Temporal Alterations in Mitochondrial β-Oxidation and Oxidative Stress Aggravate Chronic Kidney Disease Development in 5/6 Nephrectomy Induced Renal Damage

 , , ,
, , ,  and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
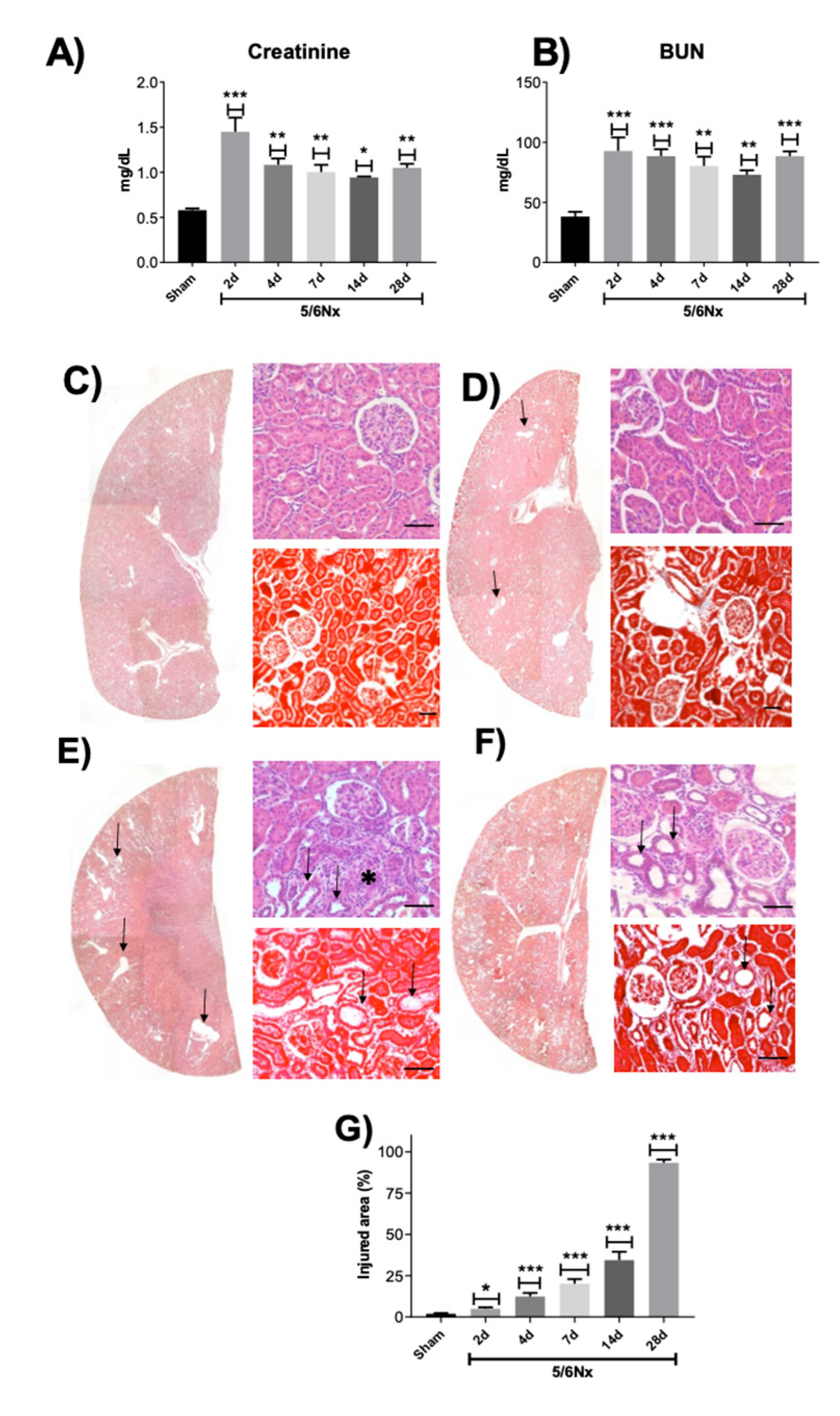
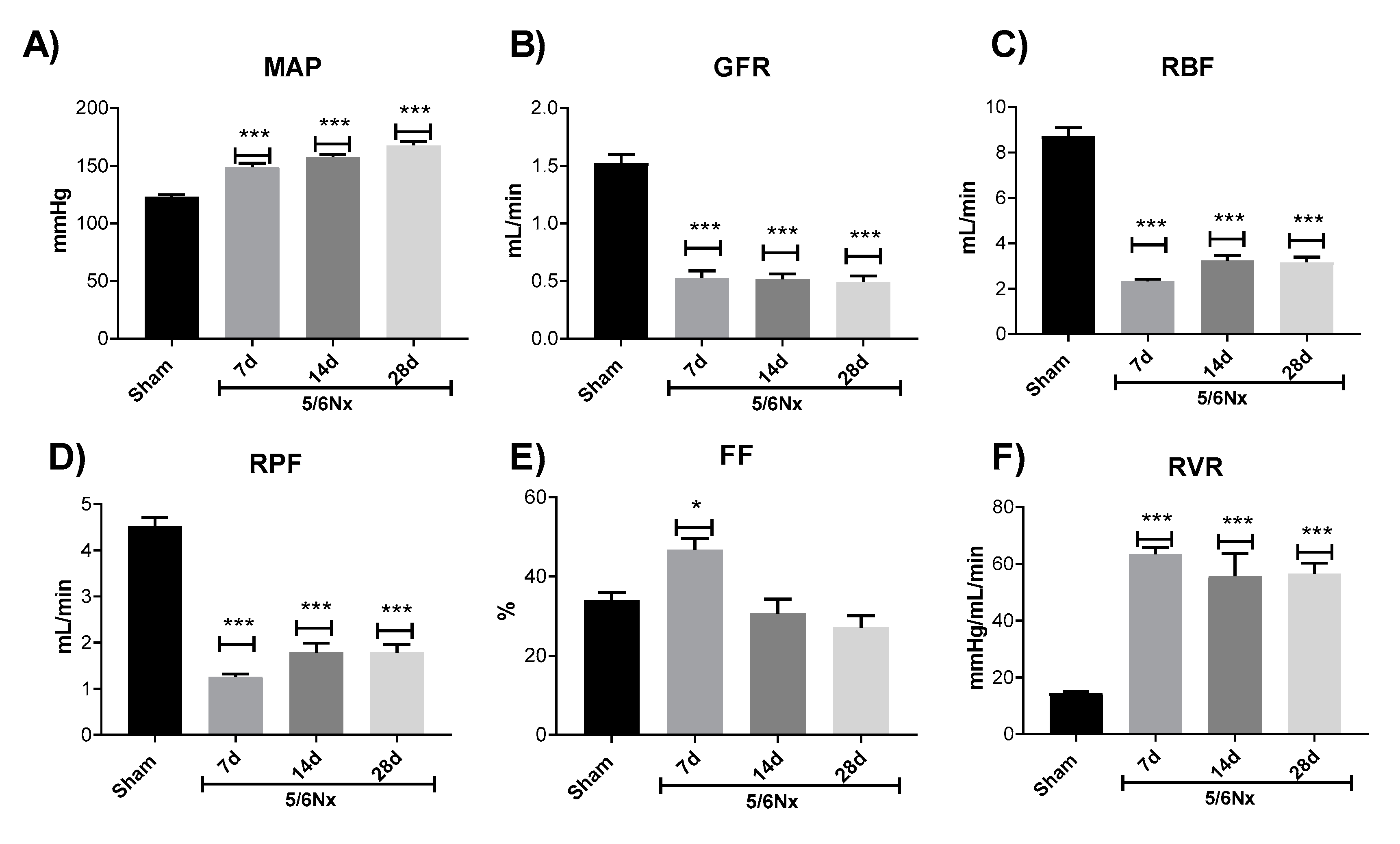
2.1. Renal Function Loss Induced by 5/6 Nephrectomy
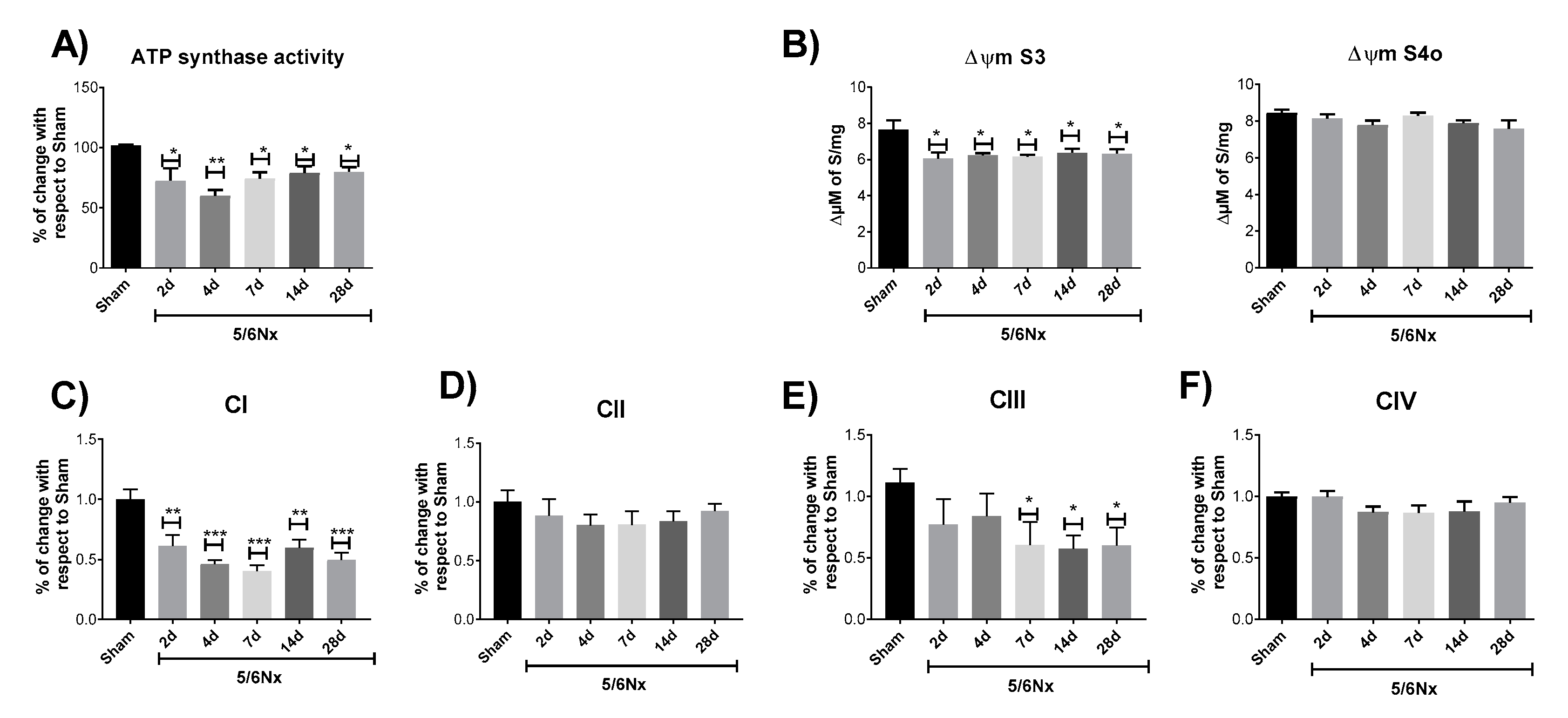
2.2. 5/6Nx Induces a Permanent Decrease in Oxidative Phosphorylation (OXPHOS) Capacity in CI-Linked Respiration and a Progressive Decoupling in Mitochondrial β-Oxidation
2.3. 5/6Nx-Induced Decrease in OXPHOS Capacity is Related to Mitochondrial Depolarization by CI and CIII Activity Decrease
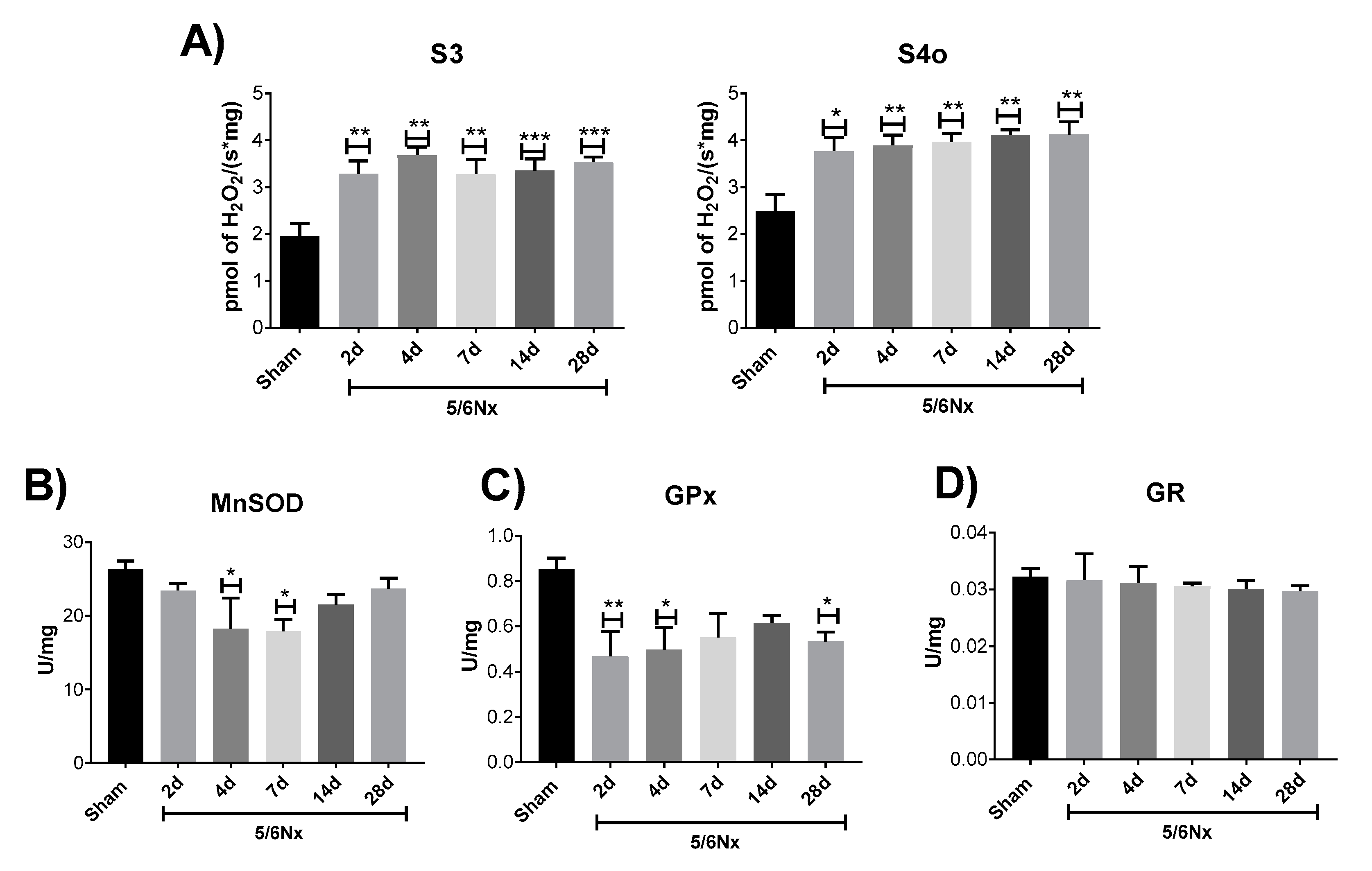
2.4. Mitochondrial Decoupling Induced by 5/6Nx is Related to a Pro-oxidative State in Mitochondria
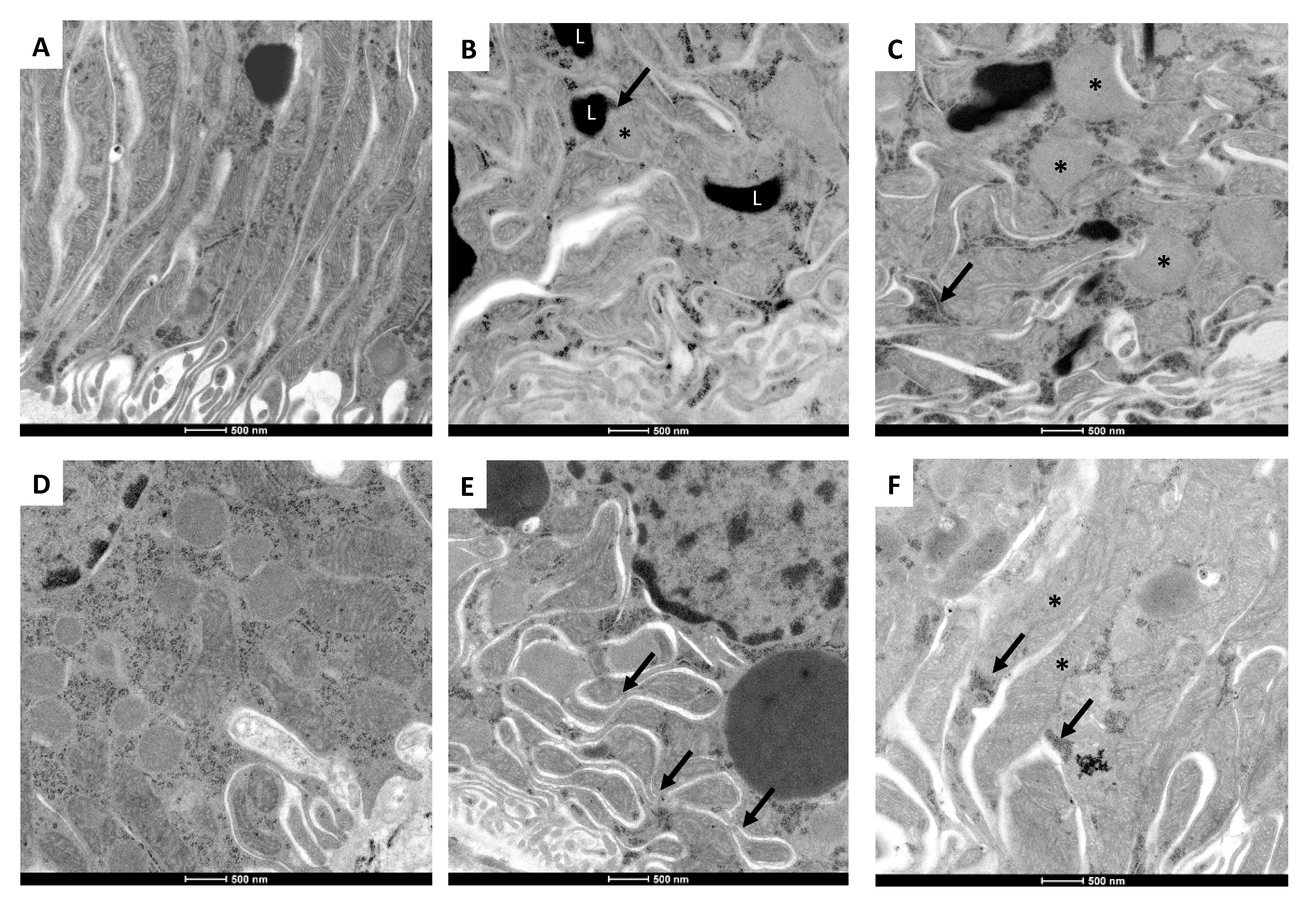
2.5. 5/6Nx-Induced Temporal Alterations in Mitochondrial Ultrastructure
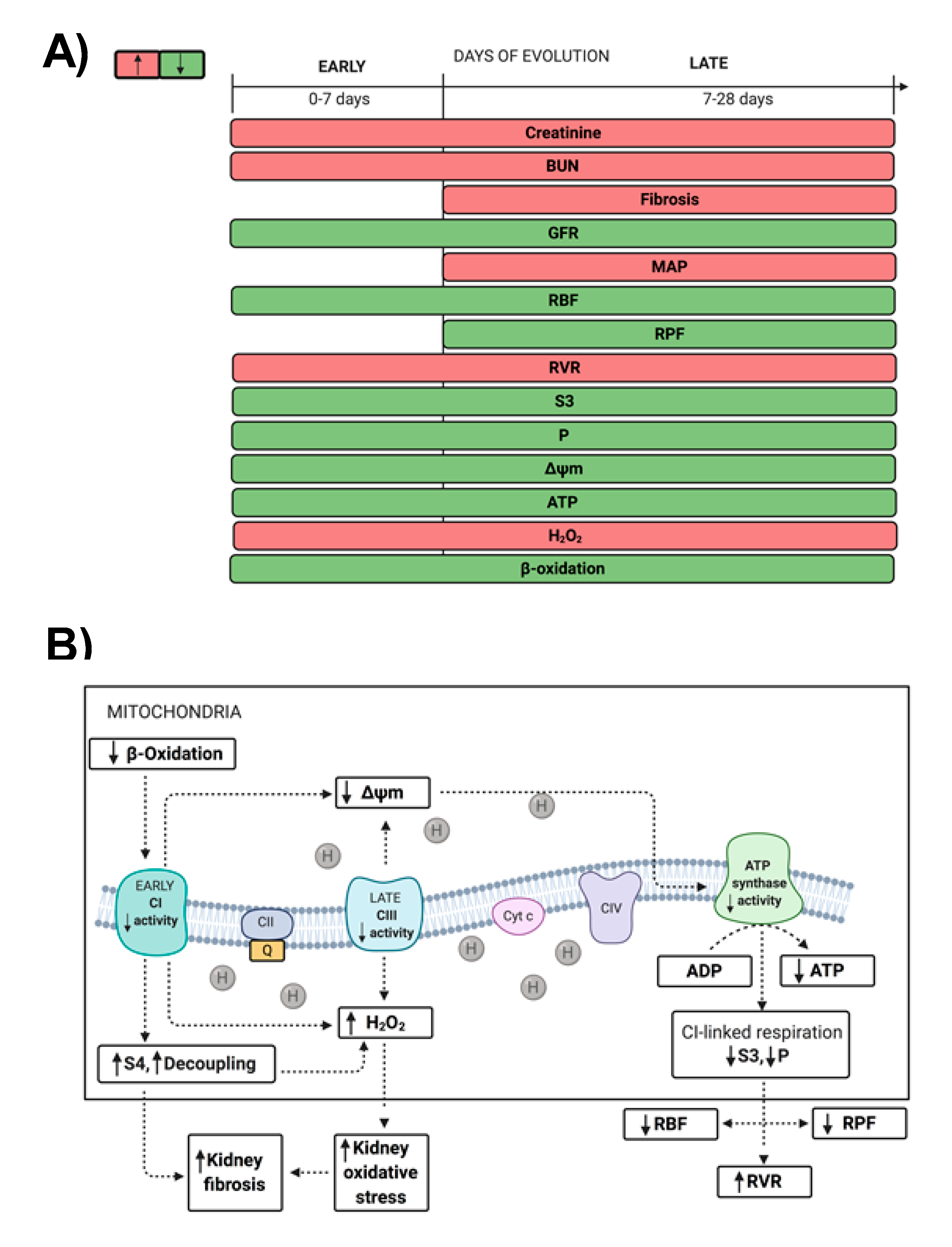
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Experimental Design
4.3. Biochemical Markers of Renal Damage and Hemodynamic Parameters
4.4. Histology Evaluation and Electron Microscopy
4.5. Renal Mitochondria Isolation
4.6. Mitochondrial O2 Consumption and ΔΨm
4.7. ATP Synthase and Mitochondrial Respiratory Complexes’ Activities
4.8. Mitochondrial H2O2 Production
4.9. Activity of Antioxidant Enzymes in Mitochondria
