Prodigiosin Modulates the Immune Response and Could Promote a Stable Atherosclerotic Lession in C57bl/6 Ldlr-/- Mice

 , ,
, ,  ,
,  and
and
Abstract
1. Introduction
2. Results
2.1. Effect of PG or UPG on Viability and Cell Cycle of HUVEC (Human Umbilical Vascular Endothelial Cell), SMC and THP-1 Derived Macrophage-Like Cells
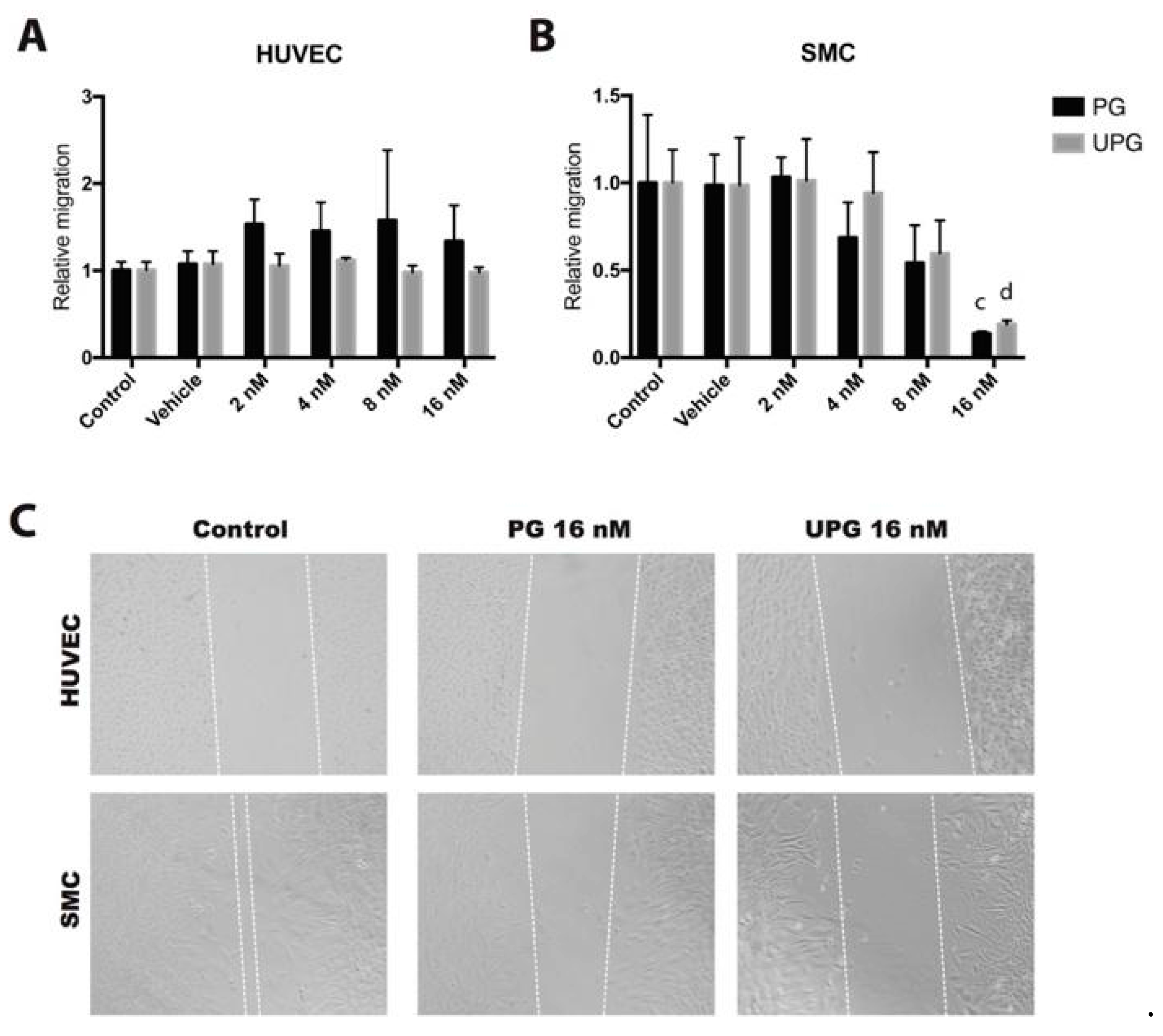
2.2. Effect of PG or UPG on Migration of HUVEC and SMC
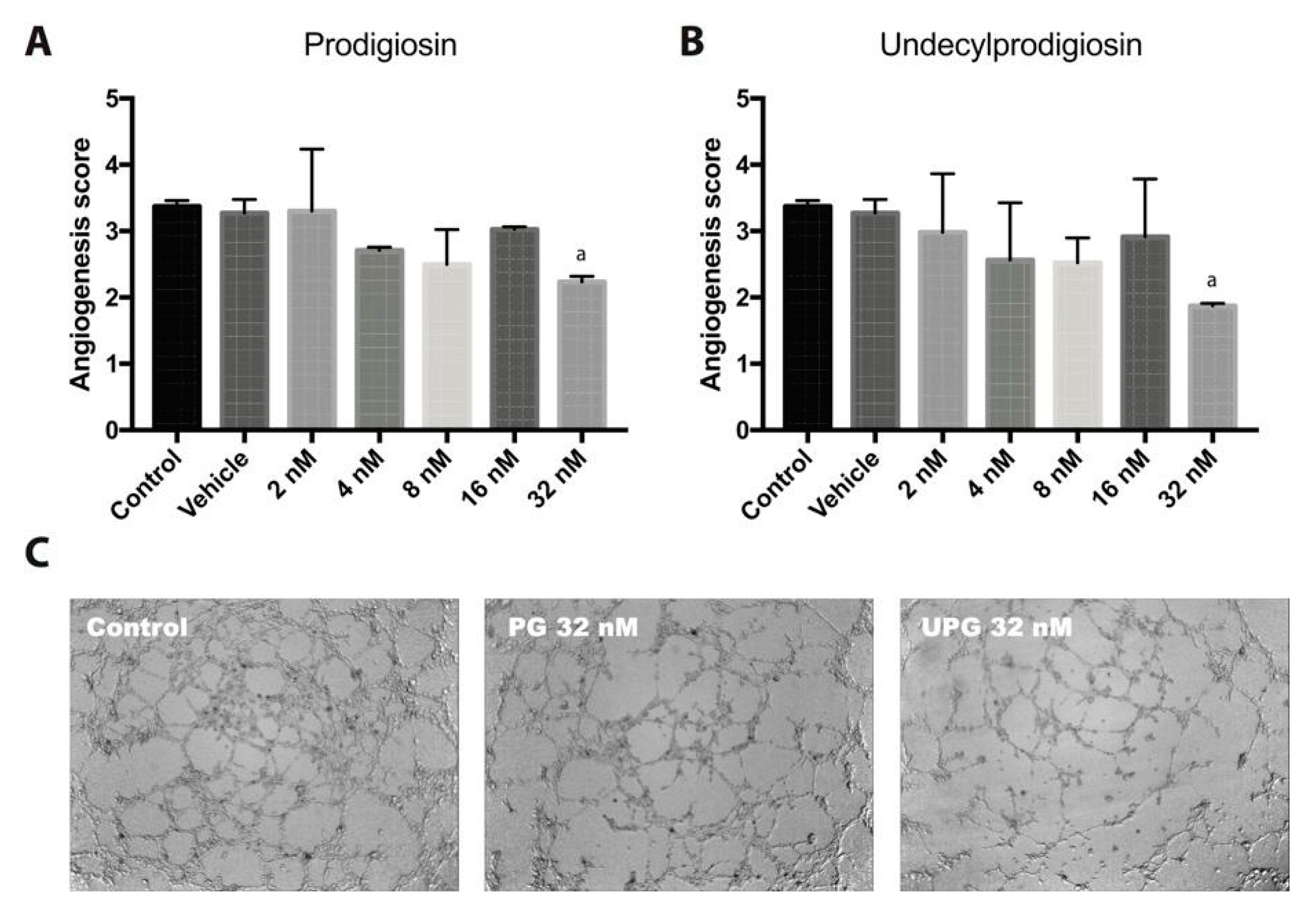
2.3. Effect of PG or UPG on In Vitro Angiogenesis
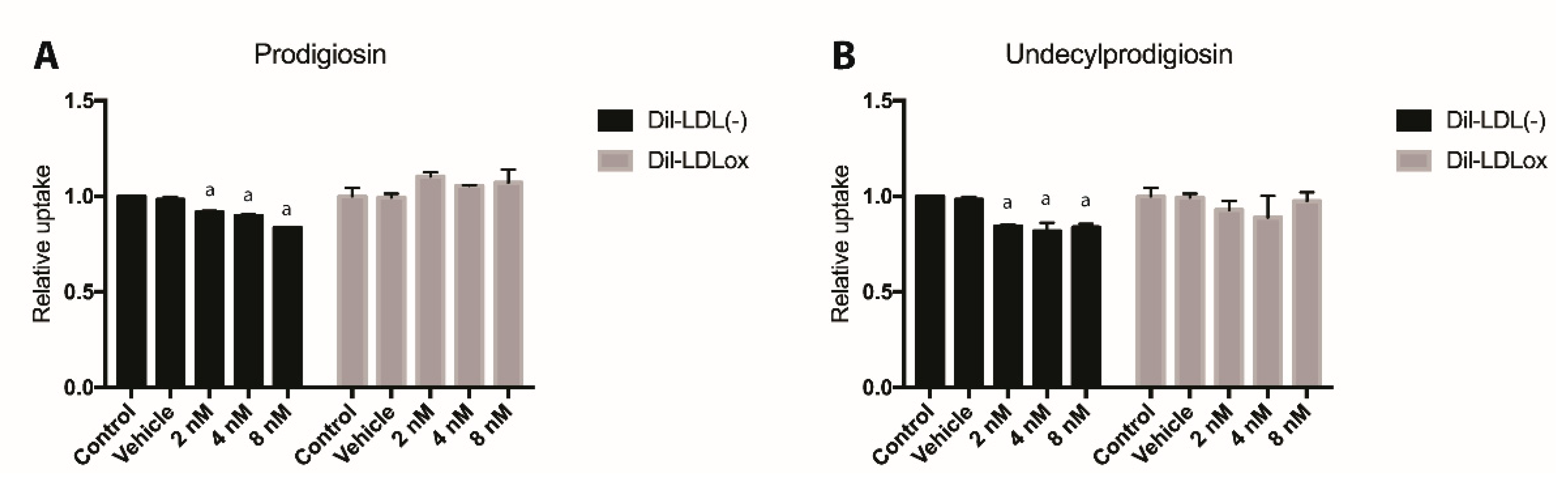
2.4. Effect of PG or UPG on oxLDL and LDL(-) Uptake
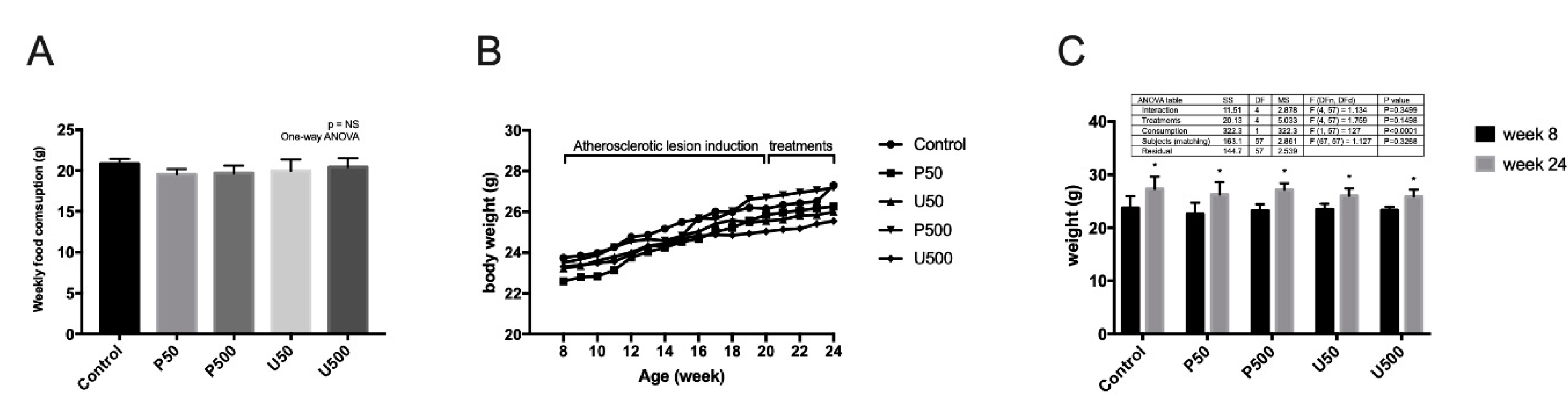
2.5. Effects of PDGs on Food Consumption, Animal Growth and Lipid Profile
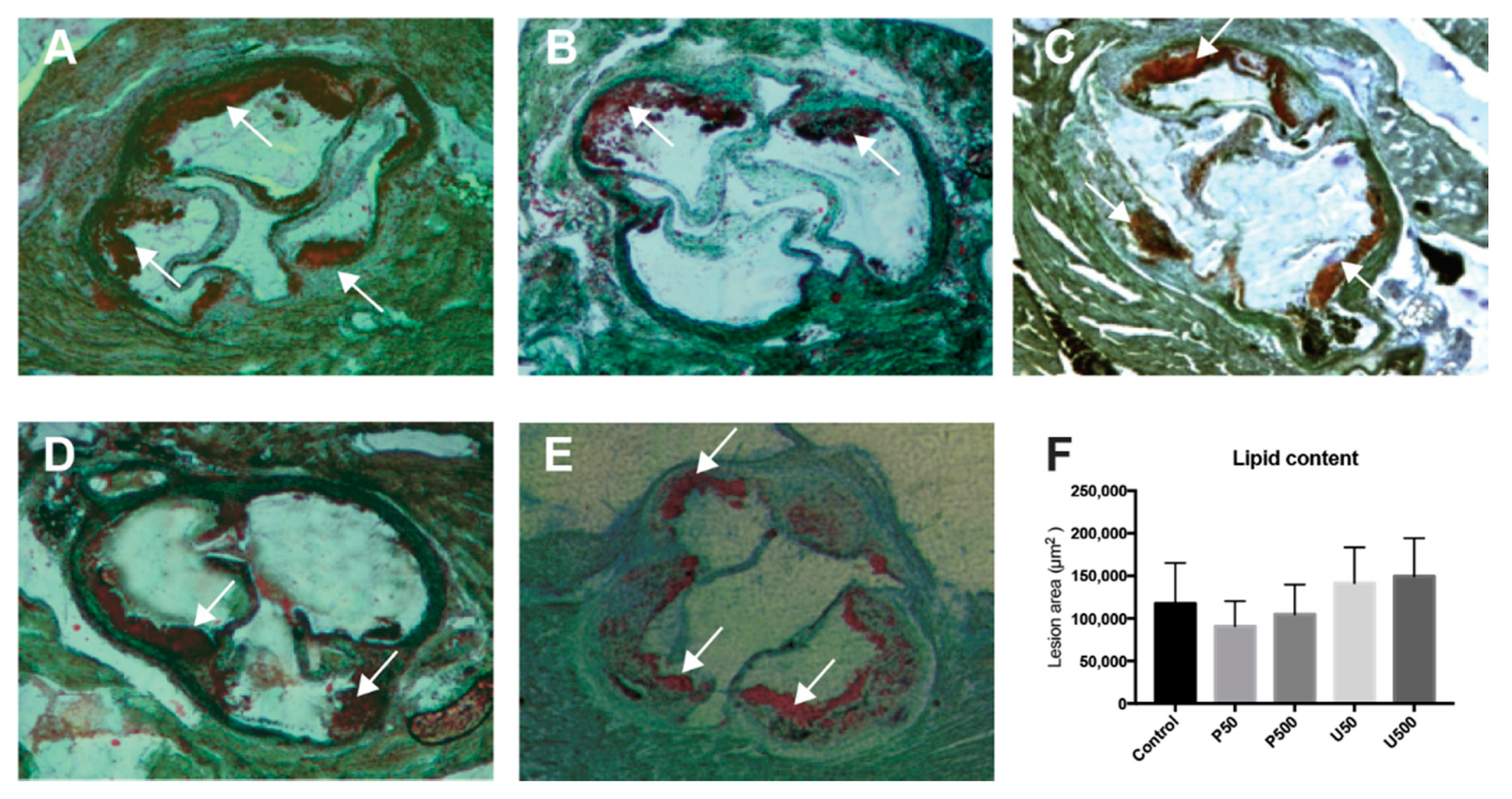
2.6. PDGs Do not Modify the Size of Atherosclerotic Lesions
2.7. PDGs Modify the Expression of Genes Associated with Plaque Stability and Inflammatory Processes in the Aortic Sinus
2.8. Effect of PG and UPG on Signature Transcripts of Different Th Cell Subsets from Paraaortic Lymph Nodes
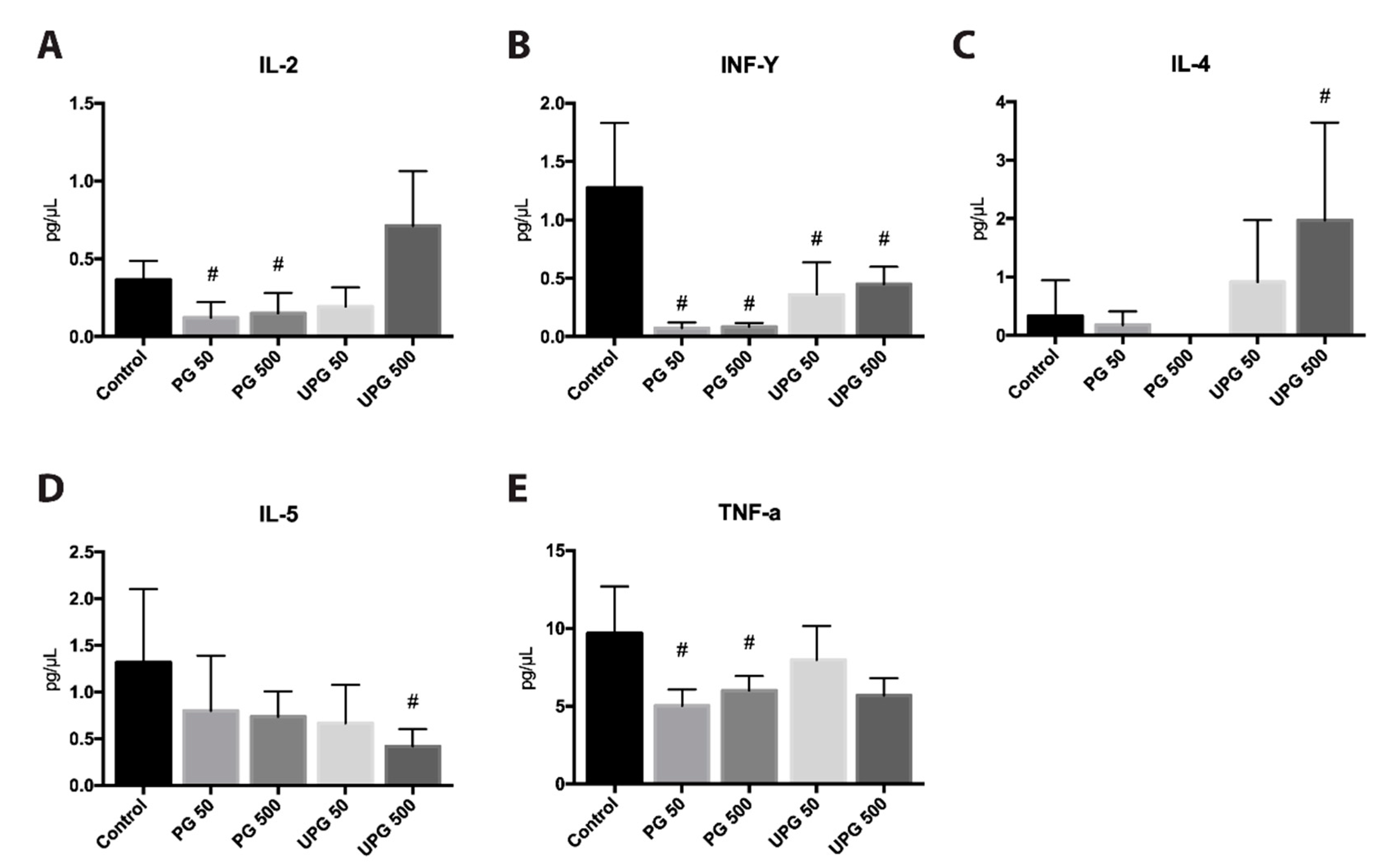
2.9. Effect of PG and UPG on Th1- and Th2-Related Cytokines in Serum
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Annexin V-Fluorescein Isothiocyanate (FITC)/Propidium Iodide (PI) Staining Assay
4.3. Cell Cycle Analysis
4.4. Migration Assay
4.5. Tube Formation Assay
4.6. DiI-OxLDL/Dil-LDL(-) Uptake Assays
4.7. Animals and Treatments
4.8. Serum Lipid Profile Analysis
4.9. Morphometric Analysis of Atherosclerotic Plaques
4.10. Gene Expression Analysis
4.11. Serum Cytokine Quantification
4.12. Statistical Analysis
Abbreviation
| LDL | Low-density lipoprotein |
| Ldlr-/- | LDL receptor null |
| SMC | Smooth muscle cell |
| TNF-α | Tumor necrosis factor alpha |
| IL | Interleukin |
| IFN-γ | Interferon-gamma |
| Ccl2 | chemokine (C-C motif) ligand 2 |
| Teff | Effector T cell |
| Treg | Regularoty T cell |
| CD25 | Interleukin-2 receptor alpha chain |
| FoxP3 | transcription element fork-head box P3 |
| PDG | Prodigiosin-like pigment |
| UPG | Undecylprodigiosin |
| CTL | Cytolytic T lymphocytes |
| NO | Nitric oxide |
| iNOS | Inducible nitric oxide synthase |
