Analyzing Gene Expression Profiles from Ataxia and Spasticity Phenotypes to Reveal Spastic Ataxia Related Pathways

 , , and
, , and 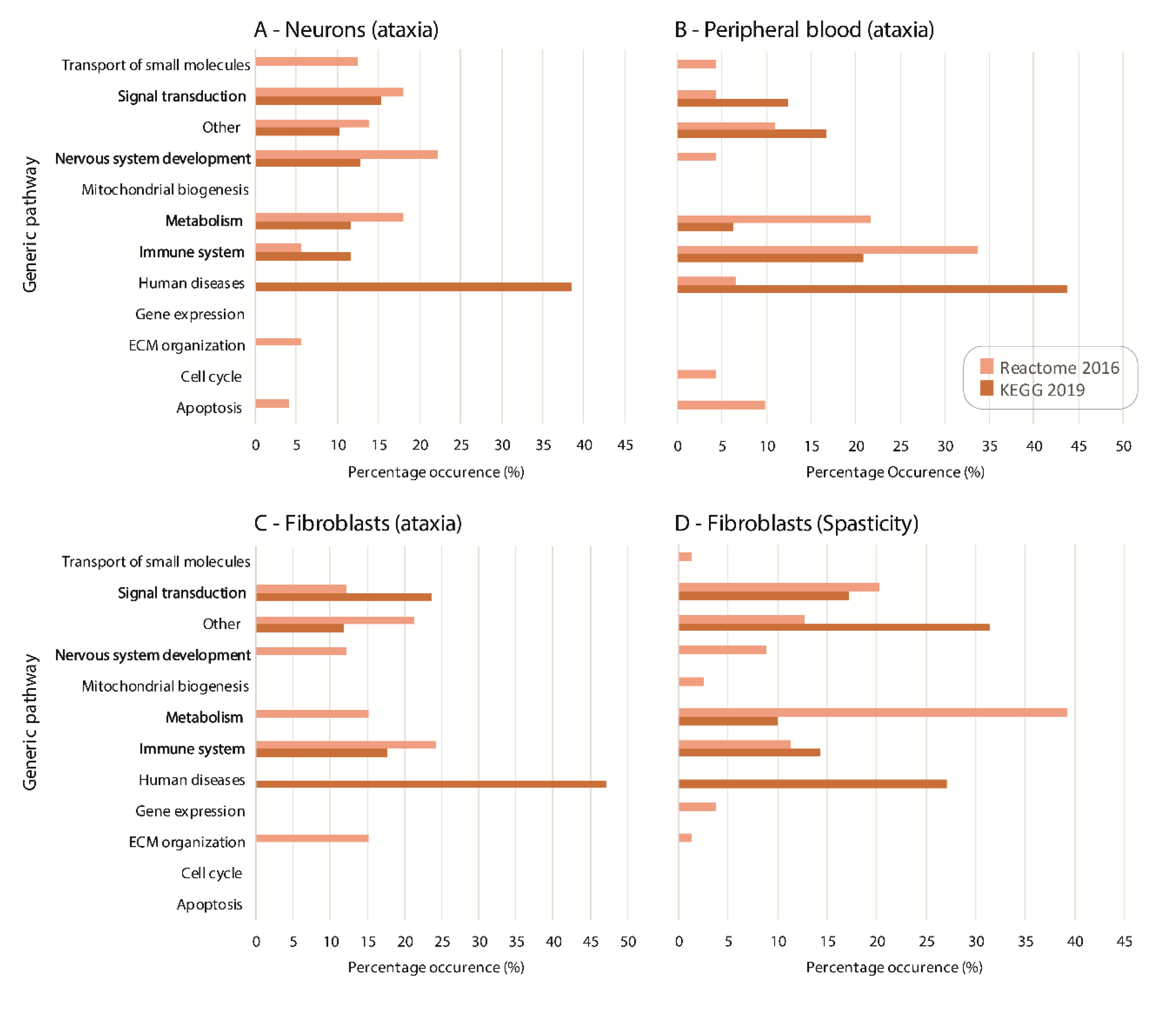{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Pathway Analysis of Differentially Expressed Genes
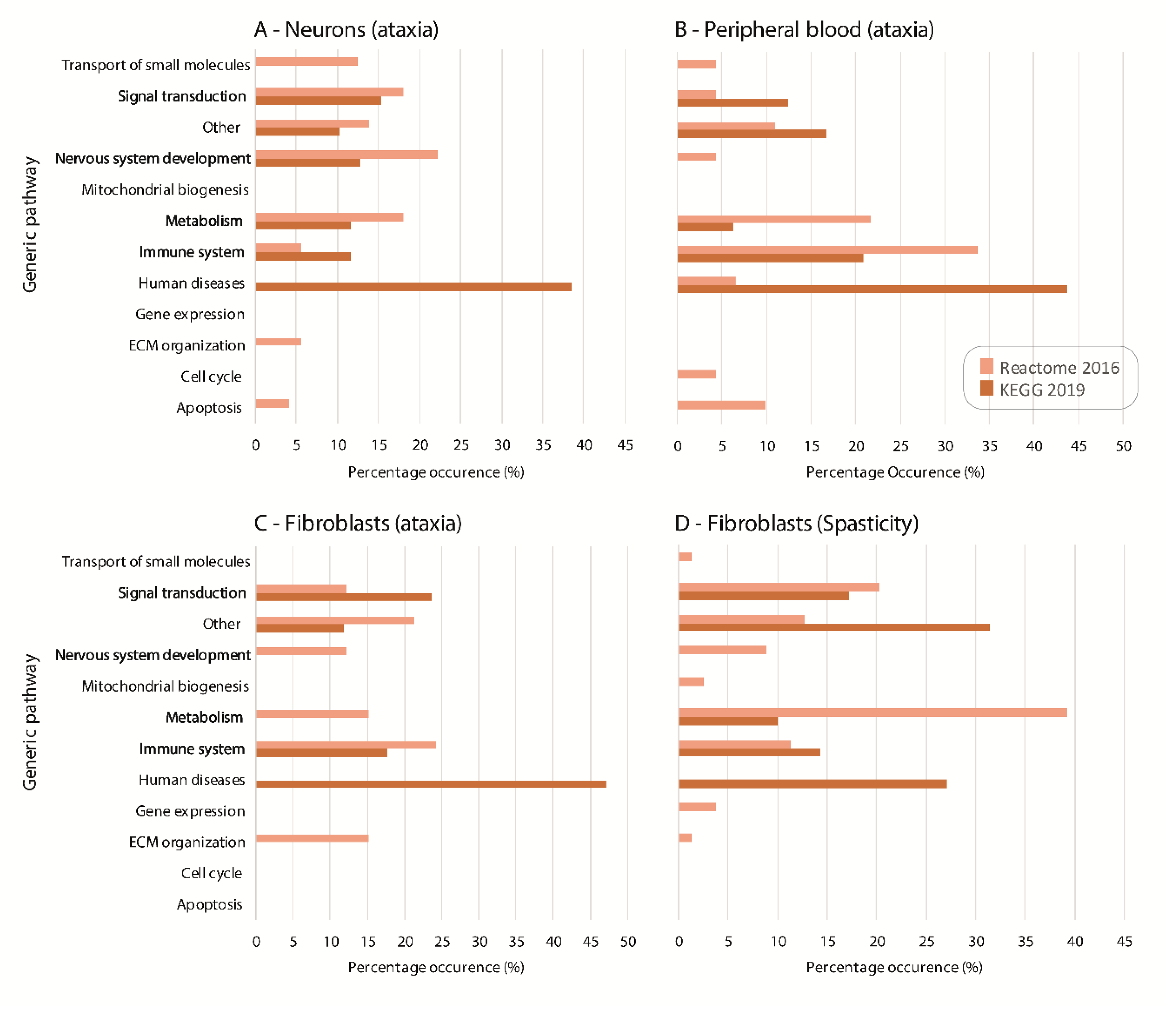
2.2. Gene Ontology Analysis of Differentially Expressed Genes
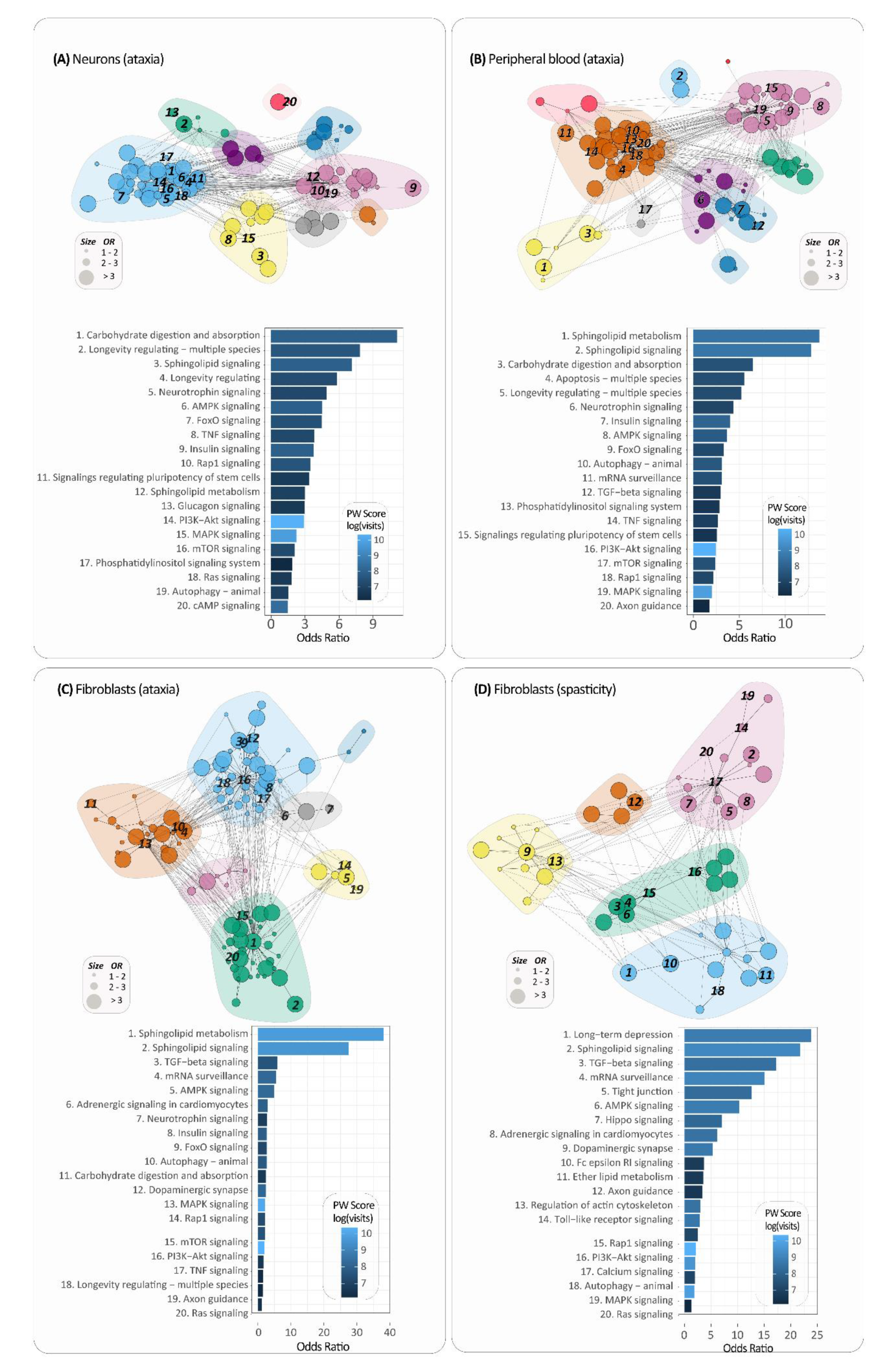
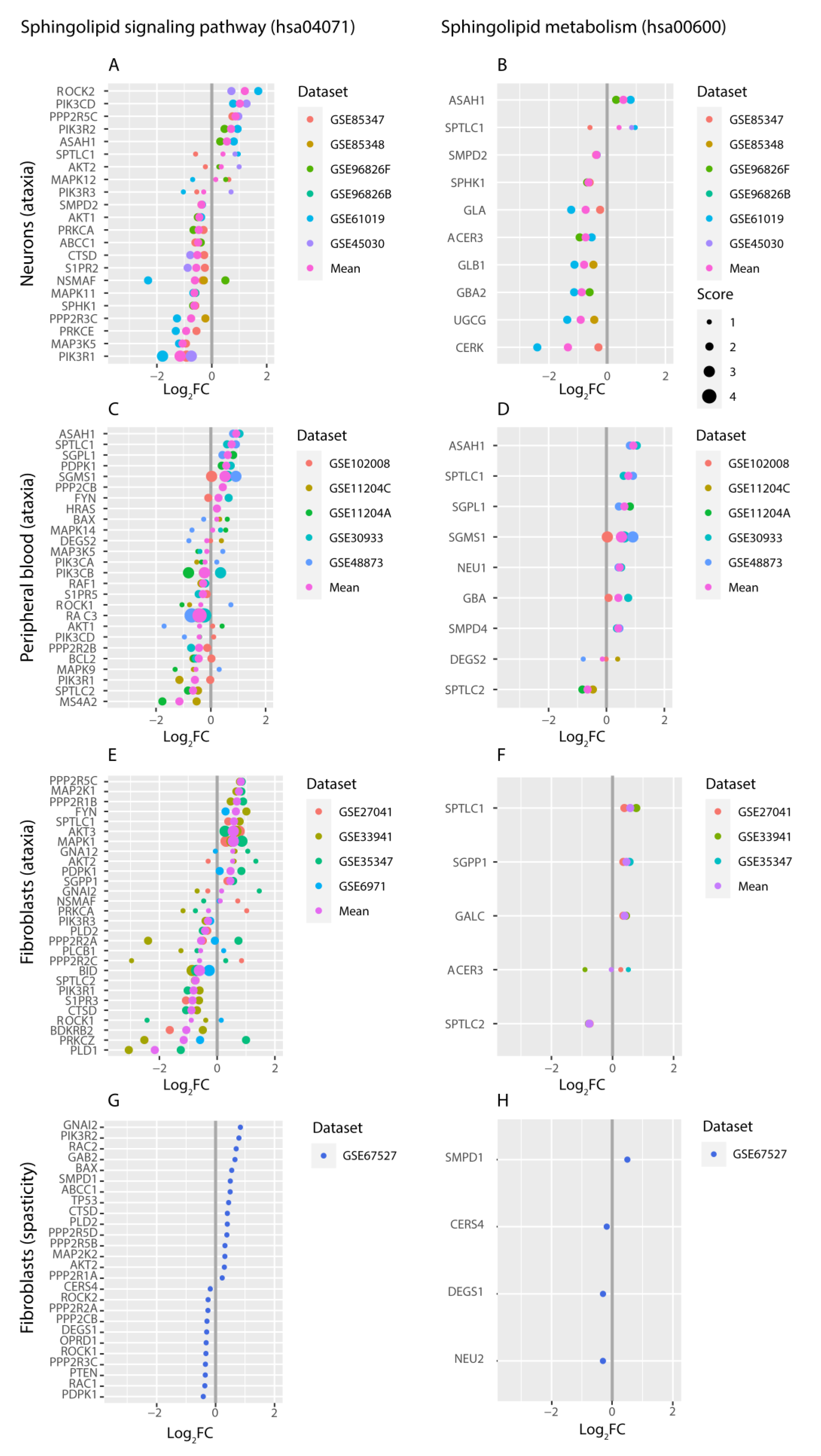
2.3. Targeted Expression Analysis of Sphingolipid Pathways
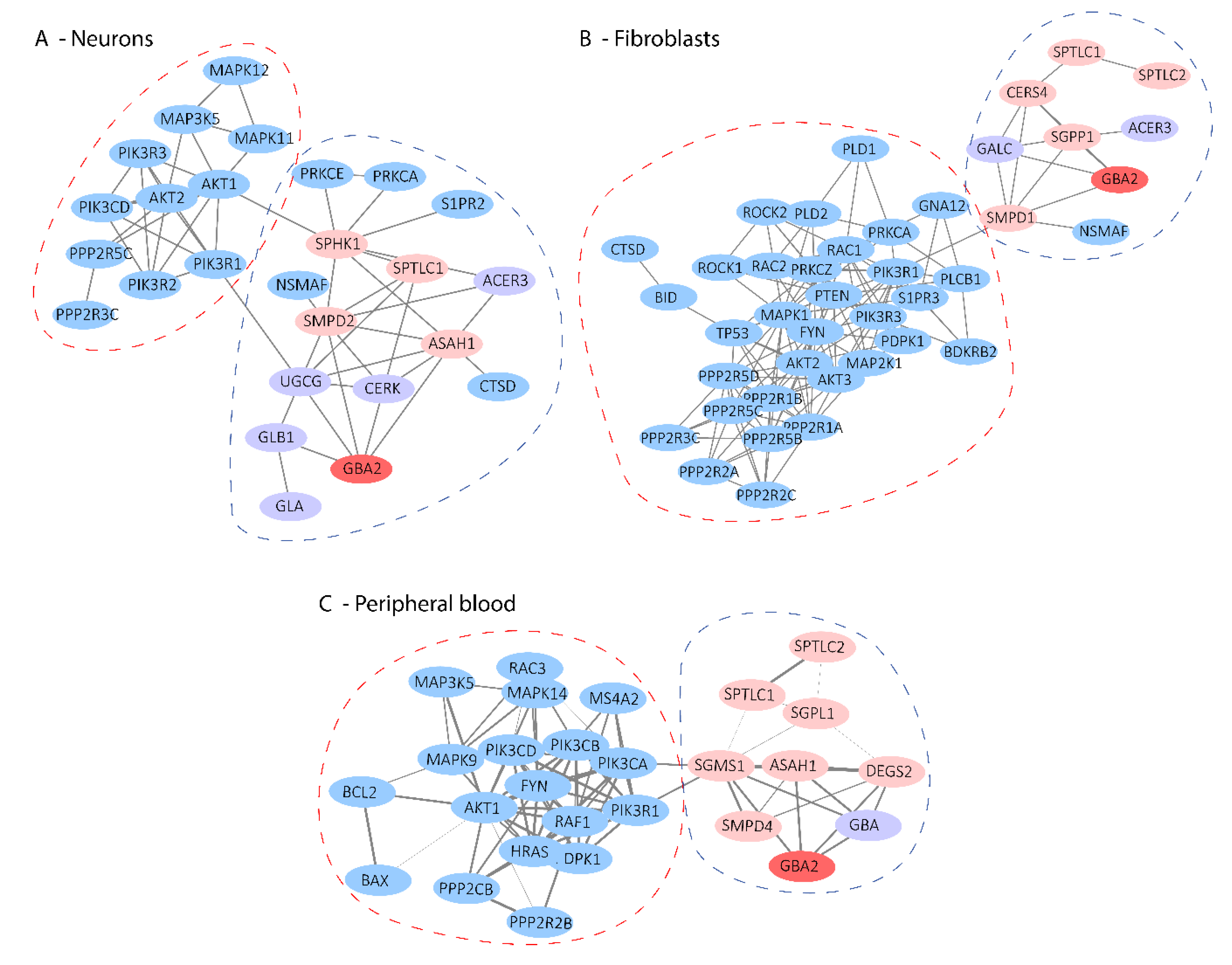
2.4. Highlighting Pathway Communities around Sphingolipid Pathways
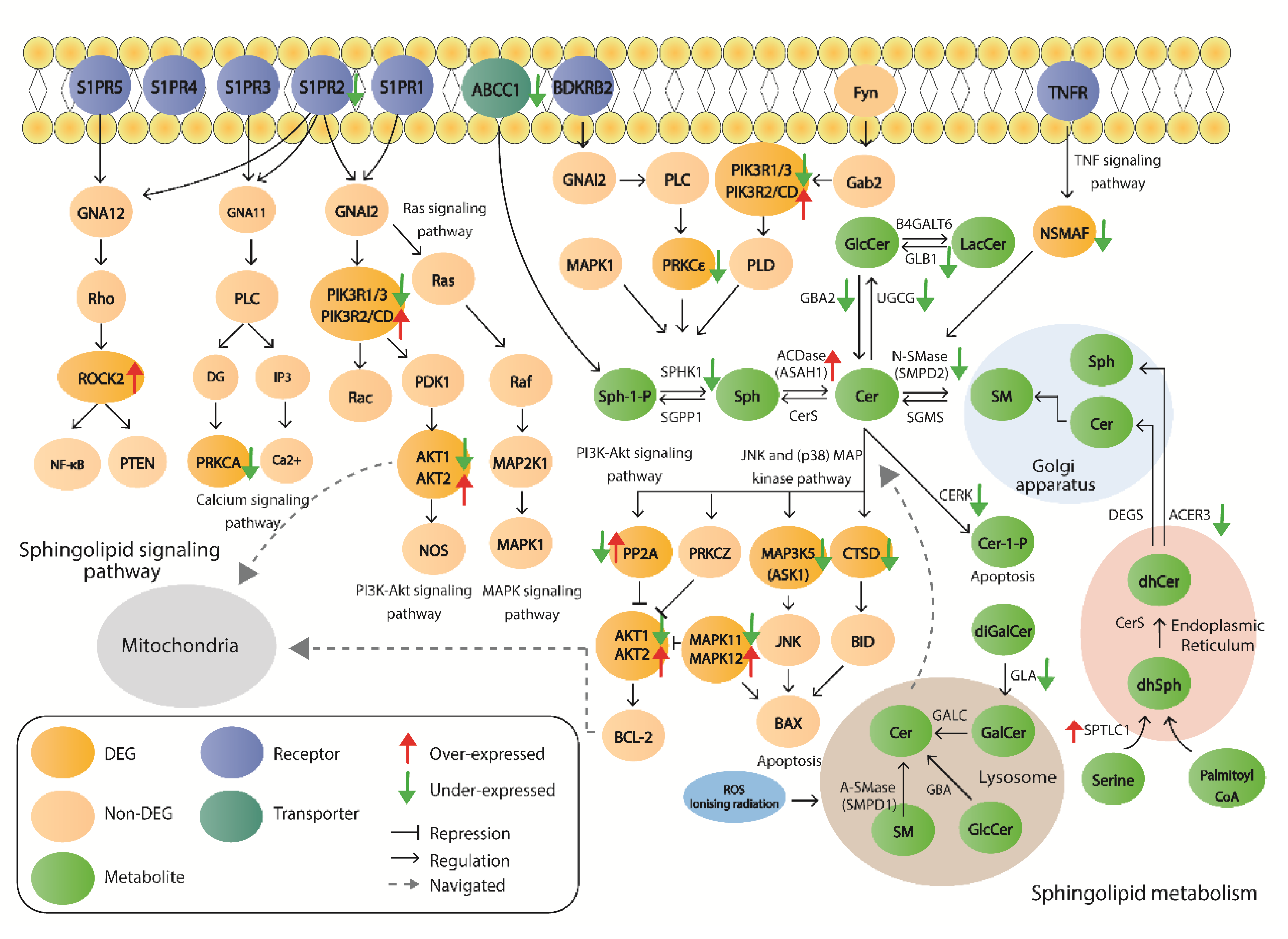
2.5. Mapping of Sphingolipid-Related Degs to the Protein–Protein Interaction Network
3. Discussion
4. Materials and Methods
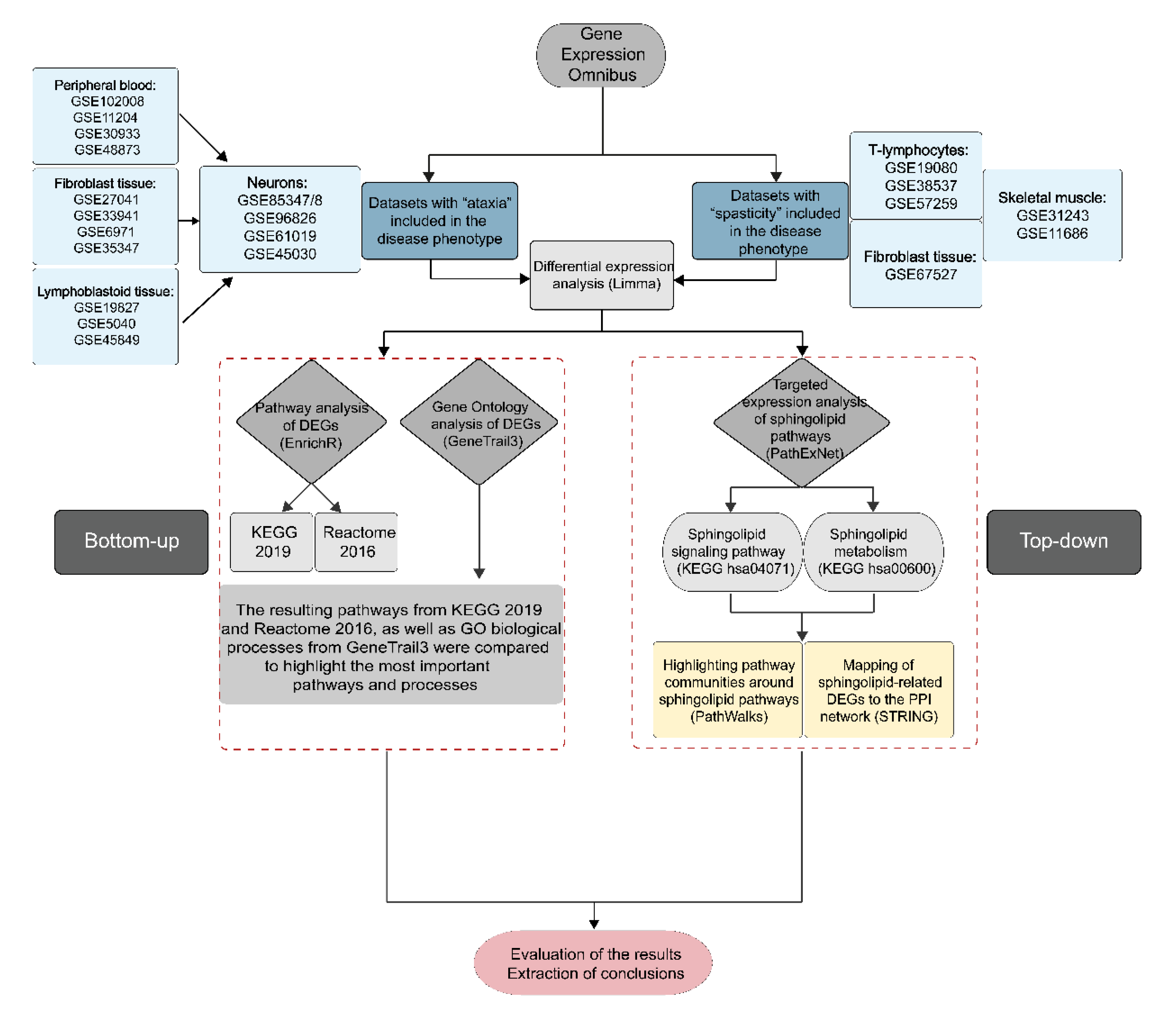
4.1. Collection of Gene Expression Datasets from Gene Expression Omnibus
4.2. Differential Expression Analysis of Microarray Datasets
4.3. Pathway Analysis
4.4. Network Construction
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACER3 | Alkaline ceramidase 3 |
| ASAH1 | Acid ceramidase |
| Cer | Ceramide |
| Cer-1-P | Ceramide-1-phosphate |
| CerS | Ceramide synthase |
| DEGS | Dihydroceramide desaturase |
| dhCer | Dihydroceramides |
| dhSph | Dihydrosphingosine |
| diGalCer | Digalactosylceramide |
| GalCer | Galactosylceramide |
| GlcCer | Glucosylceramide |
| LacCer | Lactosylceramide |
| NOS | Nitric oxide synthase |
| SGMS | Sphingomyelin synthase |
| SGPP1 | Sphingosine-1-phosphate phosphatase 1 |
| SM | sphingomyelin |
| SMPD2 | Sphingomyelin phosphodiesterase 2 |
| Sph | Sphingosine |
| Sph-1-P | Sphingosine-1-phosphate |
| SPHK | Sphingosine kinase |
