The Ubiquitin Proteasome System in Neuromuscular Disorders: Moving Beyond Movement

 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
1.1. Neuromuscular System
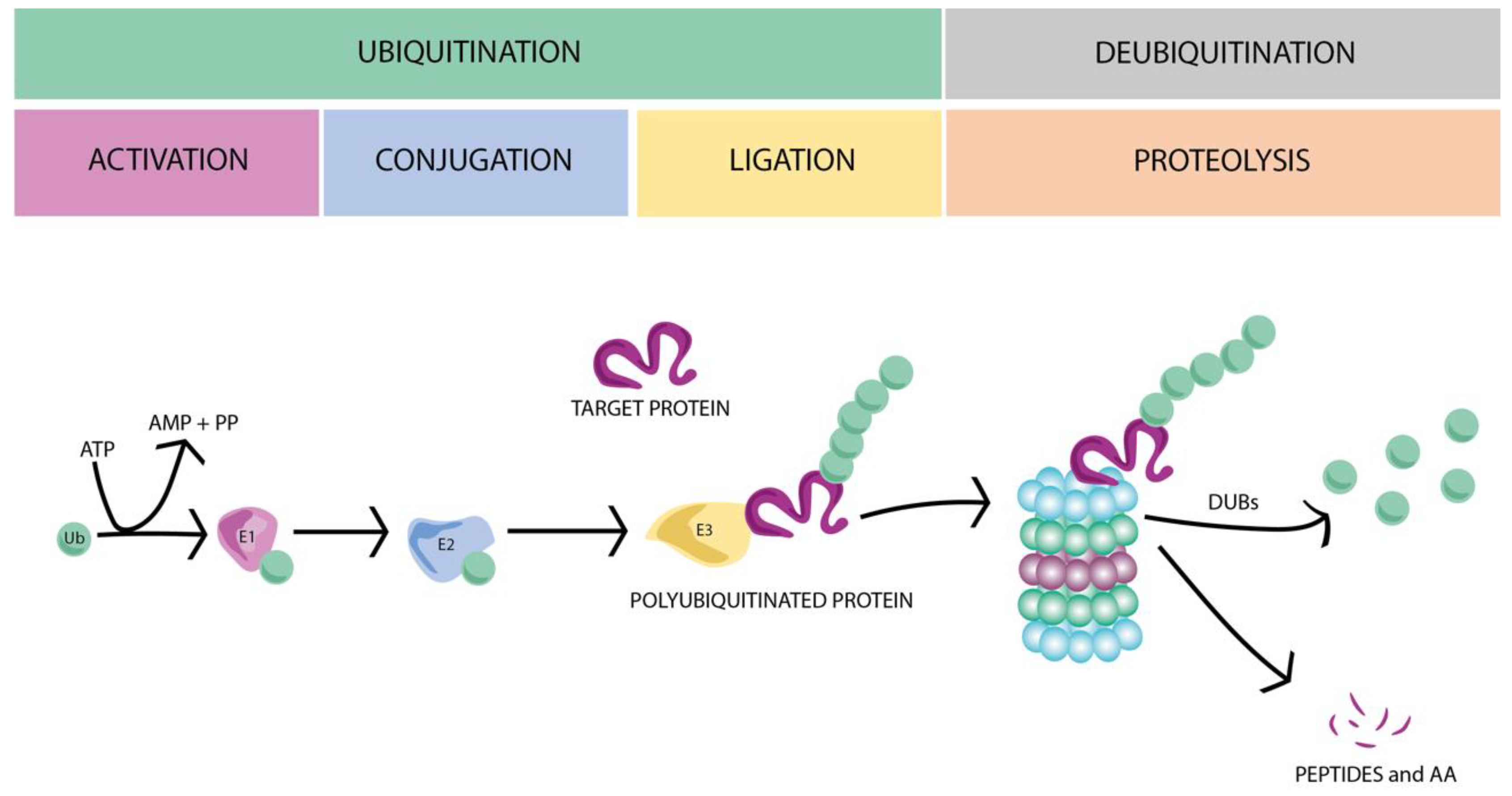
1.2. The Ubiquitin–Proteasome System (UPS)
1.3. UPS and Neuromuscular System
2. UPS in Neuromuscular Disorders (NMDs)
2.1. Motor Neuron Diseases
2.1.1. Spinal Muscular Atrophy (SMA)
2.1.2. Amyotrophic Lateral Sclerosis (ALS)
2.2. Peripheral Nerve Diseases
2.2.1. Charcot–Marie–Tooth Disease (CMT)
2.2.2. Friedreich Ataxia (FRDA)
2.3. Neuromuscular Junction
2.4. Muscle
2.4.1. Duchenne Muscular Dystrophy (DMD)
2.4.2. Other Muscular Dystrophies
3. Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Camacho, A.; Esteban, J.; Paradas, C. Report by the Spanish Foundation for the Brain on the social impact of amyotrophic lateral sclerosis and other neuromuscular disorders. Neurol. (Engl. Ed.) 2018, 33, 35–46. [Google Scholar] [CrossRef]
- Emery, A.E. Population frequencies of inherited neuromuscular diseases—A world survey. Neuromuscul. Disord. 1991, 1, 19–29. [Google Scholar] [CrossRef]
- Deenen, J.C.; Horlings, C.G.; Verschuuren, J.J.; Verbeek, A.L.; Van Engelen, B.G. The Epidemiology of Neuromuscular Disorders: A Comprehensive Overview of the Literature. J. Neuromuscul. Dis. 2015, 2, 73–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Q.; Huang, T.; Zhang, L.; Zhou, Y.; Luo, H.; Xu, H.; Wang, X. Dysregulation of Ubiquitin-Proteasome System in Neurodegenerative Diseases. Front. Aging Neurosci. 2016, 8, 303. [Google Scholar] [CrossRef]
- Deng, H.X.; Chen, W.; Hong, S.T.; Boycott, K.Y.; Gorrie, G.H.; Siddique, N.; Faisal Fecto, Y.Y.; Hong Zhai, Y.S.; Makito Hirano, H.J.; Rampersaud, E.; et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature 2011, 477, 211–215. [Google Scholar] [CrossRef] [Green Version]
- Dlamini, N.; Josifova, D.J.; Paine, S.M.; Wraige, E.; Pitt, M.; Murphy, A.J.; King, A.; Buk, S.; Smith, F.; Abbs, S.; et al. Clinical and neuropathological features of X-linked spinal muscular atrophy (SMAX2) associated with a novel mutation in the UBA1 gene. Neuromuscul. Disord. 2013, 23, 391–398. [Google Scholar] [CrossRef]
- Ramser, J.; Ahearn, M.E.; Lenski, C.; Yariz, K.O.; Hellebrand, H.; Von Rhein, M.; Clark, R.D.; Schmutzler, R.K.; Lichtner, P.; Hoffman, E.P.; et al. Rare Missense and Synonymous Variants in UBE1 Are Associated with X-Linked Infantile Spinal Muscular Atrophy. Am. J. Hum. Genet. 2008, 82, 188–193. [Google Scholar] [CrossRef] [Green Version]
- Rusmini, P.; Sau, D.; Crippa, V.; Palazzolo, I.; Simonini, F.; Onesto, E.; Martini, L.; Poletti, A. Aggregation and proteasome: The case of elongated polyglutamine aggregation in spinal and bulbar muscular atrophy. Neurobiol. Aging 2007, 28, 1099–1111. [Google Scholar] [CrossRef]
- Sarvestany, A.A.; Hunter, G.; Tavendale, A.; Lamont, D.J.; Hurtado, M.L.; Graham, L.C.; Wishart, T.M.; Gillingwater, T.H. Label-Free Quantitative Proteomic Profiling Identifies Disruption of Ubiquitin Homeostasis As a Key Driver of Schwann Cell Defects in Spinal Muscular Atrophy. J. Proteome Res. 2014, 13, 4546–4557. [Google Scholar] [CrossRef]
- Bogdanik, L.P.; Sleigh, J.N.; Tian, C.; Samuels, M.E.; Bedard, K.; Seburn, K.L.; Burgess, R.W. Loss of the E3 ubiquitin ligase LRSAM1 sensitizes peripheral axons to degeneration in a mouse model of Charcot-Marie-Tooth disease. Dis. Model. Mech. 2013, 6, 780–792. [Google Scholar] [CrossRef] [Green Version]
- Saifi, G.M.; Szigeti, K.; Wiszniewski, W.; Shy, M.E.; Krajewski, K.; Hausmanowa-Petrusewicz, I.; Kochanski, A.; Reeser, S.; Mancias, P.; Butler, I.; et al. SIMPLE mutations in Charcot-Marie-Tooth disease and the potential role of its protein product in protein degradation. Hum. Mutat. 2005, 25, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Ylikallio, E.; Pöyhönen, R.; Zimoń, M.; De Vriendt, E.; Hilander, T.; Paetau, A.; Jordanova, A.; Lönnqvist, T.; Tyynismaa, H. Deficiency of the E3 ubiquitin ligase TRIM2 in early-onset axonal neuropathy. Hum. Mol. Genet. 2013, 22, 2975–2983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugiura, Y.; Lin, W. Neuron–glia interactions: The roles of Schwann cells in neuromuscular synapse formation and function. Biosci. Rep. 2011, 31, 295–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegde, A.N.; Upadhya, S.C. The ubiquitin–proteasome pathway in health and disease of the nervous system. Trends Neurosci. 2007, 30, 587–595. [Google Scholar] [CrossRef]
- Lu, Z.; Je, H.S.; Young, P.; Gross, J.; Lu, B.; Feng, G. Regulation of synaptic growth and maturation by a synapse-associated E3 ubiquitin ligase at the neuromuscular junction. J. Cell Boil. 2007, 177, 1077–1089. [Google Scholar] [CrossRef] [Green Version]
- Okano, H.J.; Tada, H.; Okano, H. Fbxo45, a novel ubiquitin ligase, regulates synaptic activity. Neurosci. Res. 2010, 68, e60. [Google Scholar] [CrossRef] [Green Version]
- Van Tijn, P.; Hol, E.M.; Van Leeuwen, F.W.; Fischer, D.F. The neuronal ubiquitin-proteasome system: Murine models and their neurological phenotype. Prog. Neurobiol. 2008, 85, 176–193. [Google Scholar] [CrossRef]
- Dantuma, N.P.; Bott, L.C. The ubiquitin-proteasome system in neurodegenerative diseases: Precipitating factor, yet part of the solution. Front. Mol. Neurosci. 2014, 7, 70. [Google Scholar] [CrossRef] [Green Version]
- Glickman, M.H.; Ciechanover, A. The Ubiquitin-Proteasome Proteolytic Pathway: Destruction for the Sake of Construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef]
- Dikic, I. Proteasomal and Autophagic Degradation Systems. Annu. Rev. Biochem. 2017, 86, 193–224. [Google Scholar] [CrossRef]
- Gao, T.; Liu, Z.-X.; Wang, Y.; Cheng, H.; Yang, Q.; Guo, A.; Ren, J.; Xue, Y. UUCD: A family-based database of ubiquitin and ubiquitin-like conjugation. Nucleic Acids Res. 2012, 41, D445–D451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, K.P.; Greer, S.F. Proteolytic and non-proteolytic roles of ubiquitin and the ubiquitin proteasome system in transcriptional regulation. Biochim. Biophys. Acta (BBA) Bioenerg. 2011, 1809, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.L.; Cascio, P.; Sarica, T.; Rock, K.L. The importance of the proteasome and subsequent proteolytic steps in the generation of antigenic peptides. Mol. Immunol. 2002, 39, 147–164. [Google Scholar] [CrossRef]