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shafi, T.; Coresh, J. Chronic kidney disease: Definition, epidemiology, cost, and outcomes. In Chronic Kidney Disease, Dialysis, and Transplantation; Elsevier: Amsterdam, The Netherlands, 2010; pp. 3–21. [Google Scholar] [CrossRef]
- Carrero, J.J.; Hecking, M.; Ulasi, I.; Sola, L.; Thomas, B. Chronic Kidney Disease, Gender, and Access to Care: A Global Perspective, Semin. Nephrology 2017, 37, 296–308. [Google Scholar] [CrossRef]
- Khwaja, A.; El Kossi, M.; Floege, J.; El Nahas, M. The management of CKD: A look into the future. Kidney Int. 2007, 72, 1316–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aparicio-Trejo, O.E.; Tapia, E.; Sánchez-Lozada, L.G.; Pedraza-Chaverri, J. Pedraza-Chaverri, Mitochondrial bioenergetics, redox state, dynamics and turnover alterations in renal mass reduction models of chronic kidney diseases and their possible implications in the progression of this illness. Pharmacol. Res. 2018, 135, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Meng, R.; Huang, B.; Bi, Y.; Shen, S.; Zhu, D. Silymarin protects against renal injury through normalization of lipid metabolism and mitochondrial biogenesis in high fat-fed mice. Free Radic. Biol. Med. 2017, 110, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Tapia, E.; Zatarain-Barrón, Z.L.; Hernández-Pando, R.; Zarco-Márquez, G.; Molina-Jijón, E.; Cristóbal-García, M.; Pedraza-Chaverri, J. Curcumin reverses glomerular hemodynamic alterations and oxidant stress in 5/6 nephrectomized rats. Phytomedicine 2013, 20, 359–366. [Google Scholar] [CrossRef]
