1. Introduction
Multiple sclerosis (MS) is a chronic, inflammatory, demyelinating, autoimmune disease of the central nervous system (CNS), which affects more than 2.3 million people worldwide, mostly young adults. The pathological features of MS include the formation of plaques (lesions) in the brain and spinal cord, where inflammation, demyelination, gliosis (scarring), axonal injury, and axonal loss occur [
1]. Although it is not considered as a fatal disease, it is associated with lower life expectancy and disability that affects the quality of life [
2]. A new therapeutic paradigm for the treatment of MS was introduced in 2010 and featured a modulation of the sphingosine 1-phosphate receptor 1 (S1P
1) signaling by using fingolimod (FTY720, Gilenya
®). Fingolimod desensitizes S1P
1 in T cells, blocking their egress from lymph nodes and leading to lymphopenia [
3]. After the approval of fingolimod, which is a non-selective S1PR modulator, the search for structures with better selectivity, potency, pharmacokinetic properties, effectiveness for other autoimmune and inflammatory diseases, and fewer adverse effects was boosted [
4]. At the beginning of 2020, two additional drugs, siponimod (BAF312, Mayzent
®) and ozanimod (RPC-1063, Zeposia
®), were approved by the European Medicines Agency for oral treatment of patients with relapsing–remitting MS (ozanimod) or the active secondary progressive form of MS (siponimod). Interestingly, siponimod and ozanimod, compared to fingolimod, exhibit selective affinity for S1P
1 and S1P
5, leading to a lower risk of adverse events [
5], which proves the rationale for selective targeting of the S1P receptors.
The five S1P receptor subtypes belong to the family of G-protein-coupled receptors (GPCRs), which are present on the cell surface where they bind with high affinity to secreted S1P and mediate an “inside-out” signaling [
6]. Versatile cellular responses are elicited upon binding of the receptors with S1P as their cognate ligand, including proliferation, survival, invasion, adhesion, and migration, pertaining to the indispensability of S1P for the normal functioning of the cardiovascular, nervous, immune, and other systems [
4]. S1P and S1P receptors also regulate pathophysiological processes, including inflammatory mediator synthesis, tissue remodeling, and angiogenesis, contributing to the pathogenesis of various diseases; thus, they may serve as targets for prevention or treatment [
4].
S1P
1, S1P
2, and S1P
3 are ubiquitously expressed, while S1P
4 and S1P
5 have a restricted expression in the immune compartments and in the CNS, respectively. Lack of S1P
2/3/4 or S1P
5 in mice does not produce an apparent phenotype; however, lack of S1P
1 in mice results in embryonic lethality due to an inefficient process of vascular maturation and consequent hemorrhage, pointing to a non-redundant role of S1P
1 in the vascular system [
7]. S1P
1 also plays a significant physiological role as a key regulator of lymphocyte trafficking, and the main pharmacodynamic effect of fingolimod and siponimod lays in their ability to induce a persistent internalization of S1P
1 in lymphocytes and subsequent lymphopenia [
8].
Critical for the lymphocyte trafficking is a S1P gradient, which exists among the plasma, the interstitial space of secondary lymphoid tissues, and the lymph, and which is sensed by the lymphocytes through S1P
1 [
9]. As they move from spaces with high to low S1P concentrations, their desensitized S1P
1 is recycled back to the cell surface and can become again responsive to S1P [
10]. Fingolimod and siponimod also induce S1P
1 internalization; however, in contrast to S1P, the receptor is irreversibly degraded in the proteasome and does not recycle back to the cell surface which reduces its membrane expression, a phenomenon known as “functional antagonism” [
11]. That way, the egress of lymphocytes, particularly naïve and central memory T cells, is hindered, while effector memory T cells are spared, resulting in an overall preferential reduction of the pathogenic immune responses and sparing large parts of protective immunity in multiple sclerosis patients [
12]. As autoaggressive T cells are involved in the pathology of many autoimmune diseases in addition to MS, such as Crohn’s disease, ulcerative colitis, and rheumatoid arthritis, a wider therapeutic use for S1PR modulators may be envisioned.
