Metabolism Regulation and Redox State: Insight into the Role of Superoxide Dismutase 1



 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Redox Regulation of Metabolism
1.1. Cellular Sources of ROS and Antioxidant Systems
1.2. Impact of Nutrients on ROS Metabolism
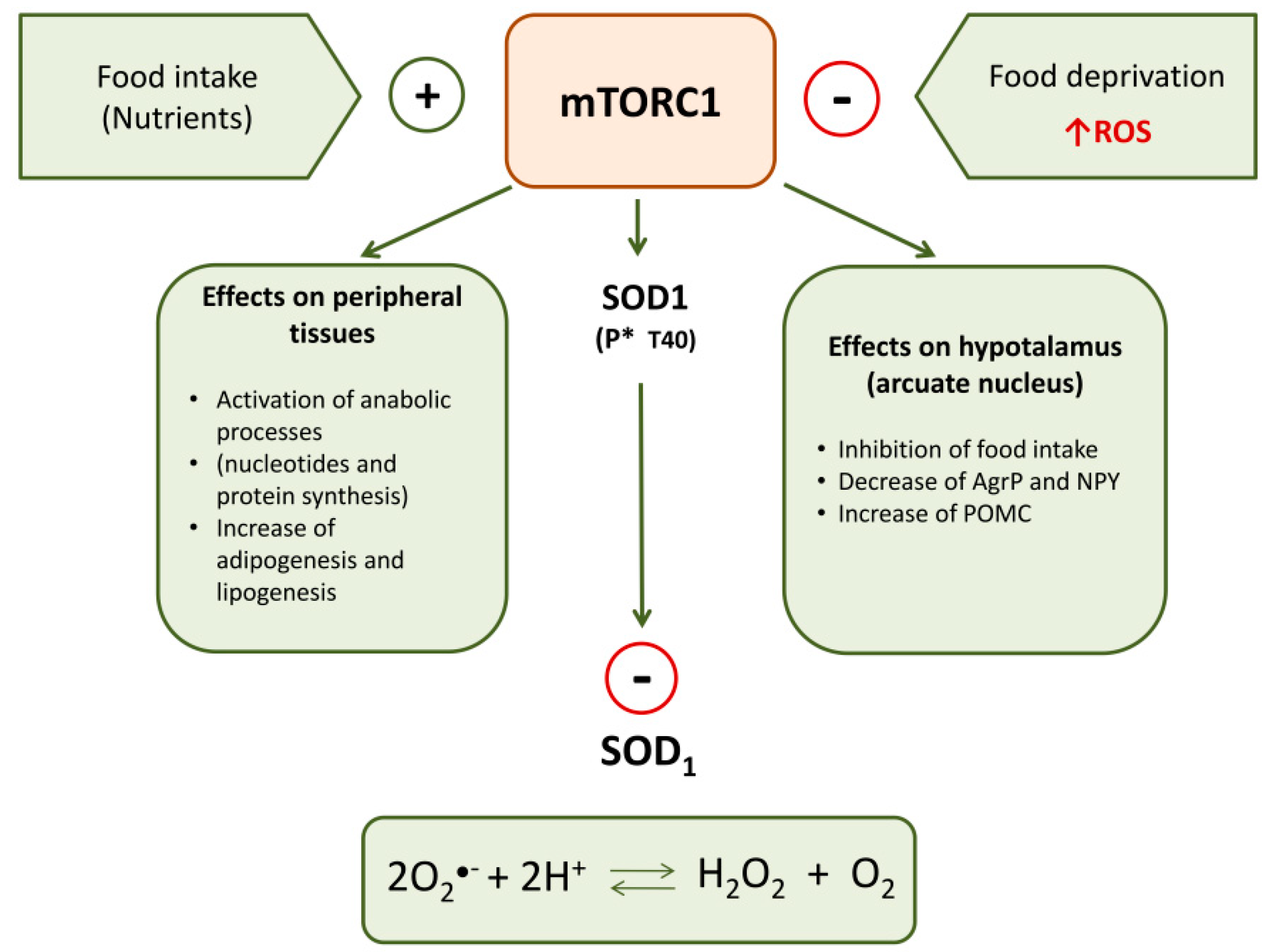
2. Superoxide Dismutase 1 and mTOR Signaling
2.1. mTOR Complexes
2.2. mTOR in the Hypothalamic Control of Food Intake and Energy Balance
2.3. Modulation of SOD1 Activity by mTORC1
3. SOD1, Diet and Cholesterol Homeostasis
3.1. SOD1 as Target of Dietary Interventions
3.2. Presence of SOD1 in Serum Lipoprotein
3.3. Effects of SOD1 on HMGCoA Reductase and LDL Receptor
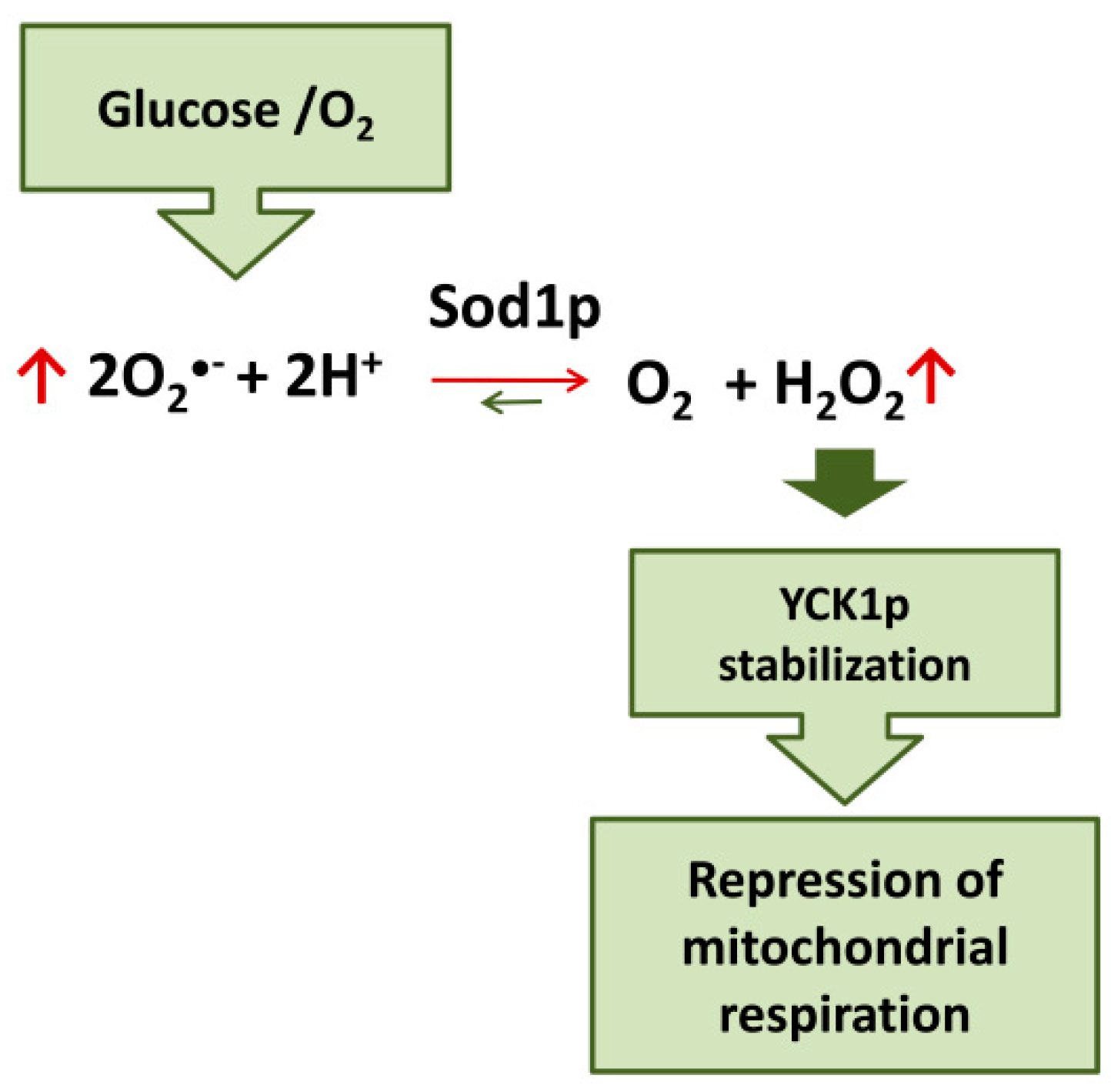
4. SOD1-Mediated Repression of Mitochondrial Respiration
5. Redox and Metabolic Dysregulation in Mutant SOD1 Linked Familial Amyotrophic Lateral Sclerosis
6. SOD1 in T Cell Activation
7. SOD1 Functions beyond Its Role as Superoxide Scavenger
8. Concluding Remarks and Future Directions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Acb1 | Acyl-CoA binding protein |
| ACC1 and ACC2 | Acetyl-CoA carboxylases |
| AgRP | Agouti-related peptide |
| Akt | Protein kinase B |
| ALS | Amyotrophic lateral sclerosis |
| AMPK | 5′ adenosine monophosphate-activated protein kinase |
| ARC | Arcuate nucleus |
| Bcl-2 | B-cell lymphoma 2 |
| CART | Cocaine- and amphetamine-regulated transcript |
| Cebpα | Transcriptional Repressor of T-Cell |
| CK1γ | Casein kinase 1-gamma |
| CoQH2 | Reduced coenzyme Q |
| DUOX | Dual oxidases |
| ERK1-2 | extracellular signal-regulated kinase |
| ETC | Electron transport chain |
| fALS | Familial ALS |
| FFA | Free fatty acid |
| GSH-Px | Glutathione peroxidase |
| HDL | High density lipoproteins |
| HepG2 | Human hepatocarcinoma cell line |
| HFD | High-fat-diet |
| HMGCoA | 3-hydroxy-3-methylglutaryl-CoA |
| HMG-CoA | Microsomal enzyme 3-hydroxy-3-methylglutaryl CoA |
| IGF-1 | Insulin/insulin-like growth factor-1 |
| LDL | Low density lipoproteins |
| LTP | Long term potentiation |
| MBH | Medial-basal hypothalamus |
| MEFs | Mouse embryonic fibroblasts |
| MnSOD | Manganese superoxide dismutase |
| mTORC1 | Mechanistic target-of-rapamycin complex 1 |
| NOXs | NADPH oxidase enzymes |
| NPY | Neuropeptide Y |
| NSC-34 | Mouse Motor Neuron-Like Hybrid Cell Line |
| OXYPHOS | Oxidative phosphorylation |
| PGC-1α | Peroxisome proliferative activated receptor, gamma, coactivator 1 |
| PI3K | Phosphoinositide 3-kinases |
| PKC | Protein kinase C |
| POMC | Anorexigenic neurons coexpressing proopiomelanocortin |
| PPAR | Peroxisome proliferator-activated receptor |
| RET | Reverse electron transport |
| ROS | Reactive oxygen species |
| sALS | Sporadic ALS |
| SGK1 | Serum-and glucocorticoid-induced protein kinase-1 |
| SIRT3 | Sirtuin3 |
| SK-N-BE | Human neuroblastoma cell line |
| SOD | Superoxide dismutase |
| TCR | T cell receptor |
| UCP | Mitochondrial uncoupling protein |
References
- Squadrito, G.L.; Pryor, W.A. Oxidative chemistry of nitric oxide: The roles of superoxide, peroxynitrite, and carbon dioxide. Free Radic. Biol. Med. 1998, 25, 392–403. [Google Scholar] [CrossRef]
- Drougard, A.; Fournel, A.; Valet, P.; Knauf, C. Impact of hypothalamic reactive oxygen species in the regulation of energy metabolism and food intake. Front. Neurosci. 2015, 9, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Accetta, R.; Damiano, S.; Morano, A.; Mondola, P.; Paternò, R.; Avvedimento, E.V.; Santillo, M. Reactive Oxygen Species Derived from NOX3 and NOX5 Drive Differentiation of Human Oligodendrocytes. Front. Cell. Neurosci. 2016, 10, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxid. Med. Cell. Longev. 2016, 2016, 4350965. [Google Scholar] [CrossRef] [Green Version]
- Damiano, S.; Sasso, A.; Accetta, R.; Monda, M.; De Luca, B.; Pavone, L.M.; Belfiore, A.; Santillo, M.; Mondola, P. Effect of Mutated Cu, Zn Superoxide Dismutase (SOD1(G93A)) on Modulation of Transductional Pathway Mediated by M1 Muscarinic Receptor in SK-N-BE and NSC-34 Cells. Front. Physiol. 2018, 9, 611. [Google Scholar] [CrossRef] [Green Version]
- De Felice, B.; Damiano, S.; Montanino, C.; del Buono, A.; La Rosa, G.; Guida, B.; Santillo, M. Effect of beta- and alpha-glucans on immune modulating factors expression in enterocyte-like Caco-2 and goblet-like LS 174T cells. Int. J. Biol. Macromol. 2020, 153, 600–607. [Google Scholar] [CrossRef]
- Brieger, K.; Schiavone, S.; Miller, F.J., Jr.; Krause, K.H. Reactive oxygen species: From health to disease. Swiss Med. Wkly. 2012, 142, w13659. [Google Scholar] [CrossRef]
- Panieri, E.; Santoro, M.M. ROS homeostasis and metabolism: A dangerous liason in cancer cells. Cell Death Dis. 2016, 7, e2253. [Google Scholar] [CrossRef]
- Damiano, S.; Sasso, A.; de Felice, B.; Terrazzano, G.; Bresciamorra, V.; Carotenuto, A.; Orefice, N.S.; Orefice, G.; Vacca, G.; Belfiore, A.; et al. The IFN-β 1b effect on Cu Zn superoxide dismutase (SOD1) in peripheral mononuclear blood cells of relapsing-remitting multiple sclerosis patients and in neuroblastoma SK-N-BE cells. Brain Res. Bull. 2015, 118, 1–6. [Google Scholar] [CrossRef]
