differentiate Toward Functionally Active Neurons Under Appropriate Environmental Cues. Stem Cells 2008, 26, 1787–1795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, F.; Sloan, A.; Song, B. Dental pulp stem cells and their potential roles in central nervous system regeneration and repair. J. Neurosci. Res. 2013, 91, 1383–1393. [Google Scholar] [CrossRef] [PubMed]
- Pisciotta, A.; Carnevale, G.; Meloni, S.; Riccio, M.; De Biasi, S.; Gibellini, L.; Ferrari, A.; Bruzzesi, G.; De Pol, A. Human Dental pulp stem cells (hDPSCs): Isolation, enrichment and comparative differentiation of two sub-populations Integrative control of development. BMC Dev. Biol. 2015, 15, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gervois, P.; Struys, T.; Hilkens, P.; Bronckaers, A.; Ratajczak, J.; Politis, C.; Brône, B.; Lambrichts, I.; Martens, W. Neurogenic Maturation of Human Dental Pulp Stem Cells Following Neurosphere Generation Induces Morphological and Electrophysiological Characteristics of Functional Neurons. Stem Cells Dev. 2015, 24, 296–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianco, P.; Cao, X.; Frenette, P.S.; Mao, J.J.; Robey, P.G.; Simmons, P.J.; Wang, C.Y. The meaning, the sense and the significance: Translating the science of mesenchymal stem cells into medicine. Nat. Med. 2013, 19, 35–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leong, W.K.; Henshall, T.L.; Arthur, A.; Kremer, K.L.; Lewis, M.D.; Helps, S.C.; Field, J.; Hamilton-Bruce, M.A.; Warming, S.; Manavis, J.; et al. Human Adult Dental Pulp Stem Cells Enhance Poststroke Functional Recovery Through Non-Neural Replacement Mechanisms. Stem Cells Transl. Med. 2012, 1, 177–187. [Google Scholar] [CrossRef]
- Nagpal, A.; Kremer, K.L.; HamiltonBruce, M.A.; Kaidonis, X.; Milton, A.G.; Levi, C.; Shi, S.; Carey, L.; Hillier, S.; Rose, M.; et al. TOOTH (The Open study Of dental pulp stem cell Therapy in Humans): Study protocol for evaluating safety and feasibility of autologous human adult dental pulp stem cell therapy in patients with chronic disability after stroke. Int. J. Stroke 2016, 11, 575585. [Google Scholar] [CrossRef]
- Bryniarska, N.; Kubiak, A.; Labȩdz-Maslowska, A.; Zuba-Surma, E. Impact of developmental origin, niche mechanics and oxygen availability on osteogenic differentiation capacity of mesenchymal stem/stromal cells. Acta Biochim. Pol. 2019, 66, 491–498. [Google Scholar] [CrossRef] [Green Version]
- Kini, U.; Nandeesh, B.N. Physiology of bone formation, remodeling, and metabolism. In Radionuclide and Hybrid Bone Imaging; Springer: Berlin\Heidelberg, Germany, 2012; Volume 9783642024, pp. 29–57. ISBN 9783642024009. [Google Scholar]
- Theveneau, E.; Mayor, R. Neural crest migration: Interplay between chemorepellents, chemoattractants, contact inhibition, epithelial-mesenchymal transition, and collective cell migration. Wiley Interdiscip. Rev. Dev. Biol. 2012, 1, 435–445. [Google Scholar] [CrossRef]
- Baranova, J.; Büchner, D.; Götz, W.; Schulze, M.; Tobiasch, E. Tooth Formation: Are the Hardest Tissues of Human Body Hard to Regenerate? Int. J. Mol. Sci. 2020, 21, 4031. [Google Scholar] [CrossRef] [PubMed]