Apart from S1P
1, fingolimod also targets S1P
3, S1P
4, and S1P
5. While S1P
5 is thought to mediate beneficial effects in the CNS, S1P
3 is rather involved in adverse reactions’ signaling [
4], such as vasoconstrictive events and hypertension [
13]. The role of S1P
4 in physiological processes is still poorly understood and, therefore, the effects of fingolimod mediated by S1P
4 are vague. This highlights the need for developing compounds with a narrow S1PR selectivity profile which will allow tackling a specific pathology while having a more predictable spectrum of adverse reactions.
In this study, we present two novel compounds derived from fingolimod, ST-1893 and ST-1894, which chemically feature slight alterations in the polar head group by introducing a substituted morpholino ring (
Figure 1), and this results in a selectivity toward S1P
1. Both compounds were full agonists at S1P
1, although less potent than S1P, and they also exhibited functional antagonistic activity as shown by a sustained internalization in a cell culture of S1P
1-overexpressing Chinese hamster ovary (CHO)-K1 cells. In vivo, they exerted a lymphopenic effect in mice and were efficacious in an experimental antigen-induced encephalomyelitis (EAE) model, where they reduced the motor deficits, improved the body weight, and prevented the development of disease with the highest clinical score. Due to their S1P
1-selective nature, they possibly exhibit fewer adverse effects and have an advantage over fingolimod and siponimod, not only in MS but also in other autoimmune diseases.
2. Results
To evaluate the S1P receptor subtype activation profile of ST-1893 and ST-1894, CHO-K1 cells, that stably overexpress the different S1P receptor subtypes, were used, and phosphorylation of p42/p44- mitogen-activated protein kinase (MAPK) was measured as an early read-out of receptor activation [
14,
15]. Ten minutes of stimulation with ST-1893 or ST-1894 concentration-dependently increased phospho-p42/p44-MAPK in CHO-S1P
1, while none of the other receptors (S1P
2, 3, 4 or S1P
5) were activated (
Figure 2A).
We further studied whether ST-1893 and ST-1894 could act as antagonists. For this, cells were stimulated with a fixed concentration of S1P in the presence of increasing concentrations of either ST-1893 or ST-1894. However, no reduction of S1P-stimulated p42/p44-MAPK activation was detected, excluding a direct antagonistic effect of ST-1893 and ST-1894 (
Figure 2A). Furthermore, ST-1893- and ST-1894-stimulated p42/p44-MAPK activation was completely abolished by pretreatment with the competitive S1P
1 antagonist NIBR-0213 (
Figure 3A) [
16] and by pertussis toxin (
Figure 3B), thus confirming the reported signaling of S1P
1 and the involvement of G
i/o.
As lymphocytes traffic between the circulation and secondary lymphoid tissues, they encounter substantially different S1P concentrations, which are very high in the blood and low in the tissue. High concentrations of S1P are supposed to induce desensitization of S1P
1, and, as the concentration of S1P drops, S1P
1 is recycled back to the cell surface and becomes again sensitive to S1P [
10]. The initial phase of fingolimod-induced internalization resembles that of S1P; however, the subsequent mechanism is unique for fingolimod as it does not dissociate from the receptor, and the whole fingolimod–S1P
1 receptor complex is sorted for degradation, resulting in receptor depletion on the surface [
17]. Since fingolimod is known to induce sustained internalization and, thus, desensitization of S1P
1, described as functional antagonism, we further tested whether ST-1893 and ST-1894 exert the same functional antagonism. To this end, an in situ ELISA was established to detect cell surface-expressed Myc-tagged S1P
1. Stimulation of CHO-S1P
1 cells for 3 h, with either S1P or the ST compounds, resulted in a 50–75% reduction of cell surface S1P
1 (
Figure 4A). When cells were stimulated for 3 h and a subsequent washout period of 22 h, a complete recovery of S1P
1 in the S1P-stimulated group was observed, pointing to an efficient recycling process (
Figure 4B). Conversely, in ST-1893- or ST-1894-treated samples, cell surface S1P
1 remained low, suggesting a persistent internalization.
To further prove that sustained S1P
1 internalization also leads to a sustained downregulation of S1P
1 signaling, after the washout period, cells were re-stimulated with a new pulse of S1P for 10 min and taken for MAPK activation. Data showed that prolonged pre-treatment with ST-1893 or ST-1894, but not S1P, caused a downregulation of S1P signaling. As seen in
Figure 4C, S1P
1 is similarly activated in the S1P-treated group as in the control, suggesting that, within the 22 h of recovery period, S1P
1 was recycled to the cell surface and could be stimulated again by S1P. On the other hand, due to the long-term internalization and desensitization, without receptor recycling, ST-1893 and ST-1894 groups had decreased p42/p44-MAPK activation after a new pulse of S1P stimulation.