| LPS | Lipopolysaccharide |
| MAPK | mitogen-activated protein kinase |
| JNK | c-Jun N-terminal kinase |
| NF-κB | nuclear factor-kappa B |
| COX-2 | cyclooxygenase-2 |
| Th1 | T helper cell 1 |
| HUVEC | Human umbilical vascular endothelial cell |
| PCR | Polymerase chain reaction |
| Vcam-1 | vascular cell adhesion molecule 1 |
| Aoc3 | amine oxidase, copper containing 3 |
| Pecam-1 | platelet endothelial cell adhesion molecule |
| Abca1 | ATP-binding cassette transporter A |
Author Contributions
Funding
Conflicts of Interest
References
- Castellano, J.M.; Narula, J.; Castillo, J.; Fuster, V. Promoting cardiovascular health worldwide: Strategies, challenges, and opportunities. Rev. Esp. Cardiol. (Engl. Ed.) 2014, 67, 724–730. [Google Scholar] [CrossRef] [PubMed]
- Pastrana, J.L.; Sha, X.; Virtue, A.; Mai, J.; Cueto, R.; Lee, I.A.; Wang, H.; Yang, X. Regulatory T Cells and Atherosclerosis. J. Clin. Exp. Cardiolog. 2013, 1. [Google Scholar] [CrossRef] [PubMed]
- Ait-Oufella, H.; Taleb, S.; Mallat, Z.; Tedgui, A. Recent Advances on the Role of Cytokines in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 969–979. [Google Scholar] [CrossRef] [PubMed]
- Moroni, F.; Ammirati, E.; Norata, G.D.; Magnoni, M.; Camici, P.G. The Role of Monocytes and Macrophages in Human Atherosclerosis, Plaque Neoangiogenesis, and Atherothrombosis. Mediators Inflamm. 2019, 2019, 7434376. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, N.; Yamashita, T.; Takeda, M.; Hirata, K. Regulatory T cells in atherogenesis. J. Atheroscler. Thromb. 2012, 19, 503–515. [Google Scholar] [CrossRef]
- Amin, H.Z.; Sasaki, N.; Hirata, K.-I. Regulatory T Cell Immunity in Atherosclerosis. Acta Med. Indones. 2017, 49, 63–68. [Google Scholar]
- Kawasaki, T.; Sakurai, F.; Nagatsuka, S.Y.; Hayakawa, Y. Prodigiosin biosynthesis gene cluster in the roseophilin producer Streptomyces griseoviridis. J. Antibiot. 2009, 62, 271–276. [Google Scholar] [CrossRef]
- Staric, N.; Danevcic, T.; Stopar, D. Vibrio sp. DSM 14379 pigment production—A competitive advantage in the environment? Microb. Ecol. 2010, 60, 592–598. [Google Scholar] [CrossRef]