| SPTLC1 | Serine palmitoyltransferase 1 |
| SPTLC2 | Serine palmitoyltransferase 2 |
References
- De Bot, S.T.; Willemsen, M.A.A.P.; Vermeer, S.; Kremer, H.P.H.; Van De, B.P.C. Warrenburg Reviewing the genetic causes of spastic-ataxias. Neurology 2012, 79, 1507–1514. [Google Scholar] [CrossRef]
- Bereznyakova, O.; Dupré, N. Spastic ataxias. In Handbook of Clinical Neurology; Elsevier B.V.: Amsterdam, The Netherlands, 2018; Volume 155, pp. 191–203. [Google Scholar]
- Hersheson, J.; Haworth, A.; Houlden, H. The inherited ataxias: Genetic heterogeneity, mutation databases, and future directions in research and clinical diagnostics. Hum. Mutat. 2012, 33, 1324–1332. [Google Scholar] [CrossRef]
- Noreau, A.; Dupré, N.; Bouchard, J.P.; Dion, P.A.; Rouleau, G.A. Autosomal recessive cerebellar Ataxias. In Handbook of the Cerebellum and Cerebellar Disorders; Springer: Dordrecht, The Netherlands, 2013; pp. 2177–2192. ISBN 9789400713338. [Google Scholar]
- Votsi, C.; Zamba-Papanicolaou, E.; Middleton, L.T.; Pantzaris, M.; Christodoulou, K. A Novel GBA2 Gene Missense Mutation in Spastic Ataxia. Ann. Hum. Genet. 2014, 78, 13–22. [Google Scholar] [CrossRef]
- Minnerop, M.; Kurzwelly, D.; Wagner, H.; Soehn, A.S.; Reichbauer, J.; Tao, F.; Rattay, T.W.; Peitz, M.; Rehbach, K.; Giorgetti, A.; et al. Hypomorphic mutations in POLR3A are a frequent cause of sporadic and recessive spastic ataxia. Brain 2017, 140, 1561–1578. [Google Scholar] [CrossRef] [Green Version]
- Chelban, V.; Patel, N.; Vandrovcova, J.; Zanetti, M.N.; Lynch, D.S.; Ryten, M.; Botía, J.A.; Bello, O.; Tribollet, E.; Efthymiou, S.; et al. Mutations in NKX6-2 Cause Progressive Spastic Ataxia and Hypomyelination. Am. J. Hum. Genet. 2017, 100, 969–977. [Google Scholar] [CrossRef] [Green Version]