In a further step to analyze the binding of ST compounds to the ligand-binding pocket of S1P
1 and to determine the possibly interacting amino acids, a homologous S1P
1 model was developed on the basis of the X-ray structure [
18]. To generate an activated S1P
1 model, the adrenergic receptor was replaced by the homologous S1P
1 using an established β2-adrenergic receptor–arrestin complex (
Figure S1, Supplementary Materials). This approach allows the development of homologous models of activated GPCR proteins [
19,
20]. The ligands were charged by applying the molecular operating environment (MOE) program and docking experiments were performed. Indeed, both molecules, ST-1893 (
Figure S2A, Supplementary Materials) and ST-1894 (
Figure S2B, Supplementary Materials) fitted well into the binding pocket as shown in the two- (
Figure S2, Supplementary Materials) and the three-dimensional models (
Figure S3, Supplementary Materials). Moreover, the docking results and energy minimization calculations predicted an interaction with S1P
1 (
Table S1, Supplementary Materials). The morpholino-group of ST-1893 and ST-1894 and their hydroxymethyl substituents interacted with Lys-34 (H-donor), Tyr-29 (H-donor), Glu-121 (H-acceptor), and Glu-294 (H-acceptor). These interactions with specific amino-acid residues suggested that both ST-1893 and ST-1894 may exert S1P
1 agonist activity. The docking score for ST-1893 corresponds approximately to ST-1071 [
15]; however, due to the larger volume of ST-1893, we predicted that dissociation from the receptor would be more hampered, leading to a higher activity.
Interestingly, docking experiments and calculation of binding energies indicated that a more favorable interaction with S1P
1 occurs when both substances, ST-1893 and ST-1894, are phosphorylated (
Table S1, Supplementary Materials). The binding energy of ST-1893 increased from −112.80 kcal/mol of the non-phosphorylated form to −150.15 kcal/mol when the molecule was phosphorylated. In analogy, ST-1894 shows an enhanced binding energy of the phosphorylated molecule as there was an increase from −128.09 kcal/mol to −156.47 kcal/mol. FTY720-phosphate showed in the same docking experiments a binding energy of −141.98 kcal/mol. The oxygen and a carbon from the morpholino-group from ST-1893-phosphate and ST-1894-phosphate both interacted with Asn-101 (
Figure S2A,B, Supplementary Materials). The phosphorylated head of both compounds interacts with the amino acids Tyr-29 (H-donor), Lys-34 (H-donor), and Arg-120 (H-donor) similar to S1P. With the amino acid Leu-297 there is an aren-H interaction with the phenyl ring. ST-1893-phosphate interacts additionally with Phe-125 (aren-H).
The functional antagonism of fingolimod in vivo manifests as blocked egress from secondary lymphoid tissues and a subsequent drop in the number of lymphocytes in the blood. To confirm the functional antagonistic properties of ST-1893 and ST-1894 on S1P
1 in vivo, wild-type C57BL/6J mice were injected intraperitoneally with 5 mg/kg of compounds or dimethyl sulfoxide (DMSO) and, after 24 h, the number of lymphocytes in blood was measured. Results show that, at the dose of 5 mg/kg of ST-1893 or ST-1894, a drop in blood lymphocytes of 58% and 64%, respectively, occurred, confirming their immunosuppressive potential (
Figure 5).
In a next step, to evaluate the efficacy of ST-1893 and ST-1894 in a model of autoimmune inflammatory disease, we conducted an active myelin oligodendrocyte glycoprotein (MOG)-induced EAE experiment in C57BL/6J mice, in which ST-1893 and ST-1894 were given prophylactically and daily starting at day 5 post immunization. In vehicle-treated control mice, the first motor symptom, i.e., a limp tail, appeared at approximately day 10, and, as the disease progressed, the motor signs in the vehicle-treated group aggravated and mice acquired hind leg paraplegia and loss of lower body control. ST-1893- and ST-1894-treated mice showed a much milder clinical picture, with reduced motor deficits (
Figure 6A), the highest reached clinical score (
Figure 6B), and improved body weight (
Figure 6C). In these observations, there was a slight advantage of ST-1894 over ST-1893 at equal dose, which could be associated with the more potent effect on the S1P
1 internalization and lymphocyte count. At day 21 post immunization, mice were sacrificed, and lymphocyte analysis in blood showed only a pattern of decrease in the ST-1893- and ST-1894-treated groups (
Figure 6D), which contrasts the data obtained from healthy mice, where the depletion of blood lymphocytes was apparent and statistically significant. We attribute this discrepancy to the already low level of lymphocytes in the control group, which is in agreement with previous studies and the fact that the development of EAE itself is associated with a marked reduction in peripheral blood lymphocyte count [
16,
21].