- Bennett, J.W.; Bentley, R. Seeing red: The story of prodigiosin. Adv. Appl. Microbiol. 2000, 47, 1–32. [Google Scholar]
- Perez-Tomas, R.; Vinas, M. New insights on the antitumoral properties of prodiginines. Curr. Med. Chem. 2010, 17, 2222–2231. [Google Scholar] [CrossRef]
- Magae, J.; Miller, M.W.; Nagai, K.; Shearer, G.M. Effect of metacycloprodigiosin, an inhibitor of killer T cells on murine skin and heart transplants. J. Antibiot. 1996, 49, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Kataoka, T.; Honjo, N.; Magae, J.; Nagai, K. In vivo rapid reduction of alloantigen-activated CD8+ mature cytotoxic T cells by inhibitors of acidification of intracellular organelles, prodigiosin 25-C and concanamycin B. Immunology 2000, 99, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.H.; Yamashita, M.; Tsuji, R.F.; Yamasaki, M.; Kataoka, T.; Magae, J.; Nagai, K. Suppression of T cell stimulating function of allogeneic antigen presenting cells by prodigiosin 25-C. J. Antibiot. 1998, 51, 92–94. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Huh, J.E.; Yim, J.H.; Lee, H.K.; Moon, E.Y.; Rhee, D.K.; Pyo, S. Prodigiosin isolated from Hahella chejuensis suppresses lipopolysaccharide-induced NO production by inhibiting p38 MAPK, JNK and NF-kappaB activation in murine peritoneal macrophages. Int. Immunopharmacol. 2007, 7, 1825–1833. [Google Scholar] [CrossRef]
- Han, S.B.; Park, S.H.; Jeon, Y.J.; Kim, Y.K.; Kim, H.M.; Yang, K.H. Prodigiosin blocks T cell activation by inhibiting interleukin-2Ralpha expression and delays progression of autoimmune diabetes and collagen-induced arthritis. J. Pharmacol. Exp. Ther. 2001, 299, 415–425. [Google Scholar]
- Lee, M.H.; Kataoka, T.; Magae, J.; Nagai, K. Prodigiosin 25-C suppression of cytotoxic T cells in vitro and in vivo similar to that of concanamycin B, a specific inhibitor of vacuolar type H(+)-ATPase. Biosci. Biotechnol. Biochem. 1995, 59, 1417–1421. [Google Scholar] [CrossRef][Green Version]
- Krishna, P.S.; Vani, K.; Prasad, M.R.; Samatha, B.; Bindu, N.S.; Charya, M.A.; Reddy Shetty, P. In -silico molecular docking analysis of prodigiosin and cycloprodigiosin as COX-2 inhibitors. Springerplus 2013, 2, 172. [Google Scholar] [CrossRef]