- Taal, M.W.; Brenner, B.M. Adaptation to Nephron Loss and Mechanisms of Progression in Chronic Kidney Disease. In Brenner & Rector’s The Kidney, 9th ed.; Skorecki, K., Chertow, G.M., Marsden, P.A., Taal, M.W., Yu, A.S.L., Eds.; Elsevier: Amsterdam, The Netherlands, 2012; pp. 1918–1971. [Google Scholar] [CrossRef]
- Ishimoto, Y.; Inagi, R. Mitochondria: A therapeutic target in acute kidney injury. Nephrol. Dial. Transplant. 2015, 31, 1062–1069. [Google Scholar] [CrossRef]
- Che, R.; Yuan, Y.; Huang, S.; Zhang, A. Mitochondrial dysfunction in the pathophysiology of renal diseases. Am. J. Physiol. Physiol. 2014, 306, F367–F378. [Google Scholar] [CrossRef]
- Aparicio-Trejo, O.E.; Avila-Rojas, S.H.; Tapia, E.; Rojas-Morales, P.; León-Contreras, J.C.; Martínez-Klimova, E.; Hernández-Pando, R.; GabrielaSánchez-Lozada, L.; Pedraza-Chaverri, J. Pedraza-Chaverri, Chronic impairment of mitochondrial bioenergetics and β-oxidation promotes experimental AKI-to-CKD transition induced by folic acid. Free Radic. Biol. Med. 2020, 154, 18–32. [Google Scholar] [CrossRef]
- Tamaki, M.; Miyashita, K.; Wakino, S.; Mitsuishi, M.; Hayashi, K.; Itoh, H. Chronic kidney disease reduces muscle mitochondria and exercise endurance and its exacerbation by dietary protein through inactivation of pyruvate dehydrogenase. Kidney Int. 2014, 85, 1330–1339. [Google Scholar] [CrossRef] [Green Version]