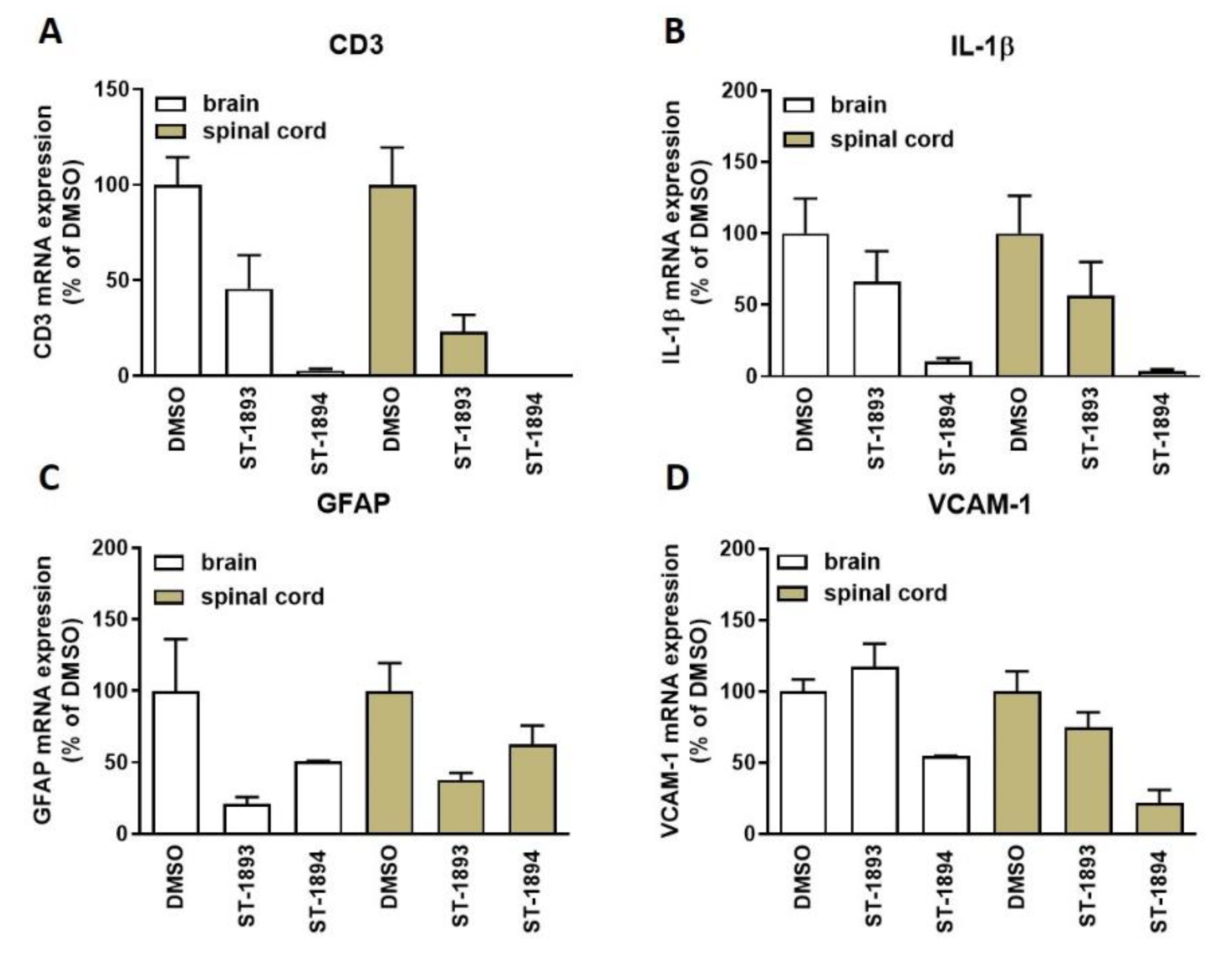
Nevertheless, tissue analysis showed a reduced expression of the T-cell marker cluster of differentiation 3 (CD3) (
Figure 7A) in the CNS of ST-1893- and ST-1894-treated mice, as well as reduced messenger RNA (mRNA) expression of the inflammatory markers interleukin (IL)-1β (
Figure 7B) and vascular cell adhesion molecule (VCAM)-1 (
Figure 7D), as well as of the astroglial marker of scarring, glial fibrillary acidic protein (GFAP) (
Figure 7C).