- Potenza, N.; Mosca, N.; Mondola, P.; Damiano, S.; Russo, A.; de Felice, B. Human miR-26a-5p regulates the glutamate transporter SLC1A1 (EAAT3) expression. Relevance in multiple sclerosis. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 317–323. [Google Scholar] [CrossRef]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tahara, E.B.; Navarete, F.D.; Kowaltowski, A.J. Tissue-, substrate-, and site-specific characteristics of mitochondrial reactive oxygen species generation. Free Radic. Biol. Med. 2009, 46, 1283–1297. [Google Scholar] [CrossRef] [PubMed]
- Collins, Y.; Chouchani, E.T.; James, A.M.; Menger, K.E.; Cochemé, H.M.; Murphy, M.P. Mitochondrial redox signalling at a glance. J. Cell Sci. 2012, 125, 801–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sena, L.A.; Li, S.; Jairaman, A.; Prakriya, M.; Ezponda, T.; Hildeman, D.A.; Wang, C.R.; Schumacker, P.T.; Licht, J.D.; Perlman, H.; et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 2013, 38, 225–236. [Google Scholar] [CrossRef] [Green Version]
- Radi, R.; Cassina, A.; Hodara, R.; Quijano, C.; Castro, L. Peroxynitrite reactions and formation in mitochondria. Free Radic. Biol. Med. 2002, 33, 1451–1464. [Google Scholar] [CrossRef]
- Castro, L.; Demicheli, V.; Tórtora, V.; Radi, R. Mitochondrial protein tyrosine nitration. Free Radic. Res. 2011, 45, 37–52. [Google Scholar] [CrossRef]
- Koenitzer, J.R.; Freeman, B.A. Redox signaling in inflammation: Interactions of endogenous electrophiles and mitochondria in cardiovascular disease. Ann. N. Y. Acad. Sci. 2010, 1203, 45–52. [Google Scholar] [CrossRef] [Green Version]
- Nadtochiy, S.M.; Baker, P.R.; Freeman, B.A.; Brookes, P.S. Mitochondrial nitroalkene formation and mild uncoupling in ischaemic preconditioning: Implications for cardioprotection. Cardiovasc. Res. 2009, 82, 333–340. [Google Scholar] [CrossRef] [Green Version]
- Botti, H.; Trostchansky, A.; Batthyány, C.; Rubbo, H. Reactivity of peroxynitrite and nitric oxide with LDL. Iubmb Life 2005, 57, 407–412. [Google Scholar] [CrossRef]
- Botti, H.; Trujillo, M.; Batthyány, C.; Rubbo, H.; Ferrer-Sueta, G.; Radi, R. Homolytic pathways drive peroxynitrite-dependent Trolox C oxidation. Chem. Res. Toxicol. 2004, 17, 1377–1384. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, J.D.; Kawahara, T.; Diebold, B. Regulation of Nox and Duox enzymatic activity and expression. Free Radic. Biol. Med. 2007, 43, 319–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Deken, X.; Wang, D.; Many, M.C.; Costagliola, S.; Libert, F.; Vassart, G.; Dumont, J.E.; Miot, F. Cloning of two human thyroid cDNAs encoding new members of the NADPH oxidase family. J. Biol. Chem. 2000, 275, 23227–23233. [Google Scholar] [CrossRef] [Green Version]
- Harper, R.W.; Xu, C.; Eiserich, J.P.; Chen, Y.; Kao, C.Y.; Thai, P.; Setiadi, H.; Wu, R. Differential regulation of dual NADPH oxidases/peroxidases, Duox1 and Duox2, by Th1 and Th2 cytokines in respiratory tract epithelium. FEBS Lett. 2005, 579, 4911–4917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Vliet, A. NADPH oxidases in lung biology and pathology: Host defense enzymes, and more. Free Radic. Biol. Med. 2008, 44, 938–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, Y.S.; Choi, M.K.; Lee, W.J. Dual oxidase in mucosal immunity and host-microbe homeostasis. Trends Immunol. 2010, 31, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Santillo, M.; Mondola, P.; Serù, R.; Annella, T.; Cassano, S.; Ciullo, I.; Tecce, M.F.; Iacomino, G.; Damiano, S.; Cuda, G.; et al. Opposing functions of Ki- and Ha-Ras genes in the regulation of redox signals. Curr. Biol. 2001, 11, 614–619. [Google Scholar] [CrossRef] [Green Version]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [Green Version]
- Chan, E.C.; Jiang, F.; Peshavariya, H.M.; Dusting, G.J. Regulation of cell proliferation by NADPH oxidase-mediated signaling: Potential roles in tissue repair, regenerative medicine and tissue engineering. Pharmacol. Ther. 2009, 122, 97–108. [Google Scholar] [CrossRef]
- Ris-Stalpers, C. Physiology and pathophysiology of the DUOXes. Antioxid. Redox Signal. 2006, 8, 1563–1572. [Google Scholar] [CrossRef]
- Damiano, S.; Morano, A.; Ucci, V.; Accetta, R.; Mondola, P.; Paternò, R.; Avvedimento, V.E.; Santillo, M. Dual oxidase 2 generated reactive oxygen species selectively mediate the induction of mucins by epidermal growth factor in enterocytes. Int. J. Biochem. Cell Biol. 2015, 60, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Santillo, M.; Colantuoni, A.; Mondola, P.; Guida, B.; Damiano, S. NOX signaling in molecular cardiovascular mechanisms involved in the blood pressure homeostasis. Front. Physiol. 2015, 6, 194. [Google Scholar] [CrossRef] [PubMed]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS Sources in Physiological and Pathological Conditions. Oxid. Med. Cell. Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Bienert, G.P.; Schjoerring, J.K.; Jahn, T.P. Membrane transport of hydrogen peroxide. Biochim. Biophys. Acta 2006, 1758, 994–1003. [Google Scholar] [CrossRef] [Green Version]
- Marinho, H.S.; Real, C.; Cyrne, L.; Soares, H.; Antunes, F. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol. 2014, 2, 535–562. [Google Scholar] [CrossRef] [Green Version]
- McCord, J.M.; Fridovich, I. Superoxide dismutase. An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar]
- Weisiger, R.A.; Fridovich, I. Mitochondrial superoxide simutase. Site of synthesis and intramitochondrial localization. J. Biol. Chem. 1973, 248, 4793–4796. [Google Scholar]