- Rinetti, G.V.; Schweizer, F.E. Ubiquitination acutely regulates presynaptic neurotransmitter release in mammalian neurons. J. Neurosci. 2010, 30, 3157–3166. [Google Scholar] [CrossRef] [PubMed]
- Limanaqi, F.; Busceti, C.L.; Biagioni, F.; Cantini, F.; Lenzi, P.; Fornai, F. Cell-Clearing Systems Bridging Repeat Expansion Proteotoxicity and Neuromuscular Junction Alterations in ALS and SBMA. Int. J. Mol. Sci. 2020, 21, 4021. [Google Scholar] [CrossRef]
- Sambataro, F.; Pennuto, M. Post-translational Modifications and Protein Quality Control in Motor Neuron and Polyglutamine Diseases. Front. Mol. Neurosci. 2017, 10, 82. [Google Scholar] [CrossRef] [Green Version]
- Bax, M.; McKenna, J.; Do-Ha, D.; Stevens, C.H.; Higginbottom, S.; Balez, R.; Cabral-Da-Silva, M.C.; Farrawell, N.E.; Engel, M.; Poronnik, P.; et al. The Ubiquitin Proteasome System Is a Key Regulator of Pluripotent Stem Cell Survival and Motor Neuron Differentiation. Cells 2019, 8, 581. [Google Scholar] [CrossRef] [Green Version]
- Pinto, M.J.; Alves, P.L.; Martins, L.; Pedro, J.R.; Ryu, H.R.; Jeon, N.L.; Taylor, A.M.; Almeida, R.D. The proteasome controls presynaptic differentiation through modulation of an on-site pool of polyubiquitinated conjugates. J. Cell Boil. 2016, 212, 789–801. [Google Scholar] [CrossRef]
- Jiang, X.; Litkowski, P.E.; Taylor, A.A.; Lin, Y.; Snider, B.J.; Moulder, K.L. A role for the ubiquitin-proteasome system in activity-dependent presynaptic silencing. J. Neurosci. 2010, 30, 1798–1809. [Google Scholar] [CrossRef]
- Montenegro-Venegas, C.; Annamneedi, A.; Hoffmann-Conaway, S.; Gundelfinger, E.D.; Garner, C.C. BSN (bassoon) and PRKN/parkin in concert control presynaptic vesicle autophagy. Autophagy 2020, 1–2. [Google Scholar] [CrossRef]
- Lee, H.K.; Shin, Y.K.; Jung, J.; Park, H.T.; Seo, S.-Y.; Baek, S.-Y. Proteasome inhibition suppresses Schwann cell dedifferentiationin vitroandin vivo. Glia 2009, 57, 1825–1834. [Google Scholar] [CrossRef] [PubMed]
- Polge, C.; Cabantous, S.; Deval, C.; Claustre, A.; Hauvette, A.; Bouchenot, C.; Aniort, J.; Béchet, D.; Combaret, L.; Attaix, D.; et al. A muscle-specific MuRF1-E2 network requires stabilization of MuRF1-E2 complexes by telethonin, a newly identified substrate. J. Cachex- Sarcopenia Muscle 2017, 9, 129–145. [Google Scholar] [CrossRef] [PubMed]
- Polge, C.; Aniort, J.; Armani, A.; Claustre, A.; Coudy-Gandilhon, C.; Tournebize, C.; Deval, C.; Combaret, L.; Bechet, D.M.; Sandri, M.; et al. UBE2E1 Is Preferentially Expressed in the Cytoplasm of Slow-Twitch Fibers and Protects Skeletal Muscles from Exacerbated Atrophy upon Dexamethasone Treatment. Cells 2018, 7, 214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krivoi, I.I.; Petrov, A.M. Cholesterol and the Safety Factor for Neuromuscular Transmission. Int. J. Mol. Sci. 2019, 20, 1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitt, F.; Hussain, G.; Dupuis, L.; Loeffler, J.-P.; Henriques, A. A plural role for lipids in motor neuron diseases: Energy, signaling and structure. Front. Cell. Neurosci. 2014, 8, 25. [Google Scholar] [CrossRef]
- Shorrock, H.K.; van der Hoorn, D.; Boyd, P.J.; Llavero Hurtado, M.; Lamont, D.J.; Wirth, B.; Sleigh, J.N.; Schiavo, G.; Wishart, T.M.; Groen, E.J.N.; et al. UBA1/GARS-dependent pathways drive sensory-motor connectivity defects in spinal muscular atrophy. Brain 2018, 141, 2878–2894. [Google Scholar] [CrossRef] [Green Version]
- Lambert-Smith, I.A.; Saunders, D.N.; Yerbury, J.J. The pivotal role of ubiquitin-activating enzyme E1 (UBA1) in neuronal health and neurodegeneration. Int. J. Biochem. Cell Boil. 2020, 123, 105746. [Google Scholar] [CrossRef]
- Schulze, E.; Altmann, M.E.; Adham, I.M.; Schulze, B.; Fröde, S.; Engel, W. The maintenance of neuromuscular function requires UBC-25 in Caenorhabditis elegans. Biochem. Biophys. Res. Commun. 2003, 305, 691–699. [Google Scholar] [CrossRef]
- Van Roessel, P.; Elliott, D.A.; Robinson, I.M.; Prokop, A.; Brand, A.H. Independent Regulation of Synaptic Size and Activity by the Anaphase-Promoting Complex. Cell 2004, 119, 707–718. [Google Scholar] [CrossRef]
- Kowalski, J.R.; Dube, H.; Touroutine, D.; Rush, K.M.; Goodwin, P.R.; Carozza, M.; Didier, Z.; Francis, M.M.; Juo, P. The Anaphase-Promoting Complex (APC) ubiquitin ligase regulates GABA transmission at the C. elegans neuromuscular junction. Mol. Cell. Neurosci. 2013, 58, 62–75. [Google Scholar] [CrossRef] [Green Version]
- Bachiller, S.; Roca-Ceballos, M.A.; García-Domínguez, I.; Pérez-Villegas, E.M.; Martos-Carmona, D.; Pérez-Castro, M.A.; Real, L.M.; Rosa, J.L.; Tabares, L.; Venero, J.L.; et al. HERC1 Ubiquitin Ligase Is Required for Normal Axonal Myelination in the Peripheral Nervous System. Mol. Neurobiol. 2018, 55, 8856–8868. [Google Scholar] [CrossRef] [PubMed]
- Bachiller, S.; Rybkina, T.; Porras-Garcia, E.; Pérez-Villegas, E.; Tabares, L.; Armengol, J.A.; Carrion, A.M.; Ruiz, R. The HERC1 E3 Ubiquitin Ligase is essential for normal development and for neurotransmission at the mouse neuromuscular junction. Cell. Mol. Life Sci. 2015, 72, 2961–2971. [Google Scholar] [CrossRef] [PubMed]
- Wan, H.I.; DiAntonio, A.; Fetter, R.D.; Bergstrom, K.; Strauss, R.; Goodman, C.S. Highwire Regulates Synaptic Growth in Drosophila. Neuron 2000, 26, 313–329. [Google Scholar] [CrossRef] [Green Version]
- Burgess, R.W.; Peterson, K.A.; Johnson, M.J.; Roix, J.J.; Welsh, I.C.; O’Brien, T.P. Evidence for a Conserved Function in Synapse Formation Reveals Phr1 as a Candidate Gene for Respiratory Failure in Newborn Mice. Mol. Cell. Boil. 2004, 24, 1096–1105. [Google Scholar] [CrossRef] [Green Version]
- Zhen, M.; Huang, X.; Bamber, B.; Jin, Y. Regulation of Presynaptic Terminal Organization by C. elegans RPM-1, a Putative Guanine Nucleotide Exchanger with a RING-H2 Finger Domain. Neuron 2000, 26, 331–343. [Google Scholar] [CrossRef] [Green Version]
- Ding, M.; Chao, D.; Wang, G.; Shen, K. Spatial Regulation of an E3 Ubiquitin Ligase Directs Selective Synapse Elimination. Science 2007, 317, 947–951. [Google Scholar] [CrossRef]
- Loria, P.M.; Hodgkin, J.; Hobert, O. A Conserved Postsynaptic Transmembrane Protein Affecting Neuromuscular Signaling in Caenorhabditis elegans. J. Neurosci. 2004, 24, 2191–2201. [Google Scholar] [CrossRef]