Since fingolimod is a prodrug and is known to strictly depend on phosphorylation by sphingosine kinase (SK)-2, but not SK-1, for therapeutic activity, we further tested whether ST-1893 and ST-1894 would also require phosphorylation by SK-2 for activity.
To this end, in vitro assays were first conducted in CHO-S1P
1 cells pretreated with the SK-2 inhibitor SLM6031434, after which they were stimulated with ST-1893 or ST-1894. S1P and fingolimod were used as positive controls for biotransformation by SK-2. Data in
Figure 8 show that inhibition of SK-2 completely abolishes the activation of p42/p44-MAPK through the S1P
1 receptor by fingolimod and confirms that preceding phosphorylation is needed for agonistic activity. In contrast, since S1P is already active, it is not affected by the inhibition of SK-2 and demonstrates no difference in the activation of S1P
1 in the presence or absence of SLM6031434. ST-1893 and ST-1894 behaved like fingolimod and lost their S1P
1-activating abilities in the presence of the inhibitor.
To investigate further the possible prodrug characteristics of ST-1893 and ST-1894, we used SK-2 knockout mice and analyzed for lymphopenia reaction after injection of ST-1893 and ST-1894. The number of lymphocytes in peripheral blood after 24 h was similar between the groups, with a tendency of decrease by ST-1894, although not significant (
Figure 9). These data support the modeling study confirming that the phosphorylated structures are more potent.
Finally, in an EAE experiment using SK-2 knockout mice, ST-1893-treated mice conferred no protection from developing the disease, while ST-1894-treated mice bore a partial protection. This was observed not only in terms of their mean daily clinical score (
Figure 10A), but also in their weight (
Figure 10B) and maximal clinical score (
Figure 10C). Altogether, these data point to a transformation of ST-1893 and ST-1894 by SK-2 in order to reach their full immunomodulatory effectiveness in vivo.
3. Discussions
One decade after fingolimod gained access to the market as the first S1P receptor modulator for the treatment of multiple sclerosis, the therapeutic potential of the S1P–S1PR axis signaling took center stage in sphingolipid research. Fingolimod is an unselective modulator of four of the five S1P receptors; therefore, one of the focuses of second-generation modulators is placed on structures with narrower S1P receptor targeting, thereby achieving fewer unspecific adverse effects.
In this study, we report the synthesis and validation of two novel selective modulators of the S1P
1 receptor, ST-1893 and ST-1894, which chemically are morpholino-derivatives of fingolimod (
Figure 1). Based on their chemical structures, namely, the presence of a polar head group and a voluminous aryl chain in the
para-position [
22], we hypothesized that they would fit in the ligand-binding pocket of the S1P
1 receptor and exert potent agonistic conformational change. Indeed, molecular modeling using a homology of S1P
1 in its active form showed that the docking results and binding strengths fully correspond to the theory of S1P–S1P
1 interactions (
Figure S1, Supplementary Materials). On the basis of the good establishment of hydrogen bonds between the substituted morpholino-moiety on one hand and the surrounding amino-acid residues from the binding pocket on the other hand, a good S1P
1 agonistic activity was predicted. Next, we fully characterized these compounds for agonistic and antagonistic activity on each of the S1P receptor subtypes and applied them in vivo in healthy mice with EAE as a model of MS. Both substances showed in vivo efficacy in the EAE model where they reduced the motor deficits, improved the body weight, and prevented the development of EAE with the highest clinical score (
Figure 6A–C). This was accompanied by reduced T-cell infiltration in the CNS and reduced expression of several mediators which have an aggravating role in the neuroinflammatory process, which explains the ameliorated phenotype (
Figure 7). In vitro, they potently activated the S1P
1 receptor and acted as agonists at early time-points (
Figure 2), while, later on, they downregulated the surface expression of the receptor (
Figure 4A–C), which accounts for their lymphopenic effect in mice (
Figure 5).