- Marklund, S.L. Human copper-containing superoxide dismutase of high molecular weight. Proc. Natl. Acad. Sci. USA 1982, 79, 7634–7638. [Google Scholar] [CrossRef] [Green Version]
- Okado-Matsumoto, A.; Fridovich, I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu,Zn-SOD in mitochondria. J. Biol. Chem. 2001, 276, 38388–38393. [Google Scholar] [CrossRef] [Green Version]
- Papa, L.; Hahn, M.; Marsh, E.L.; Evans, B.S.; Germain, D. SOD2 to SOD1 switch in breast cancer. J. Biol. Chem. 2014, 289, 5412–5416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slot, J.W.; Geuze, H.J.; Freeman, B.A.; Crapo, J.D. Intracellular localization of the copper-zinc and manganese superoxide dismutases in rat liver parenchymal cells. Lab. Investig. 1986, 55, 363–371. [Google Scholar] [PubMed]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dechant, R.; Peter, M. Nutrient signals driving cell growth. Curr. Opin. Cell Biol. 2008, 20, 678–687. [Google Scholar] [CrossRef]
- Jorgensen, P.; Rupes, I.; Sharom, J.R.; Schneper, L.; Broach, J.R.; Tyers, M. A dynamic transcriptional network communicates growth potential to ribosome synthesis and critical cell size. Genes Dev. 2004, 18, 2491–2505. [Google Scholar] [CrossRef] [Green Version]
- Chae, Y.C.; Angelin, A.; Lisanti, S.; Kossenkov, A.V.; Speicher, K.D.; Wang, H.; Powers, J.F.; Tischler, A.S.; Pacak, K.; Fliedner, S.; et al. Landscape of the mitochondrial Hsp90 metabolome in tumours. Nat. Commun. 2013, 4, 2139. [Google Scholar] [CrossRef] [Green Version]
- Liang, Q.; Benavides, G.A.; Vassilopoulos, A.; Gius, D.; Darley-Usmar, V.; Zhang, J. Bioenergetic and autophagic control by Sirt3 in response to nutrient deprivation in mouse embryonic fibroblasts. Biochem. J. 2013, 454, 249–257. [Google Scholar] [CrossRef]
- Liu, Z.; Sun, Y.; Tan, S.; Liu, L.; Hu, S.; Huo, H.; Li, M.; Cui, Q.; Yu, M. Nutrient deprivation-related OXPHOS/glycolysis interconversion via HIF-1α/C-MYC pathway in U251 cells. Tumour Biol. 2016, 37, 6661–6671. [Google Scholar] [CrossRef]
- Nisoli, E.; Tonello, C.; Cardile, A.; Cozzi, V.; Bracale, R.; Tedesco, L.; Falcone, S.; Valerio, A.; Cantoni, O.; Clementi, E.; et al. Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science 2005, 310, 314–317. [Google Scholar] [CrossRef]
- Tebay, L.E.; Robertson, H.; Durant, S.T.; Vitale, S.R.; Penning, T.M.; Dinkova-Kostova, A.T.; Hayes, J.D. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic. Biol. Med. 2015, 88, 108–146. [Google Scholar] [CrossRef] [Green Version]
- Faraonio, R.; Vergara, P.; Marzo, D.D.; Napolitano, M.; Russo, T.; Cimino, F. Transcription regulation in NIH3T3 cell clones resistant to diethylmaleate-induced oxidative stress and apoptosis. Antioxid. Redox Signal. 2006, 8, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Sihvola, V.; Levonen, A.L. Keap1 as the redox sensor of the antioxidant response. Arch. Biochem. Biophys. 2017, 617, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Park, E.Y.; Rho, H.M. The transcriptional activation of the human copper/zinc superoxide dismutase gene by 2,3,7,8-tetrachlorodibenzo-p-dioxin through two different regulator sites, the antioxidant responsive element and xenobiotic responsive element. Mol. Cell. Biochem. 2002, 240, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Hallows, W.C.; Yu, W.; Smith, B.C.; Devries, M.K.; Ellinger, J.J.; Someya, S.; Shortreed, M.R.; Prolla, T.; Markley, J.L.; Smith, L.M.; et al. Sirt3 promotes the urea cycle and fatty acid oxidation during dietary restriction. Mol. Cell 2011, 41, 139–149. [Google Scholar] [CrossRef] [Green Version]
- Shi, T.; Wang, F.; Stieren, E.; Tong, Q. SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. J. Biol. Chem. 2005, 280, 13560–13567. [Google Scholar] [CrossRef] [Green Version]
- Schwer, B.; Verdin, E. Conserved metabolic regulatory functions of sirtuins. Cell Metab. 2008, 7, 104–112. [Google Scholar] [CrossRef] [Green Version]
- Qiu, X.; Brown, K.; Hirschey, M.D.; Verdin, E.; Chen, D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010, 12, 662–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greer, E.L.; Brunet, A. Different dietary restriction regimens extend lifespan by both independent and overlapping genetic pathways in C. elegans. Aging Cell 2009, 8, 113–127. [Google Scholar] [CrossRef] [Green Version]
- Greer, E.L.; Dowlatshahi, D.; Banko, M.R.; Villen, J.; Hoang, K.; Blanchard, D.; Gygi, S.P.; Brunet, A. An AMPK-FOXO pathway mediates longevity induced by a novel method of dietary restriction in C. elegans. Curr. Biol. 2007, 17, 1646–1656. [Google Scholar] [CrossRef] [Green Version]
- Johnson, E.C.; Kazgan, N.; Bretz, C.A.; Forsberg, L.J.; Hector, C.E.; Worthen, R.J.; Onyenwoke, R.; Brenman, J.E. Altered metabolism and persistent starvation behaviors caused by reduced AMPK function in Drosophila. PLoS ONE 2010, 5, 0012799. [Google Scholar] [CrossRef] [Green Version]
- Schulz, T.J.; Zarse, K.; Voigt, A.; Urban, N.; Birringer, M.; Ristow, M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab. 2007, 6, 280–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.C.; Hardie, D.G. AMPK: Sensing Glucose as well as Cellular Energy Status. Cell Metab. 2018, 27, 299–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.L.; Kim, S.J.; Lee, K.T.; Kim, J.; Mu, J.; Birnbaum, M.J.; Soo Kim, S.; Ha, J. The regulation of AMP-activated protein kinase by H(2)O(2). Biochem. Biophys. Res. Commun. 2001, 287, 92–97. [Google Scholar] [CrossRef]