- McCabe, B.D.; Hom, S.; Aberle, H.; Fetter, R.D.; Marques, G.; E Haerry, T.; Wan, H.; O’Connor, M.B.; Goodman, C.S.; Haghighi, A.P. Highwire regulates presynaptic BMP signaling essential for synaptic growth. Neuron 2004, 41, 891–905. [Google Scholar] [CrossRef] [Green Version]
- Bodine, S.C. Identification of Ubiquitin Ligases Required for Skeletal Muscle Atrophy. Science 2001, 294, 1704–1708. [Google Scholar] [CrossRef]
- Glass, D.J. Molecular mechanisms modulating muscle mass. Trends Mol. Med. 2003, 9, 344–350. [Google Scholar] [CrossRef]
- Rudolf, R.; Bogomolovas, J.; Strack, S.; Choi, K.-R.; Khan, M.M.; Wagner, A.; Brohm, K.; Hanashima, A.; Gasch, A.; Labeit, D.; et al. Regulation of nicotinic acetylcholine receptor turnover by MuRF1 connects muscle activity to endo/lysosomal and atrophy pathways. AGE 2012, 35, 1663–1674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Y.; Shtineman-Kotler, A.; Nguyen, L.; Iliadi, K.G.; Boulianne, G.L.; Rotin, D. A Splice Isoform of DNedd4, DNedd4-Long, Negatively Regulates Neuromuscular Synaptogenesis and Viability in Drosophila. PLoS ONE 2011, 6, e27007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ing, B.; Shteiman-Kotler, A.; Castelli, M.; Henry, P.; Pak, Y.; Stewart, B.; Boulianne, G.L.; Rotin, D. Regulation of Commissureless by the Ubiquitin Ligase DNedd4 Is Required for Neuromuscular Synaptogenesis in Drosophila melanogaster. Mol. Cell. Boil. 2006, 27, 481–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Oppenheim, R.W.; Sugiura, Y.; Lin, W. Abnormal development of the neuromuscular junction in Nedd4-deficient mice. Dev. Boil. 2009, 330, 153–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safi, F.; Shteiman-Kotler, A.; Zhong, Y.; Iliadi, K.G.; Boulianne, G.L.; Rotin, D. Drosophila Nedd4-long reduces Amphiphysin levels in muscles and leads to impaired T-tubule formation. Mol. Boil. Cell 2016, 27, 907–918. [Google Scholar] [CrossRef] [PubMed]
- Blondelle, J.; Tallapaka, K.; Seto, J.; Ghassemian, M.; Clark, M.; Laitila, J.M.; Bournazos, A.M.; Singer, J.D.; Lange, S. Cullin-3 dependent deregulation of ACTN1 represents a new pathogenic mechanism in nemaline myopathy. JCI Insight 2019, 5, 5. [Google Scholar] [CrossRef]
- Arribat, Y.; Mysiak, K.S.; Lescouzères, L.; Boizot, A.; Ruiz, M.; Rossel, M.; Bomont, P. Sonic Hedgehog repression underlies gigaxonin mutation–induced motor deficits in giant axonal neuropathy. J. Clin. Investig. 2019, 129, 5312–5326. [Google Scholar] [CrossRef] [Green Version]
- Berg, E.L.; Pride, M.C.; Petkova, S.P.; Lee, R.D.; Copping, N.A.; Shen, Y.; Adhikari, A.; Fenton, T.A.; Pedersen, L.R.; Noakes, L.S.; et al. Translational outcomes in a full gene deletion of ubiquitin protein ligase E3A rat model of Angelman syndrome. Transl. Psychiatry 2020, 10, 1–16. [Google Scholar] [CrossRef]
- Vatsa, N.; Jana, N.R. UBE3A and Its Link With Autism. Front. Mol. Neurosci. 2018, 11, 448. [Google Scholar] [CrossRef]
- Assereto, S.; Piccirillo, R.; Baratto, S.; Scudieri, P.; Fiorillo, C.; Massacesi, M.; Traverso, M.; Galietta, L.J.; Bruno, C.; Minetti, C.; et al. The ubiquitin ligase tripartite-motif-protein 32 is induced in Duchenne muscular dystrophy. Lab. Investig. 2016, 96, 862–871. [Google Scholar] [CrossRef]
- Meroni, G. TRIM E3 Ubiquitin Ligases in Rare Genetic Disorders. Adv. Exp. Med. Biol. 2020, 311–325. [Google Scholar] [CrossRef]
- Panicucci, C.; Traverso, M.; Baratto, S.; Romeo, C.; Iacomino, M.; Gemelli, C.; Tagliafico, A.; Broda, P.; Zara, F.; Bruno, C.; et al. Novel TRIM32 mutation in sarcotubular myopathy. Acta Myol 2019, 38, 8–12. [Google Scholar]
- Servián-Morilla, E.; Cabrera-Serrano, M.; Rivas-Infante, E.; Carvajal, A.; Lamont, P.J.; Pelayo-Negro, A.L.; Ravenscroft, G.; Junckerstorff, R.; Dyke, J.M.; Fletcher, S.; et al. Altered myogenesis and premature senescence underlie human TRIM32-related myopathy. Acta Neuropathol. Commun. 2019, 7, 30. [Google Scholar] [CrossRef] [Green Version]
- Kwon, D.Y.; Dimitriadi, M.; Terzic, B.; Cable, C.; Hart, A.C.; Chitnis, A.; Fischbeck, K.H.; Burnett, B.G. The E3 ubiquitin ligase mind bomb 1 ubiquitinates and promotes the degradation of survival of motor neuron protein. Mol. Boil. Cell 2013, 24, 1863–1871. [Google Scholar] [CrossRef] [PubMed]
- Bendotti, C.; Marino, M.; Cheroni, C.; Fontana, E.; Crippa, V.; Poletti, A.; De Biasi, S. Dysfunction of constitutive and inducible ubiquitin-proteasome system in amyotrophic lateral sclerosis: Implication for protein aggregation and immune response. Prog. Neurobiol. 2012, 97, 101–126. [Google Scholar] [CrossRef] [PubMed]
- Wadosky, K.M.; Li, L.; Bs, J.E.R.; Min, J.-N.; Bogan, D.; Bs, J.G.; Patterson, C.; Kornegay, J.N.; Willis, M.S. Regulation of the calpain and ubiquitin-proteasome systems in a canine model of muscular dystrophy. Muscle Nerve 2011, 44, 553–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nyamsuren, O.; Faggionato, D.; Loch, W.; Schulze, E.; Baumeister, R. A mutation in CHN-1/CHIP suppresses muscle degeneration in Caenorhabditis elegans. Dev. Biol. 2007, 312, 193–202. [Google Scholar] [CrossRef] [Green Version]
- Ballinger, C.A.; Connell, P.; Wu, Y.; Hu, Z.; Thompson, L.J.; Yin, L.-Y.; Patterson, C. Identification of CHIP, a Novel Tetratricopeptide Repeat-Containing Protein That Interacts with Heat Shock Proteins and Negatively Regulates Chaperone Functions. Mol. Cell. Boil. 1999, 19, 4535–4545. [Google Scholar] [CrossRef] [Green Version]
- Benini, M.; Fortuni, S.; Condò, I.; Alfedi, G.; Malisan, F.; Toschi, N.; Serio, D.; Massaro, D.S.; Arcuri, G.; Testi, R.; et al. E3 Ligase RNF126 Directly Ubiquitinates Frataxin, Promoting Its Degradation: Identification of a Potential Therapeutic Target for Friedreich Ataxia. Cell Rep. 2017, 18, 2007–2017. [Google Scholar] [CrossRef] [Green Version]
- Watabe, K.; Kato, Y.; Sakuma, M.; Murata, M.; Niida-Kawaguchi, M.; Takemura, T.; Hanagata, N.; Tada, M.; Kakita, A.; Shibata, N. Praja1 RING -finger E3 ubiquitin ligase suppresses neuronal cytoplasmic TDP -43 aggregate formation. Neuropathology 2020. [Google Scholar] [CrossRef]