Our group previously characterized oxazolo–oxazolo-derivatives of fingolimod, denoted ST-968 and ST-1071, which have a dual S1P
1+3 selectivity and turned out to be as efficient as fingolimod in vitro in cell cultures, as well as in vivo in reducing disease symptoms of EAE in mice [
15]. The primary mechanism of action, for our novel morpholino-derivatives, as well as for the oxazolo–oxazolo-derivatives and fingolimod itself, relies on engaging the S1P
1 receptor, thereby inducing lymphocyte sequestration [
3]. On the other hand, it is believed that the stimulation of S1P
3, which is ubiquitously expressed throughout the body, may be associated with some adverse reactions from fingolimod medication, such as headache, hypertension, stroke [
23], cough, dyspnea, and macular edema [
24]. On the other hand, the physiological role of S1P
4, which is expressed in the immune and hematopoietic compartments, is still poorly understood and, therefore, the effects of fingolimod mediated by S1P
4, whether beneficial or detrimental, are vague. Furthermore, the net effect of the drug binding is dependent on many factors, which include the S1P receptor repertoire and predominant expression of a particular receptor in a cell, the collection of signaling pathways within a cell, variations in the expression of both S1P receptors and downstream signaling proteins, the convergence of signaling pathways initiated by different G proteins, the localization of S1P receptors on cell membrane vs. internalized/nuclearized, etc. [
25,
26]. All these factors bring another level of complexity to S1P receptor signaling. Obviously, the novel structures ST-1893 and ST-1894 are devoid of unspecific effects through any of the other S1P receptor subtypes and, therefore, have an additional advantage.
Although another selective S1P
1 agonist, SEW2871, does not target other S1P receptors; it binds and activates S1P
1 through a combination of hydrophobic and ion–dipole interactions in the binding pocket [
27], rendering it less potent at downstream G-protein binding and kinase activation. This tetraaromatic compound is a weak agonist that induces S1P
1 internalization but then allows, similar to S1P, receptor recycling instead of receptor degradation, which translates into rapidly reversible lymphopenia [
28]. ST-1893 and ST-1894 establish hydrogen bonds with surrounding amino-acid residues from the receptor and likely induce a potent signal transduction which is then evidenced by a strong reduction in the number of circulating lymphocytes in vivo. Other factors include the molecular volume, which is larger for ST-1893 as compared to the oxazolo–oxazole compound ST-1071 [
15]. This might hamper the dissociation from the receptor and translate into a higher activity as predicted computationally, even though both had similar docking score.
S1P
1 antagonism is also a valid approach to achieve a block of lymphocyte egress, and, through the induction of lymphopenia, several competitive antagonists were demonstrated to be effective in EAE, collagen-induced arthritis, traumatic brain injury, and cardiac allograft rejection animal models [
16,
29,
30,
31]. However, S1P
1 antagonism is associated with a disturbance of the endothelial barrier integrity, especially in the lungs and skin, promoting vascular leakage, edema, and chronic remodeling with functional impairments [
32,
33]. ST-1893 and ST-1894 had no effect on S1P-stimulated receptor activation on any of the five S1P receptors, excluding any antagonism in the presence of S1P (
Figure 2A), indicating that the lymphopenic effect is solely due to a functional antagonism.
A prerequisite for the lymphocyte-depleting activity of fingolimod is the reversible and stereoselective transformation by SK-2, in order to yield the active phospho-metabolite [
34]. Contrariwise, our previously described oxazolo–oxazole substances have an advantage over fingolimod, as they do not require SK-2-mediated phosphorylation in order to become active. Notably, the SK-2 inhibitor ABC294640 (Yeliva
®) received an orphan drug designation by the Food and Drug Administration (FDA) due to its anticancer properties in several clinical trials (NCT01488513, NCT02757326, NCT02229981, NCT03377179), although some of the effects observed in in vivo studies may be attributed to off-target effects due to its low potency at SK-2 [
35]. ABC294640, as well as other SK-2 inhibitors, such as SLP120701 and ROMe, are being increasingly explored in inflammatory models, giving promising anti-inflammatory results in acute and chronic models of ulcerative colitis, viral and bacterial infections, renal inflammation/fibrosis, and EAE [
36,
37,
38,
39,
40]. Co-medication of fingolimod with an SK-2 inhibitor, in theory, seems like a synergistic anti-inflammatory combination, but would clearly result in therapeutic incompatibility, due to the lack of a biotransformation step for fingolimod. In this context, using SK-2 independent structures would be mandatory, and ST-968 and ST-1071 fulfill this criterion.