- Shaw, R.J.; Kosmatka, M.; Bardeesy, N.; Hurley, R.L.; Witters, L.A.; DePinho, R.A.; Cantley, L.C. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl. Acad. Sci. USA 2004, 101, 3329–3335. [Google Scholar] [CrossRef] [Green Version]
- Woods, A.; Dickerson, K.; Heath, R.; Hong, S.P.; Momcilovic, M.; Johnstone, S.R.; Carlson, M.; Carling, D. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005, 2, 21–33. [Google Scholar] [CrossRef] [Green Version]
- Hawley, S.A.; Ross, F.A.; Chevtzoff, C.; Green, K.A.; Evans, A.; Fogarty, S.; Towler, M.C.; Brown, L.J.; Ogunbayo, O.A.; Evans, A.M.; et al. Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab. 2010, 11, 554–565. [Google Scholar] [CrossRef] [Green Version]
- Jeon, S.M.; Chandel, N.S.; Hay, N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 2012, 485, 661–665. [Google Scholar] [CrossRef] [Green Version]
- Rabinovitch, R.C.; Samborska, B.; Faubert, B.; Ma, E.H.; Gravel, S.P.; Andrzejewski, S.; Raissi, T.C.; Pause, A.; St-Pierre, J.; Jones, R.G. AMPK Maintains Cellular Metabolic Homeostasis through Regulation of Mitochondrial Reactive Oxygen Species. Cell Rep. 2017, 21, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bournat, J.C.; Brown, C.W. Mitochondrial dysfunction in obesity. Curr. Opin. Endocrinol. Diabetes. Obes. 2010, 17, 446–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liesa, M.; Shirihai, O.S. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013, 17, 491–506. [Google Scholar] [CrossRef] [Green Version]
- Kozakiewicz, M.; Kornatowski, M.; Krzywińska, O.; Kędziora-Kornatowska, K. Changes in the blood antioxidant defense of advanced age people. Clin. Interv. Aging 2019, 14, 763–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damiano, S.; Muscariello, E.; La Rosa, G.; Di Maro, M.; Mondola, P.; Santillo, M. Dual Role of Reactive Oxygen Species in Muscle Function: Can Antioxidant Dietary Supplements Counteract Age-Related Sarcopenia? Int. J. Mol. Sci. 2019, 20, 3815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, W.J.; Jacinto, E. mTOR complex 2 signaling and functions. Cell Cycle 2011, 10, 2305–2316. [Google Scholar] [CrossRef]
- Sacks, D.; Baxter, B.; Campbell, B.C.V.; Carpenter, J.S.; Cognard, C.; Dippel, D.; Eesa, M.; Fischer, U.; Hausegger, K.; Hirsch, J.A.; et al. Multisociety Consensus Quality Improvement Revised Consensus Statement for Endovascular Therapy of Acute Ischemic Stroke. Int. J. Stroke 2018, 13, 612–632. [Google Scholar] [CrossRef] [Green Version]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [Green Version]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef] [Green Version]
- Feng, Z.; Hu, W.; de Stanchina, E.; Teresky, A.K.; Jin, S.; Lowe, S.; Levine, A.J. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: Stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007, 67, 3043–3053. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Nicholatos, J.; Dreier, J.R.; Ricoult, S.J.; Widenmaier, S.B.; Hotamisligil, G.S.; Kwiatkowski, D.J.; Manning, B.D. Coordinated regulation of protein synthesis and degradation by mTORC1. Nature 2014, 513, 440–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caron, A.; Richard, D.; Laplante, M. The Roles of mTOR Complexes in Lipid Metabolism. Annu. Rev. Nutr. 2015, 35, 321–348. [Google Scholar] [CrossRef] [PubMed]
- Ben-Sahra, I.; Hoxhaj, G.; Ricoult, S.J.H.; Asara, J.M.; Manning, B.D. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 2016, 351, 728–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betz, C.; Hall, M.N. Where is mTOR and what is it doing there? J. Cell Biol. 2013, 203, 563–574. [Google Scholar] [CrossRef] [Green Version]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [Green Version]
- García-Martínez, J.M.; Alessi, D.R. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). Biochem. J. 2008, 416, 375–385. [Google Scholar] [CrossRef] [Green Version]
- Yuan, M.; Pino, E.; Wu, L.; Kacergis, M.; Soukas, A.A. Identification of Akt-independent regulation of hepatic lipogenesis by mammalian target of rapamycin (mTOR) complex 2. J. Biol. Chem. 2012, 287, 29579–29588. [Google Scholar] [CrossRef] [Green Version]
- Cybulski, N.; Hall, M.N. TOR complex 2: A signaling pathway of its own. Trends Biochem. Sci. 2009, 34, 620–627. [Google Scholar] [CrossRef]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef] [Green Version]
- Martins, L.; Fernández-Mallo, D.; Novelle, M.G.; Vázquez, M.J.; Tena-Sempere, M.; Nogueiras, R.; López, M.; Diéguez, C. Hypothalamic mTOR signaling mediates the orexigenic action of ghrelin. PLoS ONE 2012, 7, e46923. [Google Scholar] [CrossRef] [Green Version]
- Thomanetz, V.; Angliker, N.; Cloëtta, D.; Lustenberger, R.M.; Schweighauser, M.; Oliveri, F.; Suzuki, N.; Rüegg, M.A. Ablation of the mTORC2 component rictor in brain or Purkinje cells affects size and neuron morphology. J. Cell Biol. 2013, 201, 293–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proulx, K.; Cota, D.; Woods, S.C.; Seeley, R.J. Fatty acid synthase inhibitors modulate energy balance via mammalian target of rapamycin complex 1 signaling in the central nervous system. Diabetes 2008, 57, 3231–3238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villanueva, E.C.; Münzberg, H.; Cota, D.; Leshan, R.L.; Kopp, K.; Ishida-Takahashi, R.; Jones, J.C.; Fingar, D.C.; Seeley, R.J.; Myers, M.G., Jr. Complex regulation of mammalian target of rapamycin complex 1 in the basomedial hypothalamus by leptin and nutritional status. Endocrinology 2009, 150, 4541–4551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cota, D.; Proulx, K.; Smith, K.A.; Kozma, S.C.; Thomas, G.; Woods, S.C.; Seeley, R.J. Hypothalamic mTOR signaling regulates food intake. Science 2006, 312, 927–930. [Google Scholar] [CrossRef] [Green Version]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008, 320, 1496–1501. [Google Scholar] [CrossRef] [Green Version]
