The Emerging Relevance of AIM2 in Liver Disease

Abstract
:1. Introduction
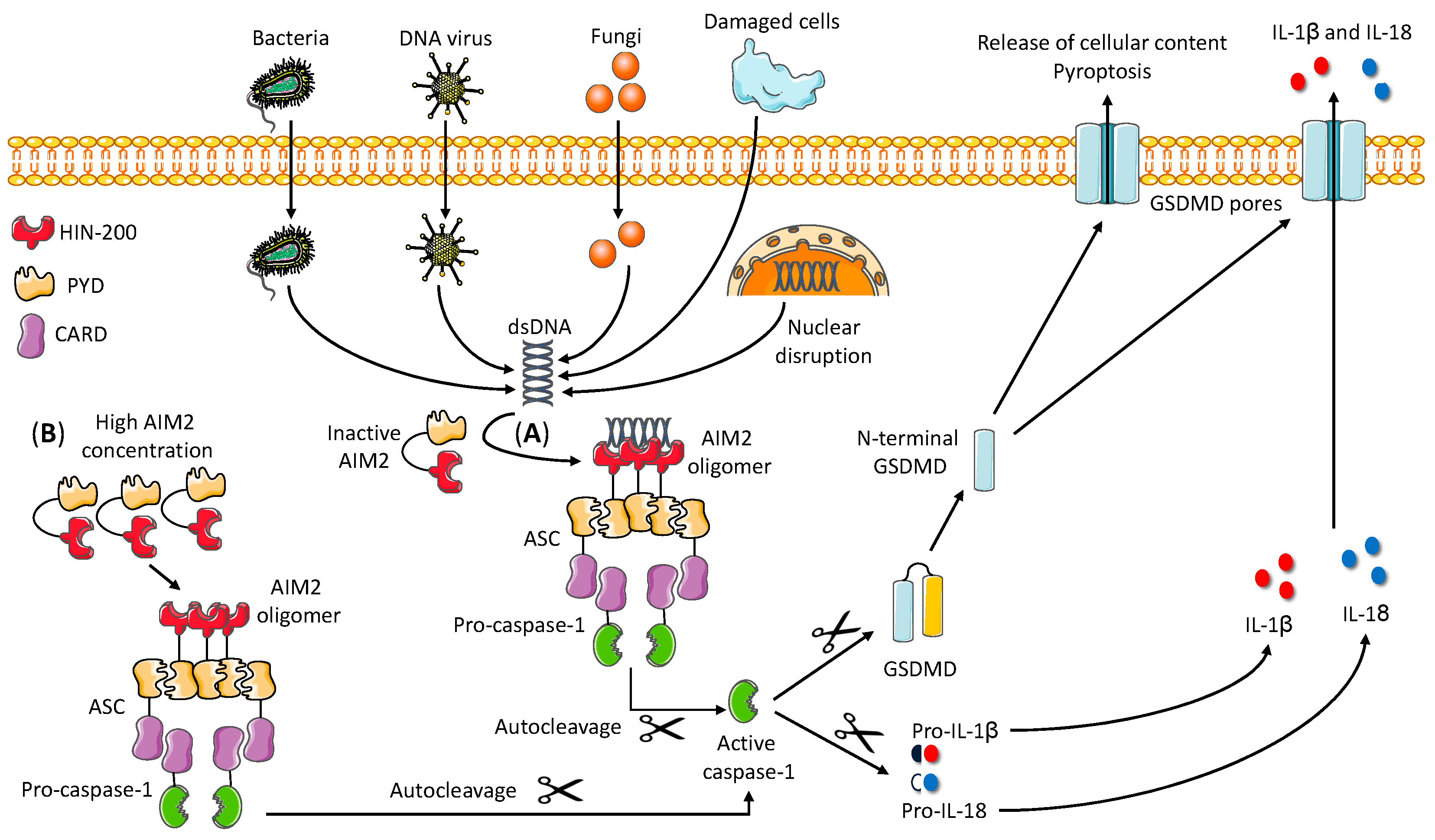
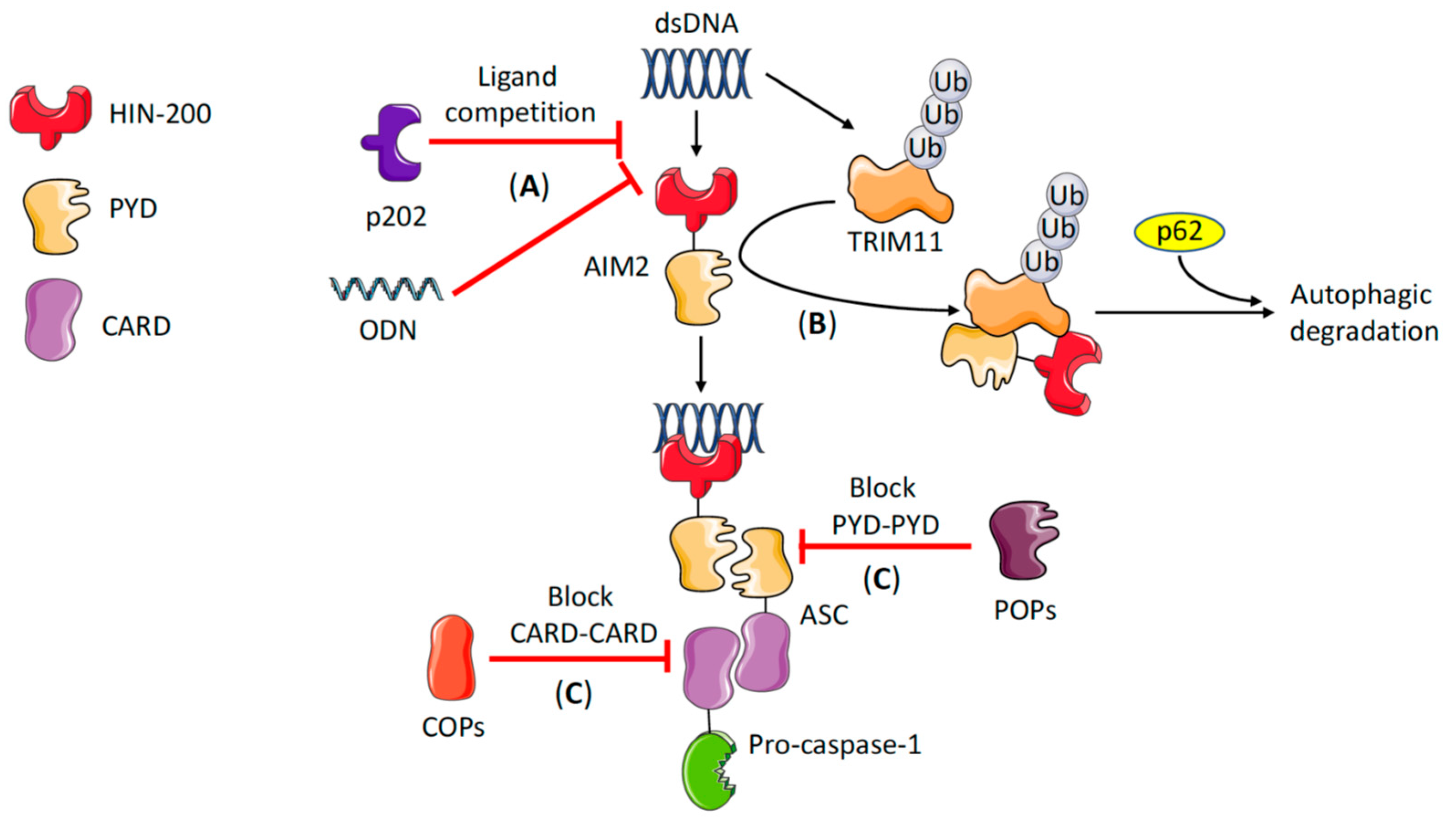
2. Brief Overview of AIM2 Inflammasome Activation and Regulation
2.1. Mechanism of AIM2 Inflammasome Assembly
2.2. AIM2 Inflammasome Regulation
3. AIM2 in Liver Disease
3.1. NAFLD and NASH
3.2. HBV Infection
3.3. Liver Fibrosis and Cirrhosis
3.4. Hepatocellular Carcinoma (HCC)
3.5. Acute Liver Failure (ALF)
4. Inflammasome-Independent Role of AIM2 in Health and Disease
5. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AHB | Acute hepatitis B |
| AIM2 | Absent in melanoma 2 |
| Akt | Protein kinase B |
| ALF | Acute liver failure |
| ALR | AIM2-like receptor |
| ASC | Apoptosis-associated speck-like protein containing a CARD |
| CARD | Caspase recruitment domain |
| cGAS | Cyclic GMP-AMP synthase |
| CHB | Chronic hepatitis B |
| CHC | Chronic hepatitis C |
| CLR | C-type lectin receptor |
| ConA | Concanavalin A |
| COP | CARD-only protein |
| CRC | Colorectal cancer |
| CXCL16 | C-X-C motif chemokine ligand 16 |
| DAMP | Damage-associated molecular pattern |
| DEN | Diethylnitrosamine |
| DNA-PK | DNA protein kinase |
| GSDMD | Gasdermin D |
| HBeAg | Hepatitis B e antigen |
| HBV | Hepatitis B virus |
| HCV | Hepatitis C virus |
| HCC | Hepatocellular carcinoma |
| HFD | High fat diet |
| HIN | Hematopoietic expression, interferon-inducible and nuclear localization |
| HMGB1 | High mobility group box 1 |
| HSC | Hepatic stellate cells |
| IFI16 | IFNγ-inducible protein 16 |
| IFN | Interferon |
| IKKα | IκB kinase alpha |
| IL | Interleukin |
| KC | Kupffer cell |
| MCD | Methionine-choline deficient diet |
| MyD88 | Myeloid differentiation factor 88 |
| NAFLD | Non-alcoholic fatty liver disease |
| NASH | Non-alcoholic steatohepatitis |
| NLR | NOD-like receptor |
| NLRC4 | NLR family, CARD domain-containing 4 |
| NLRP3 | NLR family, pyrin domain-containing 3 |
| NOD | Nucleotide oligomerization domain |
| ODN | Oligodeoxynucleotide |
| PBMC | Peripheral blood mononuclear cells |
| PAMP | Pathogen-associated molecular pattern |
| PI3K | Phosphatidylinositol 3 kinase |
| POP | PYD-only protein |
| PRR | Pattern recognition receptor |
| PYD | Pyrin domain |
| RLR | RIG-1 like receptor |
| STAT1 | Signal transducer and activator of transcription 1 |
| STING | Stimulator of interferon response cGAMP interactor 1 |
| TLR | Toll-like receptor |
| TRIM11 | Tripartite motif containing 11 |
| WT | Wild type |
References
- Fitzgerald, K.A.; Kagan, J.C. Toll-like Receptors and the Control of Immunity. Cell 2020, 180, 1044–1066. [Google Scholar] [CrossRef]
- Szabo, G.; Petrasek, J. Inflammasome activation and function in liver disease. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 387–400. [Google Scholar] [CrossRef]
- Shi, C.; Yang, H.; Zhang, Z. Involvement of Nucleotide-Binding Oligomerization Domain-Like Receptor Family Pyrin Domain Containing 3 Inflammasome in the Pathogenesis of Liver Diseases. Front. Cell Dev. Biol. 2020, 8, 139. [Google Scholar] [CrossRef] [Green Version]
- Xu, T.; Du, Y.; Fang, X.-B.; Chen, H.; Zhou, D.-D.; Wang, Y.; Zhang, L. New insights into Nod-like receptors (NLRs) in liver diseases. Int. J. Physiol. Pathophysiol. Pharm. 2018, 10, 1–16. [Google Scholar]