- Boot, R.G.; Verhoek, M.; Donker-Koopman, W.; Strijland, A.; Van Marle, J.; Overkleeft, H.S.; Wennekes, T.; Aerts, J.M.F.G. Identification of the non-lysosomal glucosylceramidase as β-glucosidase 2. J. Biol. Chem. 2007, 282, 1305–1312. [Google Scholar] [CrossRef] [Green Version]
- Sultana, S.; Reichbauer, J.; Schüle, R.; Mochel, F.; Synofzik, M.; van der Spoel, A.C. Lack of enzyme activity in GBA2 mutants associated with hereditary spastic paraplegia/cerebellar ataxia (SPG46). Biochem. Biophys. Res. Commun. 2015, 465, 35–40. [Google Scholar] [CrossRef]
- Malekkou, A.; Samarani, M.; Drousiotou, A.; Votsi, C.; Aureli, M.; Loberto, N.; Christodoulou, K. Biochemical Characterization of the GBA2 c. 1780G > C Missense Mutation in Lymphoblastoid Cells from Patients with Spastic Ataxia. Int. J. Mol. Sci. 2018, 19, 3099. [Google Scholar] [CrossRef] [Green Version]
- Gatchel, J.; Watase, K.; Thaller, C.; Carson, J.; Jafar-Nejad, P. The insulin-like growth factor pathway is altered in spinocerebellar ataxia type 1 and type 7. Proc. Natl. Acad. Sci. USA 2008, 105, 1291–1296. [Google Scholar] [CrossRef] [Green Version]
- Ingram, M.; Wozniak, E.A.L.; Duvick, L.; Yang, R.; Bergmann, P.; Carson, R.; OCallaghan, B.; Zoghbi, H.Y.; Henzler, C.; Orr, H.T. Cerebellar Transcriptome Profiles of ATXN1 Transgenic Mice Reveal SCA1 Disease Progression and Protection Pathways. Neuron 2016, 89, 1194–1207. [Google Scholar] [CrossRef] [Green Version]
- Serra, H.; Byam, C.; Lande, J.; Tousey, S.; Zoghbi, H. Gene profiling links SCA1 pathophysiology to glutamate signaling in Purkinje cells of transgenic mice. Hum. Mol. Genet. 2004, 13, 2535–2543. [Google Scholar] [CrossRef]