- Layman, D.K.; Walker, D.A. Potential importance of leucine in treatment of obesity and the metabolic syndrome. J. Nutr. 2006, 136, 319s–323s. [Google Scholar] [CrossRef]
- Morrison, C.D.; Xi, X.; White, C.L.; Ye, J.; Martin, R.J. Amino acids inhibit Agrp gene expression via an mTOR-dependent mechanism. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E165–E171. [Google Scholar] [CrossRef] [Green Version]
- Cota, D.; Matter, E.K.; Woods, S.C.; Seeley, R.J. The role of hypothalamic mammalian target of rapamycin complex 1 signaling in diet-induced obesity. J. Neurosci. 2008, 28, 7202–7208. [Google Scholar] [CrossRef] [Green Version]
- Hu, F.; Xu, Y.; Liu, F. Hypothalamic roles of mTOR complex I: Integration of nutrient and hormone signals to regulate energy homeostasis. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E994–E1002. [Google Scholar] [CrossRef] [Green Version]
- Tsang, C.K.; Chen, M.; Cheng, X.; Qi, Y.; Chen, Y.; Das, I.; Li, X.; Vallat, B.; Fu, L.W.; Qian, C.N.; et al. SOD1 Phosphorylation by mTORC1 Couples Nutrient Sensing and Redox Regulation. Mol. Cell 2018, 70, 502–515. [Google Scholar] [CrossRef] [Green Version]
- Di Renzo, L.; Cioccoloni, G.; Bernardini, S.; Abenavoli, L.; Aiello, V.; Marchetti, M.; Cammarano, A.; Alipourfard, I.; Ceravolo, I.; Gratteri, S. A Hazelnut-Enriched Diet Modulates Oxidative Stress and Inflammation Gene Expression without Weight Gain. Oxid. Med. Cell. Longev. 2019, 2019, 4683723. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Khare, P.; Kumar, A.; Chunduri, V.; Kumar, A.; Kapoor, P.; Mangal, P.; Kondepudi, K.K.; Bishnoi, M.; Garg, M. Anthocyanin-Biofortified Colored Wheat Prevents High Fat Diet-Induced Alterations in Mice: Nutrigenomics Studies. Mol. Nutr. Food Res. 2020, 64, e1900999. [Google Scholar] [CrossRef] [PubMed]
- Nakao, R.; Abe, T.; Yamamoto, S.; Oishi, K. Ketogenic diet induces skeletal muscle atrophy via reducing muscle protein synthesis and possibly activating proteolysis in mice. Sci. Rep. 2019, 9, 19652. [Google Scholar] [CrossRef] [Green Version]
- Barter, P.; Gotto, A.M.; LaRosa, J.C.; Maroni, J.; Szarek, M.; Grundy, S.M.; Kastelein, J.J.; Bittner, V.; Fruchart, J.C. HDL cholesterol, very low levels of LDL cholesterol, and cardiovascular events. N. Engl. J. Med. 2007, 357, 1301–1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahotupa, M. Oxidized lipoprotein lipids and atherosclerosis. Free Radic. Res. 2017, 51, 439–447. [Google Scholar] [CrossRef]
- Mondola, P.; Bifulco, M.; Serù, R.; Annella, T.; Ciriolo, M.R.; Santillo, M. Presence of CuZn superoxide dismutase in human serum lipoproteins. FEBS Lett. 2000, 467, 57–60. [Google Scholar] [CrossRef]
- Holvoet, P.; Macy, E.; Landeloos, M.; Jones, D.; Jenny, N.S.; Van de Werf, F.; Tracy, R.P. Analytical performance and diagnostic accuracy of immunometric assays for the measurement of circulating oxidized LDL. Clin. Chem. 2006, 52, 760–764. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. A receptor-mediated pathway for cholesterol homeostasis. Science 1986, 232, 34–47. [Google Scholar] [CrossRef] [Green Version]
- Brown, M.S.; Goldstein, J.L. Multivalent feedback regulation of HMG CoA reductase, a control mechanism coordinating isoprenoid synthesis and cell growth. J. Lipid Res. 1980, 21, 505–517. [Google Scholar]
- Miller, S.J.; Parker, R.A.; Gibson, D.M. Phosphorylation and degradation of HMG CoA reductase. Adv. Enzym. Regul. 1989, 28, 65–77. [Google Scholar] [CrossRef]
- Mondola, P.; Serù, R.; Santillo, M.; Damiano, S.; Bifulco, M.; Laezza, C.; Formisano, P.; Rotilio, G.; Ciriolo, M.R. Effect of Cu,Zn superoxide dismutase on cholesterol metabolism in human hepatocarcinoma (HepG2) cells. Biochem. Biophys. Res. Commun. 2002, 295, 603–609. [Google Scholar] [CrossRef]
- De Felice, B.; Santillo, M.; Serù, R.; Damiano, S.; Matrone, G.; Wilson, R.R.; Mondola, P. Modulation of 3-hydroxy-3-methylglutaryl-CoA reductase gene expression by CuZn superoxide dismutase in human fibroblasts and HepG2 cells. Gene Expr. 2004, 12, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [Green Version]
- Zaman, S.; Lippman, S.I.; Zhao, X.; Broach, J.R. How Saccharomyces responds to nutrients. Annu. Rev. Genet. 2008, 42, 27–81. [Google Scholar] [CrossRef] [PubMed]
- Sehati, S.; Clement, M.H.; Martins, J.; Xu, L.; Longo, V.D.; Valentine, J.S.; Gralla, E.B. Metabolic alterations in yeast lacking copper-zinc superoxide dismutase. Free Radic. Biol. Med. 2011, 50, 1591–1598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Thornton, J.; Spírek, M.; Butow, R.A. Activation of the SPS amino acid-sensing pathway in Saccharomyces cerevisiae correlates with the phosphorylation state of a sensor component, Ptr3. Mol. Cell. Biol. 2008, 28, 551–563. [Google Scholar] [CrossRef] [Green Version]