- Saigoh, K.; Wang, Y.-L.; Suh, J.-G.; Yamanishi, T.; Sakai, Y.; Kiyosawa, H.; Harada, T.; Ichihara, N.; Wakana, S.; Kikuchi, T.; et al. Intragenic deletion in the gene encoding ubiquitin carboxy-terminal hydrolase in gad mice. Nat. Genet. 1999, 23, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Sugiura, Y.; Myers, K.G.; Liu, Y.; Lin, W. Ubiquitin carboxyl-terminal hydrolase L1 is required for maintaining the structure and function of the neuromuscular junction. Proc. Natl. Acad. Sci. USA 2010, 107, 1636–1641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genç, B.; Jara, J.H.; Schultz, M.C.; Manuel, M.; Stanford, M.J.; Gautam, M.; Klessner, J.L.; Sekerková, G.; Heller, D.B.; Cox, G.A.; et al. Absence of UCHL 1 function leads to selective motor neuropathy. Ann. Clin. Transl. Neurol. 2016, 3, 331–345. [Google Scholar] [CrossRef]
- Reinicke, A.T.; Laban, K.; Sachs, M.; Kraus, V.; Walden, M.; Damme, M.; Sachs, W.; Reichelt, J.; Schweizer, M.; Janiesch, P.C.; et al. Ubiquitin C-terminal hydrolase L1 (UCH-L1) loss causes neurodegeneration by altering protein turnover in the first postnatal weeks. Proc. Natl. Acad. Sci. USA 2019, 116, 7963–7972. [Google Scholar] [CrossRef] [Green Version]
- Ng, A.S.; Tan, Y.J.; Lu, Z.; Ng, E.Y.; Ng, S.Y.E.; Chia, N.S.Y.; Setiawan, F.; Xu, Z.; Keong, N.C.H.; Tay, K.Y.; et al. Plasma ubiquitin C-terminal hydrolase L1 levels reflect disease stage and motor severity in Parkinson’s disease. Aging 2020, 12, 1488–1495. [Google Scholar] [CrossRef]
- DiAntonio, A.; Haghighi, A.P.; Portman, S.L.; Lee, J.D.; Amaranto, A.M.; Goodman, C.S. Ubiquitination-dependent mechanisms regulate synaptic growth and function. Nature 2001, 412, 449–452. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.M.; Bhattacharyya, B.; Rachel, R.A.; Coppola, V.; Tessarollo, L.; Householder, D.B.; Fletcher, C.F.; Miller, R.J.; Copeland, N.G.; Jenkins, N.A. Synaptic defects in ataxia mice result from a mutation in Usp14, encoding a ubiquitin-specific protease. Nat. Genet. 2002, 32, 420–425. [Google Scholar] [CrossRef]
- Anderson, C.; Crimmins, S.; Wilson, J.A.; Korbel, G.A.; Ploegh, H.L.; Wilson, S.M. Loss of Usp14 results in reduced levels of ubiquitin in ataxia mice. J. Neurochem. 2005, 95, 724–731. [Google Scholar] [CrossRef]
- Chen, P.-C.; Qin, L.-N.; Li, X.-M.; Walters, B.J.; Wilson, J.A.; Mei, L.; Wilson, S.M. The proteasome-associated deubiquitinating enzyme Usp14 is essential for the maintenance of synaptic ubiquitin levels and the development of neuromuscular junctions. J. Neurosci. 2009, 29, 10909–10919. [Google Scholar] [CrossRef] [Green Version]
- Crimmins, S.; Jin, Y.; Wheeler, C.; Huffman, A.K.; Chapman, C.; Dobrunz, L.E.; Levey, A.; Roth, K.A.; Wilson, J.A.; Wilson, S.M. Transgenic Rescue of ataxia Mice with Neuronal-Specific Expression of Ubiquitin-Specific Protease 14. J. Neurosci. 2006, 26, 11423–11431. [Google Scholar] [CrossRef]
- Vaden, J.H.; Bhattacharyya, B.J.; Chen, P.-C.; A Watson, J.; Marshall, A.; Phillips, S.E.; A Wilson, J.; King, G.D.; Miller, R.J.; Wilson, S.M. Ubiquitin-specific protease 14 regulates c-Jun N-terminal kinase signaling at the neuromuscular junction. Mol. Neurodegener. 2015, 10, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liddell, E.G.T.; Sherrington, C.S. Recruitment and some other features of reflex inhibition. Proc. R. Soc. London. Ser. B, Boil. Sci. 1925, 97, 488–518. [Google Scholar] [CrossRef]
- Feinstein, B.; Lindegård, B.; Nyman, E.; Wohlfart, G. MORPHOLOGIC STUDIES OF MOTOR UNITS IN NORMAL HUMAN MUSCLES. Cells Tissues Organs 1955, 23, 127–142. [Google Scholar] [CrossRef]
- Ruiz, R.; Casañas, J.J.; Torres-Benito, L.; Cano, R.; Tabares, L. Altered Intracellular Ca2+ Homeostasis in Nerve Terminals of Severe Spinal Muscular Atrophy Mice. J. Neurosci. 2010, 30, 849–857. [Google Scholar] [CrossRef] [Green Version]
- Crawford, T.O.; Pardo, C.A. The Neurobiology of Childhood Spinal Muscular Atrophy. Neurobiol. Dis. 1996, 3, 97–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeGuise, M.-O.; Baranello, G.; Mastella, C.; Beauvais, A.; Michaud, J.; Leone, A.; De Amicis, R.; Battezzati, A.; Dunham, C.; Selby, K.; et al. Abnormal fatty acid metabolism is a core component of spinal muscular atrophy. Ann. Clin. Transl. Neurol. 2019, 6, 1519–1532. [Google Scholar] [CrossRef] [Green Version]
- Lefebvre, S.; Burglen, L.; Reboullet, S.; Clermont, O.; Burlet, P.; Viollet, L.; Bénichou, B.; Cruaud, C.; Millasseau, P.; Zeviani, M.; et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Bergin, A.; Kim, G.; Price, D.L.; Sisodia, S.S.; Lee, M.; Rabin, B.A. Identification and characterization of a mouse homologue of the Spinal Muscular Atrophy-determining gene, survival motor neuron. Gene 1997, 204, 47–53. [Google Scholar] [CrossRef]
- Le, T.T.; Pham, L.T.; Butchbach, M.E.; Zhang, H.L.; Monani, U.R.; Coovert, D.D.; Gavrilina, T.O.; Xing, L.; Bassell, G.J.; Burghes, A.H.M. SMNΔ7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum. Mol. Genet. 2005, 14, 845–857. [Google Scholar] [CrossRef]
- Burghes, A.H.M.; Beattie, C.E. Spinal muscular atrophy: Why do low levels of survival motor neuron protein make motor neurons sick? Nat. Rev. Neurosci. 2009, 10, 597–609. [Google Scholar] [CrossRef] [Green Version]
- Ackermann, B.; Kröber, S.; Torres-Benito, L.; Borgmann, A.; Peters, M.; Barkooie, S.M.H.; Tejero, R.; Jakubik, M.; Schreml, J.; Milbradt, J.; et al. Plastin 3 ameliorates spinal muscular atrophy via delayed axon pruning and improves neuromuscular junction functionality. Hum. Mol. Genet. 2012, 22, 1328–1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oprea, G.E.; Kröber, S.; McWhorter, M.L.; Rossol, W.; Müller, S.; Krawczak, M.; Bassell, G.J.; Beattie, C.E.; Wirth, B. Plastin 3 Is a Protective Modifier of Autosomal Recessive Spinal Muscular Atrophy. Science 2008, 320, 524–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ning, K.; Drepper, C.; Valori, C.F.; Ahsan, M.; Wyles, M.; Higginbottom, A.; Herrmann, T.; Shaw, C.E.; Azzouz, M.; Sendtner, M. PTEN depletion rescues axonal growth defect and improves survival in SMN-deficient motor neurons. Hum. Mol. Genet. 2010, 19, 3159–3168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, G.; Dury, A.Y.; Murray, L.M.; Biondi, O.; Tadesse, H.; El Fatimy, R.; Kothary, R.K.; Charbonnier, F.; Khandjian, E.W.; Côté, J. A novel function for the survival motoneuron protein as a translational regulator. Hum. Mol. Genet. 2012, 22, 668–684. [Google Scholar] [CrossRef] [Green Version]