- Driessen, T.M.; Lee, P.J.; Lim, J. Molecular pathway analysis towards understanding tissue vulnerability in spinocerebellar ataxia type 1. Elife 2018, 7, e39981. [Google Scholar] [CrossRef]
- Napierala, J.S.; Li, Y.; Lu, Y.; Lin, K.; Hauser, L.A.; Lynch, D.R.; Napierala, M. Comprehensive analysis of gene expression patterns in Friedreich’s ataxia fibroblasts by RNA sequencing reveals altered levels of protein synthesis factors and solute carriers. DMM Dis. Model. Mech. 2017, 10, 1353–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toonen, L.J.A.; Overzier, M.; Evers, M.M.; Leon, L.G.; Van Der Zeeuw, S.A.J.; Mei, H.; Kielbasa, S.M.; Goeman, J.J.; Hettne, K.M.; Magnusson, O.T.; et al. Transcriptional profiling and biomarker identification reveal tissue specific effects of expanded ataxin-3 in a spinocerebellar ataxia type 3 mouse model. Mol. Neurodegener. 2018, 13, 1–18. [Google Scholar] [CrossRef]
- Gerstner, N.; Kehl, T.; Lenhof, K.; Müller, A.; Mayer, C.; Eckhart, L.; Grammes, N.L.; Diener, C.; Hart, M.; Hahn, O.; et al. GeneTrail 3: Advanced high-throughput enrichment analysis. Nucleic Acids Res. 2020. [Google Scholar] [CrossRef]
- Karatzas, E.; Zachariou, M.; Bourdakou, M.M.; Minadakis, G.; Oulas, A.; Kolios, G.; Delis, A.; Spyrou, G.M. PathWalks: Identifying pathway communities using a disease-related map of integrated information. Bioinformatics 2020. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software Environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Riboldi, G.M.; Di Fonzo, A.B. GBA, Gaucher Disease, and Parkinson’s Disease: From Genetic to Clinic to New Therapeutic Approaches. Cells 2019, 8, 364. [Google Scholar] [CrossRef] [Green Version]