- Moriya, H.; Johnston, M. Glucose sensing and signaling in Saccharomyces cerevisiae through the Rgt2 glucose sensor and casein kinase I. Proc. Natl. Acad. Sci. USA 2004, 101, 1572–1577. [Google Scholar] [CrossRef] [Green Version]
- Reddi, A.R.; Culotta, V.C. SOD1 integrates signals from oxygen and glucose to repress respiration. Cell 2013, 152, 224–235. [Google Scholar] [CrossRef] [Green Version]
- Banks, C.J.; Rodriguez, N.W.; Gashler, K.R.; Pandya, R.R.; Mortenson, J.B.; Whited, M.D.; Soderblom, E.J.; Thompson, J.W.; Moseley, M.A.; Reddi, A.R.; et al. Acylation of Superoxide Dismutase 1 (SOD1) at K122 Governs SOD1-Mediated Inhibition of Mitochondrial Respiration. Mol. Cell. Biol. 2017, 37, 00354-17. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Xu, W.; Jiang, W.; Yu, W.; Lin, Y.; Zhang, T.; Yao, J.; Zhou, L.; Zeng, Y.; Li, H.; et al. Regulation of cellular metabolism by protein lysine acetylation. Science 2010, 327, 1000–1004. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Palmer, A.K.; Ding, H.; Weivoda, M.M.; Pirtskhalava, T.; White, T.A.; Sepe, A.; Johnson, K.O.; Stout, M.B.; Giorgadze, N.; et al. Targeting senescent cells enhances adipogenesis and metabolic function in old age. Elife 2015, 4, e12997. [Google Scholar] [CrossRef]
- Cristancho, A.G.; Lazar, M.A. Forming functional fat: A growing understanding of adipocyte differentiation. Nat. Rev. Mol. Cell Biol. 2011, 12, 722–734. [Google Scholar] [CrossRef] [PubMed]
- Galic, S.; Oakhill, J.S.; Steinberg, G.R. Adipose tissue as an endocrine organ. Mol. Cell. Endocrinol. 2010, 316, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Fernando, R.; Wardelmann, K.; Deubel, S.; Kehm, R.; Jung, T.; Mariotti, M.; Vasilaki, A.; Gladyshev, V.N.; Kleinridders, A.; Grune, T.; et al. Low steady-state oxidative stress inhibits adipogenesis by altering mitochondrial dynamics and decreasing cellular respiration. Redox Biol. 2020, 32, 101507. [Google Scholar] [CrossRef]
- Robberecht, W.; Philips, T. The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci. 2013, 14, 248–264. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O‘Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Dupuis, L.; Pradat, P.F.; Ludolph, A.C.; Loeffler, J.P. Energy metabolism in amyotrophic lateral sclerosis. Lancet Neurol. 2011, 10, 75–82. [Google Scholar] [CrossRef]
- Dupuis, L.; Corcia, P.; Fergani, A.; Gonzalez De Aguilar, J.L.; Bonnefont-Rousselot, D.; Bittar, R.; Seilhean, D.; Hauw, J.J.; Lacomblez, L.; Loeffler, J.P.; et al. Dyslipidemia is a protective factor in amyotrophic lateral sclerosis. Neurology 2008, 70, 1004–1009. [Google Scholar] [CrossRef]
- Bandres-Ciga, S.; Noyce, A.J.; Hemani, G.; Nicolas, A.; Calvo, A.; Mora, G.; Tienari, P.J.; Stone, D.J.; Nalls, M.A.; Singleton, A.B.; et al. Shared polygenic risk and causal inferences in amyotrophic lateral sclerosis. Ann. Neurol. 2019, 85, 470–481. [Google Scholar] [CrossRef]
- Dupuis, L.; Oudart, H.; René, F.; Gonzalez de Aguilar, J.L.; Loeffler, J.P. Evidence for defective energy homeostasis in amyotrophic lateral sclerosis: Benefit of a high-energy diet in a transgenic mouse model. Proc. Natl. Acad. Sci. USA 2004, 101, 11159–11164. [Google Scholar] [CrossRef] [Green Version]
- Tan, W.; Pasinelli, P.; Trotti, D. Role of mitochondria in mutant SOD1 linked amyotrophic lateral sclerosis. Biochim. Biophys. Acta 2014, 1842, 1295–1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattiazzi, M.; D‘Aurelio, M.; Gajewski, C.D.; Martushova, K.; Kiaei, M.; Beal, M.F.; Manfredi, G. Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J. Biol. Chem. 2002, 277, 29626–29633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkinezos, I.G.; Bacman, S.R.; Hernandez, D.; Oca-Cossio, J.; Arias, L.J.; Perez-Pinzon, M.A.; Bradley, W.G.; Moraes, C.T. Cytochrome c association with the inner mitochondrial membrane is impaired in the CNS of G93A-SOD1 mice. J. Neurosci. 2005, 25, 164–172. [Google Scholar] [CrossRef] [Green Version]
- Damiano, M.; Starkov, A.A.; Petri, S.; Kipiani, K.; Kiaei, M.; Mattiazzi, M.; Flint Beal, M.; Manfredi, G. Neural mitochondrial Ca2+ capacity impairment precedes the onset of motor symptoms in G93A Cu/Zn-superoxide dismutase mutant mice. J. Neurochem. 2006, 96, 1349–1361. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.; Xu, Z. Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J. Neurosci. 1998, 18, 3241–3250. [Google Scholar] [CrossRef]
- Kawamata, H.; Manfredi, G. Mitochondrial dysfunction and intracellular calcium dysregulation in ALS. Mech. Ageing Dev. 2010, 131, 517–526. [Google Scholar] [CrossRef] [Green Version]
- Jung, C.; Higgins, C.M.; Xu, Z. Mitochondrial electron transport chain complex dysfunction in a transgenic mouse model for amyotrophic lateral sclerosis. J. Neurochem. 2002, 83, 535–545. [Google Scholar] [CrossRef]
- Pasinelli, P.; Belford, M.E.; Lennon, N.; Bacskai, B.J.; Hyman, B.T.; Trotti, D.; Brown, R.H., Jr. Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron 2004, 43, 19–30. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Lillo, C.; Jonsson, P.A.; Vande Velde, C.; Ward, C.M.; Miller, T.M.; Subramaniam, J.R.; Rothstein, J.D.; Marklund, S.; Andersen, P.M.; et al. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron 2004, 43, 5–17. [Google Scholar] [CrossRef] [Green Version]
- Pickles, S.; Destroismaisons, L.; Peyrard, S.L.; Cadot, S.; Rouleau, G.A.; Brown, R.H., Jr.; Julien, J.P.; Arbour, N.; Vande Velde, C. Mitochondrial damage revealed by immunoselection for ALS-linked misfolded SOD1. Hum. Mol. Genet. 2013, 22, 3947–3959. [Google Scholar] [CrossRef]