- Lukomska, B.; Stanaszek, L.; Zuba-Surma, E.; Legosz, P.; Sarzynska, S.; Drela, K. Challenges and Controversies in Human Mesenchymal Stem Cell Therapy. Stem Cells Int. 2019, 37, 855–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, J.M.; Przybyla, L.; Weaver, V.M. Tissue mechanics regulate brain development, homeostasis and disease. J. Cell Sci. 2017, 130, 71–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Westhrin, M.; Xie, M.; Olderøy, M.; Sikorski, P.; Strand, B.L.; Standal, T. Osteogenic differentiation of human mesenchymal stem cells in mineralized alginate matrices. PLoS ONE 2015, 10, e0120374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riccio, M.; Resca, E.; Maraldi, T.; Pisciotta, A.; Ferrari, A.; Bruzzesi, G.; de Pol, A. Human dental pulp stem cells produce mineralized matrix in 2D and 3D cultures. Eur. J. Histochem. 2010, 54, 205–213. [Google Scholar] [CrossRef] [Green Version]
- Mohyeldin, A.; Garzón-Muvdi, T.; Quiñones-Hinojosa, A. Oxygen in Stem Cell Biology: A Critical Component of the Stem Cell Niche. Cell Stem Cell 2010, 7, 150–161. [Google Scholar] [CrossRef] [Green Version]
- Kakudo, N.; Morimoto, N.; Ogawa, T.; Taketani, S.; Kusumoto, K. Hypoxia enhances proliferation of human adipose-derived stem cells via HIF-1α activation. PLoS ONE 2015, 10, e0139890. [Google Scholar] [CrossRef] [Green Version]
- Grayson, W.L.; Zhao, F.; Bunnell, B.; Ma, T. Hypoxia enhances proliferation and tissue formation of human mesenchymal stem cells. Biochem. Biophys. Res. Commun. 2007, 358, 948–953. [Google Scholar] [CrossRef]
- Sakdee, J.B.; White, R.R.; Pagonis, T.C.; Hauschka, P. V Hypoxia-amplified Proliferation of Human Dental Pulp Cells. J. Endod. 2009, 35, 818–823. [Google Scholar] [CrossRef]
- Kumar, S.; Vaidya, M. Hypoxia inhibits mesenchymal stem cell proliferation through HIF1α-dependent regulation of P27. Mol. Cell. Biochem. 2016, 415, 29–38. [Google Scholar] [CrossRef]
- Hsu, S.-H.; Chen, C.-T.; Wei, Y.-H. Inhibitory effects of hypoxia on metabolic switch and osteogenic differentiation of human mesenchymal stem cells. Stem Cells 2013, 31, 2779–2788. [Google Scholar] [CrossRef] [PubMed]
- Noda, S.; Kawashima, N.; Yamamoto, M.; Hashimoto, K.; Nara, K.; Sekiya, I.; Okiji, T. Effect of cell culture density on dental pulp-derived mesenchymal stem cells with reference to osteogenic differentiation. Sci. Rep. 2019, 9, 5430. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Lu, Y.; Li, F.; Qiao, L.; Wang, Q.; Li, N.; Borgia, J.A.; Deng, Y.; Lei, G.; Zheng, Q. Identification and characterization of the novel Col10a1 regulatory mechanism during chondrocyte hypertrophic differentiation. Cell Death Dis. 2014, 5, e1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bilezikan, J.P.; Raisz, L.G.; Martin, T.J. Principles of Bone Biology, 3rd ed. Am. J. Neuroradiol. 2009, 30, E139. [Google Scholar] [CrossRef] [Green Version]
- Nam, S.; Chaudhuri, O. Mitotic cells generate protrusive extracellular forces to divide in three-dimensional microenvironments. Nat. Phys. 2018, 14, 621–628. [Google Scholar] [CrossRef]
- De Fusco, C.; Messina, A.; Monda, V.; Viggiano, E.; Moscatelli, F.; Valenzano, A.; Esposito, T.; Chieffi, S.; Cibelli, G.; Monda, M.; et al. Osteopontin: Relation between Adipose Tissue and Bone Homeostasis. Stem Cells Int. 2017, 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, X.; Sun, Z.; Chu, C.; Boltze, J.; Li, S. Dental pulp stem cells: An attractive alternative for cell therapy in ischemic stroke. Front. Neurol. 2019, 10, 824. [Google Scholar] [CrossRef]
- Sakai, K.; Yamamoto, A.; Matsubara, K.; Nakamura, S.; Naruse, M.; Yamagata, M.; Sakamoto, K.; Tauchi, R.; Wakao, N.; Imagama, S.; et al. Human dental pulp-derived stem cells promote locomotor recovery after complete transection of the rat spinal cord by multiple neuro-regenerative mechanisms. J. Clin. Investig. 2012, 122, 80–90. [Google Scholar] [CrossRef]