- Szabo, G.; Csak, T. Inflammasomes in liver diseases. J. Hepatol. 2012, 57, 642–654. [Google Scholar] [CrossRef] [Green Version]
- Luan, J.; Ju, D. Inflammasome: A Double-Edged Sword in Liver Diseases. Front. Immunol. 2018, 9, 2201. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Kayagaki, N.; Wong, M.T.; Stowe, I.B.; Ramani, S.R.; Gonzalez, L.C.; Akashi-Takamura, S.; Miyake, K.; Zhang, J.; Lee, W.P.; Muszyński, A.; et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 2013, 341, 1246–1249. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef] [Green Version]
- Albrecht, M.; Choubey, D.; Lengauer, T. The HIN domain of IFI-200 proteins consists of two OB folds. Biochem. Biophys. Res. Commun. 2005, 327, 679–687. [Google Scholar] [CrossRef]
- Flynn, R.L.; Zou, L. Oligonucleotide/oligosaccharide-binding fold proteins: A growing family of genome guardians. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 266–275. [Google Scholar] [CrossRef] [Green Version]
- Martinon, F.; Hofmann, K.; Tschopp, J. The pyrin domain: A possible member of the death domain-fold family implicated in apoptosis and inflammation. Curr. Biol. 2001, 11, 118–120. [Google Scholar] [CrossRef] [Green Version]
- Lugrin, J.; Martinon, F. The AIM2 inflammasome: Sensor of pathogens and cellular perturbations. Immunol. Rev. 2018, 281, 99–114. [Google Scholar] [CrossRef]
- Jin, T.; Perry, A.; Jiang, J.; Smith, P.; Curry, J.A.; Unterholzner, L.; Jiang, Z.; Horvath, G.; Rathinam, V.A.; Johnstone, R.W.; et al. Structures of the HIN domain:DNA complexes reveal ligand binding and activation mechanisms of the AIM2 inflammasome and IFI16 receptor. Immunity 2012, 36, 561–571. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Yin, Q. AIM2 inflammasome activation and regulation: A structural perspective. J. Struct. Biol. 2017, 200, 279–282. [Google Scholar] [CrossRef]
- Morrone, S.R.; Matyszewski, M.; Yu, X.; Delannoy, M.; Egelman, E.H.; Sohn, J. Assembly-driven activation of the AIM2 foreign-dsDNA sensor provides a polymerization template for downstream ASC. Nat. Commun. 2015, 6, 7827. [Google Scholar] [CrossRef]
- Fernandes-Alnemri, T.; Yu, J.-W.; Datta, P.; Wu, J.; Alnemri, E.S. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 2009, 458, 509–513. [Google Scholar] [CrossRef] [Green Version]
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; Bauernfeind, F.; Horvath, G.; Caffrey, D.R.; Latz, E.; Fitzgerald, K.A. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009, 458, 514–518. [Google Scholar] [CrossRef] [Green Version]
- Dick, M.S.; Sborgi, L.; Rühl, S.; Hiller, S.; Broz, P. ASC filament formation serves as a signal amplification mechanism for inflammasomes. Nat. Commun. 2016, 7, 11929. [Google Scholar] [CrossRef] [Green Version]
- Hoss, F.; Rodriguez-Alcazar, J.F.; Latz, E. Assembly and regulation of ASC specks. Cell. Mol. Life Sci. 2017, 74, 1211–1229. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Fang, R.; Hara, H.; Sakai, S.; Hernandez-Cuellar, E.; Mitsuyama, M.; Kawamura, I.; Tsuchiya, K. Type I interferon signaling regulates activation of the absent in melanoma 2 inflammasome during Streptococcus pneumoniae infection. Infect. Immun. 2014, 82, 2310–2317. [Google Scholar] [CrossRef] [Green Version]