- Andersson, J.; Libby, P.; Hansson, G.K. Adaptive immunity and atherosclerosis. Clin. Immunol. 2010, 134, 33–46. [Google Scholar] [CrossRef]
- Whitman, S.C.; Ravisankar, P.; Elam, H.; Daugherty, A. Exogenous interferon-gamma enhances atherosclerosis in apolipoprotein E-/- mice. Am. J. Pathol. 2000, 157, 1819–1824. [Google Scholar] [CrossRef]
- Daugherty, A.; Rateri, D.L. T lymphocytes in atherosclerosis: The yin-yang of Th1 and Th2 influence on lesion formation. Circ. Res. 2002, 90, 1039–1040. [Google Scholar] [CrossRef]
- Zhou, X.; Robertson, A.K.; Rudling, M.; Parini, P.; Hansson, G.K. Lesion development and response to immunization reveal a complex role for CD4 in atherosclerosis. Circ. Res. 2005, 96, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, T.; Hucko, T.; Schneemann, C.; Neumann, M.; Menrad, A.; Willuda, J.; Atrott, K.; Stibenz, D.; Fleck, E.; Graf, K.; et al. Local delivery of IL-2 reduces atherosclerosis via expansion of regulatory T cells. Atherosclerosis 2012, 220, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Oppi, S.; Lüscher, T.F.; Stein, S. Mouse Models for Atherosclerosis Research—Which Is My Line? Front. Cardiovasc. Med. 2019, 6, 46. [Google Scholar] [CrossRef] [PubMed]
- Sanchis-Gomar, F.; Perez-Quilis, C.; Leischik, R.; Lucia, A. Epidemiology of coronary heart disease and acute coronary syndrome. Ann. Transl. Med. 2016, 4, 256. [Google Scholar] [CrossRef]
- Levitan, I.; Volkov, S.; Subbaiah, P. V Oxidized LDL: Diversity, patterns of recognition, and pathophysiology. Antioxid. Redox Signal. 2010, 13, 39–75. [Google Scholar] [CrossRef]
- Parasassi, T.; Bittolo-Bon, G.; Brunelli, R.; Cazzolato, G.; Krasnowska, E.K.; Mei, G.; Sevanian, A.; Ursini, F. Loss of apoB-100 secondary structure and conformation in hydroperoxide rich, electronegative LDL(-). Free Radic. Biol. Med. 2001, 31, 82–89. [Google Scholar] [CrossRef]
- Estruch, M.; Bancells, C.; Beloki, L.; Sanchez-Quesada, J.L.; Ordonez-Llanos, J.; Benitez, S. CD14 and TLR4 mediate cytokine release promoted by electronegative LDL in monocytes. Atherosclerosis 2013, 229, 356–362. [Google Scholar] [CrossRef]