- Forbes, J.M.; Thorburn, D.R. Mitochondrial dysfunction in diabetic kidney disease. Nat. Rev. Nephrol. 2018, 14, 291–312. [Google Scholar] [CrossRef]
- Mafra, D.; Gidlund, E.K.; Borges, N.A.; Magliano, D.C.; Lindholm, B.; Stenvinkel, P.; von Walden, F. Bioactive food and exercise in chronic kidney disease: Targeting the mitochondria. Eur. J. Clin. Investig. 2018, 48, e13020. [Google Scholar] [CrossRef] [Green Version]
- Correa, F.; Buelna-Chontal, M.; Hernández-Reséndiz, S.; García-Niño, W.R.; Roldán, F.J.; Soto, V.; Silva-Palacios, A.; Amador, A.; Pedraza-Chaverrí, J.; Tapia, E.; et al. Curcumin maintains cardiac and mitochondrial function in chronic kidney disease. Free Radic. Biol. Med. 2013, 61, 119–129. [Google Scholar] [CrossRef]
- Lash, L.H.; Putt, D.A.; Horky, S.J.; Zalups, R.K. Functional and toxicological characteristics of isolated renal mitochondria: Impact of compensatory renal growth. Biochem. Pharmacol. 2001, 62, 383–395. [Google Scholar] [CrossRef]
- Thomas, J.L.; Pham, H.; Li, Y.; Hall, E.; Perkins, G.A.; Ali, S.S.; Patel, H.H.; Singh, P. Hypoxia-inducible factor-1α activation improves renal oxygenation and mitochondrial function in early chronic kidney disease. Am. J. Physiol. Ren. Physiol. 