- González-Navajas, J.M.; Lee, J.; David, M.; Raz, E. Immunomodulatory functions of type I interferons. Nat. Rev. Immunol. 2012, 12, 125–135. [Google Scholar] [CrossRef] [Green Version]
- Adamiak, M.; Ciechanowicz, A.; Skoda, M.; Cymer, M.; Tracz, M.; Xu, B.; Ratajczak, M.Z. Novel Evidence that Purinergic Signaling—Nlrp3 Inflammasome Axis Regulates Circadian Rhythm of Hematopoietic Stem/Progenitor Cells Circulation in Peripheral Blood. Stem Cell Rev. Rep. 2020, 16, 335–343. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Liu, Z.; Xiao, T.S. Post-translational regulation of inflammasomes. Cell. Mol. Immunol. 2017, 14, 65–79. [Google Scholar] [CrossRef] [Green Version]
- Hara, H.; Tsuchiya, K.; Kawamura, I.; Fang, R.; Hernandez-Cuellar, E.; Shen, Y.; Mizuguchi, J.; Schweighoffer, E.; Tybulewicz, V.; Mitsuyama, M. Phosphorylation of the adaptor ASC acts as a molecular switch that controls the formation of speck-like aggregates and inflammasome activity. Nat. Immunol. 2013, 14, 1247–1255. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.-C.; Huang, D.-Y.; Wang, J.-S.; Lin, Y.-L.; Hsieh, S.-L.; Huang, K.-C.; Lin, W.-W. Syk is involved in NLRP3 inflammasome-mediated caspase-1 activation through adaptor ASC phosphorylation and enhanced oligomerization. J. Leukoc. Biol. 2015, 97, 825–835. [Google Scholar] [CrossRef]
- Martin, B.N.; Wang, C.; Willette-Brown, J.; Herjan, T.; Gulen, M.F.; Zhou, H.; Bulek, K.; Franchi, L.; Sato, T.; Alnemri, E.S.; et al. IKKα negatively regulates ASC-dependent inflammasome activation. Nat. Commun. 2014, 5, 4977. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Tang, Q.; Liu, K.; Xie, W.; Liu, X.; Wang, H.; Wang, R.-F.; Cui, J. TRIM11 Suppresses AIM2 Inflammasome by Degrading AIM2 via p62-Dependent Selective Autophagy. Cell Rep. 2016, 16, 1988–2002. [Google Scholar] [CrossRef] [Green Version]
- Dorfleutner, A.; Chu, L.; Stehlik, C. Inhibiting the inflammasome: One domain at a time. Immunol. Rev. 2015, 265, 205–216. [Google Scholar] [CrossRef] [Green Version]
- Stehlik, C.; Krajewska, M.; Welsh, K.; Krajewski, S.; Godzik, A.; Reed, J.C. The PAAD/PYRIN-only protein POP1/ASC2 is a modulator of ASC-mediated nuclear-factor-kappa B and pro-caspase-1 regulation. Biochem. J. 2003, 373, 101–113. [Google Scholar] [CrossRef]
- Dorfleutner, A.; Bryan, N.B.; Talbott, S.J.; Funya, K.N.; Rellick, S.L.; Reed, J.C.; Shi, X.; Rojanasakul, Y.; Flynn, D.C.; Stehlik, C. Cellular pyrin domain-only protein 2 is a candidate regulator of inflammasome activation. Infect. Immun. 2007, 75, 1484–1492. [Google Scholar] [CrossRef] [Green Version]
- Khare, S.; Ratsimandresy, R.A.; de Almeida, L.; Cuda, C.M.; Rellick, S.L.; Misharin, A.V.; Wallin, M.C.; Gangopadhyay, A.; Forte, E.; Gottwein, E.; et al. The PYRIN domain-only protein POP3 inhibits ALR inflammasomes and regulates responses to infection with DNA viruses. Nat. Immunol. 2014, 15, 343–353. [Google Scholar] [CrossRef]