- Estruch, M.; Sanchez-Quesada, J.L.; Ordonez-Llanos, J.; Benitez, S. Inflammatory intracellular pathways activated by electronegative LDL in monocytes. Biochim. Biophys. Acta. 2016, 1861, 963–969. [Google Scholar] [CrossRef]
- Espona-Fiedler, M.; Soto-Cerrato, V.; Hosseini, A.; Lizcano, J.M.; Guallar, V.; Quesada, R.; Gao, T.; Pérez-Tomás, R. Identification of dual mTORC1 and mTORC2 inhibitors in melanoma cells: Prodigiosin vs. obatoclax. Biochem. Pharmacol. 2012, 83, 489–496. [Google Scholar] [CrossRef]
- Yu, M.; Kang, X.; Xue, H.; Yin, H. Toll-like receptor 4 is up-regulated by mTOR activation during THP-1 macrophage foam cells formation. Acta Biochim. Biophys. Sin. (Shanghai) 2011, 43, 940–947. [Google Scholar] [CrossRef]
- Karar, J.; Maity, A. PI3K/AKT/mTOR Pathway in Angiogenesis. Front. Mol. Neurosci. 2011, 4, 51. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular Smooth Muscle Cells in Atherosclerosis. Circ. Res. 2016, 118, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Nowak-Sliwinska, P.; Alitalo, K.; Allen, E.; Anisimov, A.; Aplin, A.C.; Auerbach, R.; Augustin, H.G.; Bates, D.O.; van Beijnum, J.R.; Bender, R.H.F.; et al. Consensus guidelines for the use and interpretation of angiogenesis assays. Angiogenesis 2018, 21, 425–532. [Google Scholar] [CrossRef] [PubMed]
- Tedesco, S.; De Majo, F.; Kim, J.; Trenti, A.; Trevisi, L.; Fadini, G.P.; Bolego, C.; Zandstra, P.W.; Cignarella, A.; Vitiello, L. Convenience versus Biological Significance: Are PMA-Differentiated THP-1 Cells a Reliable Substitute for Blood-Derived Macrophages When Studying in Vitro Polarization? Front. Pharmacol. 2018, 9, 71. [Google Scholar] [CrossRef]
- Harvey, E.J.; Ramji, D.P. Interferon-gamma and atherosclerosis: Pro- or anti-atherogenic? Cardiovasc. Res. 2005, 67, 11–20. [Google Scholar] [CrossRef]
- Voloshyna, I.; Littlefield, M.J.; Reiss, A.B. Atherosclerosis and interferon-gamma: New insights and therapeutic targets. Trends Cardiovasc. Med. 2014, 24, 45–51. [Google Scholar] [CrossRef]
- McLaren, J.E.; Ramji, D.P. Interferon gamma: A master regulator of atherosclerosis. Cytokine Growth Factor Rev. 2009, 20, 125–135. [Google Scholar] [CrossRef]