- Cheng, P.H.; Snyder, B.; Fillos, D.; Ibegbu, C.C.; Huang, A.H.C.; Chan, A.W.S. Postnatal stem/progenitor cells derived from the dental pulp of adult chimpanzee. BMC Cell Biol. 2008, 9, 20. [Google Scholar] [CrossRef] [Green Version]
- Király, M.; Porcsalmy, B.; Pataki, Á.; Kádár, K.; Jelitai, M.; Molnár, B.; Hermann, P.; Gera, I.; Grimm, W.D.; Ganss, B.; et al. Simultaneous PKC and cAMP activation induces differentiation of human dental pulp stem cells into functionally active neurons. Neurochem. Int. 2009, 55, 323–332. [Google Scholar] [CrossRef]
- Davis, J. Tissue Regeneration—From Basic Biology to Clinical Application; IntechOpen: London, UK, 2012; ISBN 978-953-51-0387-5. [Google Scholar] [CrossRef] [Green Version]
- Ibarretxe, G.; Crende, O.; Aurrekoetxea, M.; García-Murga, V.; Etxaniz, J.; Unda, F. Neural crest stem cells from dental tissues: A new hope for dental and neural regeneration. Stem Cells Int. 2012, 2012, 103503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellis, K.M.; O’Carroll, D.C.; Lewis, M.D.; Rychkov, G.Y.; Koblar, S.A. Neurogenic potential of dental pulp stem cells isolated from murine incisors. Stem Cell Res. Ther. 2014, 5, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camci-Unal, G.; Laromaine, A.; Hong, E.; Derda, R.; Whitesides, G.M. Biomineralization Guided by Paper Templates. Sci. Rep. 2016, 6, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Koussounadis, A.; Langdon, S.P.; Um, I.H.; Harrison, D.J.; Smith, V.A. Relationship between differentially expressed mRNA and mRNA-protein correlations in a xenograft model system. Sci. Rep. 2015, 5, 10775. [Google Scholar] [CrossRef] [Green Version]
- Fujii, Y.; Kawase-Koga, Y.; Hojo, H.; Yano, F.; Sato, M.; Chung, U.-I.; Ohba, S.; Chikazu, D. Bone regeneration by human dental pulp stem cells using a helioxanthin derivative and cell-sheet technology. Stem Cell Res. Ther. 2018, 9, 215. [Google Scholar] [CrossRef] [Green Version]
- Rizzatti, V.; Boschi, F.; Pedrotti, M.; Zoico, E.; Sbarbati, A.; Zamboni, M. Lipid droplets characterization in adipocyte differentiated 3T3-L1 cells: Size and optical density distribution. Eur. J. Histochem. 2013, 57. [Google Scholar] [CrossRef] [Green Version]
- Armiñán, A.; Gandía, C.; Bartual, M.; García-Verdugo, J.M.; Lledó, E.; Mirabet, V.; Llop, M.; Barea, J.; Montero, J.A.; Sepúlveda, P. Cardiac differentiation is driven by nkx2.5 and gata4 nuclear translocation in tissue-specific mesenchymal stem cells. Stem Cells Dev. 2009, 18, 907–917. [Google Scholar] [CrossRef]
- Abbott, A. Biology’s new dimension. Nature 2003, 424, 870–872. [Google Scholar] [CrossRef]
- Spencer, J.A.; Ferraro, F.; Roussakis, E.; Klein, A.; Wu, J.; Runnels, J.M.; Zaher, W.; Mortensen, L.J.; Alt, C.; Turcotte, R.; et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature 2014, 508, 269–273. [Google Scholar] [CrossRef] [Green Version]
- Wenger, R.; Kurtcuoglu, V.; Scholz, C.; Marti, H.; Hoogewijs, D. Frequently asked questions in hypoxia research. Hypoxia 2015, 3, 35–43. [Google Scholar] [CrossRef] [Green Version]