The novel morpholino-derivatives, ST-1893 and ST-1894, have a different dependence on SK-2 for their in vivo activity (
Figure 10). ST-1893 fully depends on SK-2, and, although ST-1894 decreases the motor deficits in the absence of SK-2, the full immunomodulatory effectiveness in vivo was observed in wild-type mice where SK-2 is normally expressed. This finding points to an inherent difference in the oxazolo–oxazole and morpholine functional groups, which dictates whether the structure will become a substrate of SK-2 or not. Since the crystal structure of SK-2 is still missing, predictions of substrate binding and structure-based drug design by molecular docking studies remain vague [
35].
It is also well conceivable that the ether moiety in the aliphatic chain (present in ST-1894, but not in ST-1893) may influence the prodrug properties, as we noticed that it relatively increases the S1P1 activation, internalization rate, and lymphopenic effect in equivalent doses. Finally, the in vivo effects depend also on other modes of biotransformation, the stability and elimination, the volume of distribution in the secondary lymphoid tissues, etc., which warrants further studies.
In summary, here, we present two novel structures derived from fingolimod, ST-1893 and ST-1894, which possess a selective S1P1 activation profile. Both compounds are functional antagonists that induce sustained S1P1 internalization in cell culture and a profound lymphopenia in mice. As they demonstrated in vivo efficacy in the EAE model, and due to their S1P1-selective nature, we speculate that they will exhibit fewer adverse effects and have an advantage over fingolimod and siponimod, not only in MS but also in other autoimmune diseases.
4. Materials and Methods
4.1. Chemicals and Chemical Synthesis of ST-1893 and ST-1894
4.2. Cell Culturing
Chinese hamster ovary (CHO)-K1 cells, which stably overexpress different human S1P1–5 receptor subtypes, were kindly provided by Dr. Danilo Guerini (Novartis Institutes for Biomedical Research, Basel, Switzerland). CHO-S1P1/4/5 cells were maintained in alpha minimum essential medium Eagle (αMEM), while CHO-S1P2/3 were cultivated in Roswell Park Memorial Institute (RPMI) medium. Both media were supplemented with 10% fetal bovine serum (FBS), 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 50 µg/mL gentamycin, and 0.5 mg/mL G418 as a selection antibiotic. Cells were cultured in an environment of 37 °C with 5% CO2 and were passaged 2–3 times a week, using Trypsin-ethylenediaminetetraacetic acid (EDTA) 0.25% for detachment. Prior to stimulation, cells were rendered serum-free overnight with a medium consisting of Dulbecco’s modified Eagle medium (DMEM), 0.1 mg/mL bovine serum albumin (BSA), and 10 mM HEPES (later on referred to as serum-free medium).
4.3. Western Blot Analysis
Western blot analysis was performed as previously described [
41] by incubating membranes with antibodies for 16 h at 4 °C. Commercially available antibodies used were as follows: phospho-Thr
202/Tyr
204 p42/p44-MAPK (Cell Signaling, cat. no. 4377, 1:1000 dilution) and Myc-tag (Cell Signaling, cat. no. 2276, 1:1000 dilution). IRDye
® or horseradish peroxidase (HRP)-linked secondary antibodies were used, and bands were detected and analyzed by the LI-COR Image Studio
TM (v. 3.1; LI-COR Biotechnology GmbH, Bad Homburg, Germany). Signal intensity was evaluated by ImageJ (National Institutes of Health, USA). Polyclonal antisera against p42-MAPK and p44-MAPK were previously generated and characterized [
42] and used in a dilution of 1:3000.
4.4. RNA Extraction and Quantitative PCR Analysis
RNA extraction, complementary DNA (cDNA) synthesis, and quantitative PCR analysis were performed as previously described [
43]. Primer sequences of mouse CD3, VCAM-1, and 18S RNA were as previously described [
15,
44]. The sequences for other primers were as follows: GFAP, forward: CCAGCTTCGAGCCAAGGA, reverse: GAAGCTCCGCCTGGTAGACA; IL-1β, forward: TGTCCTGTGTAATGAAAGACGG, reverse: TCTTGTGACCCTGAGCGAC.