- Andersen, J.L.; Kornbluth, S. The tangled circuitry of metabolism and apoptosis. Mol. Cell 2013, 49, 399–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.J.; Lee, L.M.; Lai, H.L.; Chern, Y. Aberrant activation of AMP-activated protein kinase contributes to the abnormal distribution of HuR in amyotrophic lateral sclerosis. FEBS Lett. 2015, 589, 432–439. [Google Scholar] [CrossRef] [PubMed]
- Sui, Y.; Zhao, Z.; Liu, R.; Cai, B.; Fan, D. Adenosine monophosphate-activated protein kinase activation enhances embryonic neural stem cell apoptosis in a mouse model of amyotrophic lateral sclerosis. Neural Regen. Res. 2014, 9, 1770–1778. [Google Scholar] [PubMed]
- Dupuis, L.; di Scala, F.; Rene, F.; de Tapia, M.; Oudart, H.; Pradat, P.F.; Meininger, V.; Loeffler, J.P. Up-regulation of mitochondrial uncoupling protein 3 reveals an early muscular metabolic defect in amyotrophic lateral sclerosis. FASEB J. 2003, 17, 2091–2093. [Google Scholar] [CrossRef]
- Echaniz-Laguna, A.; Zoll, J.; Ponsot, E.; N‘Guessan, B.; Tranchant, C.; Loeffler, J.P.; Lampert, E. Muscular mitochondrial function in amyotrophic lateral sclerosis is progressively altered as the disease develops: A temporal study in man. Exp. Neurol. 2006, 198, 25–30. [Google Scholar] [CrossRef]
- Madji Hounoum, B.; Mavel, S.; Coque, E.; Patin, F.; Vourc‘h, P.; Marouillat, S.; Nadal-Desbarats, L.; Emond, P.; Corcia, P.; Andres, C.R.; et al. Wildtype motoneurons, ALS-Linked SOD1 mutation and glutamate profoundly modify astrocyte metabolism and lactate shuttling. Glia 2017, 65, 592–605. [Google Scholar] [CrossRef]
- Grosskreutz, J.; Haastert, K.; Dewil, M.; Van Damme, P.; Callewaert, G.; Robberecht, W.; Dengler, R.; Van Den Bosch, L. Role of mitochondria in kainate-induced fast Ca2+ transients in cultured spinal motor neurons. Cell Calcium 2007, 42, 59–69. [Google Scholar] [CrossRef]
- Irvin, C.W.; Kim, R.B.; Mitchell, C.S. Seeking homeostasis: Temporal trends in respiration, oxidation, and calcium in SOD1 G93A Amyotrophic Lateral Sclerosis mice. Front. Cell. Neurosci. 2015, 9, 248. [Google Scholar] [CrossRef] [Green Version]
- Bond, L.; Bernhardt, K.; Madria, P.; Sorrentino, K.; Scelsi, H.; Mitchell, C.S. A Metadata Analysis of Oxidative Stress Etiology in Preclinical Amyotrophic Lateral Sclerosis: Benefits of Antioxidant Therapy. Front. Neurosci. 2018, 12, 10. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, C.S.; Hollinger, S.K.; Goswami, S.D.; Polak, M.A.; Lee, R.H.; Glass, J.D. Antecedent Disease is Less Prevalent in Amyotrophic Lateral Sclerosis. Neurodegener. Dis. 2015, 15, 109–113. [Google Scholar] [CrossRef] [Green Version]
- Körner, S.; Kollewe, K.; Ilsemann, J.; Müller-Heine, A.; Dengler, R.; Krampfl, K.; Petri, S. Prevalence and prognostic impact of comorbidities in amyotrophic lateral sclerosis. Eur. J. Neurol. 2013, 20, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Hollinger, S.K.; Okosun, I.S.; Mitchell, C.S. Antecedent Disease and Amyotrophic Lateral Sclerosis: What Is Protecting Whom? Front. Neurol. 2016, 7, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franchina, D.G.; Dostert, C.; Brenner, D. Reactive Oxygen Species: Involvement in T Cell Signaling and Metabolism. Trends Immunol. 2018, 39, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.; Perl, A. Metabolism as a Target for Modulation in Autoimmune Diseases. Trends Immunol. 2018, 39, 562–576. [Google Scholar] [CrossRef] [PubMed]
- Sekkat, C.; Dornand, J.; Gerber, M. Oxidative phenomena are implicated in human T-cell stimulation. Immunology 1988, 63, 431–437. [Google Scholar]
- Williams, M.S.; Henkart, P.A. Role of reactive oxygen intermediates in TCR-induced death of T cell blasts and hybridomas. J. Immunol. 1996, 157, 2395–2402. [Google Scholar]
- Tatla, S.; Woodhead, V.; Foreman, J.C.; Chain, B.M. The role of reactive oxygen species in triggering proliferation and IL-2 secretion in T cells. Free Radic. Biol. Med. 1999, 26, 14–24. [Google Scholar] [CrossRef]
- Los, M.; Schenk, H.; Hexel, K.; Baeuerle, P.A.; Dröge, W.; Schulze-Osthoff, K. IL-2 gene expression and NF-kappa B activation through CD28 requires reactive oxygen production by 5-lipoxygenase. EMBO J. 1995, 14, 3731–3740. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.H.; Devadas, S.; Kwon, J.; Pinto, L.A.; Williams, M.S. T cells express a phagocyte-type NADPH oxidase that is activated after T cell receptor stimulation. Nat. Immunol. 2004, 5, 818–827. [Google Scholar] [CrossRef]
- Rhee, S.G.; Bae, Y.S.; Lee, S.R.; Kwon, J. Hydrogen peroxide: A key messenger that modulates protein phosphorylation through cysteine oxidation. Sci. Stke. 2000, 2000, pe1. [Google Scholar] [CrossRef]
- Reth, M. Hydrogen peroxide as second messenger in lymphocyte activation. Nat. Immunol. 2002, 3, 1129–1134. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G. Cell signaling. H2O2, a necessary evil for cell signaling. Science 2006, 312, 1882–1883. [Google Scholar] [CrossRef] [PubMed]
- Pani, G.; Colavitti, R.; Borrello, S.; Galeotti, T. Redox regulation of lymphocyte signaling. IUBMB Life 2000, 49, 381–389. [Google Scholar]