- Czubowicz, K.; Strosznajder, R. Ceramide in the Molecular Mechanisms of Neuronal Cell Death. The Role of Sphingosine-1-Phosphate. Mol. Neurobiol. 2014, 50, 26–37. [Google Scholar] [CrossRef] [Green Version]
- Lin, G.; Wang, L.; Marcogliese, P.C.; Bellen, H.J. Sphingolipids in the Pathogenesis of Parkinson’s Disease and Parkinsonism. Trends Endocrinol. Metab. 2019, 30, 106–117. [Google Scholar] [CrossRef] [PubMed]
- KEGG: Kyoto Encyclopedia of Genes and Genomes. Available online: http://www.genome.jp/kegg/ (accessed on 1 February 2020).
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Taha, T.A.; Mullen, T.D.; Obeid, L.M. A house divided: Ceramide, sphingosine, and sphingosine-1-phosphate in programmed cell death. Biochim. Biophys. Acta Biomembr. 2006, 1758, 2027–2036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coant, N.; Sakamoto, W.; Mao, C.; Hannun, Y.A. Ceramidases, roles in sphingolipid metabolism and in health and disease. Adv. Biol. Regul. 2017, 63, 122–131. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.B.; Ye, K. Multiple functions of Phosphoinositide-3 Kinase Enhancer (PIKE). Sci. World J. 2010, 10, 613–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuesto, G.; Enriquez-Barreto, L.; Caramés, C.; Cantarero, M.; Gasull, X.; Sandi, C.; Ferrús, A.; Acebes, Á.; Morales, M. Phosphoinositide-3-kinase activation controls synaptogenesis and spinogenesis in hippocampal neurons. J. Neurosci. 2011, 31, 2721–2733. [Google Scholar] [CrossRef] [Green Version]
- Bruel-Jungerman, E.; Veyrac, A.; Dufour, F.; Horwood, J.; Laroche, S.; Davis, S. Inhibition of PI3K-Akt signaling blocks exercise-mediated enhancement of adult neurogenesis and synaptic plasticity in the dentate gyrus. PLoS ONE 2009, 4, e7901. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Alegría, K.; Flores-León, M.; Avila-Muñoz, E.; Rodríguez-Corona, N.; Arias, C. PI3K signaling in neurons: A central node for the control of multiple functions. Int. J. Mol. Sci. 2018, 19, 3725. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Qiu, J.; Liang, M.; Golinski, J.; Van Leyen, K.; Jung, J.E.; You, Z.; Lo, E.H.; Degterev, A.; Whalen, M.J. Akt and mTOR mediate programmed necrosis in neurons. Cell Death Dis. 2014, 5, e1084. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, M.; Rosenstock, T.R.; Oliveira, A.M.; Oliveira, C.R.; Rego, A.C. Insulin and IGF-1 improve mitochondrial function in a PI-3K/Akt-dependent manner and reduce mitochondrial generation of reactive oxygen species in Huntington’s disease knock-in striatal cells. Free Radic. Biol. Med. 2014, 74, 129–144. [Google Scholar] [CrossRef]
- Bezprozvanny, I. Calcium signaling and neurodegenerative diseases. Trends Mol. Med. 2009, 15, 89–100. [Google Scholar] [CrossRef] [Green Version]
- Sultana, S.; Stewart, J.; van der Spoel, A.C. Truncated mutants of beta-glucosidase 2 (GBA2) are localized in the mitochondrial matrix and cause mitochondrial fragmentation. PLoS ONE 2020, 15, e0233856. [Google Scholar] [CrossRef]
- Criscuolo, C.; Procaccini, C.; Meschini, M.C.; Cianflone, A.; Carbone, R.; Doccini, S.; Devos, D.; Nesti, C.; Vuillaume, I.; Pellegrino, M.; et al. Powerhouse failure and oxidative damage in autosomal recessive spastic ataxia of Charlevoix-Saguenay. J. Neurol. 2015, 262, 2755–2763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seong, E.; Insolera, R.; Dulovic, M.; Kamsteeg, E.J.; Trinh, J.; Brüggemann, N.; Sandford, E.; Li, S.; Ozel, A.B.; Li, J.Z.; et al. Mutations in VPS13D lead to a new recessive ataxia with spasticity and mitochondrial defects. Ann. Neurol. 2018, 83, 1075–1088. [Google Scholar] [CrossRef] [PubMed]