- Roberts, T.L.; Idris, A.; Dunn, J.A.; Kelly, G.M.; Burnton, C.M.; Hodgson, S.; Hardy, L.L.; Garceau, V.; Sweet, M.J.; Ross, I.L.; et al. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science 2009, 323, 1057–1060. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Wang, J.; Wang, J.; Cao, L.-S.; Wang, Z.-X.; Wu, J.-W. Structural mechanism of DNA recognition by the p202 HINa domain: Insights into the inhibition of Aim2-mediated inflammatory signalling. Acta Cryst. F Struct. Biol. Commun. 2014, 70, 21–29. [Google Scholar] [CrossRef]
- Yin, Q.; Sester, D.P.; Tian, Y.; Hsiao, Y.-S.; Lu, A.; Cridland, J.A.; Sagulenko, V.; Thygesen, S.J.; Choubey, D.; Hornung, V.; et al. Molecular mechanism for p202-mediated specific inhibition of AIM2 inflammasome activation. Cell Rep. 2013, 4, 327–339. [Google Scholar] [CrossRef] [Green Version]
- Kaminski, J.J.; Schattgen, S.A.; Tzeng, T.-C.; Bode, C.; Klinman, D.M.; Fitzgerald, K.A. Synthetic oligodeoxynucleotides containing suppressive TTAGGG motifs inhibit AIM2 inflammasome activation. J. Immunol. 2013, 191, 3876–3883. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef]
- Ballestri, S.; Nascimbeni, F.; Romagnoli, D.; Lonardo, A. The independent predictors of non-alcoholic steatohepatitis and its individual histological features.: Insulin resistance, serum uric acid, metabolic syndrome, alanine aminotransferase and serum total cholesterol are a clue to pathogenesis and candidate. Hepatol. Res. 2016, 46, 1074–1087. [Google Scholar] [CrossRef]
- Eckel, R.H.; Grundy, S.M.; Zimmet, P.Z. The metabolic syndrome. Lancet 2005, 365, 1415–1428. [Google Scholar] [CrossRef]
- Diehl, A.M.; Farpour-Lambert, N.J.; Zhao, L.; Tilg, H. Why we need to curb the emerging worldwide epidemic of nonalcoholic fatty liver disease. Nat. Metab. 2019, 1, 1027–1029. [Google Scholar] [CrossRef] [PubMed]
- Witek, R.P.; Stone, W.C.; Karaca, F.G.; Syn, W.-K.; Pereira, T.A.; Agboola, K.M.; Omenetti, A.; Jung, Y.; Teaberry, V.; Choi, S.S.; et al. Pan-caspase inhibitor VX-166 reduces fibrosis in an animal model of nonalcoholic steatohepatitis. Hepatology 2009, 50, 1421–1430. [Google Scholar] [CrossRef] [PubMed]
- Csak, T.; Ganz, M.; Pespisa, J.; Kodys, K.; Dolganiuc, A.; Szabo, G. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology 2011, 54, 133–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandanmagsar, B.; Youm, Y.-H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef]
- Stienstra, R.; Saudale, F.; Duval, C.; Keshtkar, S.; Groener, J.E.M.; van Rooijen, N.; Staels, B.; Kersten, S.; Müller, M. Kupffer cells promote hepatic steatosis via interleukin-1β-dependent suppression of peroxisome proliferator-activated receptor α activity. Hepatology 2010, 51, 511–522. [Google Scholar] [CrossRef]
- Miura, K.; Kodama, Y.; Inokuchi, S.; Schnabl, B.; Aoyama, T.; Ohnishi, H.; Olefsky, J.M.; Brenner, D.A.; Seki, E. Toll-Like Receptor 9 Promotes Steatohepatitis by Induction of Interleukin-1β in Mice. Gastroenterology 2010, 139, 323–334.e7. [Google Scholar] [CrossRef] [Green Version]
- Csak, T.; Pillai, A.; Ganz, M.; Lippai, D.; Petrasek, J.; Park, J.-K.; Kodys, K.; Dolganiuc, A.; Kurt-Jones, E.A.; Szabo, G. Both bone marrow-derived and non-bone marrow-derived cells contribute to AIM2 and NLRP3 inflammasome activation in a MyD88-dependent manner in dietary steatohepatitis. Liver Int. 2014, 34, 1402–1413. [Google Scholar] [CrossRef] [Green Version]