4.5. S1P Receptor Activation Studies
Confluent CHO-S1P1–5 cells were incubated in serum-free medium for 20 h and then stimulated with the indicated compounds for 10 min at 37 °C. Cell monolayers were washed with phosphate-buffered saline (PBS) and lysed in lysis buffer at 4 °C and further processed for protein separation by SDS-PAGE, transfer to nitrocellulose membranes, and Western blot analysis.
4.6. S1P1 Internalization Studies by ELISA
Confluent CHO-S1P1 cells in 24-well plates were starved for 20 h prior to stimulation with 3 μM of compounds in serum-free medium. In the first experimental set-up, the stimulation was ceased after 3 h, whereas, in the second set-up, the long-term effect on receptor internalization was assessed by stimulation for 3 h, washing, and placing in serum-free conditions for an additional 21 h. Cells were washed three times with ice-cold PBS, fixed with 4% paraformaldehyde in PBS at room temperature for 20 min, and blocked in 6 % (w/v) non-fat dry milk in PBS (below labeled as blocking buffer) for 1 h. They were incubated with anti-Myc Tag antibody (1:1000 in blocking buffer) and later on with anti-mouse HRP-linked antibody (1:5000 in blocking buffer), both for 1 h, with washing steps in between (8 min × 3 times in PBS). After 3,3′,5,5′-tetramethylbenzidine (TMB) was added as a substrate for the HRP, the reaction was stopped with 1 N H2SO4, and the absorbance was measured, subtracting the blank where the primary antibody was omitted. In order to normalize with the number of cells, we proceeded with washing, staining with 0.1% crystal violet solution in 20% methanol, and solubilizing with 5% acetic acid. Absorbance was measured, normalized with the values from the ELISA, and calculated to the DMSO control being 100%.
4.7. Lymphocyte Count
C57BL/6J mice were injected intraperitoneally with 5 mg/kg of ST-1893, ST-1894, or DMSO in PBS, and, after 24 h, blood was collected in EDTA-K3-coated microvettes (Sarstedt; Sevelen, Switzerland) with cardiac puncture under deep terminal isoflurane-induced anesthesia. The number of lymphocytes was assessed with a Scil Vet ABCTM Hemocytometer (Scil animal care company GmbH; Viernheim, Germany).
4.8. Experimental Antigen-Induced Encephalomyelitis Experiments
All mice experiments were in accordance with the Swiss animal experimentation legislation and under the license number BE50/17. Active EAE was induced in C57BL/6J mice with MOG
aa35–55 emulsified in complete Freund’s adjuvant (CFA) and pertussis toxin as described previously by our group [
15]. The overall wellbeing, weight, and signs of disease were evaluated twice daily using a four-point scoring system. Disease sign scores were as follows: 0.5: limp tail, 1: hind leg weakness and unsteady gait, 2: hind leg paraplegia, 3: hind leg paraplegia, incontinence, and loss of lower body control. From day 5 until day 20 post immunization, they received daily 5 mg/kg of ST-1893, ST-1894, or DMSO in PBS via intraperitoneal injection. On day 21, blood was collected, the mice were sacrificed and perfused with ice-cold PBS, and the CNS was extracted and snap-frozen in liquid nitrogen until further analysis.
4.9. Organ Homogenization
The snap-frozen tissues in liquid nitrogen were homogenized using Sartorius Mikro-Dismembrator S (Göttingen, Germany) at 3000 rpm for 1 min each. The pulverized tissues were distributed in Eppendorf tubes; then, RNA-Solv® reagent was added and vortexed for 5 min to aid the dissolution. Samples were centrifuged at 13,000 rpm for 10 min at 4 °C, and the supernatant was used for RNA extraction and quantitative PCR analysis as described above.
4.11. Statistical Analysis
GraphPad Prism 6 Software (San Diego, CA, USA) was used for statistical analysis and graph representations. The statistical significance was calculated with ordinary one-way ANOVA without matching or unpaired t-test where applicable. The level of significance was calculated with Bonferroni correction for multiple comparisons. For the EAE experiments, the nonparametric Kruskal–Wallis test for the area under the curve (AUC) was used. Data are depicted as means ± SD for n number of independent experiments unless otherwise stated.

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}