- Saren, P.; Welgus, H.G.; Kovanen, P.T. TNF-alpha and IL-1beta selectively induce expression of 92-kDa gelatinase by human macrophages. J. Immunol. 1996, 157, 4159–4165. [Google Scholar]
- Madjid, M.; Willerson, J.T. Inflammatory markers in coronary heart disease. Br. Med. Bull 2011, 100, 23–38. [Google Scholar] [CrossRef]
- Xiao, N.; Yin, M.; Zhang, L.; Qu, X.; Du, H.; Sun, X.; Mao, L.; Ren, G.; Zhang, C.; Geng, Y.; et al. Tumor necrosis factor-alpha deficiency retards early fatty-streak lesion by influencing the expression of inflammatory factors in apoE-null mice. Mol. Genet. Metab. 2009, 96, 239–244. [Google Scholar] [CrossRef]
- Puz, P.; Lasek-Bal, A.; Ziaja, D.; Kazibutowska, Z.; Ziaja, K. Inflammatory markers in patients with internal carotid artery stenosis. Arch. Med. Sci. 2013, 9, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Taleb, S. Inflammation in atherosclerosis. Arch. Cardiovasc. Dis. 2016, 109, 708–715. [Google Scholar] [CrossRef]
- Raphael, I.; Nalawade, S.; Eagar, T.N.; Forsthuber, T.G. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine 2015, 74, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.W.; Lee, W.H.; Kim, P.H. Role of NADPH oxidase in interleukin-4-induced monocyte chemoattractant protein-1 expression in vascular endothelium. Inflamm. Res. 2010, 59, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.W.; Eum, S.Y.; Chen, K.C.; Hennig, B.; Toborek, M. Gene expression profile in interleukin-4-stimulated human vascular endothelial cells. Mol. Med. 2004, 10, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Boyle, J.J. Macrophage activation in atherosclerosis: Pathogenesis and pharmacology of plaque rupture. Curr. Vasc. Pharmacol. 2005, 3, 63–68. [Google Scholar] [CrossRef]
- Ikeda, U.; Matsui, K.; Murakami, Y.; Shimada, K. Monocyte chemoattractant protein-1 and coronary artery disease. Clin. Cardiol. 2002, 25, 143–147. [Google Scholar] [CrossRef]
- Pannecoeck, R.; Serruys, D.; Benmeridja, L.; Delanghe, J.R.; Geel, N.V.; Speeckaert, R.; Speeckaert, M.M. Vascular adhesion protein-1: Role in human pathology and application as a biomarker. Crit. Rev. Clin. Lab. Sci. 2015, 52, 284–300. [Google Scholar] [CrossRef]