- Sims, G.P.; Rowe, D.C.; Rietdijk, S.T.; Herbst, R.; Coyle, A.J. HMGB1 and RAGE in inflammation and cancer. Annu. Rev. Immunol. 2010, 28, 367–388. [Google Scholar] [CrossRef]
- Ganz, M.; Csak, T.; Szabo, G. High fat diet feeding results in gender specific steatohepatitis and inflammasome activation. World J. Gastroenterol. 2014, 20, 8525–8534. [Google Scholar] [CrossRef]
- Gong, Z.; Zhang, X.; Su, K.; Jiang, R.; Sun, Z.; Chen, W.; Forno, E.; Goetzman, E.S.; Wang, J.; Dong, H.H.; et al. Deficiency in AIM2 induces inflammation and adipogenesis in white adipose tissue leading to obesity and insulin resistance. Diabetologia 2019, 62, 2325–2339. [Google Scholar] [CrossRef]
- Trépo, C.; Chan, H.L.Y.; Lok, A. Hepatitis B virus infection. Lancet 2014, 384, 2053–2063. [Google Scholar] [CrossRef]
- Wu, D.-L.; Xu, G.-H.; Lu, S.-M.; Ma, B.-L.; Miao, N.-Z.; Liu, X.-B.; Cheng, Y.; Feng, J.-H.; Liu, Z.-G.; Ding, F.; et al. Correlation of AIM2 expression in peripheral blood mononuclear cells from humans with acute and chronic hepatitis B. Hum. Immunol. 2013, 74, 514–521. [Google Scholar] [CrossRef]
- Croagh, C.M.N.; Lubel, J.S. Natural history of chronic hepatitis B: Phases in a complex relationship. World J. Gastroenterol. 2014, 20, 10395–10404. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; He, G.; Chen, Y.; Zhang, X.; Wu, S. Differential Activation of NLRP3, AIM2, and IFI16 Inflammasomes in Humans with Acute and Chronic Hepatitis B. Viral Immunol. 2018, 31, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Chen, Z.; Hou, R.; Yan, D.; Liu, C.; Chen, S.; Li, X.; Du, W. Expression of AIM2 is correlated with increased inflammation in chronic hepatitis B patients. Virol. J. 2015, 12, 129. [Google Scholar] [CrossRef] [Green Version]
- Pan, X.; Xu, H.; Zheng, C.; Li, M.; Zou, X.; Cao, H.; Xu, Q. Human hepatocytes express absent in melanoma 2 and respond to hepatitis B virus with interleukin-18 expression. Virus Genes 2016, 52, 445–452. [Google Scholar] [CrossRef]
- Du, W.; Zhen, J.; Zheng, Z.; Ma, S.; Chen, S. Expression of AIM2 is high and correlated with inflammation in hepatitis B virus associated glomerulonephritis. J. Inflamm. (Lond.) 2013, 10, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellicoro, A.; Ramachandran, P.; Iredale, J.P.; Fallowfield, J.A. Liver fibrosis and repair: Immune regulation of wound healing in a solid organ. Nat. Rev. Immunol. 2014, 14, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Mederacke, I.; Hsu, C.C.; Troeger, J.S.; Huebener, P.; Mu, X.; Dapito, D.H.; Pradere, J.-P.; Schwabe, R.F. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat. Commun. 2013, 4, 2823. [Google Scholar] [CrossRef] [Green Version]
- George, J.; Tsuchishima, M.; Tsutsumi, M. Metabolism of N-nitrosodimethylamine, methylation of macromolecules, and development of hepatic fibrosis in rodent models. J. Mol. Med. 2020, 98, 1203–1213. [Google Scholar] [CrossRef]