- Eshraghi, M.; Gombar, R.; De Repentigny, Y.; Vacratsis, P.O.; Kothary, R. Pathologic Alterations in the Proteome of Synaptosomes from a Mouse Model of Spinal Muscular Atrophy. J. Proteome Res. 2019, 18, 3042–3051. [Google Scholar] [CrossRef]
- Chaytow, H.; Huang, Y.-T.; Gillingwater, T.H.; Faller, K. The role of survival motor neuron protein (SMN) in protein homeostasis. Cell. Mol. Life Sci. 2018, 75, 3877–3894. [Google Scholar] [CrossRef] [Green Version]
- Hicke, L. Protein regulation by monoubiquitin. Nat. Rev. Mol. Cell Boil. 2001, 2, 195–201. [Google Scholar] [CrossRef]
- Chang, H.-C.; Hung, W.-C.; Chuang, Y.-J.; Jong, Y.-J. Degradation of survival motor neuron (SMN) protein is mediated via the ubiquitin/proteasome pathway. Neurochem. Int. 2004, 45, 1107–1112. [Google Scholar] [CrossRef]
- Abera, M.B.; Xiao, J.; Nofziger, J.H.; Titus, S.; Southall, N.; Zheng, W.; Moritz, K.E.; Ferrer, M.; Cherry, J.J.; Androphy, E.J.; et al. ML372 blocks SMN ubiquitination and improves spinal muscular atrophy pathology in mice. JCI Insight 2016, 1, 88427. [Google Scholar] [CrossRef] [Green Version]
- Wishart, T.M.; Mutsaers, C.A.; Riessland, M.; Reimer, M.M.; Hunter, G.; Hannam, M.L.; Eaton, S.L.; Fuller, H.R.; Roche, S.L.; Somers, E.; et al. Dysregulation of ubiquitin homeostasis and beta-catenin signaling promote spinal muscular atrophy. J. Clin. Investig. 2014, 124, 1821–1834. [Google Scholar] [CrossRef] [Green Version]
- Han, K.-J.; Foster, D.G.; Zhang, N.-Y.; Kanisha, K.; Dzieciatkowska, M.; Sclafani, R.A.; Hansen, K.C.; Peng, J.; Liu, C.-W. Ubiquitin-specific Protease 9x Deubiquitinates and Stabilizes the Spinal Muscular Atrophy Protein-Survival Motor Neuron. J. Boil. Chem. 2012, 287, 43741–43752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Upadhyay, V.; Katz, M.; Kiernan, M.; Henderson, R. Review of the revised amyotrophic lateral sclerosis diagnostic criteria. Clin. Neurophysiol. 2020, 131, 1767–1768. [Google Scholar] [CrossRef] [PubMed]
- Riva, N.; Clarelli, F.; Domi, T.; Cerri, F.; Gallia, F.; Trimarco, A.; Brambilla, P.; Lunetta, C.; Lazzerini, A.; Lauria, G.; et al. Unraveling gene expression profiles in peripheral motor nerve from amyotrophic lateral sclerosis patients: Insights into pathogenesis. Sci. Rep. 2016, 6, 39297. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, E.C.; Shah, N.; Gomez, M.; Casalena, G.; Zhao, D.; Kenny, T.C.; Guariglia, S.R.; Manfredi, G.; Germain, D. Proteasome mapping reveals sexual dimorphism in tissue-specific sensitivity to protein aggregations. EMBO Rep. 2020, 21, 48978. [Google Scholar] [CrossRef]
- Blasco, H.; Mavel, S.; Corcia, P.; Gordon, P.H. The glutamate hypothesis in ALS: Pathophysiology and drug development. Curr. Med. Chem. 2014, 21, 3551–3575. [Google Scholar] [CrossRef]
- Haase, G.; Rabouille, C. Golgi Fragmentation in ALS Motor Neurons. New Mechanisms Targeting Microtubules, Tethers, and Transport Vesicles. Front. Mol. Neurosci. 2015, 9, 448. [Google Scholar] [CrossRef]
- Renaud, L.; Picher-Martel, V.; Codron, P.; Julien, J.-P. Key role of UBQLN2 in pathogenesis of amyotrophic lateral sclerosis and frontotemporal dementia. Acta Neuropathol. Commun. 2019, 7, 1–11. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [Green Version]
- Ghasemi, M.; Brown, R.H. Genetics of Amyotrophic Lateral Sclerosis. Cold Spring Harb. Perspect. Med. 2017, 8, a024125. [Google Scholar] [CrossRef]
- Sau, D.; De Biasi, S.; Vitellaro-Zuccarello, L.; Riso, P.; Guarnieri, S.; Porrini, M.; Simeoni, S.; Crippa, V.; Onesto, E.; Palazzolo, I.; et al. Mutation of SOD1 in ALS: A gain of a loss of function. Hum. Mol. Genet. 2007, 16, 1604–1618. [Google Scholar] [CrossRef] [Green Version]
- Rosen, D.R. Mutations in cu/zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 364, 362. [Google Scholar] [CrossRef]
- Niwa, J.-I.; Ishigaki, S.; Hishikawa, N.; Yamamoto, M.; Murata, S.; Tanaka, K.; Sobue, G.; Doyu, M.; Taniguchi, N. Dorfin Ubiquitylates Mutant SOD1 and Prevents Mutant SOD1-mediated Neurotoxicity. J. Boil. Chem. 2002, 277, 36793–36798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meacham, G.C.; Patterson, C.; Zhang, W.; Younger, J.M.; Cyr, D.M. The Hsc70 co-chaperone CHIP targets immature CFTR for proteasomal degradation. Nature 2000, 3, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Ying, Z.; Wang, H.; Fan, H.; Zhu, X.; Zhou, J.; Fei, E.; Wang, G. Gp78, an ER associated E3, promotes SOD1 and ataxin-3 degradation. Hum. Mol. Genet. 2009, 18, 4268–4281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazaki, K.; Fujita, T.; Ozaki, T.; Kato, C.; Kurose, Y.; Sakamoto, M.; Kato, S.; Goto, T.; Itoyama, Y.; Aoki, M.; et al. NEDL1, a Novel Ubiquitin-protein Isopeptide Ligase for Dishevelled-1, Targets Mutant Superoxide Dismutase-1. J. Boil. Chem. 2003, 279, 11327–11335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cozzolino, M.; Pesaresi, M.G.; Amori, I.; Crosio, C.; Ferri, A.; Nencini, M.; Carrı, M.T. Oligomerization of Mutant SOD1 in Mitochondria of Motoneuronal Cells Drives Mitochondrial Damage and Cell Toxicity. Antioxidants Redox Signal. 2009, 11, 1547–1558. [Google Scholar] [CrossRef] [Green Version]
- Halloran, M.; Ragagnin, A.M.G.; Vidal, M.; Parakh, S.; Yang, S.; Heng, B.; Grima, N.; Shahheydari, H.; Soo, K.-Y.; Blair, I.; et al. Amyotrophic lateral sclerosis-linked UBQLN2 mutants inhibit endoplasmic reticulum to Golgi transport, leading to Golgi fragmentation and ER stress. Cell. Mol. Life Sci. 2019, 1–15. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [Green Version]
- Scharnagl, H.; März, W.; Böhm, M.; Luger, T.A.; Fracassi, F.; Diana, A.; Frieling, T.; Mac, S.; Donoghue, D.; Kohl, S.; et al. Amyotrophic Lateral Sclerosis. Encycl. Mol. Mech. Dis. 2009, 3, 78–79. [Google Scholar] [CrossRef]
- Suk, T.R.; Rousseaux, M.W.C. The role of TDP-43 mislocalization in amyotrophic lateral sclerosis. Mol. Neurodegener. 2020, 15, 1–16. [Google Scholar] [CrossRef]