- Terrazzano, G.; Rubino, V.; Damiano, S.; Sasso, A.; Petrozziello, T.; Ucci, V.; Palatucci, A.T.; Giovazzino, A.; Santillo, M.; De Felice, B.; et al. T cell activation induces CuZn superoxide dismutase (SOD)-1 intracellular re-localization, production and secretion. Biochim. Biophys. Acta 2014, 1843, 265–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beers, D.R.; Appel, S.H. Immune dysregulation in amyotrophic lateral sclerosis: Mechanisms and emerging therapies. Lancet Neurol. 2019, 18, 211–220. [Google Scholar] [CrossRef]
- Mondola, P.; Annella, T.; Santillo, M.; Santangelo, F. Evidence for secretion of cytosolic CuZn superoxide dismutase by Hep G2 cells and human fibroblasts. Int. J. Biochem. Cell Biol. 1996, 28, 677–681. [Google Scholar] [CrossRef]
- Mondola, P.; Annella, T.; Serù, R.; Santangelo, F.; Iossa, S.; Gioielli, A.; Santillo, M. Secretion and increase of intracellular CuZn superoxide dismutase content in human neuroblastoma SK-N-BE cells subjected to oxidative stress. Brain Res. Bull. 1998, 45, 517–520. [Google Scholar] [CrossRef]
- Mondola, P.; Ruggiero, G.; Serù, R.; Damiano, S.; Grimaldi, S.; Garbi, C.; Monda, M.; Greco, D.; Santillo, M. The Cu,Zn superoxide dismutase in neuroblastoma SK-N-BE cells is exported by a microvesicles dependent pathway. Brain Res. Mol. Brain Res. 2003, 110, 45–51. [Google Scholar] [CrossRef]
- Cimini, V.; Ruggiero, G.; Buonomo, T.; Seru, R.; Sciorio, S.; Zanzi, C.; Santangelo, F.; Mondola, P. CuZn-superoxide dismutase in human thymus: Immunocytochemical localisation and secretion in thymus-derived epithelial and fibroblast cell lines. Histochem. Cell Biol. 2002, 118, 163–169. [Google Scholar] [CrossRef]
- Turner, B.J.; Atkin, J.D.; Farg, M.A.; Zang, D.W.; Rembach, A.; Lopes, E.C.; Patch, J.D.; Hill, A.F.; Cheema, S.S. Impaired extracellular secretion of mutant superoxide dismutase 1 associates with neurotoxicity in familial amyotrophic lateral sclerosis. J. Neurosci. 2005, 25, 108–117. [Google Scholar] [CrossRef]
- Santillo, M.; Secondo, A.; Serù, R.; Damiano, S.; Garbi, C.; Taverna, E.; Rosa, P.; Giovedì, S.; Benfenati, F.; Mondola, P. Evidence of calcium- and SNARE-dependent release of CuZn superoxide dismutase from rat pituitary GH3 cells and synaptosomes in response to depolarization. J. Neurochem. 2007, 102, 679–685. [Google Scholar] [CrossRef] [PubMed]
- Gomes, C.; Keller, S.; Altevogt, P.; Costa, J. Evidence for secretion of Cu,Zn superoxide dismutase via exosomes from a cell model of amyotrophic lateral sclerosis. Neurosci. Lett. 2007, 428, 43–46. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Garcia, D.; Brouwers, N.; Duran, J.M.; Mora, G.; Curwin, A.J.; Malhotra, V. A diacidic motif determines unconventional secretion of wild-type and ALS-linked mutant SOD1. J. Cell Biol. 2017, 216, 2691–2700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz-Garcia, D.; Malhotra, V.; Curwin, A.J. Unconventional protein secretion triggered by nutrient starvation. Semin. Cell Dev. Biol. 2018, 83, 22–28. [Google Scholar] [CrossRef]
- Mondola, P.; Damiano, S.; Sasso, A.; Santillo, M. The Cu, Zn Superoxide Dismutase: Not Only a Dismutase Enzyme. Front. Physiol. 2016, 7, 594. [Google Scholar] [CrossRef] [Green Version]
- Damiano, S.; Petrozziello, T.; Ucci, V.; Amente, S.; Santillo, M.; Mondola, P. Cu-Zn superoxide dismutase activates muscarinic acetylcholine M1 receptor pathway in neuroblastoma cells. Mol. Cell. Neurosci. 2013, 52, 31–37. [Google Scholar] [CrossRef]
- Mondola, P.; Santillo, M.; Serù, R.; Damiano, S.; Alvino, C.; Ruggiero, G.; Formisano, P.; Terrazzano, G.; Secondo, A.; Annunziato, L. Cu,Zn superoxide dismutase increases intracellular calcium levels via a phospholipase C-protein kinase C pathway in SK-N-BE neuroblastoma cells. Biochem. Biophys. Res. Commun. 2004, 324, 887–892. [Google Scholar] [CrossRef]
- Secondo, A.; De Mizio, M.; Zirpoli, L.; Santillo, M.; Mondola, P. The Cu-Zn superoxide dismutase (SOD1) inhibits ERK phosphorylation by muscarinic receptor modulation in rat pituitary GH3 cells. Biochem. Biophys. Res. Commun. 2008, 376, 143–147. [Google Scholar] [CrossRef]
- Viggiano, A.; Serù, R.; Damiano, S.; De Luca, B.; Santillo, M.; Mondola, P. Inhibition of long-term potentiation by CuZn superoxide dismutase injection in rat dentate gyrus: Involvement of muscarinic M1 receptor. J. Cell. Physiol. 2012, 227, 3111–3115. [Google Scholar] [CrossRef]
- Tsang, C.K.; Liu, Y.; Thomas, J.; Zhang, Y.; Zheng, X.F. Superoxide dismutase 1 acts as a nuclear transcription factor to regulate oxidative stress resistance. Nat. Commun. 2014, 5, 3446. [Google Scholar] [CrossRef] [PubMed] [Green Version]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Damiano, S.; Sozio, C.; La Rosa, G.; Guida, B.; Faraonio, R.; Santillo, M.; Mondola, P. Metabolism Regulation and Redox State: Insight into the Role of Superoxide Dismutase 1. Int. J. Mol. Sci. 2020, 21, 6606. https://doi.org/10.3390/ijms21186606
Damiano S, Sozio C, La Rosa G, Guida B, Faraonio R, Santillo M, Mondola P. Metabolism Regulation and Redox State: Insight into the Role of Superoxide Dismutase 1. International Journal of Molecular Sciences. 2020; 21(18):6606. https://doi.org/10.3390/ijms21186606
Chicago/Turabian StyleDamiano, Simona, Concetta Sozio, Giuliana La Rosa, Bruna Guida, Raffaella Faraonio, Mariarosaria Santillo, and Paolo Mondola. 2020. "Metabolism Regulation and Redox State: Insight into the Role of Superoxide Dismutase 1" International Journal of Molecular Sciences 21, no. 18: 6606. https://doi.org/10.3390/ijms21186606