- Ge, P.S.; Runyon, B.A. Treatment of Patients with Cirrhosis. N. Engl. J. Med. 2016, 375, 767–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alegre, F.; Pelegrin, P.; Feldstein, A.E. Inflammasomes in Liver Fibrosis. Semin. Liver Dis. 2017, 37, 119–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozano-Ruiz, B.; Bachiller, V.; García-Martínez, I.; Zapater, P.; Gómez-Hurtado, I.; Moratalla, A.; Giménez, P.; Bellot, P.; Francés, R.; Such, J.; et al. Absent in melanoma 2 triggers a heightened inflammasome response in ascitic fluid macrophages of patients with cirrhosis. J. Hepatol. 2015, 62, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Arriola Benitez, P.C.; Pesce Viglietti, A.I.; Gomes, M.T.R.; Oliveira, S.C.; Quarleri, J.F.; Giambartolomei, G.H.; Delpino, M.V. Brucella abortus Infection Elicited Hepatic Stellate Cell-Mediated Fibrosis Through Inflammasome-Dependent IL-1β Production. Front. Immunol. 2019, 10, 3036. [Google Scholar] [CrossRef]
- Chen, T.T.W.; Cheng, P.C.; Chang, K.C.; Cao, J.P.; Feng, J.L.; Chen, C.C.; Lam, H.Y.P.; Peng, S.Y. Activation of the NLRP3 and AIM2 inflammasomes in a mouse model of Schistosoma mansoni infection. J. Helminthol. 2019, 94, 72. [Google Scholar] [CrossRef]
- Andrade, Z.A. Schistosomiasis and liver fibrosis. Parasite Immunol. 2009, 31, 656–663. [Google Scholar] [CrossRef]
- Llovet, J.M.; Zucman-Rossi, J.; Pikarsky, E.; Sangro, B.; Schwartz, M.; Sherman, M.; Gores, G. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2016, 2, 16018. [Google Scholar] [CrossRef]
- Ringelhan, M.; Pfister, D.; O’Connor, T.; Pikarsky, E.; Heikenwalder, M. The immunology of hepatocellular carcinoma. Nat. Immunol. 2018, 19, 222–232. [Google Scholar] [CrossRef]
- DeYoung, K.L.; Ray, M.E.; Su, Y.A.; Anzick, S.L.; Johnstone, R.W.; Trapani, J.A.; Meltzer, P.S.; Trent, J.M. Cloning a novel member of the human interferon-inducible gene family associated with control of tumorigenicity in a model of human melanoma. Oncogene 1997, 15, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Farshchian, M.; Nissinen, L.; Siljamäki, E.; Riihilä, P.; Piipponen, M.; Kivisaari, A.; Kallajoki, M.; Grénman, R.; Peltonen, J.; Peltonen, S.; et al. Tumor cell-specific AIM2 regulates growth and invasion of cutaneous squamous cell carcinoma. Oncotarget 2017, 8, 45825–45836. [Google Scholar] [CrossRef]
- Man, S.M.; Zhu, Q.; Zhu, L.; Liu, Z.; Karki, R.; Malik, A.; Sharma, D.; Li, L.; Malireddi, R.K.S.; Gurung, P.; et al. Critical Role for the DNA Sensor AIM2 in Stem Cell Proliferation and Cancer. Cell 2015, 162, 45–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, J.E.; Petrucelli, A.S.; Chen, L.; Koblansky, A.A.; Truax, A.D.; Oyama, Y.; Rogers, A.B.; Brickey, W.J.; Wang, Y.; Schneider, M.; et al. Inflammasome-independent role of AIM2 in suppressing colon tumorigenesis via DNA-PK and Akt. Nat. Med. 2015, 21, 906–913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dihlmann, S.; Tao, S.; Echterdiek, F.; Herpel, E.; Jansen, L.; Chang-Claude, J.; Brenner, H.; Hoffmeister, M.; Kloor, M. Lack of Absent in Melanoma 2 (AIM2) expression in tumor cells is closely associated with poor survival in colorectal cancer patients. Int. J. Cancer 2014, 135, 2387–2396. [Google Scholar] [CrossRef]