- Goodier, J.L.; Soares, A.O.; Pereira, G.C.; Devine, L.R.; Sanchez, L.; Cole, R.N.; García-Pérez, J.L. C9orf72-associated SMCR8 protein binds in the ubiquitin pathway and with proteins linked with neurological disease. Acta Neuropathol. Commun. 2020, 8, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Sunderland, S. THE INTRANEURAL TOPOGRAPHY OF THE RADIAL, MEDIAN AND ULNAR NERVES. Brain 1945, 68, 243–298. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.; Olzmann, J.A.; Chin, L.-S.; Li, L. Mutations associated with Charcot-Marie-Tooth disease cause SIMPLE protein mislocalization and degradation by the proteasome and aggresome-autophagy pathways. J. Cell Sci. 2011, 124, 3319–3331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barisic, N.; Claeys, K.G.; Löfgren, A.; Nelis, E.; De Jonghe, P.; Timmerman, V.; Sirotković-Skerlev, M. Charcot-Marie-Tooth Disease: A Clinico-genetic Confrontation. Ann. Hum. Genet. 2008, 72, 416–441. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Bazick, H.; Chittoor-Vinod, V.; Al Salihi, M.O.; Xia, G.; Notterpek, L. Elevated Peripheral Myelin Protein 22, Reduced Mitotic Potential, and Proteasome Impairment in Dermal Fibroblasts from Charcot-Marie-Tooth Disease Type 1A Patients. Am. J. Pathol. 2018, 188, 728–738. [Google Scholar] [CrossRef] [Green Version]
- Naef, R.; Suter, U. Many facets of the peripheral myelin protein PMP22 in myelination and disease. Microsc. Res. Tech. 1998, 41, 359–371. [Google Scholar] [CrossRef]
- Zhou, Y.; Borchelt, D.; Bauson, J.C.; Fazio, S.; Miles, J.R.; Tavori, H.; Notterpek, L. Subcellular diversion of cholesterol by gain- and loss-of-function mutations in PMP22. Glia 2020. [Google Scholar] [CrossRef]
- Ryan, M.C.; Shooter, E.M.; Notterpek, L. Aggresome formation in neuropathy models based on peripheral myelin protein 22 mutations. Neurobiol. Dis. 2002, 10, 109–118. [Google Scholar] [CrossRef] [Green Version]
- Fortun, J.; Li, J.; Go, J.; Fenstermaker, A.; Fletcher, B.S.; Notterpek, L. Impaired proteasome activity and accumulation of ubiquitinated substrates in a hereditary neuropathy model. J. Neurochem. 2005, 92, 1531–1541. [Google Scholar] [CrossRef]
- Nishimura, T.; Yoshikawa, H.; Fujimura, H.; Sakoda, S.; Yanagihara, T. Accumulation of peripheral myelin protein 22 in onion bulbs and Schwann cells of biopsied nerves from patients with Charcot-Marie-Tooth disease type 1A. Acta Neuropathol. 1996, 92, 454–460. [Google Scholar] [CrossRef]
- Madorsky, I.; Opalach, K.; Waber, A.; Verrier, J.D.; Solmo, C.; Foster, T.; Dunn, W.A.; Notterpek, L. Intermittent fasting alleviates the neuropathic phenotype in a mouse model of Charcot-Marie-Tooth disease. Neurobiol. Dis. 2009, 34, 146–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuanalo-Contreras, K.; Mukherjee, A.; Soto, C. Role of Protein Misfolding and Proteostasis Deficiency in Protein Misfolding Diseases and Aging. Int. J. Cell Boil. 2013, 2013, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chandran, V.; Gao, K.; Swarup, V.; Versano, R.; Dong, H.; Jordan, M.C.; Geschwind, D. Inducible and reversible phenotypes in a novel mouse model of Friedreich’s Ataxia. eLife 2017, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Rufini, A.; Cavallo, F.; Condò, I.; Fortuni, S.; De Martino, G.; Incani, O.; Di Venere, A.; Benini, M.; Massaro, D.S.; Arcuri, G.; et al. Highly specific ubiquitin-competing molecules effectively promote frataxin accumulation and partially rescue the aconitase defect in Friedreich ataxia cells. Neurobiol. Dis. 2014, 75, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, V.; Montermini, L.; Moltó, M.D.; Pianese, L.; Cossee, M.; Cavalcanti, F.; Monros, E.; Rodius, F.; Duclos, F.; Monticelli, A.; et al. Friedreich’s Ataxia: Autosomal Recessive Disease Caused by an Intronic GAA Triplet Repeat Expansion. Science 1996, 271, 1423–1427. [Google Scholar] [CrossRef] [PubMed]
- Yandim, C.; Natisvili, T.; Festenstein, R. Gene regulation and epigenetics in Friedreich’s ataxia. J. Neurochem. 2013, 126, 21–42. [Google Scholar] [CrossRef]
- Parkinson, M.H.; Boesch, S.; Nachbauer, W.; Mariotti, C.; Giunti, P. Clinical features of Friedreich’s ataxia: Classical and atypical phenotypes. J. Neurochem. 2013, 126, 103–117. [Google Scholar] [CrossRef]
- Anzovino, A.; Lane, D.J.R.; Huang, M.L.-H.; Richardson, D.R. Fixing frataxin: ‘ironing out’ the metabolic defect in Friedreich’s ataxia. Br. J. Pharmacol. 2014, 171, 2174–2190. [Google Scholar] [CrossRef] [Green Version]
- Pastore, A.; Puccio, H. Frataxin: A protein in search for a function. J. Neurochem. 2013, 126, 43–52. [Google Scholar] [CrossRef]
- Koeppen, A.H.; Kuntzsch, E.C.; Bjork, S.T.; Ramirez, R.L.; Mazurkiewicz, J.E.; Feustel, P.J. Friedreich ataxia: Metal dysmetabolism in dorsal root ganglia. Acta Neuropathol. Commun. 2013, 1, 26. [Google Scholar] [CrossRef]
- Tamarit, J.; Obis, È.; Ros, J. Oxidative stress and altered lipid metabolism in Friedreich ataxia. Free. Radic. Boil. Med. 2016, 100, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Turchi, R.; Tortolici, F.; Guidobaldi, G.; Iacovelli, F.; Falconi, M.; Rufini, S.; Faraonio, R.; Casagrande, V.; Federici, M.; De Angelis, L.; et al. Frataxin deficiency induces lipid accumulation and affects thermogenesis in brown adipose tissue. Cell Death Dis. 2020, 11, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abeti, R.; Parkinson, M.H.; Hargreaves, I.P.; Angelova, P.R.; Sandi, C.; A Pook, M.; Giunti, P.; Abramov, A.Y. Mitochondrial energy imbalance and lipid peroxidation cause cell death in Friedreich’s ataxia. Cell Death Dis. 2016, 7, e2237. [Google Scholar] [CrossRef] [PubMed]
- Condò, I.; Ventura, N.; Malisan, F.; Rufini, A.; Tomassini, B.; Testi, R. In vivo maturation of human frataxin. Hum. Mol. Genet. 2007, 16, 1534–1540. [Google Scholar] [CrossRef] [Green Version]
- Koutnikova, H.; Campuzano, V.; Koenig, M. Maturation of wild-type and mutated frataxin by the mitochondrial processing peptidase. Hum. Mol. Genet. 1998, 7, 1485–1489. [Google Scholar] [CrossRef]
- Rufini, A.; Fortuni, S.; Arcuri, G.; Condò, I.; Serio, D.; Incani, O.; Malisan, F.; Ventura, N.; Testi, R. Preventing the ubiquitin–proteasome-dependent degradation of frataxin, the protein defective in Friedreich’s ataxia. Hum. Mol. Genet. 2011, 20, 1253–1261. [Google Scholar] [CrossRef]
- Nabhan, J.F.; Gooch, R.L.; Chekler, E.L.P.; Pierce, B.; Bulawa, C.E. Perturbation of cellular proteostasis networks identifies pathways that modulate precursor and intermediate but not mature levels of frataxin. Sci. Rep. 2015, 5, 18251. [Google Scholar] [CrossRef]