- Crespo-Barreto, J.; Fryer, J.D.; Shaw, C.A.; Orr, H.T.; Zoghbi, H.Y. Partial Loss of Ataxin-1 Function Contributes to Transcriptional Dysregulation in Spinocerebellar Ataxia Type 1 Pathogenesis. PLoS Genet. 2010, 6, e1001021. [Google Scholar] [CrossRef]
- Almeida, A.; Delgado-Esteban, M.; Bolaños, J.P.; Medina, J.M. Oxygen and glucose deprivation induces mitochondrial dysfunction and oxidative stress in neurones but not in astrocytes in primary culture. J. Neurochem. 2002, 81, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.G.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 1240–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, J.W.; Golovoy, D.; Vincent, A.M.; Mahendru, P.; Olzmann, J.A.; Mentzer, A.; Feldman, E.L. High glucose-induced oxidative stress and mitochondrial dysfunction in neurons. FASEB J. 2002, 16, 1738–1748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jęśko, H.; Stępień, A.; Lukiw, W.J.; Strosznajder, R.P. The Cross-Talk between Sphingolipids and Insulin-Like Growth Factor Signaling: Significance for Aging and Neurodegeneration. Mol. Neurobiol. 2019, 56, 3501–3521. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Smyth, G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csardi, G. The Igraph Package Title Routines for Simple Graphs, Network Analysis; Department of Medical Genetics, University of Lausanne: Lausanne, Switzerland, 2008. [Google Scholar]