- Woerner, S.M.; Kloor, M.; Schwitalle, Y.; Youmans, H.; von Doeberitz, M.K.; Gebert, J.; Dihlmann, S. The putative tumor suppressor AIM2 is frequently affected by different genetic alterations in microsatellite unstable colon cancers. Genes Chromosomes Cancer 2007, 46, 1080–1089. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.-F.; Ou-Yang, F.; Hung, J.-Y.; Liu, J.-C.; Wang, H.; Wang, S.-C.; Hou, M.-F.; Hortobagyi, G.N.; Hung, M.-C. AIM2 suppresses human breast cancer cell proliferation in vitro and mammary tumor growth in a mouse model. Mol. Cancer Ther. 2006, 5, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Guo, P.; Qiu, Y.; Mu, K.; Zhu, L.; Zhao, W.; Li, T.; Han, L. Loss of AIM2 expression promotes hepatocarcinoma progression through activation of mTOR-S6K1 pathway. Oncotarget 2016, 7, 36185–36197. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.-L.; Liu, L.-L.; Lu, S.-X.; Luo, R.-Z.; Wang, C.-H.; Wang, H.; Cai, S.-H.; Yang, X.; Xie, D.; Zhang, C.Z.; et al. HBx-mediated decrease of AIM2 contributes to hepatocellular carcinoma metastasis. Mol. Oncol. 2017, 11, 1225–1240. [Google Scholar] [CrossRef] [PubMed]
- Sonohara, F.; Inokawa, Y.; Kanda, M.; Nishikawa, Y.; Yamada, S.; Fujii, T.; Sugimoto, H.; Kodera, Y.; Nomoto, S. Association of Inflammasome Components in Background Liver with Poor Prognosis After Curatively-resected Hepatocellular Carcinoma. Anticancer Res. 2017, 37, 293–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Cardona, C.; Lozano-Ruiz, B.; Bachiller, V.; Peiró, G.; Algaba-Chueca, F.; Gómez-Hurtado, I.; Such, J.; Zapater, P.; Francés, R.; González-Navajas, J.M. AIM2 deficiency reduces the development of hepatocellular carcinoma in mice. Int. J. Cancer 2018, 143, 2997–3007. [Google Scholar] [CrossRef] [Green Version]
- Macek Jilkova, Z.; Kurma, K.; Decaens, T. Animal Models of Hepatocellular Carcinoma: The Role of Immune System and Tumor Microenvironment. Cancers (Basel) 2019, 11, 1487. [Google Scholar] [CrossRef] [Green Version]
- Rovegno, M.; Vera, M.; Ruiz, A.; Benítez, C. Current concepts in acute liver failure. Ann. Hepatol. 2019, 18, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Stravitz, R.T.; Lee, W.M. Acute liver failure. Lancet 2019, 394, 869–881. [Google Scholar] [CrossRef]
- Woolbright, B.L.; Jaeschke, H. Role of the inflammasome in acetaminophen-induced liver injury and acute liver failure. J. Hepatol. 2017, 66, 836–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Lou, G.; Zhou, X.; Zheng, M.; He, J.; Chen, Z. MicroRNA-223 acts as an important regulator to Kupffer cells activation at the early stage of Con A-induced acute liver failure via AIM2 signaling pathway. Cell. Physiol. Biochem. 2014, 34, 2137–2152. [Google Scholar] [CrossRef]
- Yuan, X.; Berg, N.; Lee, J.W.; Le, T.-T.; Neudecker, V.; Jing, N.; Eltzschig, H. MicroRNA miR-223 as regulator of innate immunity. J. Leukoc. Biol. 2018, 104, 515–524. [Google Scholar] [CrossRef]
- Neudecker, V.; Haneklaus, M.; Jensen, O.; Khailova, L.; Masterson, J.C.; Tye, H.; Biette, K.; Jedlicka, P.; Brodsky, K.S.; Gerich, M.E.; et al. Myeloid-derived miR-223 regulates intestinal inflammation via repression of the NLRP3 inflammasome. J. Exp. Med. 2017, 214, 1737–1752. [Google Scholar] [CrossRef]
- Chen, J.; Wang, Z.; Yu, S. AIM2 regulates viability and apoptosis in human colorectal cancer cells via the PI3K/Akt pathway. Onco Targets Ther. 2017, 10, 811–817. [Google Scholar] [CrossRef] [Green Version]