- Chen, P.-C.; Bhattacharyya, B.J.; Hanna, J.; Minkel, H.; Wilson, J.A.; Finley, D.; Miller, R.J.; Wilson, S.M. Ubiquitin homeostasis is critical for synaptic development and function. J. Neurosci. 2011, 31, 17505–17513. [Google Scholar] [CrossRef] [Green Version]
- Kowalski, J.R.; Juo, P. The Role of Deubiquitinating Enzymes in Synaptic Function and Nervous System Diseases. Neural Plast. 2012, 2012, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Vincent, A.; Palace, J.; Hilton-Jones, D. Myasthenia gravis. Lancet 2001, 357, 2122–2128. [Google Scholar] [CrossRef]
- Flanigan, K.M. Duchenne and Becker Muscular Dystrophies. Neurol. Clin. 2014, 32, 671–688. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Ms, C.S.; Leslie, N.D.; Flanigan, K.M.; Al-Dahhak, R.; Gastier-Foster, J.; Kneile, K.; Dunn, D.M.; Duval, B.; Bs, A.A.; et al. Evidence-based path to newborn screening for duchenne muscular dystrophy. Ann. Neurol. 2012, 71, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Blake, D.J.; Kröger, S. The neurobiology of Duchenne muscular dystrophy: Learning lessons from muscle? Trends Neurosci. 2000, 23, 92–99. [Google Scholar] [CrossRef]
- Bladen, C.L.; Salgado, D.; Monges, S.; Foncuberta, M.E.; Kekou, K.; Kosma, K.; Dawkins, H.; Lamont, L.; Roy, A.J.; Chamova, T.; et al. The TREAT-NMD DMD Global Database: Analysis of More than 7000 Duchenne Muscular Dystrophy Mutations. Hum. Mutat. 2015, 36, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Rybakova, I.N.; Patel, J.R.; Ervasti, J.M. The Dystrophin Complex Forms a Mechanically Strong Link between the Sarcolemma and Costameric Actin. J. Cell Boil. 2000, 150, 1209–1214. [Google Scholar] [CrossRef]
- Kumamoto, T.; Fujimoto, S.; Ito, T.; Horinouchi, H.; Ueyama, H.; Tsuda, T. Proteasome expression in the skeletal muscles of patients with muscular dystrophy. Acta Neuropathol. 2000, 100, 595–602. [Google Scholar] [CrossRef]
- Hammers, D.W.; Hart, C.C.; Patsalos, A.; Matheny, M.K.; Wright, L.A.; Nagy, L.; Sweeney, H.L. Glucocorticoids counteract hypertrophic effects of myostatin inhibition in dystrophic muscle. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [Green Version]
- Gazzerro, E.; Assereto, S.; Bonetto, A.; Sotgia, F.; Scarfì, S.; Pistorio, A.; Bonuccelli, G.; Cilli, M.; Bruno, C.; Zara, F.; et al. Therapeutic Potential of Proteasome Inhibition in Duchenne and Becker Muscular Dystrophies. Am. J. Pathol. 2010, 176, 1863–1877. [Google Scholar] [CrossRef] [Green Version]
- Bonuccelli, G.; Sotgia, F.; Capozza, F.; Gazzerro, E.; Minetti, C.; Lisanti, M.P. Localized Treatment with a Novel FDA-Approved Proteasome Inhibitor Blocks the Degradation of Dystrophin and Dystrophin-Associated Proteins in mdx Mice. Cell Cycle 2007, 6, 1242–1248. [Google Scholar] [CrossRef] [Green Version]
- McCourt, J.L.; Talsness, D.M.; Lindsay, A.; Arpke, R.W.; Chatterton, P.D.; Nelson, D.M.; Chamberlain, C.M.; Olthoff, J.T.; Belanto, J.J.; McCourt, P.M.; et al. Mouse models of two missense mutations in actin-binding domain 1 of dystrophin associated with Duchenne or Becker muscular dystrophy. Hum. Mol. Genet. 2018, 27, 451–462. [Google Scholar] [CrossRef] [Green Version]
- Udd, B.; Krahe, R. The myotonic dystrophies: Molecular, clinical, and therapeutic challenges. Lancet Neurol. 2012, 11, 891–905. [Google Scholar] [CrossRef]
- Vignaud, A.; Ferry, A.; Huguet, A.; Baraibar, M.A.; Trollet, C.; Hyzewicz, J.; Butler-Browne, G.; Puymirat, J.; Gourdon, G.; Furling, D. Progressive skeletal muscle weakness in transgenic mice expressing CTG expansions is associated with the activation of the ubiquitin–proteasome pathway. Neuromuscul. Disord. 2010, 20, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Lemmers, R.J.L.F.; Van Der Vliet, P.J.; Klooster, R.; Sacconi, S.; Camaño, P.; Dauwerse, J.G.; Snider, L.; Straasheijm, K.R.; Van Ommen, G.J.; Padberg, G.W.; et al. A Unifying Genetic Model for Facioscapulohumeral Muscular Dystrophy. Science 2010, 329, 1650–1653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homma, S.; Beermann, M.L.; Boyce, F.M.; Miller, J.B. Expression of FSHD-related DUX4-FL alters proteostasis and induces TDP-43 aggregation. Ann. Clin. Transl. Neurol. 2015, 2, 151–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Proteins | Organism | Function and Molecular Consequences Linked to the Dysregulation | Neuromuscular disorders and Motor Impairments | Refs. |
|---|---|---|---|---|
| Ubiquitin-Activating Enzymes (E1S) | ||||
| Uba1 | Mus musculus | Modulation of ubiquitin homeostasis and regulation of sensory motor connectivity. Its mutation causes the degeneration of lower motor neurons in the anterior horn of the spinal cord. | Spinal muscular atrophy (SMA) and X-linked SMA | [36,37] |
| Ubiquitin-Conjugating Enzymes (E2S) | ||||
| Ubc-25 | C. elegans | Maintenance of neuromuscular functions. The loss of function induces uncoordinated mode of locomotion. | Severe signs of a progressive paralysis | [38] |
| E2e1 | Mus Musculus | Control of contractile and metabolic properties of the skeletal muscles through interaction with MuRF1 and telethonin. Its knockdown aggravates the atrophying process in Dex-treated mice. | Muscle atrophy | [33] |
| Ubiquitin Ligases (E3S) | ||||
| Apc | C. elegans D. melanogaster | Regulation of axonal morphogenesis, synaptic size, and activity. The loss of function increases muscle excitation at the neuromuscular junction (NMJ). | Convulsions | [39,40] |
| Herc1 | Mus musculus | Important in motor function, neuromuscular transmission and peripheral myelination. Gly483Glu substitution induces protein overexpression that alters the NMJ structure. | Severe ataxia, uncoordinated gait, irregular hindlimb posture, and trembling. Decreased synaptic release, altered non-myelinating Schwann cells at NMJ, and anomalous myelination | [41,42] |
| Hiw Rpm-1 Phr1 | D. melanogaster C.elegans Mus musculus | Presynaptic regulators of synapse formation and growth. The loss of function increases the number of NMJ boutons, organizes the presynaptic terminals at GABAergic NMJs, and a sprouting of nerve terminals. | Synaptic release defects and altered NMJ development | [43,44,45,46,47,48] |