- Kwon, S.Y.; Chun, S.Y.; Ha, Y.S.; Kim, D.H.; Kim, J.; Song, P.H.; Kim, H.T.; Yoo, E.S.; Kim, B.S.; Kwon, T.G. Hypoxia Enhances Cell Properties of Human Mesenchymal Stem Cells. Tissue Eng. Regen. Med. 2017, 14, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Iida, K.; Takeda-Kawaguchi, T.; Tezuka, Y.; Kunisada, T.; Shibata, T.; Tezuka, K.I. Hypoxia enhances colony formation and proliferation but inhibits differentiation of human dental pulp cells. Arch. Oral Biol. 2010, 55, 648–654. [Google Scholar] [CrossRef] [PubMed]
- Werle, S.B.; Chagastelles, P.; Pranke, P.; Casagrande, L. The effects of hypoxia on in vitro culture of dental-derived stem cells. Arch. Oral Biol. 2016, 68, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Ikezoe, T.; Daar, E.S.; Hisatake, J.I.; Taguchi, H.; Koeffler, H.P. HIV-1 protease inhibitors decrease proliferation and induce differentiation of human myelocytic leukemia cells. Blood 2000, 96, 3553–3559. [Google Scholar] [CrossRef]
- Cukierman, E.; Pankov, R.; Yamada, K.M. Cell interactions with three-dimensional matrices. Curr. Opin. Cell Biol. 2002, 14, 633–640. [Google Scholar] [CrossRef]
- Valorani, M.G.; Montelatici, E.; Germani, A.; Biddle, A.; D’Alessandro, D.; Strollo, R.; Patrizi, M.P.; Lazzari, L.; Nye, E.; Otto, W.R.; et al. Pre-culturing human adipose tissue mesenchymal stem cells under hypoxia increases their adipogenic and osteogenic differentiation potentials. Cell Prolif. 2012, 45, 225–238. [Google Scholar] [CrossRef]
- Basciano, L.; Nemos, C.; Foliguet, B.; de Isla, N.; de Carvalho, M.; Tran, N.; Dalloul, A. Long term culture of mesenchymal stem cells in hypoxia promotes a genetic program maintaining their undifferentiated and multipotent status. BMC Cell Biol. 2011, 12, 12. [Google Scholar] [CrossRef] [Green Version]
- Buravkova, L.B.; Andreeva, E.R.; Gogvadze, V.; Zhivotovsky, B. Mesenchymal stem cells and hypoxia: Where are we ? Mitochondrion Mesenchymal stem cells and hypoxia: Where are we? MITOCH 2014, 19, 105–112. [Google Scholar] [CrossRef]
- Persson, M.; Lehenkari, P.P.; Berglin, L.; Turunen, S.; Finnilä, M.A.J.; Risteli, J.; Skrifvars, M.; Tuukkanen, J. Osteogenic Differentiation of Human Mesenchymal Stem cells in a 3D Woven Scaffold. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef]
- Diomede, F.; Marconi, G.D.; Fonticoli, L.; Pizzicanella, J.; Merciaro, I.; Bramanti, P.; Mazzon, E.; Trubiani, O. Functional relationship between osteogenesis and angiogenesis in tissue regeneration. Int. J. Mol. Sci. 2020, 21, 3242. [Google Scholar] [CrossRef]
- Rapino, M.; Di Valerio, V.; Zara, S.; Gallorini, M.; Marconi, G.D.; Sancilio, S.; Marsich, E.; Ghinassi, B.; Di Giacomo, V.; Cataldi, A. Chitlac-coated thermosets enhance osteogenesis and angiogenesis in a co-culture of dental pulp stem cells and endothelial cells. Nanomaterials 2019, 9, 928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.; Zhou, J.; Xu, C.T.; Zhang, J.; Jin, Y.J.; Sun, G.L. Derivation and growth characteristics of dental pulp stem cells from patients of different ages. Mol. Med. Rep. 2015, 12, 5127–5134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yildirim, S.; Zibandeh, N.; Genc, D.; Ozcan, E.M.; Goker, K.; Akkoc, T. The comparison of the immunologic properties of stem cells isolated from human exfoliated deciduous teeth, dental pulp, and dental follicles. Stem Cells Int. 2016, 2016. [Google Scholar] [CrossRef]
- Monterubbianesi, R.; Bencun, M.; Pagella, P.; Woloszyk, A.; Orsini, G.; Mitsiadis, T.A. A comparative in vitro study of the osteogenic and adipogenic potential of human dental pulp stem cells, gingival fibroblasts and foreskin fibroblasts. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lekka, M. Discrimination Between Normal and Cancerous Cells Using AFM. Bionanoscience 2016, 6, 65–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sequences |