2017, 313, F282–F290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tapia, E.; Soto, V.; Ortiz-Vega, K.M.; Zarco-Márquez, G.; Molina-Jijón, E.; Cristóbal-García, M.; Santamaría, J.; García-Niño, W.R.; Correa, F.; Zazueta, C.; et al. Curcumin induces Nrf2 nuclear translocation and prevents glomerular hypertension, hyperfiltration, oxidant stress, and the decrease in antioxidant enzymes in 5/6 nephrectomized rats. Oxid. Med. Cell. Longev. 2013, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Aparicio-Trejo, O.E.; Tapia, E.; Molina-Jijón, E.; Medina-Campos, O.N.; Macías-Ruvalcaba, N.A.; León-Contreras, J.C.; Hernández-Pando, R.; García-Arroyo, F.E.; Cristóbal, M.; Sánchez-Lozada, L.G.; et al. Curcumin prevents mitochondrial dynamics disturbances in early 5/6 nephrectomy: Relation to oxidative stress and mitochondrial bioenergetics. BioFactors 2017, 43, 293–310. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.F.; Liu, H.; Ni, H.F.; Lv, L.L.; Zhang, M.H.; Zhang, A.H.; Tang, R.N.; Chen, P.S.; Liu, B.C. Improved mitochondrial function underlies the protective effect of pirfenidone against tubulointerstitial fibrosis in 5/6 nephrectomized rats. PLoS ONE 2013, 8, 1–15. [Google Scholar] [CrossRef]
- Zhao, H.; Liu, Y.J.; Liu, Z.R.; Tang, D.D.; Chen, X.W.; Chen, Y.H.; Zhou, R.N.; Chen, S.Q.; Niu, H.X. Role of mitochondrial dysfunction in renal fibrosis promoted by hypochlorite-modified albumin in a remnant kidney model and protective effects of antioxidant peptide SS-31. Eur. J. Pharmacol. 2017, 804, 57–67. [Google Scholar] [CrossRef]