- Kleemann, R.; Zadelaar, S.; Kooistra, T. Cytokines and atherosclerosis: A comprehensive review of studies in mice. Cardiovasc. Res. 2008, 79, 360–376. [Google Scholar] [CrossRef]
- Toth, P.P.; Farnier, M.; Tomassini, J.E.; Foody, J.M.; Tershakovec, A.M. Statin combination therapy and cardiovascular risk reduction. Future Cardiol. 2016, 12, 289–315. [Google Scholar] [CrossRef]
- Yakala, G.K.; Cabrera-Fuentes, H.A.; Crespo-Avilan, G.E.; Rattanasopa, C.; Burlacu, A.; George, B.L.; Anand, K.; Mayan, D.C.; Corlianò, M.; Hernández-Reséndiz, S.; et al. FURIN inhibition reduces vascular remodeling and atherosclerotic lesion progression in mice. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 387–401. [Google Scholar] [CrossRef]
- Cuevas, A.; Saavedra, N.; Rudnicki, M.; Abdalla, D.S.; Salazar, L.A. ERK1/2 and HIF1alpha Are Involved in Antiangiogenic Effect of Polyphenols-Enriched Fraction from Chilean Propolis. Evid. Based Complement Altern. Med. 2015, 2015, 187575. [Google Scholar] [CrossRef]
- Geback, T.; Schulz, M.M.; Koumoutsakos, P.; Detmar, M. TScratch: A novel and simple software tool for automated analysis of monolayer wound healing assays. Biotechniques 2009, 46, 265–274. [Google Scholar] [CrossRef]
- Ponce, M.L. In vitro matrigel angiogenesis assays. Methods Mol. Med. 2001, 46, 205–209. [Google Scholar] [CrossRef]
- Aranda, E.; Owen, G.I. A semi-quantitative assay to screen for angiogenic compounds and compounds with angiogenic potential using the EA.hy926 endothelial cell line. Biol. Res. 2009, 42, 377–389. [Google Scholar] [CrossRef]
- LASEC Sample Size and Power Calculator. Available online: http://www.lasec.cuhk.edu.hk/uploads/1/3/5/2/13529582/power_calculator_14_may_2014.xls (accessed on 20 June 2016).
- Matfiiiiiiiild, M. Handbook of laboratory animal management and welfare. Trends Pharmacol. Sci. 1995, 16, 174. [Google Scholar] [CrossRef]
- GraphPad, QuickCalcs. Available online: https://www.graphpad.com/quickcalcs/randomSelect1/ (accessed on 20 June 2016).
- Paigen, B.; Morrow, A.; Holmes, P.A.; Mitchell, D.; Williams, R.A. Quantitative assessment of atherosclerotic lesions in mice. Atherosclerosis 1987, 68, 231–240. [Google Scholar] [CrossRef]