- El-Zaatari, M.; Bishu, S.; Zhang, M.; Grasberger, H.; Hou, G.; Haley, H.; Humphries, B.; Syu, L.-J.; Dlugosz, A.A.; Luker, K.; et al. Aim2-mediated/IFN-β-independent regulation of gastric metaplastic lesions via CD8+ T cells. JCI Insight 2020, 5, 94035. [Google Scholar] [CrossRef]
- Furrer, A.; Hottiger, M.O.; Valaperti, A. Absent in Melanoma 2 (AIM2) limits pro-inflammatory cytokine transcription in cardiomyocytes by inhibiting STAT1 phosphorylation. Mol. Immunol. 2016, 74, 47–58. [Google Scholar] [CrossRef] [Green Version]
- Qi, M.; Dai, D.; Liu, J.; Li, Z.; Liang, P.; Wang, Y.; Cheng, L.; Zhan, Y.; An, Z.; Song, Y.; et al. AIM2 promotes the development of non-small cell lung cancer by modulating mitochondrial dynamics. Oncogene 2020, 39, 2707–2723. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Disease | AIM2 Expression and Function | References |
|---|---|---|
| NAFLD and NASH | Increased AIM2 expression in MCD and HFD models. Associated with NASH in MCD model | [47,49] |
| Aim2−/− mice show signs of spontaneous metabolic disease | [50] | |
| HBV infection | High AIM2 expression in PBMCs during acute hepatitis B and during the clearance phase of chronic hepatitis B | [52,54] |
| Expressed in liver cells of chronic hepatitis B patients. Higher expression correlates with more severe liver inflammation | [55,56] | |
| HBV genome triggers AIM2 inflammasome activation in HepG2 cell line | [56] | |
| Expressed in kidney tissue of patients with HBV-associated glomerulonephritis | [57] | |
| Liver fibrosis and cirrhosis | High AIM2 expression and activation in macrophages from ascitic fluid of patients with advanced cirrhosis | [63] |
| High AIM2 activation in ascitic fluid macrophages correlates with severity of cirrhosis | [63] | |
| B. abortus triggers AIM2-dependent IL-1β production by human HSCs | [64] | |
| Aim2−/− mice develop reduced liver fibrosis after B. abortus infection. AIM2 expression is elevated in the liver of mice infected with S. mansoni | [64,65] | |
| HCC | Reduced AIM2 expression in HCC tissue compared with non-cancerous liver tissue of the same patients | [76,77,78] |
| Lower expression correlates with more advanced HCC features | [76,77] | |
| DEN-induced liver damage potentiates AIM2 inflammasome activation in KCs | [79] | |
| Aim2−/− mice show lower inflammasome activation and reduced HCC development in the DEN model | [79] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lozano-Ruiz, B.; González-Navajas, J.M. The Emerging Relevance of AIM2 in Liver Disease. Int. J. Mol. Sci. 2020, 21, 6535. https://doi.org/10.3390/ijms21186535
Lozano-Ruiz B, González-Navajas JM. The Emerging Relevance of AIM2 in Liver Disease. International Journal of Molecular Sciences. 2020; 21(18):6535. https://doi.org/10.3390/ijms21186535
Chicago/Turabian StyleLozano-Ruiz, Beatriz, and José M. González-Navajas. 2020. "The Emerging Relevance of AIM2 in Liver Disease" International Journal of Molecular Sciences 21, no. 18: 6535. https://doi.org/10.3390/ijms21186535
APA StyleLozano-Ruiz, B., & González-Navajas, J. M. (2020). The Emerging Relevance of AIM2 in Liver Disease. International Journal of Molecular Sciences, 21(18), 6535. https://doi.org/10.3390/ijms21186535