- Freund, D.M.; Prenni, J.E.; Curthoys, N.P. Proteomic profiling of the mitochondrial inner membrane of rat renal proximal convoluted tubules. Proteomics 2013, 13, 2495–2499. [Google Scholar] [CrossRef] [Green Version]
- Fedorova, L.V.; Tamirisa, A.; Kennedy, D.J.; Haller, S.T.; Budnyy, G.; Shapiro, J.I.; Malhotra, D. Mitochondrial impairment in the five-sixth nephrectomy model of chronic renal failure: Proteomic approach. BMC Nephrol. 2013, 14, 209–230. Available online: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3851543&tool=pmcentrez&rendertype=abstract (accessed on 3 June 2020). [CrossRef] [Green Version]
- Hui, Y.; Lu, M.; Han, Y.; Zhou, H.; Liu, W.; Li, L.; Jin, R. Resveratrol improves mitochondrial function in the remnant kidney from 5/6 nephrectomized rats. Acta Histochem. 2017, 119, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Yuan, Q.; Xu, T.; Yao, L.; Feng, J.; Ma, J.; Wang, L.; Lu, C.; Wang, D. Pioglitazone Improves Mitochondrial Function in the Remnant Kidney and Protects against Renal Fibrosis in 5/6 Nephrectomized Rats. Front. Pharmacol. 2017, 8, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gnaiger, E. Mitochondrial Pathways and Respiratory Control an Introduction to OXPHOS Analysis. 2014. Available online: http://wiki.oroboros.at/images/f/fc/Gnaiger_2014_Mitochondr_Physiol_Network_MitoPathways.pdf (accessed on 6 June 2020).