- Wang, J.; Zhong, J.; Chen, G.; Li, M.; Wu, F.X.; Pan, Y. ClusterViz: A Cytoscape APP for Cluster Analysis of Biological Network. IEEE/ACM Trans. Comput. Biol. Bioinforma. 2015, 12, 815–822. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, J.; Chen, J. A fast agglomerate algorithm for mining functional modules in protein interaction networks. In BioMedical Engineering and Informatics: New Development and the Future, Proceedings of the 1st International Conference on BioMedical Engineering and Informatics, BMEI, Sanya, China, 27–30 May 2008; IEEE: Piscataway Township, NJ, USA, 2008. [Google Scholar]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kakouri, A.C.; Votsi, C.; Tomazou, M.; Minadakis, G.; Karatzas, E.; Christodoulou, K.; Spyrou, G.M. Analyzing Gene Expression Profiles from Ataxia and Spasticity Phenotypes to Reveal Spastic Ataxia Related Pathways. Int. J. Mol. Sci. 2020, 21, 6722. https://doi.org/10.3390/ijms21186722
Kakouri AC, Votsi C, Tomazou M, Minadakis G, Karatzas E, Christodoulou K, Spyrou GM. Analyzing Gene Expression Profiles from Ataxia and Spasticity Phenotypes to Reveal Spastic Ataxia Related Pathways. International Journal of Molecular Sciences. 2020; 21(18):6722. https://doi.org/10.3390/ijms21186722
Chicago/Turabian StyleKakouri, Andrea C., Christina Votsi, Marios Tomazou, George Minadakis, Evangelos Karatzas, Kyproula Christodoulou, and George M. Spyrou. 2020. "Analyzing Gene Expression Profiles from Ataxia and Spasticity Phenotypes to Reveal Spastic Ataxia Related Pathways" International Journal of Molecular Sciences 21, no. 18: 6722. https://doi.org/10.3390/ijms21186722
APA StyleKakouri, A. C., Votsi, C., Tomazou, M., Minadakis, G., Karatzas, E., Christodoulou, K., & Spyrou, G. M. (2020). Analyzing Gene Expression Profiles from Ataxia and Spasticity Phenotypes to Reveal Spastic Ataxia Related Pathways. International Journal of Molecular Sciences, 21(18), 6722. https://doi.org/10.3390/ijms21186722