|---|---|
| β2-microglobulin | (F) 5′ AATGCGGCATCTTCAAACCT 3′ |
| (R) 5′ TGACTTTGTCACAGCCCAAGATA 3′ | |
| ACAN | (F) 5′ AGGCAGCGTGATCCTTACC 3′ |
| (R) 5′ GGCCTCTCCAGTCTCATTCTC 3′ | |
| Sox-9 | (F) 5′ TGGGCAAGCTCTGGAGACTTC 3′ |
| (R) 5′ ATCCGGGTGGTCCTTCTTGTG 3′ | |
| Col10A1-F | (F) 5′ GCAACTAAGGGCCTCAATGG 3′ |
| (R) 5′ CTCAGGCATGACTGCTTGAC 3′ | |
| Col2A1 | (F) 5′ CGTCCAGATGACCTTCCTACG 3′ |
| (R) 5′ TGAGCAGGGCCTTCTTGAG 3′ | |
| Osteocalcin | (F) 5′ CGCTGGTCTCTTCACTAC 3′ |
| (R) 5′ CTCACACTCCTCGCCCTATT 3′ | |
| Osteopontin | (F) 5′ ACTCGAACGACTCTGATGATGT 3′ |
| (R) 5′ GTCAGGTCTGCGAAACTTCTTA 3′ | |
| Runx2 | (F) 5′ GGAGTGGACGAGGCAAGAGTTT 3′ |
| (R) 5′ AGCTTCTGTCTGTGCCTTCTGG 3′ | |
| PPARγ | (F) 5′ AGGCGAGGGCGATCTTGACAG 3′ |
| (R) 5′ GATGCGGATGGCCACCTCTTT 3′ | |
| CEBPα | (F) 5′ AGGTTTCCTGCCTCCTTCC 3′ |
| (R) 5′ CCCAAGTCCCTATGTTTCCA 3′ | |
| GATA-4 | (F) 5′ AACGACGGCAACAACGATAAT 3′ |
| (R) 5′ GTTTTTTCCCCTTTGATTTTTGATC 3′ | |
| Nkx2.5 | (F) 5′ TGCTGCTCACAGGGCCCGATACTTC 3′ |
| (R) 5′ TCCTTTCGAGCTCAGTGCACCACAAAAC 3′ | |
| hMyl2A-F | (F) 5′ GGGCCCCATCAACTTCACCGTCTTCC 3′ |
| (R) 5′ TGTAGTCGATGTTCCCCGCCAGGTCC 3′ | |
| Tie-2 | (F) 5′ TCCCGAGGTCAAGAGGTGTA 3′ |
| (R) 5′ AGGGTGTGCCTCCTAAGCTA 3′ | |
| GATA-2 | (F) 5′ GCTCGTTCCTGTTCAGAAGG 3′ |
| (R) 5′ GCCATAAGGTGGTGGTTGTC 3′ | |
| VE-cadherin | (F) 5′ TTTTCCAGCAGCCTTTCTACCA 3′ |
| (R) 5′ GCGGATGGAGTATCCAATGCTA 3′ |
| 3D Cell Encapsulation | |||
|---|---|---|---|
| Type of Cell Culture Plate | Volume of Mixture: 0.15% Hydrogel + 10% Sucrose (Per Well) | Number of Encapsulated Cells | Volume of Added Cell Culture Media (for Gelation) |
| 96-well | 50 µL | 2.0 × 103 | 100 µL |
| 24-well | 250 µL | 5.0 × 104 | 500 µL |
| 12-well | 400 µL | 1.0 × 105 | 800 µL |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Labedz-Maslowska, A.; Bryniarska, N.; Kubiak, A.; Kaczmarzyk, T.; Sekula-Stryjewska, M.; Noga, S.; Boruczkowski, D.; Madeja, Z.; Zuba-Surma, E. Multilineage Differentiation Potential of Human Dental Pulp Stem Cells—Impact of 3D and Hypoxic Environment on Osteogenesis In Vitro. Int. J. Mol. Sci. 2020, 21, 6172. https://doi.org/10.3390/ijms21176172
Labedz-Maslowska A, Bryniarska N, Kubiak A, Kaczmarzyk T, Sekula-Stryjewska M, Noga S, Boruczkowski D, Madeja Z, Zuba-Surma E. Multilineage Differentiation Potential of Human Dental Pulp Stem Cells—Impact of 3D and Hypoxic Environment on Osteogenesis In Vitro. International Journal of Molecular Sciences. 2020; 21(17):6172. https://doi.org/10.3390/ijms21176172
Chicago/Turabian StyleLabedz-Maslowska, Anna, Natalia Bryniarska, Andrzej Kubiak, Tomasz Kaczmarzyk, Malgorzata Sekula-Stryjewska, Sylwia Noga, Dariusz Boruczkowski, Zbigniew Madeja, and Ewa Zuba-Surma. 2020. "Multilineage Differentiation Potential of Human Dental Pulp Stem Cells—Impact of 3D and Hypoxic Environment on Osteogenesis In Vitro" International Journal of Molecular Sciences 21, no. 17: 6172. https://doi.org/10.3390/ijms21176172
APA StyleLabedz-Maslowska, A., Bryniarska, N., Kubiak, A., Kaczmarzyk, T., Sekula-Stryjewska, M., Noga, S., Boruczkowski, D., Madeja, Z., & Zuba-Surma, E. (2020). Multilineage Differentiation Potential of Human Dental Pulp Stem Cells—Impact of 3D and Hypoxic Environment on Osteogenesis In Vitro. International Journal of Molecular Sciences, 21(17), 6172. https://doi.org/10.3390/ijms21176172