- Sekine, T.; Endou, H. Solute Transport, Energy Consumption, and Production in the Kidney. In Seldin and Geibisch’s The Kidney, 5th ed.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 143–175. [Google Scholar] [CrossRef]
- Kang, P.T.; Zhang, L.; Chen, C.L.; Chen, J.; Green, K.B.; Chen, Y.R. Protein thiyl radical mediates S-glutathionylation of complex I. Free Radic. Biol. Med. 2012, 53, 962–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forkink, M.; Basit, F.; Teixeira, J.; Swarts, H.G.; Koopman, W.J.H.; Willems, P.H.G.M. Complex I and complex III inhibition specifically increase cytosolic hydrogen peroxide levels without inducing oxidative stress in HEK293 cells. Redox Biol. 2015, 6, 607–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aparicio-Trejo, O.E.; Reyes-Fermín, L.M.; Briones-Herrera, A.; Tapia, E.; León-Contreras, J.C.; Hernández-Pando, R.; Sánchez-Lozada, L.G.; Pedraza-Chaverri, J. Protective effects of N-acetyl-cysteine in mitochondria bioenergetics, oxidative stress, dynamics and S-glutathionylation alterations in acute kidney damage induced by folic acid. Free Radic. Biol. Med. 2019, 130, 379–396. [Google Scholar] [CrossRef]
- Quinlan, C.L.; Gerencser, A.A.; Treberg, J.R.; Brand, M.D. The mechanism of superoxide production by the antimycin-inhibited mitochondrial Q-cycle. J. Biol. Chem. 2011, 286, 31361–31372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Orr, A.L.; Brand, M.D. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 2013, 1, 304–312. [Google Scholar] [CrossRef] [Green Version]
- Hostetter, T.H.; Olson, J.L.; Renke, H.G.; Venkatachalam, M.A.; Brenner, B.M. Hyperfiltration in remnant nephrons: A potentially adverse response to renal ablation. J. Am. Soc. Nephrol. 2001, 12, 1315–1325. [Google Scholar] [CrossRef] [Green Version]
- Sinuani, I.; Averbukh, Z.; Gitelman, I.; Rapoport, M.J.; Sandbank, J.; Albeck, M.; Sredni, B.; Weissgarten, J. Mesangial cells initiate compensatory renal tubular hypertrophy via IL-10-induced TGF-beta secretion: Effect of the immunomodulator AS101 on this process. Am. J. Physiol. Renal Physiol. 2006, 291, F384–F394. [Google Scholar] [CrossRef]
- Hauser, P.; Kainz, A.; Perco, P.; Bergmeister, H.; Mitterbauer, C.; Schwarz, C.; Regele, H.M.; Mayer, B.; Meyer, T.W.; Oberbauer, R. Transcriptional response in the unaffected kidney after contralateral hydronephrosis or nephrectomy. Kidney Int. 2005, 68, 2497–2507. [Google Scholar] [CrossRef] [Green Version]
- Wolf, G.; Ziyadeh, F.N. Molecular mechanisms of diabetic renal hypertrophy. Kidney Int. 1999, 56, 393–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rastogi, R.; Geng, X.; Li, F.; Ding, Y. NOX activation by subunit interaction and underlying mechanisms in disease. Front. Cell. Neurosci. 2017, 10, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Cavanagh, E.M.V.; Inserra, F.; Ferder, M.; Ferder, L. From mitochondria to disease: Role of the renin-angiotensin system. Am. J. Nephrol. 2007, 27, 545–553. [Google Scholar] [CrossRef]
- Mackenzie Walser. Progression of chronic renal failure in man. Kidney Int. 1990, 37, 1195–1210. [Google Scholar] [CrossRef] [Green Version]
- Priyadarshi, A.; Periyasamy, S.; Burke, T.J.; Britton, S.L.; Malhotra, D.; Shapiro, J.I. Effects of reduction of renal mass on renal oxygen tension and erythropoietin production in the rat. Kidney Int. 2002, 61, 542–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, N.J.; Devarajan, P.; van Why, S. Renal cellinjury: Metabolie and structural alterations. Pediatr. Res. 1994, 36, 129–136. [Google Scholar] [CrossRef] [Green Version]
- Basile, D.P.; Donohoe, D.; Roethe, K.; Osborn, J.L. Renal ischemic injury results in permanent damage to peritubular capillaries and influences long-term function. Am. J. Physiol. Ren. Physiol. 2001, 281, 887–899. [Google Scholar] [CrossRef]
- Benipal, B.; Lash, L.H. Modulation of mitochondrial glutathione status and cellular energetics in primary cultures of proximal tubular cells from remnant kidney of uninephrectomized rats. Biochem. Pharmacol. 2013, 85, 1379–1388. [Google Scholar] [CrossRef] [PubMed]
- Szeto, H.H.; Liu, S.; Soong, Y.; Alam, N.; Prusky, G.T.; Seshan, S.V. Protection of mitochondria prevents high-fat diet induced glomerulopathy and proximal tubular injury. Kidney Int. 2016, 90, 997–1011. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.A.; Han, S.H.; Chinga, F.; Park, A.S.D.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef]
- Benipal, B.; Lash, L.H. Influence of renal compensatory hypertrophy on mitochondrial energetics and redox status. Biochem. Pharmacol. 2011, 81, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Nath, K.A.; Croatt, A.J.; Hostetter, T.H. Oxygen consumption and oxidant stress in surviving nephrons. Am. J. Physiol. 1990, 258, F1354–F1362. [Google Scholar] [CrossRef] [PubMed]