| Murf1 | Mus musculus | Regulation of muscle protein degradation by the UPS. The gene deletion is involved in the resistance to atrophy. | Muscle atrophy | [49,50,51] |
| Mafbx | Mus musculus | Important in muscle maintenance. Its overexpression in myotubes is related to atrophy. | Muscle atrophy | [49] |
| Nedd4 | D. melanogaster Mus musculus | Regulation of formation and function of the NMJ. In flies, its overexpression causes defects in backward innervation and increases the number of nerve branches. In mice, the deficient mutant show aberrant innervation patterns and structure of the nerve terminals. | Flies: Abnormal larval locomotion Mice: premature embryonic lethality | [52,53,54,55] |
| Pdzrn3 | Mus musculus | Regulation of the expression of muscle-specific receptor tyrosine kinase (MuSK), an organizer of postsynaptic development at the NMJ. Its overexpression reduces MuSK expression, leading to a reduction in the NMJ size. | Defects in the growth and maturation of the neuromuscular junction | [15] |
| Cullin-3 | Mus musculus | Muscle protein breakdown. Its absence is characterized by a decreased neddylation and polyubiquitylation, as well as by an accumulation of non-muscular α-actinins in muscles, altering the normal development of the NMJ. | Nemaline myopathies | [56] |
| Gigaxonin | Danio rerio | Involved in the decision of neuronal and muscular fate in vertebrates. Its repression impaired motor neuron specification and somitogenesis, and suppressed NMJ formation and locomotion | Giant axonal neuropathy | [57] |
| UBE3A E6-AP | Mus musculus Homo sapiens Rattus | Ubiquitin ligase and transcriptional coactivator. Its deletion induces delayed development of reflexes, motor deficiencies. And fine motor skills. | Angelman syndrome | [58,59] |
| TRIM32 TRIM75 | Homo sapiens Mus musculus | Involved in the control of myogenesis. Reduced levels or abnormal functionality leads to loss of ubiquitination and accumulation of tripartite motif-containing protein (TRIM) substrates within the muscle fibers. | Altered myogenesis, premature senescence of skeletal muscle, reduced proliferation, and differentiation of myoblasts; Duchenne muscular dystrophy | [60,61,62,63] |
| Mib1 | C. elegans | Interaction and ubiquitination of survival of motorneuron protein (SMN), facilitating its degradation. It induces modifications in the UBA1 levels, knocks down Mib1 orthologues, and improves neuromuscular function in an motorneuron-deficient C. elegans model. | Abnormal larval locomotion | [64] |
| Dorfin Chip Gp78 Trapd-Nedl1 Mitol | Mus musculus | They interact and ubiquitinate mutated SOD1 proteins, leading to their degradation. The expression of these ligases is associated to a protective function. | Amyotrophic lateral sclerosis | [65] |
| Mdm2 | Canis lupus familiaris | Apoptotic inhibition by targeting p53 for degradation by the proteasome. Decreased levels in the left ventricle are related to decreased proteasome activity. | Duchenne muscular dystrophy | [66] |
| Chn-1/CHIP | C.elegans | Critical role in the ubiquitylation in the control of muscle wasting and degeneration. Its deletion decelerates the progression of muscular dystrophy. | Duchenne muscular dystrophy | [67,68] |
| Ubqln2 | Mus musculus | Member of the ubiquitin-like protein family involved in proteasome degradation. Inclusions of mutant UBQLN2 appear in ALS patients. | Amyotrophic lateral sclerosis | [5] |
| RNF126 | Homo sapiens | It participates in the ubiquitination of frataxin for degradation. Knockdown of RNF126 induces frataxin accumulation. | Friedreich ataxia | [69] |
| Praja1 (PJA1) | Rattus | It controls phosphorylation and proteosomal degradation of TDP-43. Possible mechanism for the prevention of ALS is linked to the ability to conjugate and prevent the formation of TDP-43 aggregates. | Amyotrophic lateral sclerosis | [70] |
| Deubiquitinating Enzymes (DUBs) | ||||
| UCH-L1 | Mus musculus Homo sapiens | Processing of ubiquitin precursors and ubiquitinated proteins. Its absence impairs the synaptic transmission at the NMJ, inducing profound structural defects at the presynaptic nerve terminals and denervation of the muscles. | Gracile axonal dystrophy and neurodegeneration of the peripheral nervous system | [71,72,73,74,75] |
| Fat facets (faf) | D. melanogaster | It antagonizes ubiquitin-mediated proteolysis, preventing protein degradation and controlling synapse development. Its overexpression increases the number of synaptic boutons, re-elaborates the synaptic branching pattern, and disrupts the synaptic function. | Defects in the synaptic transmission at the neuromuscular junction | [76] |
| Usp14 | Mus musculus | Crucial for synaptic development and function at NMJ. Its catalytically inactive form causes developmental deficits in the NMJ structure and synaptic transmission. Its loss causes presynaptic defects. | Severe tremors, hind limb paralysis, and postnatal lethality | [77,78,79,80,81] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bachiller, S.; Alonso-Bellido, I.M.; Real, L.M.; Pérez-Villegas, E.M.; Venero, J.L.; Deierborg, T.; Armengol, J.Á.; Ruiz, R. The Ubiquitin Proteasome System in Neuromuscular Disorders: Moving Beyond Movement. Int. J. Mol. Sci. 2020, 21, 6429. https://doi.org/10.3390/ijms21176429
Bachiller S, Alonso-Bellido IM, Real LM, Pérez-Villegas EM, Venero JL, Deierborg T, Armengol JÁ, Ruiz R. The Ubiquitin Proteasome System in Neuromuscular Disorders: Moving Beyond Movement. International Journal of Molecular Sciences. 2020; 21(17):6429. https://doi.org/10.3390/ijms21176429
Chicago/Turabian StyleBachiller, Sara, Isabel M. Alonso-Bellido, Luis Miguel Real, Eva María Pérez-Villegas, José Luis Venero, Tomas Deierborg, José Ángel Armengol, and Rocío Ruiz. 2020. "The Ubiquitin Proteasome System in Neuromuscular Disorders: Moving Beyond Movement" International Journal of Molecular Sciences 21, no. 17: 6429. https://doi.org/10.3390/ijms21176429
APA StyleBachiller, S., Alonso-Bellido, I. M., Real, L. M., Pérez-Villegas, E. M., Venero, J. L., Deierborg, T., Armengol, J. Á., & Ruiz, R. (2020). The Ubiquitin Proteasome System in Neuromuscular Disorders: Moving Beyond Movement. International Journal of Molecular Sciences, 21(17), 6429. https://doi.org/10.3390/ijms21176429