- Puig, O.; Yuan, J.; Stepaniants, S.; Zieba, R.; Zycband, E.; Morris, M.; Coulter, S.; Yu, X.; Menke, J.; Woods, J.; et al. A gene expression signature that classifies human atherosclerotic plaque by relative inflammation status. Circ. Cardiovasc. Genet. 2011, 4, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic. Acids. Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | TC (mg/dL) | HDL-C (mg/dL) | LDL-C (mg/dL) | TG (mg/dL) |
|---|---|---|---|---|
| Control | 1585 ± 400 | 35.2 ± 6.1 | 1376 ± 100.5 | 435.0 ± 114.7 |
| P50 | 1480 ± 193 | 28.7 ± 6.7 | 1367 ± 208.4 | 423.3 ± 90.0 |
| P500 | 1735 ± 238 | 22.8 ± 8.6 | 1602 ± 193.7 | 402.0 ± 64.2 |
| U50 | 1653 ± 326 | 35.3 ± 7.4 | 1535 ± 371.7 | 411.7 ± 140.3 |
| U500 | 1616 ± 391 | 28.3 ± 11.9 | 1435 ± 459.0 | 435.7 ± 68.0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuevas, A.; Saavedra, N.; Salazar, L.A.; Cavalcante, M.F.; Silva, J.C.; Abdalla, D.S.P. Prodigiosin Modulates the Immune Response and Could Promote a Stable Atherosclerotic Lession in C57bl/6 Ldlr-/- Mice. Int. J. Mol. Sci. 2020, 21, 6417. https://doi.org/10.3390/ijms21176417
Cuevas A, Saavedra N, Salazar LA, Cavalcante MF, Silva JC, Abdalla DSP. Prodigiosin Modulates the Immune Response and Could Promote a Stable Atherosclerotic Lession in C57bl/6 Ldlr-/- Mice. International Journal of Molecular Sciences. 2020; 21(17):6417. https://doi.org/10.3390/ijms21176417
Chicago/Turabian StyleCuevas, Alejandro, Nicolás Saavedra, Luis A. Salazar, Marcela F. Cavalcante, Jacqueline C. Silva, and Dulcineia S. P. Abdalla. 2020. "Prodigiosin Modulates the Immune Response and Could Promote a Stable Atherosclerotic Lession in C57bl/6 Ldlr-/- Mice" International Journal of Molecular Sciences 21, no. 17: 6417. https://doi.org/10.3390/ijms21176417
APA StyleCuevas, A., Saavedra, N., Salazar, L. A., Cavalcante, M. F., Silva, J. C., & Abdalla, D. S. P. (2020). Prodigiosin Modulates the Immune Response and Could Promote a Stable Atherosclerotic Lession in C57bl/6 Ldlr-/- Mice. International Journal of Molecular Sciences, 21(17), 6417. https://doi.org/10.3390/ijms21176417