- Granata, S.; Masola, V.; Zoratti, E.; Scupoli, M.T.; Baruzzi, A.; Messa, M.; Sallustio, F.; Gesualdo, L.; Lupo, A.; Zaza, G. NLRP3 inflammasome activation in dialyzed chronic kidney disease patients. PLoS ONE 2015, 10, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gai, Z.; Wang, T.; Visentin, M.; Kullak-Ublick, G.A.; Fu, X.; Wang, Z. Lipid accumulation and chronic kidney disease. Nutrients 2019, 11, 722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chunsun, D.; Lawrence, P.K.; Youhua, L. Animal Models of Kidney Diseases. In Sourcebook of Models for Biomedical Research; Humana Press: Totowa, NJ, USA, 2008; pp. 657–664. [Google Scholar]
- Hamzaoui, M.; Djerada, Z.; Brunel, V.; Mulder, P.; Richard, V.; Bellien, J.; Guerrot, D. 5/6 nephrectomy induces different renal, cardiac and vascular consequences in 129/Sv and C57BL/6JRj mice. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Becker, G.J.; Hewitson, T.D. Animal models of chronic kidney disease: Useful but not perfect. Nephrol. Dial. Transplant. 2013, 28, 2432–2438. [Google Scholar] [CrossRef] [Green Version]
- Hallan, S.; Sharma, K. The Role of Mitochondria in Diabetic Kidney Disease. Curr. Diab. Rep. 2016, 16, 1–9. [Google Scholar] [CrossRef]
- Pedraza-Chaverri, J.; Sánchez-Lozada, L.G.; Osorio-Alonso, H.; Tapia, E.; Scholze, A. New Pathogenic Concepts and Therapeutic Approaches to Oxidative Stress in Chronic Kidney Disease. Oxid. Med. Cell. Longev. 2016, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Afshinnia, F.; Rajendiran, T.M.; Soni, T.; Byun, J.; Wernisch, S.; Sas, K.M.; Hawkins, J.; Bellovich, K.; Gipson, D.; Michailidis, G.; et al. Impaired β -Oxidation and Altered Complex Lipid Fatty Acid Partitioning with Advancing CKD. J. Am. Soc. Nephrol. 2018, 29, 295–306. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wang, Y.; Ding, W.; Wang, Y. Mito-TEMPO Alleviates Renal Fibrosis by Reducing Inflammation, Mitochondrial Dysfunction, and Endoplasmic Reticulum Stress. Oxid. Med. Cell. Longev. 2018, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, N.A.; Armaly, Z.; Berman, S.; Jabour, A.; Aga-Mizrachi, S.; Mosenego-Ornan, E.; Avital, A. L-Carnitine improves cognitive and renal functions in a rat model of chronic kidney disease. Physiol. Behav. 2016, 164, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Morales, P.; León-Contreras, J.C.; Aparicio-Trejo, O.E.; Reyes-Ocampo, J.G.; Medina-Campos, O.N.; Jiménez-Osorio, A.S.; González-Reyes, S.; Marquina-Castillo, B.; Hernández-Pando, R.; Barrera-Oviedo, D.; et al. Fasting reduces oxidative stress, mitochondrial dysfunction and fibrosis induced by renal ischemia-reperfusion injury. Free Radic. Biol. Med. 2019, 135, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Ojuka, E.; Andrew, B.; Bezuidenhout, N.; George, S.; Maarman, G.; Madlala, H.P.; Mendham, A.; Osiki, P.O. Measurement of β-oxidation capacity of biological samples by respirometry: A review of principles and substrates. Am. J. Physiol. Metab. 2016, 310, E715–E723. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aparicio-Trejo, O.E.; Rojas-Morales, P.; Avila-Rojas, S.H.; León-Contreras, J.C.; Hernández-Pando, R.; Jiménez-Uribe, A.P.; Prieto-Carrasco, R.; Sánchez-Lozada, L.G.; Pedraza-Chaverri, J.; Tapia, E. Temporal Alterations in Mitochondrial β-Oxidation and Oxidative Stress Aggravate Chronic Kidney Disease Development in 5/6 Nephrectomy Induced Renal Damage. Int. J. Mol. Sci. 2020, 21, 6512. https://doi.org/10.3390/ijms21186512
Aparicio-Trejo OE, Rojas-Morales P, Avila-Rojas SH, León-Contreras JC, Hernández-Pando R, Jiménez-Uribe AP, Prieto-Carrasco R, Sánchez-Lozada LG, Pedraza-Chaverri J, Tapia E. Temporal Alterations in Mitochondrial β-Oxidation and Oxidative Stress Aggravate Chronic Kidney Disease Development in 5/6 Nephrectomy Induced Renal Damage. International Journal of Molecular Sciences. 2020; 21(18):6512. https://doi.org/10.3390/ijms21186512
Chicago/Turabian StyleAparicio-Trejo, Omar Emiliano, Pedro Rojas-Morales, Sabino Hazael Avila-Rojas, Juan Carlos León-Contreras, Rogelio Hernández-Pando, Alexis Paulina Jiménez-Uribe, Rodrigo Prieto-Carrasco, Laura Gabriela Sánchez-Lozada, José Pedraza-Chaverri, and Edilia Tapia. 2020. "Temporal Alterations in Mitochondrial β-Oxidation and Oxidative Stress Aggravate Chronic Kidney Disease Development in 5/6 Nephrectomy Induced Renal Damage" International Journal of Molecular Sciences 21, no. 18: 6512. https://doi.org/10.3390/ijms21186512